Back to Journals » Journal of Asthma and Allergy » Volume 11
Non-eosinophilic asthma: current perspectives
Authors Esteban-Gorgojo I, Antolín-Amérigo D, Domínguez-Ortega J ![]() , Quirce S
, Quirce S ![]()
Received 8 June 2018
Accepted for publication 20 August 2018
Published 29 October 2018 Volume 2018:11 Pages 267—281
DOI https://doi.org/10.2147/JAA.S153097
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Amrita Dosanjh
Video abstract presented by Ignacio Esteban-Gorgojo.
Views: 993
Ignacio Esteban-Gorgojo,1 Darío Antolín-Amérigo,2 Javier Domínguez-Ortega,3,4 Santiago Quirce3,4
1Department of Allergy, Hospital General de Villalba, Madrid, Spain; 2Department of Allergy, Hospital Universitario Ramón y Cajal (IRYCIS), Madrid, Spain; 3Department of Allergy, Hospital La Paz Institute for Health Research (IdiPAZ), 4CIBER de Enfermedades Respiratorias, CIBERES, Madrid, Spain
Abstract: Although non-eosinophilic asthma (NEA) is not the best known and most prevalent asthma phenotype, its importance cannot be underestimated. NEA is characterized by airway inflammation with the absence of eosinophils, subsequent to activation of non-predominant type 2 immunologic pathways. This phenotype, which possibly includes several not well-defined subphenotypes, is defined by an eosinophil count <2% in sputum. NEA has been associated with environmental and/or host factors, such as smoking cigarettes, pollution, work-related agents, infections, and obesity. These risk factors, alone or in conjunction, can activate specific cellular and molecular pathways leading to non-type 2 inflammation. The most relevant clinical trait of NEA is its poor response to standard asthma treatments, especially to inhaled corticosteroids, leading to a higher severity of disease and to difficult-to-control asthma. Indeed, NEA constitutes about 50% of severe asthma cases. Since most current and forthcoming biologic therapies specifically target type 2 asthma phenotypes, such as uncontrolled severe eosinophilic or allergic asthma, there is a dramatic lack of effective treatments for uncontrolled non-type 2 asthma. Research efforts are now focusing on elucidating the phenotypes underlying the non-type 2 asthma, and several studies are being conducted with new drugs and biologics aiming to develop effective strategies for this type of asthma, and various immunologic pathways are being scrutinized to optimize efficacy and to abolish possible adverse effects.
Keywords: asthma, non-eosinophilic asthma, asthma phenotype, asthma endotype, neutrophilic asthma
Introduction
Asthma, as described by the Global Initiative for Asthma (GINA), is a heterogeneous disease, usually characterized by chronic airway inflammation. It is defined by the history of respiratory symptoms such as wheeze, shortness of breath, chest tightness, and cough that vary over time and in intensity, together with variable expiratory airflow limitation.1 This heterogeneous syndrome exhibits marked variability in its clinical and pathological features, eliciting factors and triggers, as well as in the response to therapy, giving rise to different asthma phenotypes (ie, observable characteristics without a relation to the underlying pathology that can change over time). Several classifications of asthma have been proposed over the years, based on the etiopathogenesis (allergic or non-allergic), inflammatory pattern (eosinophilic or non-eosinophilic), and more recently, in the underlying distinctive pathophysiologic mechanism (type 2-high or type 2-low).2 This latter classification, simplified as type 2 and non-type 2 asthma, is becoming widely accepted since it is considered that it reflects better the endotype (ie, a subtype of a condition defined by a distinct pathophysiological mechanism), which has important implications for predicting responsiveness to corticosteroids3 and specific treatments such as currently available biologics.4 Although the heterogeneity of asthma is present across the whole spectrum of severity, it is particularly relevant in the most severe forms of the disease. In this patient population, inflammatory phenotyping is recommended to prescribe selective therapies.5
Non-eosinophilic airway inflammation: prevalence and mechanisms
Assessment of airway inflammation by sputum cytology (more commonly used), bronchial wash, bronchoalveolar lavage (BAL) and bronchial biopsies shows evidence of eosinophilic, neutrophilic, and mixed eosinophilic/neutrophilic and paucigranulocytic profiles.6 The neutrophilic and paucigranulocytic variants are considered to be non-type 2 asthma.
Approximately 50% of asthma patients have an eosinophilic inflammatory phenotype,7 whereas the remaining patients show a non-eosinophilic phenotype that can be neutrophilic or paucigranulocytic. Non-eosinophilic asthma (NEA) can be observed in patients with severe asthma but also in approximately half of patients with mild-to-moderate asthma.8 Gibson et al9 reported that in a group of 56 nonsmoking adults with persistent asthma treated with high-dose inhaled corticosteroids (ICS), 59% exhibited NEA associated with neutrophil influx and activation. Moreover, NEA is also common among patients with work-related asthma,10 particularly when caused by low-molecular-weight agents.11
The diagnosis of neutrophilic asthma is made when high sputum neutrophils counts (>65%) or neutrophilic infiltration in bronchial biopsies are observed.12,13 Nair et al13 have proposed that the term “neutrophilic asthma” should be limited to those patients who consistently (on at least two occasions) have a sputum neutrophil count ≥65% or ≥500 × 104/mL.
It is widely accepted that corticosteroids increase the neutrophil counts in peripheral blood and sputum due to an antiapoptotic effect on neutrophils,14 suggesting that this type of inflammation could be secondary to the treatment with these drugs. However, neutrophilic inflammation in asthma patients can be observed regardless of corticosteroid therapy, confirming that NEA represents a pathologically distinct disease phenotype, which in fact is less responsive to corticosteroid therapy.15 Using a cluster analysis from the Severe Asthma Research Program, elevated sputum neutrophil counts were found to be associated with more severe asthma phenotypes and with poor response to corticosteroids.16 Notwithstanding, mixed eosinophilic and neutrophilic inflammation of the airways are commonly found in severe asthma,17 and this mixed inflammatory pattern can be a biomarker of the most severe types of the disease.18
In adult patients with refractory asthma, airway neutrophilia has been associated with persistent airflow obstruction.19 Neutrophil recruitment and activation into the airways have been related to the stimulation of toll-like receptor signaling and activation of innate immunity, causing a shift toward Th1 and Th17 responses. This process leads to increased production of interleukin (IL)-8, IL-17A, neutrophil elastase, and a form of matrix metalloproteinase-9 (MMP-9) that shows reduced inhibition by tissue inhibitors of metalloproteinases.20 These cytokines and activated enzymes, acting together, can modify airway structures to contribute to the lower FEV1, remodeling, and fixed airway obstruction seen in adult patients with severe neutrophilic asthma.20 However, intraepithelial airway neutrophils in children with severe asthma were associated with a higher FEV1 and better symptom control, despite lower dose maintenance with inhaled steroids, which suggests a potential benefit for neutrophils in pediatric severe asthma pathophysiology.21
Interestingly, airway microbiology in neutrophilic asthma has been found to be significantly different from that seen in patients with eosinophilic asthma, and multivariate analysis showed that sputum neutrophil count was the strongest predictor of microbiota composition.22 Patients with neutrophilic asthma showed a greater frequency of pathogenic taxa at high relative abundance and reduced Streptococcus, Gemella, and Porphyromonas taxa relative abundance.22
Ntontsi et al23 investigated the functional and inflammatory characteristics of patients with paucigranulocytic asthma among 240 patients with stable asthma. Patients were categorized into inflammatory phenotypes as paucigranulocytic (47.9%), neutrophilic (5.4%) and the remaining were eosinophilic (40%) or mixed (6.7%). Patients with paucigranulocytic asthma had better lung function and less frequency of severe refractory asthma. The authors concluded that paucigranulocytic asthma most likely represents a group of patients with good response to treatment rather than a real asthma phenotype. Notwithstanding, paucigranulocytic patients who remain uncontrolled despite optimal treatment (around 15%) represent an asthmatic population that requires further study.
Diagnosis of NEA
The definition of asthma remains limited to the description of its key clinical features, with broad reference to the underlying inflammatory characteristics and heterogeneity. Although patients with asthma can be grouped into specific clusters using statistical approaches of organized data,24 the objective diagnosis of asthma does not differ in patients with different airway inflammation patterns. Identification of inflammatory asthma phenotypes requires the use of specific methods to assess the presence of inflammation in the airways, such as induced sputum, BAL, or bronchial biopsies. However, these methods to assess the type and extent of airway inflammation are not widely available, and they are not completely reliable and reproducible.
Because the upper limit of the normal range for sputum eosinophil counts in the healthy (non-asthmatic) population is ≤1.9%,25 it has been established that a percentage >2%, 2.5%, or 3%, according to different authors,26,27 is diagnostic for eosinophilic asthma. Thus, NEA has been defined as asthma with a sputum eosinophil count of less than either ≤2% or ≤3%, while neutrophilic inflammation has been defined with cut-off points varying from ≥60% to>76%.28 Sputum neutrophil cell count in healthy subjects is highly variable, usually averaging 30%–35%.29 NEA is often associated with levels of neutrophil-associated chemokines in BAL or blood. Human neutrophil lipocalin,30 leukotriene B4 (LTB4), tumor necrosis factor-alpha (TNF-α), IL-17A, IL-8, elastase, granulocyte-macrophage-colony-stimulating factor, or MMP-9 levels are detectable in sputum, BAL fluid, and plasma in patients with severe neutrophilic asthma.31 The raised concentrations in sputum of hydrogen sulfide could be used as a potential biomarker for neutrophilic asthma associated with airflow obstruction.32 IL-8 activates neutrophils and sputum IL-8 levels were higher in NEA patients while IL-8 receptors are increased in this type of asthmatics.33 However, neutrophilic inflammation in NEA is not always present. In particular, it has been previously described that there is no evidence of either neutrophilic or eosinophilic airway inflammation in over 30% of adults with asthma,34 and this is even more frequent in asthmatic adolescents.35 None of the inflammatory-phenotyping or the novel omics-endotyping strategies (metabolomics, proteomics, and transcriptomics) have been clearly translated to the clinical practice.
In everyday clinical practice, the absence of biomarkers clearly associated to type 2 asthma should be underlined. Patients with NEA are less atopic and usually have lower levels of fractional exhaled nitric oxide (FeNO), often <30 parts per billion and ≤300 eosinophils/µL in blood.36
Although blood eosinophils have been proposed as a practical alternative to predict sputum eosinophilia, sputum neutrophil count is poorly related to blood neutrophils.37 The combination of blood eosinophil count, FeNO, Asthma Control Questionnaire, medication use, nasal polyposis, aspirin sensitivity, and neutrophil/eosinophil responsiveness upon stimulation with formyl-methionyl-leucyl phenylalanine was used by Hilvering et al38 to predict the presence of eosinophilic and neutrophilic asthma. These authors compared the diagnostic value of this multivariate prediction model with sputum eosinophilia as gold standard in 115 adult asthmatic patients. They found that the number of patients correctly classified as eosinophilic asthma was 47.6%, but interestingly reached a 95.7% for NEA.
Moreover, this kind of patients usually have poor short-term response to ICS.39 The absolute sputum neutrophil counts and the percentage have been associated with low post-bronchodilator FEV1, which lends support to the hypothesis that neutrophilic airway inflammation has a role in the progression of persistent airflow limitation in asthma.40 In the Airways Disease Endotyping for Personalized Therapeutics study, non-type 2 phenotype, mostly neutrophilic, had reduced lung capacity and lower bronchodilation response, which could suggest more extensive tissue remodeling.24
Other studies have confirmed that subjects with increased neutrophils in sputum (neutrophilic asthma and mixed granulocytic asthma) were older and had increased total cell count and cell viability compared with other subtypes.34
Brooks et al41 found, in a cohort of 50 adult asthmatics, that only 8% had neutrophilic asthma, with three fourths of asthmatics classified as neutrophilic asthma at any assessment being older than 60 year of age. In a retrospective study conducted in 508 asthmatics with successful sputum induction, recruited from the University Asthma Clinic of Liege, an independent predictor of sputum neutrophilia was advanced age but not blood neutrophil count.37
Surprisingly, although previous studies associated NEA to be relatively stable over time, they also described that 52% of asthmatics changed the phenotype in response to ICS, observing at the same time, improvement in Asthma Control Questionnaire-7 score in the NEA group with optimized treatment. These facts complicate distinguishing between NEA and COPD, especially in the elderly, because both diseases have similar underlying mechanisms and show analogous patterns of simple spirometric parameters. Górska et al42 described that in COPD, the degree of systemic inflammation was independent of the level of airway neutrophilia, but the neutrophilic phenotype of COPD was associated with more severe airway obstruction and hyperinflation. Precise pulmonary function tests such as total lung capacity or the carbon monoxide diffusing capacity of the lungs (DLCO) can help distinguishing between these diseases.43 Chest images might also be helpful identifying different imaging-based phenotypes.44 Compared with healthy nonsmoker subjects, both asthma with a post-bronchodilator FEV1 <80% and COPD subjects demonstrated a decreased airway circularity especially in large and upper lobar airways, and a decreased normalized hydraulic diameter in segmental airways.45 Besides, COPD subjects had more severe emphysema and small-airway disease, smaller regional tissue fraction and lung deformation, compared with asthmatic patients.45
In summary, although the optimal diagnosis of NEA should be performed by sputum cell count, an older age of the patients, absence of atopy, low FeNO levels (considering concurrent factors), a low blood eosinophil count, in addition to a persistent airflow limitation, could guide us toward the presence of this phenotype.
Causative agents and risk factors
The causes of neutrophilic inflammation in asthma remain unclear. However, external factors such as pollution, infections, cigarette smoke, or some occupational exposures have been described as inducers of NEA.20 Host factors such as obesity seem to be related as well to this asthma phenotype.
Environmental inducers
Smoking
The prevalence of active smoking among individuals with asthma is approximately the same as in the population at large and even nonsmoking patients with asthma may have significant exposure to passive smoke. Smoking in asthma is associated with corticosteroid insensitivity and poor symptom control.46 This group of patients is at risk of developing worse asthma-specific quality of life, higher frequency of exacerbations, life-threatening asthma attacks, and an increased impact on healthcare resources due to unscheduled doctor visits and frequent hospital admissions.47 A strong association between active cigarette smoking and severity of asthma has been demonstrated, especially in those patients who smoked >20 pack-years. Shimoda et al48 evaluated the influence of cigarette smoking on airway inflammation in patients with asthma. They found that smokers with asthma showed a lower FEV1 to FVC ratio, a lower FeNO, a higher neutrophil proportion and lower eosinophil proportion, in induced sputum of smokers than in nonsmokers with asthma. They also showed that bronchial hyperresponsiveness (BHR) was increased in smokers with asthma compared with nonsmokers with asthma.48
Cigarette smoke can damage directly the epithelium and has been associated with a non-eosinophilic airway inflammation compared to never smokers with asthma. Through direct activation of macrophages, these cells produce inflammatory molecules, tissue proteases like MMP, IL-8, and other chemokines involved in the mobilization and prolonged survival of neutrophils in the lung tissue, while produce less IL-10, which leads to a non-type 2 pattern with a reduced B-cell number and lower levels of IL-4 and IL-5.49 Siew et al50 demonstrated that the expression of IL-17A, IL-6, and IL-8 and neutrophil numbers were significantly elevated in the bronchial mucosa of the asthmatic smokers compared to the nonsmokers. Moreover, the expression of IL-17A correlated with that of IL-8 and neutrophil numbers.
Pollution
Outdoor air pollution could contribute to asthma as an aggravating factor and as a cause itself of asthma. Pollutants, such as diesel exhaust particles, ozone, or nitrogen oxide, can induce neutrophilic airway inflammation and BHR.51 Probably, pollutants are capable to damage the epithelium in the airways through an oxidative mechanism. Surface macrophages and epithelial cells are involved in the activation of epithelial nuclear factor-kB (NF-kB) and generation of proinflammatory mediators such as IL-6, IL-8, TNF-α, leading to neutrophilic inflammation and remodeling in the bronchi.52 Moreover, asthmatics may be pre-disposed to the deleterious effects of pollutants like ozone, having constitutively modified host defense functions and gene signatures.52
Workplace agents
Injury to the airway epithelium is likely to play a central role in the pathogenesis of irritant-induced asthma. Inhalation of irritants induces oxidative stress causing the epithelial damage and releasing pro-neutrophilic inflammation mediators.53 Bronchial neutrophilic inflammation has also been reported in patients with occupational asthma exposed to high-molecular-weight and low-molecular weight (LMW) agents.53
In fact, NEA has been reported to be more frequent than eosinophilic asthma in patients with occupational asthma caused by LMW agents.11
A post-challenge increase in sputum neutrophil count has been documented especially after exposure to LMW agents.54 However, the interpretation of changes in sputum neutrophil counts requires further validation because increased sputum neutrophils and IL-8 levels were observed in subjects exposed to LMW agents who were diagnosed as having other respiratory diseases.55 Additionally, increased concentrations of MMP-9 and IL-1β in induced sputum were detected in COPD patients compared to occupational asthma workers.56 MMP-9 levels in induced sputum also increase in allergic occupational asthma patients who underwent specific inhalation challenge on 2 consecutive days with flour, but not changes in the levels of other MMPs have been reported.57
Work-related asthma, which is usually underdiagnosed, is an important cause of severe uncontrolled asthma with fatal or near-fatal attacks, so it is mandatory to be considered as cause of NEA.58
Infections
Several studies have associated NEA with persistent bacterial presence and potent Th1 and Th17 responses. Because it functions by recruiting neutrophils, Th17/IL-17 plays an important role in host defense and hyperimmune responses against pathogenic bacteria.59 Activation of this pathway leads to a raise in IL-8 levels in the bronchial epithelium, inducing neutrophilic inflammation. Refractory asthma has been associated with increased numbers of neutrophils and proneutrophilic biomolecules in the airways. Alan et al60 showed in 60 patients with refractory asthma that subclinical infection was present in 40%, which likely contributed to neutrophilic inflammation. Moreover, Hadebe et al61 explored, in a murine model, the effects of bacterial lipopolysaccharide (LPS) and β-glucan, two commonly encountered microbial agonists, on the pathogenesis of allergic and non-allergic respiratory responses to house dust mites. They observed that sensitization of mice with house dust mites and LPS plus β-glucan induced robust adjunctive neutrophilia, while the eosinophilic response was largely unaltered when compared to mice sensitized with house dust mites alone, driving steroid-refractory asthma. Finally, there could be an association between disordered bacterial microbiota and pathogenesis of asthma. Wood et al62 found several potentially pathogenic bacteria in the sputum from patients with stable asthma, as well as increases in sputum total cell counts, in the proportion and number of neutrophils, and IL-8 levels, suggesting the presence of a specific lung microbiota and subsequent effects on immunity.
Host factors
Obesity
The relationship between asthma and obesity has already been studied for >20 years. Camargo et al conducted a prospective study in 1999 where a strong, independent, and positive association between body mass index (BMI) and adult-onset asthma was found.63 Subsequently, cluster studies such as those carried out by Denlinger et al64 and Haldar et al65 showed a specific asthma phenotype of NEA associated with obesity, so this new phenotype was included in GINA guidelines.1 Nevertheless, other studies have not been able to verify this association, questioning if obesity could be a causal factor or just a comorbidity. Holguin et al66 studied a group of 1,049 patients with asthma differentiating between early-onset asthma and late-onset asthma, setting 12 years of age as the cutoff point. Two opposite phenotypes emerged from this study, whereas obesity in early-onset asthma was a consequence of asthma severity and acted as a comorbidity, and in late-onset asthma obesity seemed to have a causative association with asthma and its severity. Therefore, the relationship between these two factors seems to be heterogeneous, leading to a complex interplay.
Characteristics of the association between asthma and obesity have not been clarified yet. McLachlan et al67 and Sutherland68 reported that obesity in asthma patients is more prevalent among women, while Barranco et al69 and Beuther and Sutherland70 found that this association is equally distributed in both sexes or even is more prevalent among men. Obesity was not found to be associated with BHR in some studies,67,69 while other studies have found that obesity is associated with BHR in women71 and men.72 Deepening even further into these contradictions, a study specifically designed to evaluate the causative relation between obesity and asthma suggests that it is more plausible that obesity is caused by asthma and not conversely.73
Despite these controversial observations, specific molecular pathways and cellular populations have been spotted among obese asthmatics, supporting the link between asthma and obesity. Obesity-related asthma is considered to be non-Th2-related, because obesity induces a Th1 polarization among CD4 cells74 leading to a predominant NEA with no atopic or Th2-related previous responses. Nevertheless, eosinophilic asthma can be found in obese people, since obesity can also be a cofactor in other asthma phenotypes.75 A recent study by Jesus et al76 showed that obese asthmatics tend to have worse asthma control than non-obese patients. Moreover, decreased prevalence of atopy and higher basophil counts were observed among these obese asthma patients.76
The two most important biomarkers related to asthma and obesity are leptin and adiponectin. Leptin is an IL-6-like protein, overproduced in the adipose tissue of obese asthmatics, that causes BHR, pulmonary inflammation, and poor response to ICS.77–80 Inversely to leptin, adiponectin dips in obese patients, being an anti-inflammatory protein that also decreases insulin resistance.80,81 It has been suggested that adiponectin itself is not an optimal biomarker, but its ratio related to leptin seems to be a good quality biomarker.79 Other biomarkers, such as eotaxin or resistin, have also been studied in asthma and obesity, but they are not considered to be relevant nowadays.
In summary, it has been confirmed that an association between asthma and obesity exists, but the underlying mechanisms remain to be clarified.
Treatment
Non-type 2 asthma constitutes a therapeutic challenge urgently needing solution, as these patients usually have poor response to corticosteroids. However, studies assessing a step-down of ICS in this specific population were lacking until recently.33,82 Demarche et al83 withdrew or reduced the dose of ICS in two thirds of non-eosinophilic asthmatics irrespective of baseline asthma control. The best predictor of failure to stop ICS was an elevated blood eosinophil count.
The type 2 asthma-targeted medications, such as anti-IgE or anti-IL-5, are not suitable in this patient population. Although there are hypothesis of potential pathways leading to non-eosinophilic inflammation in non-type 2 asthma, currently no specific biomarkers are readily available to support diagnosis and phenotyping of these patients.82
Table 1 shows the main differences between type 2 and non-type 2 asthma.
 | Table 1 Differences between type 2 and non-type 2 asthma |
Recruitment of neutrophils into the airways is mediated by several proinflammatory neutrophil attractants generated by the asthmatic airways, including LTB4, TNF-α, IL-8, and GROα.83 In patients with moderate to severe asthma, increased expression of IL-8 correlates with raised neutrophil concentrations in sputum, which in turn correlates with an increase in the frequency of exacerbations of acute asthma.84
Neutrophil signaling pathways are complex and ligand–receptor relationships are rarely exclusive. For example, IL-8 (CXCL8) signals through both low- and high-affinity receptors designated CXCR1 and CXCR2, respectively. CXCR2 itself can respond to other chemokines beyond CXCL8, including CXCL1, 2, 3, 5, 6, and 7. In experimental models for respiratory conditions, blockade of CXCR1 and CXCR2 by specific inhibitors significantly reduced neutrophilic airway inflammation.85 Interestingly, both CXCR1 and CXCR2 are expressed in endobronchial biopsies from patients with severe COPD, but also in healthy controls.86
Neutrophils play an essential role for the antimicrobial response of the lungs, and CXCR2 inhibition, in animal models, impaired neutrophil function, especially their ability to kill infectious agents. In humans, it appears that CXCR1, rather than CXCR2, is the functionally important receptor subtype involved with neutrophil degranulation; therefore, phagocytosis may remain preserved following anti-CXCR2 use.87,88
Several authors have focused on developing strategies to regulate neutrophil function in order to modulate non-eosinophilic inflammation. Notwithstanding, most of these approaches remain investigational
Non-pharmacological interventions
Avoidance from exposure to environmental and occupational pollutants and smoking cessation may reduce neutrophilic inflammation in asthma.89,90 After cessation of exposure to occupational agents, neutrophilic inflammation was reduced in the patients whose asthma subsided or improved, as compared to those who showed no improvement.90
Inhibition of chemokine receptors
The chemokine receptor CXCR2 is a key mediator of neutrophil migration, which also plays a role in tumor development. In severe asthma, CXCR2 inhibition induced significant decreases in circulating neutrophils but had limited or no effect on pulmonary function or quality-of-life measures, the mean absolute neutrophil count in blood was reduced by 14% at the end of 4 weeks, but recovered by the fifth week.91,92
Nair et al91 evaluated the efficacy and safety of another CXCR2 antagonist, SCH 527123, in a randomized, placebo-controlled, Phase 2 trial, in severe asthmatic patients who were selected on the basis of high neutrophil counts in sputum at baseline. This drug caused a mean reduction of 36.3% in sputum neutrophil percentage compared to a 6.7% increase in the placebo arm. The mean absolute neutrophil count in blood was reduced by 14% at the end of 4 weeks, but recovered by the fifth week. The authors did not find differences in the overall rates of adverse events among the groups and it yielded a trend toward improvement in the ACQ score in the active treatment arm. No statistically significant changes were observed in FEV1, sputum myeloperoxidase, IL-8, or elastase.
O’Byrne et al92 assessed both the efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma in a multicenter, randomized, double-blind, placebo-controlled trial. The trial comprised 640 patients aged ≥18 years with uncontrolled asthma despite combination therapy with long-acting β2 agonists and medium-dose or high-dose ICS. The primary endpoint was the number of severe asthma exacerbations in 6 months. Treatment with AZD5069 was generally well tolerated, being nasopharyngitis the most commonly reported adverse event overall. However, this selective CXCR2 antagonist did not reduce the frequency of severe exacerbations in patients with uncontrolled severe asthma. In addition, the authors did not assess the precise causes of exacerbations that occurred during the study, so this limitation could cause a bias in the evaluation of these molecules.
TNF-α blockers
Wenzel et al93 assessed the safety and efficacy of golimumab in 309 patients with uncontrolled, severe, persistent asthma. Co-primary endpoints were the change from baseline through week 24 in prebronchodilator %predicted FEV1 and the number of severe asthma exacerbations through week 24, with no significant differences from placebo. Moreover, one death and all eight malignancies occurred in the active groups. The authors concluded that treatment with golimumab did not demonstrate a favorable risk–benefit profile in this study population of patients with severe, persistent asthma.
Small clinical studies with etanercept, a soluble TNF-α receptor blocker, reported beneficial effects on clinical outcomes.94
Biomarkers such as FeNO decrease in methacholine bronchial reactivity, histamine in sputum, differential cell counts, percentage of eosinophils, and levels of eosinophilic cationic protein in sputum have been assessed in several clinical trials.93,94 However, only the decrease in methacholine reactivity and histamine concentration in sputum have been found to be potentially suitable for assessing the efficacy of TNF-α blockers.93,94
Anti-IL-6
Chu et al95 observed that elevated IL-6 in sputum was associated with a lower FEV1 in patients with mixed eosinophilic-neutrophilic bronchitis. In mice, allergen exposure increased lung IL-6, and IL-6 was produced by dendritic cells and alveolar macrophages. Loss of function of IL-6 signaling abrogated elevations of eosinophil and neutrophil recruiting cytokines/chemokines and allergen-induced airway inflammation in mice. Therefore, the authors demonstrate the association of pleiotropic cellular airway inflammation with IL-6 using human and animal data. These data suggest that exacerbations of asthma, particularly those with a combined eosinophilic and neutrophilic bronchitis, may respond to therapies targeting the IL-6 pathway and therefore provide a rational basis for initiation of clinical trials to evaluate this.
In two cross-sectional studies comprising 249 and 387 patients, respectively, the IL-6 high (in plasma) asthma patients had a significantly higher average BMI, worse lung function, and more frequent asthma exacerbations than IL-6-low patients. Moreover, FEV1 values were also significantly worse in non-obese IL-6-high asthma patients than in IL6-low asthma patients.96
In a study in patients with severe eosinophilic asthma, it was found that type-2 cytokine-secreting CD8+ CRTH2+ (Tc2) cells, as well as concentrations of prostaglandin D2 (PGD2) and cysteinyl leukotriene E4 (LTE4), are increased in the airways in this group of patients. In vitro PGD2 and LTE4 function synergistically trigger Tc2 cells recruitment and activation in a T-cell receptor-independent manner. These lipids regulate diverse genes in Tc2 cells inducing other proinflammatory cytokines and chemokines, which could contribute mainly to eosinophilia.97
Anti-IL-17
Since IL-17 signaling has been implicated in the development and persistence of NEA, blocking IL-17 receptor signaling could be beneficial. Brodalumab is a human monoclonal antibody that binds with high affinity to human IL-17RA, blocking the biologic activity of IL-17A, IL-17F, IL-17A/F heterodimer, and IL-25.
Busse et al98 carried out a randomized, double-blind, placebo-controlled study in 302 patients with moderate to severe asthma who received brodalumab or placebo. The primary endpoint of the study was the ACQ total score change from baseline to week 12. FEV1, FeNO levels, and neutrophil counts were the biomarkers included in the study.
For the overall study population, no treatment differences were observed in any of the primary or secondary endpoints. However, in the high-reversibility subgroup (post-bronchodilator FEV1 improvement ≥20%; n=112), there was an ACQ significant change of uncertain clinical relevance.
IL-1β blockers
The antibody canakinumab, that blocks IL-1β, or anakinra, which is an IL-1 receptor antagonist that blocks the biologic activity of IL-1 by competitively inhibiting the binding of IL-1 to the soluble or membrane-bound IL-1 receptor, might be of benefit in neutrophilic asthma.99
In experimental respiratory disease models, including neutrophilic asthma, excessive nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome correlates with neutrophilic inflammation, disease severity, steroid resistance, and IL-1β expression.100 Treatment with anti-IL-1β, Ac-YVAD-cho, and MCC950 suppressed IL-1β responses and steroid-resistant features of disease in mice. Moreover, neutrophil depletion suppressed IL-1β-induced steroid-resistant airway hyperresponsiveness.100
Moreover, steroid insensitivity and reduced HDAC2 activity are both linked to aberrant phosphoinositide 3-kinase (PI3K) activity. Recently, infection has shown to induce increases in the levels of microRNA (miRNA)-21.101 In mouse models of Chlamydia, Haemophilus influenzae, influenza, and RSV infection–induced steroid-insensitive allergic airway disease, there was an amplification of PI3K-dependent activity, and a suppression of HDAC2. These effects were attenuated and steroid sensitivity was restored by inhibiting miRNA-21 or PI3K. Thus, a novel miRNA-21/PI3K/HDAC2 axis was identified in a previously unrecognized pathogenic role. The authors concluded that treatment with Ant-21, or the pan-PI3K inhibitor LY294002, reduced PI3K activity and restored HDAC2 levels. This led to suppression of airway hyper-responsiveness and restored steroid sensitivity to allergic airway disease.101
Kinase inhibitors
Several p38MAPK inhibitors have shown to restore corticosteroid sensitivity in PMBCs from patients with severe asthma. A post hoc analysis of a 6-month clinical trial with losmapimod (GW856553) in COPD, but not in asthma, reported a reduction in exacerbations in a subgroup of patients with a blood eosinophil count ≤2%, which may suggest a preferentially beneficial effect of p38MAPK inhibitors in non-eosinophilic inflammation.102,103
Phosphatidylinositol 3-kinase delta (PI3Kδ) and Janus-activated kinases (JAK) modulate lymphocyte activation. JAK and PI3Kδ inhibitors reduced cytokine levels by means of direct effects on T-cell activation in both asthma and healthy BAL cells. Moreover, the combination of dexamethasone with either a JAK or PI3Kδ inhibitor showed an additive anti-inflammatory effect.104
Phosphodiesterase 4 (PDE4) inhibitors
Inhibition of PDEs that increase cyclic adenosine monophosphate or cyclic guanosine monophosphate levels would result in anti-inflammatory responses. Interestingly, the PDE4 inhibitor roflumilast has shown improvements in neutrophil-mediated airway inflammation and a decrease in exacerbations in COPD patients.105
Roflumilast has shown to attenuate allergen-induced bronchoconstriction in patients with asthma. Significant reductions in allergen-induced airway inflammation, including a reduction in both eosinophil and neutrophil counts, and physiologic responses to allergen-induced challenge, achieving a significant reduction in TNF-α, have also been observed.106
FLAP inhibitor: 5-lipoxygenase-activating protein
The oral 5-lipoxygenase-activating protein (FLAP) inhibitor GSK2190915 was evaluated by Chaudhuri et al107 in 14 patients with asthma and elevated sputum neutrophils in a double-blind, cross-over, randomized design. The primary endpoint was the percentage and absolute sputum neutrophil count, averaged for end-of-treatment visits. GSK2190915 did not significantly reduce mean percentage sputum neutrophils, or mean sputum neutrophil counts. However, GSK2190915 resulted in a marked suppression (>90%) of sputum LTB4 and urine LTE4, but did not alter clinical endpoints. The authors concluded that despite suppressing the target mediator LTB4, FLAP inhibitor GSK2190915 had no short-term effect on sputum cell counts or clinical endpoints in patients with asthma and sputum neutrophilia.
Although the relative contribution of LTB4 and leukotriene D4 in improving patient outcomes in neutrophilic asthma remains unrevealed, inhibition of 5-lipoxygenase that generates LTB4 and LTD4 seems to offer therapeutic benefit in asthma associated with aspirin sensitivity and nasal polyps.31
Macrolides
Clarithromycin and azithromycin have demonstrated effectiveness in NEA.108–110 A clinical trial assessing the effect of low-dose azithromycin in 420 severe asthmatics treated for 48 weeks, the AMAZES (Asthma and Macrolides: Azithromycin Efficacy and Safety) study has shown that azithromycin reduced asthma exacerbations and significantly improved asthma-related quality of life.108 Using induced sputum, 144 (43%) patients had an eosinophilic inflammatory phenotype and 187 (57%) had a non-eosinophilic phenotype. Azithromycin reduced asthma exacerbations in both eosinophilic asthma and NEA. In NEA, patients treated with placebo (n=104) experienced 1.74 exacerbations per person-year, compared with those treated with azithromycin (n=120) who experienced 1.15 exacerbations per person-year (incidence rate ratio; IRR 0.66 [95% CI 0.47–0.93]; P=0.019). In eosinophilic asthma, patients treated with placebo (n=103) experienced 1.98 exacerbations per person-year, whereas those treated with azithromycin (n=93) experienced 0.96 exacerbations per person-year (IRR 0.52 [95% CI 0.29–0.94]; P=0.030). Moreover, azithromycin had a positive effect on lower respiratory tract infection in NEA. The findings of the AMAZES study point out that azithromycin is a potential treatment for patients with NEA, for whom there is a limited therapeutic arsenal. The most common adverse effect was diarrhea in the active group arm.
The authors concluded that azithromycin might be a useful add-on therapy in persistent asthma. The mechanism of action could be due to antibacterial or anti-inflammatory actions, which include inhibition of NF-kB and other transcription factors as well as reduction in neutrophil migration or function.108
Besides, it has been shown that macrolides may exert antiviral actions109 and an ability to restore corticosteroid sensitivity by inhibiting the PI3K pathway and restoring histone deacetylase 2 (HDAC2)110 and by attenuating TNF-α and IL-17 immune responses.111
Statins, peroxisome proliferator-activated receptor γ (PPARγ) agonist and other treatments
Statins have shown to reduce inflammatory pathways potentially relevant to the pathogenesis of asthma and smoke-induced airway diseases and might restore corticosteroid sensitivity in asthma.112
Low-dose theophylline has been shown to restore corticosteroid sensitivity in vitro by increasing HDAC-2 activity.84 Theophylline also inhibits oxidative stress-dependent PI3K-d activation and restores corticosteroid sensitivity in peripheral blood mononuclear cells from patients with COPD.84
In preclinical studies peroxisome PPARγ agonists have shown to reduce eosinophilic and neutrophilic lung infiltration in experimental animal models exposed to allergen or tobacco smoke. Inhaled PPARγ agonist analogs are under development.113
Tiotropium has demonstrated clinical efficacy in patients with fixed airway obstruction, usually associated with neutrophilic airway inflammation.114
Bronchial thermoplasty improves symptoms and reduces exacerbations in patients with severe uncontrolled asthma and chronic airflow obstruction phenotype.115
A summary of the pharmacologic treatments for NEA under development is shown in Table 2.
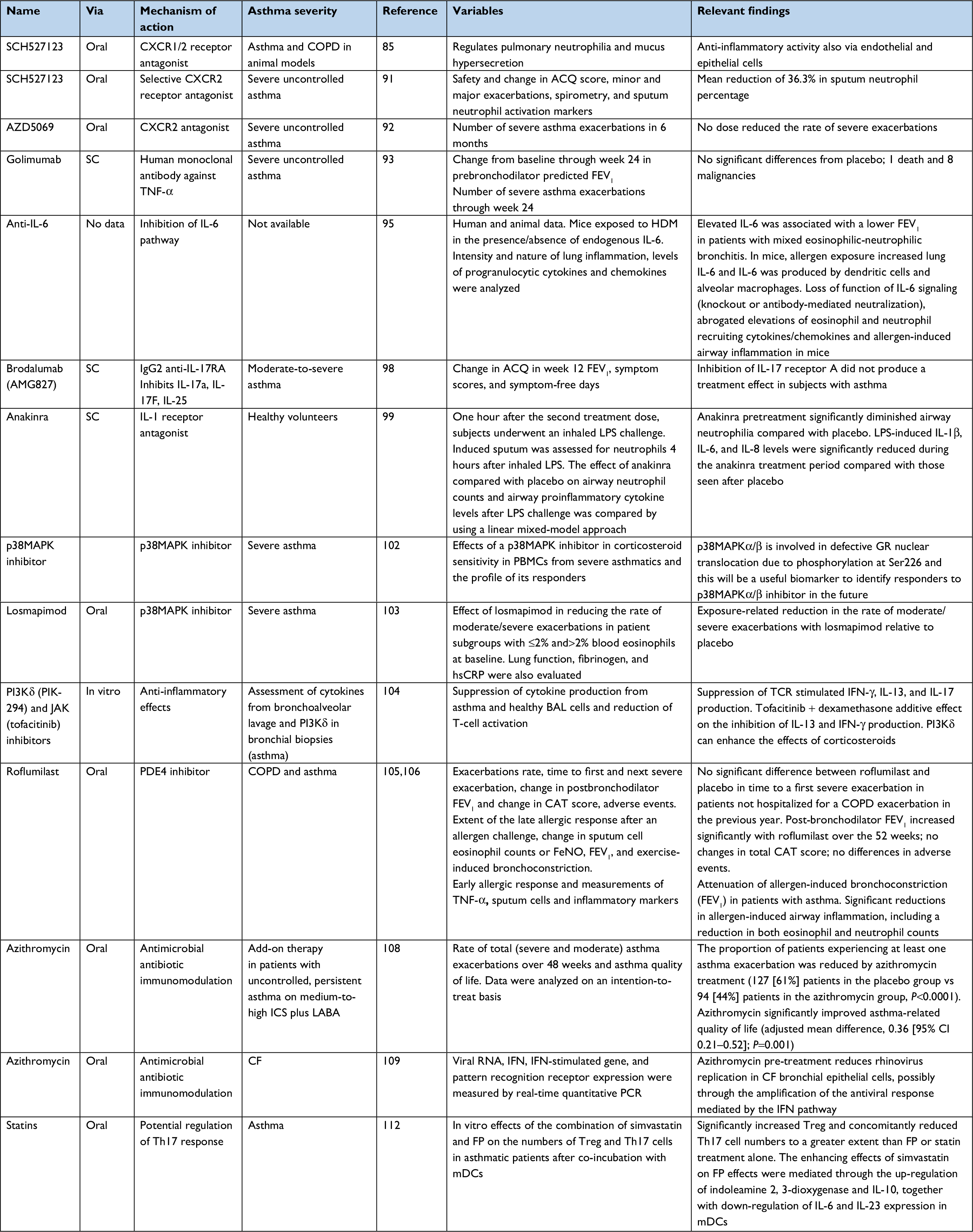 | Table 2 Pharmacologic treatments for non-eosinophilic asthma under development Abbreviations: SC, subcutaneously; TNF-α, tumor necrosis factor-α; IL, interleukin; JAK, Janus-activated kinases; LPS, lipopolysaccharide; PI3Kδ, phosphatidylinositol 3-kinase delta; PBMC, peripheral blood mononuclear cell; PDE4, phosphodiesterase 4; CAT, COPD assessment test; TNF-α, tumor necrosis factor α; ICS, inhaled corticosteroids; CF, cystic fibrosis; IFN, interferon; FP, fluticasone propionate; mDC, monocyte-derived DC; FeNO, fractional exhaled nitric oxide; ACQ, asthma control questionnaire; HDM, house dust mites; GR, glucocorticoid receptor; hsCRP, high sensitive C-reactive protein; LABA, long-acting beta-2 agonists; mDC, monocyte-derived dendritic cells; TCR, T-cell receptor. |
Comments and conclusion
Over the past 15–20 years, the study of asthma patients using different approaches has yielded several phenotypes and endotypes and has shown that in many cases, inflammation in asthma is not always characterized by eosinophilia and type 2 cytokines but could be in fact non-type 2.
NEA constitutes a differentiated phenotype among patients with asthma, with distinct clinical and pathophysiological features. Its poor response to classical treatments, particularly corticosteroids, leads to difficult-to-treat and severe asthma that frequently remains uncontrolled. Definition for NEA has been well established, showing eosinophil counts in sputum under 2%–3%. Since this is an accessible test, diagnosis of NEA should be always considered, especially in patients with difficult to control asthma who are not responding as expected to ICS. It is mandatory to rule out NEA in patients exposed to occupational triggers, infections, pollution, tobacco smoke, and in elderly or obese patients, since these are typical triggers and risk factors. Notwithstanding, it is not always feasible to completely avoid these factors, so it will be an essential concern in patients with difficult to control asthma.
New therapeutic approaches in uncontrolled asthma are mainly focused in allergic and eosinophilic asthma, due to their prevalence and known cellular and molecular targets, whereas the situation in NEA, for which there is a lack of effective specific treatments, can represent an orphan asthma endotype.
Despite the absence of substantiated specific treatment for NEA, multiple therapies have been studied to optimize management of this condition, challenging both clinicians and researchers to find the most suitable treatment for these patients.
In spite of specific treatment shortage nowadays, it is crucial that we keep on differentiating patient’s phenotypes and endotypes and that we pursue the most suitable treatment for each patient. We should never fade on our search of the ideal treatment and approach for a specific condition such as NEA, which can be of utmost relevance for a substantial number of asthma patients. Treatment approach in these patients should consider the prevailing features in each patient, such as the profile of airway inflammation (neutrophilic or paucigranulocytic), Th1 or Th17-predominant, the degree of corticosteroid insensitivity and the possible involvement of non-inflammatory pathways such as heightened BHR and airway remodeling. However, while there are several hypotheses of potential pathways leading to noneosinophilic airway inflammation, currently no specific biomarkers are readily available to support diagnosis and endotyping of these patients.
The reality is that there is a significant lack of effective treatment for these patients. The available treatments and some of the drugs under development for NEA may have failed because the wrong patients were targeted (lack of biomarker), because the wrong endpoints/outcomes were chosen, or the wrong timeframes of treatment examined. Moreover, some of the theoretical pathways that are thought to be important in NEA may be simply ineffective or irrelevant.
Therefore, the approach to this group of patients is nowadays addressed by pathophysiological hypothesis and, from a practical approach, by the evidence of efficacy of macrolides. Future clinical development of novel treatment strategies are urgently needed in order to cover these unmet needs in NEA.
Disclosure
IEG has received lecture fees from Chiesi, Leti, Shire, and Allergy Therapuetics. DA has received lecture fees from Chiesi, Novartis, GSK, Leti, ALK, and AstraZeneca and has served as a consultant to Merck. JD has served as a consultant to LETI, Mundipharma, and GSK and has received lecture fees from Chiesi, Novartis, Leti, AstraZeneca, Stallergenes, and TEVA. SQ has served as a consultant to AstraZeneca, Novartis, Sanofi, Gennetech, Teva, ALK, Mundipharma, and GSK and has received lecture fees from Chiesi, Novartis, Leti, AstraZeneca, Mundipharma, and GSK. The authors report no other conflicts of interest in this work.
References
GINA Report. Global Strategy for Asthma Management and Prevention; 2018. Available from: http://ginasthma.org/2018-gina-report-global-strategy-for-asthma-management-and-prevention/. Accessed May 2018. | ||
Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol. 2015;135(2):299–310. | ||
Woodruff PG, Modrek B, Choy DF, et al. T-helper Type 2–driven Inflammation Defines Major Subphenotypes of Asthma. Am J Respir Crit Care Med. 2009;180(5):388–395. | ||
Godar M, Blanchetot C, de Haard H, Lambrecht BN, Brusselle G. Personalized medicine with biologics for severe type 2 asthma: current status and future prospects. MAbs. 2018;10(1):34–45. | ||
Chung KF, Wenzel SE, Brozek JL, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43(2):343–373. | ||
Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: Assessment and identification using induced sputum. Respirology. 2006;11(1):54–61. | ||
Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57(7):643–648. | ||
McGrath KW, Icitovic N, Boushey HA, et al. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med. 2012;185(6):612–619. | ||
Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest. 2001;119:1329–1336. | ||
Quirce S, Campo P, Domínguez-Ortega J, et al. New developments in work-related asthma. Expert Rev Clin Immunol. 2017;13(3):271–281. | ||
Anees W, Huggins V, Pavord ID, Robertson AS, Burge PS. Occupational asthma due to low molecular weight agents: eosinophilic and non-eosinophilic variants. Thorax. 2002;57(3):231–236. | ||
Ray A, Kolls JK. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 2017;38(12):942–954. | ||
Nair P, Aziz-Ur-Rehman A, Radford K. Therapeutic implications of ‘neutrophilic asthma’. Curr Opin Pulm Med. 2015;21(1):33–38. | ||
Saffar AS, Ashdown H, Gounni AS. The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr Drug Targets. 2011;12(4):556–562. | ||
Berry M, Morgan A, Shaw DE, et al. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax. 2007;62(12):1043–1049. | ||
Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557–1563. | ||
Bobolea I, Barranco P, del Pozo V, Pozo D V, et al. Sputum periostin in patients with different severe asthma phenotypes. Allergy. 2015;70(5):540–546. | ||
Wu W, Bleecker E, Moore W, et al. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J Allergy Clin Immunol. 2014;133(5):1280–1288. | ||
Choi JS, Jang AS, Park JS, et al. Role of neutrophils in persistent airway obstruction due to refractory asthma. Respirology. 2012;17(2):322–329. | ||
Chang HS, Lee T-H, Jun JA, et al. Neutrophilic inflammation in asthma: mechanisms and therapeutic considerations. Expert Rev Respir Med. 2017;11(1):29–40. | ||
Andersson CK, Adams A, Nagakumar P, et al. Intraepithelial neutrophils in pediatric severe asthma are associated with better lung function. J Allergy Clin Immunol. 2017;139(6):e11:1819–1829. | ||
Taylor SL, Leong LEX, Choo JM, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141(1):e15:94–103. | ||
Ntontsi P, Loukides S, Bakakos P, et al. Clinical, functional and inflammatory characteristics in patients with paucigranulocytic stable asthma: Comparison with different sputum phenotypes. Allergy. 2017;72(11):1761–1767. | ||
Loza MJ, Djukanovic R, Chung KF, et al. ADEPT (Airways Disease Endotyping for Personalized Therapeutics) and U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome Consortium) investigators. Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res. 2016;17:165. | ||
Belda J, Leigh R, Parameswaran K, O’Byrne PM, Sears MR, Hargreave FE. Induced sputum cell counts in healthy adults. Am J Respir Crit Care Med. 2000;161(2):475–478. | ||
Gao J, Wu F. Association between fractional exhaled nitric oxide, sputum induction and peripheral blood eosinophil in uncontrolled asthma. Allergy Asthma Clin Immunol. 2018;14:21. | ||
Lemière C, Ernst P, Olivenstein R, et al. Airway inflammation assessed by invasive and noninvasive means in severe asthma: Eosinophilic and noneosinophilic phenotypes. J Allergy Clin Immunol. 2006;118(5):1033–1039. | ||
Chung KF. Asthma phenotyping: a necessity for improved therapeutic precision and new targeted therapies. J Intern Med. 2016;279(2):192–204. | ||
Belda J, Leigh R, Parameswaran K, O’Byrne PM, Sears MR, Hargreave FE. Induced sputum cell counts in healthy adults. Am J Respir Crit Care Med. 2000;161(2):475–478. | ||
Bakakos P, Schleich F, Alchanatis M, Louis R. Induced sputum in asthma: from bench to bedside. Curr Med Chem. 2011;18(10):1415–1422. | ||
Panettieri RA. Neutrophilic and pauci-immune phenotypes in severe asthma. Immunol PI3Kd, phosphatidylinositol 3-kinase delta Allergy Clin North Am. 2016;36(3):569–579. | ||
Saito J, Zhang Q, Hui C, et al. Sputum hydrogen sulfide as a novel biomarker of obstructive neutrophilic asthma. J Allergy Clin Immunol. 2013;131(1):232–234. | ||
Samitas K, Zervas E. Gaga M. T2-low asthma: current approach to diagnosis and therapy. Curr Opin Pulm Med. 2017;23:48–55. | ||
Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: Assessment and identification using induced sputum. Respirology. 2006;11(1):54–61. | ||
Brooks CR, van Dalen CJ, Zacharasiewicz A, et al. Absence of airway inflammation in a large proportion of adolescents with asthma. Respirology. 2016;21(3):460–466. | ||
Busse WW, Holgate ST, Wenzel SW, et al. Biomarker profiles in asthma with high vs low airway reversibility and poor disease control. Chest. 2015;148(6):1489–1496. | ||
Schleich FN, Manise M, Sele J, Henket M, Seidel L, Louis R. Distribution of sputum cellular phenotype in a large asthma cohort: predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm Med. 2013;13(1):11. | ||
Hilvering B, Vijverberg SJH, Jansen J, et al. Diagnosing eosinophilic asthma using a multivariate prediction model based on blood granulocyte responsiveness. Allergy. 2017;72(8):1202–1211. | ||
Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57(10):875–879. | ||
Shaw DE, Berry MA, Hargadon B, et al. Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest. 2007;132(6):1871–1875. | ||
Brooks CR, van Dalen CJ, Harding E, Hermans IF, Douwes J. Effects of treatment changes on asthma phenotype prevalence and airway neutrophil function. BMC Pulm Med. 2017;17(1):169. | ||
Górska K, Paplińska-Goryca M, Nejman-Gryz P, Goryca K, Krenke R. Eosinophilic and neutrophilic airway inflammation in the phenotyping of mild-to-moderate asthma and chronic obstructive pulmonary disease. COPD. 2017;14(2):181–189. | ||
Sin BA, Akkoca O, Saryal S, Oner F, Misirligil Z. Differences between asthma and COPD in the elderly. J Investig Allergol Clin Immunol. 2006;16:44–50. | ||
Choi S, Hoffman EA, Wenzel SE, et al. National Heart, Lung and Blood Institute’s Severe Asthma Research Program Quantitative computed tomographic imaging-based clustering differentiates asthmatic subgroups with distinctive clinical phenotypes. J Allergy Clin Immunol. 2017;140:690–700. | ||
Choi S, Haghighi B, Choi J, et al. Differentiation of quantitative CT imaging phenotypes in asthma versus COPD. BMJ Open Respir Res. 2017;4(1):e000252. | ||
Lazarus SC, Chinchilli VM, Rollings NJ, et al. National Heart Lung and Blood Institute’s Asthma Clinical Research Network. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am J Respir Crit Care Med. 2007;175:783–790. | ||
Polosa R, Thomson NC. Smoking and asthma: dangerous liaisons. Eur Respir J. 2013;41(3):716–726. | ||
Shimoda T, Obase Y, Kishikawa R, Iwanaga T. Influence of cigarette smoking on airway inflammation and inhaled corticosteroid treatment in patients with asthma. Allergy Asthma Proc. 2016;37(4):50–58. | ||
Polosa R, Russo C, Caponnetto P, et al. Greater severity of new onset asthma in allergic subjects who smoke: a 10-year longitudinal study. Respir Res. 2011;12(1):16. | ||
Siew LQC, Wu S-Y, Ying S, Corrigan CJ. Cigarette smoking increases bronchial mucosal IL-17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clinical & Experimental Allergy. 2017;47(6):740–750. | ||
Hekking P-PW, Bel EH. Developing and Emerging Clinical Asthma Phenotypes. J Allergy Clin Immunol. 2014;2(6):671–680. | ||
Alexis NE, Carlsten C. Interplay of air pollution and asthma immunopathogenesis: A focused review of diesel exhaust and ozone. Int Immunopharmacol. 2014;23(1):347–355. | ||
Tarlo SM, Lemiere C. Occupational Asthma. N Engl J Med Overseas Ed. 2014;370(7):640–649. | ||
Sánchez-Vidaurre S, Cruz M-J, Gómez-Ollés S, Morell F, Muñoz X. Sputum inflammatory profile before and after specific inhalation challenge in individuals with suspected occupational asthma. PLoS One. 2013;8(11):e78304. | ||
Dominguez-Ortega J, Barranco P, Rodríguez-Pérez R, Quirce S. Biomarkers in Occupational Asthma. Curr Allergy Asthma Rep. 2016;16(9):63. | ||
Kleniewska A, Walusiak-Skorupa J, Piotrowski W, Nowakowska-Świrta E, Wiszniewska M. Comparison of biomarkers in serum and induced sputum of patients with occupational asthma and chronic obstructive pulmonary disease. J Occup Health. 2016;58(4):333–339. | ||
Castano R, Miedinger D, Maghni K, et al. Matrix metalloproteinase-9 increases in the sputum from allergic occupational asthma patients after specific inhalation challenge. Int Arch Allergy Immunol. 2013;160(2):161–164. | ||
Esteban-Gorgojo I, Coman I, Rial MJ. Occupational asthma, not a trivial disorder and a source of fatal and near-fatal events. Curr Treat Options Allergy. 2018;5(1):1–10. | ||
Yang X, Jiang Y, Wang C. Does IL-17 respond to the disordered lung microbiome and contribute to the neutrophilic phenotype in asthma? Mediators Inflamm. 2016;2016(4):6470364–7. | ||
Alam R, Good J, Rollins D, et al. Airway and serum biochemical correlates of refractory neutrophilic asthma. J Allergy Clin Immunol. 2017;140(4):1004–1014. | ||
Hadebe S, Kirstein F, Fierens K, et al. Microbial ligand costimulation drives neutrophilic steroid-refractory asthma. PLoS One. 2015;10(8):e0134219. | ||
Wood LG, Simpson JL, Hansbro PM, Gibson PG. Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Radic Res. 2010;44(2):146–154. | ||
Camargo CA, Weiss ST, Zhang S, Willett WC, Speizer FE. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch Intern Med. 1999;159(21):2582–2588. | ||
Denlinger LC, Phillips BR, Ramratnam S, et al. Inflammatory and comorbid features of patients with severe asthma and frequent exacerbations. Am J Respir Crit Care Med. 2017;195(3):302–313. | ||
Haldar P, Pavord ID, Shaw DE, et al. Cluster Analysis and Clinical Asthma Phenotypes. Am J Respir Crit Care Med. 2008;178(3):218–224. | ||
Holguin F, Bleecker ER, Busse WW, et al. Obesity and asthma: An association modified by age of asthma onset. J Allergy Clin Immunol. 2011;127(6):1486–1493. | ||
Mclachlan CR, Poulton R, Car G, et al. Adiposity, asthma, and airway inflammation. J Allergy Clin Immunol. 2007;119(3):634–639. | ||
Sutherland ER. Obesity and Asthma. Immunol Allergy Clin North Am. 2008;28(3):589–602. | ||
Barranco P, García-Río F, Olivares J, et al. Asthma diagnosis is not associated with obesity in a population of adults from Madrid. J Investig Allergol Clin Immunol. 2011;21:540–545. | ||
Beuther DA. Sutherland ER. Overweight, obesity and incident asthma: a meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007;175:661–666. | ||
Sood A, Dawson BK, Eid W, et al. Obesity Is Associated with Bronchial Hyper-Responsiveness in Women. Journal of Asthma. 2005;42(10):847–852. | ||
Litonjua AA, Sparrow D, Celedon JC, Demolles D, Weiss ST. Association of body mass index with the development of methacholine airway hyperresponsiveness in men: the Normative Aging Study. Thorax. 2002;57(7):581–585. | ||
Hasler G, Gergen PJ, Ajdacic V, et al. Asthma and body weight change: a 20-year prospective community study of young adults. Int J Obes. 2006;30(7):1111–1118. | ||
Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol. 2018;141(4):1169–1179. | ||
Miethe S, Guarino M, Alhamdan F, et al. The effects of obesity on asthma: immunometabolic links. Pol Arch Intern Med. 2018. | ||
Jesus JPV, Lima-Matos AS, Almeida PCA, et al. Obesity and asthma: clinical and laboratory characterization of a common combination. Jornal Brasileiro de Pneumologia. 2018;44(3):207–212. | ||
Sood A, Ford ES, Camargo CA Jr. Association between leptin and asthma in adults. Thorax. 2006;61(4):300–305. | ||
Sierra-Honigmann MR, Nath AK, Murakami C, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281(5383):1683–1686. | ||
Nasiri Kalmarzi R, Ataee P, Mansori M, et al. Serum levels of adiponectin and leptin in asthmatic patients and its relation with asthma severity, lung function and BMI. Allergol Immunopathol. 2017;45(3):258–264. | ||
Zhang L, Yin Y, Zhang H, Zhong W, Zhang J. Association of asthma diagnosis with leptin and adiponectin: a systematic review and meta-analysis. Journal of Investigative Medicine. 2017;65(1):57–64. | ||
Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. | ||
Fahy JV. Type 2 inflammation in asthma — present in most, absent in many. Nat Rev Immunol. 2015;15(1):57–65. | ||
Demarche S, Schleich F, Henket M, Paulus V, Louis R, van Hees T. Step-down of inhaled corticosteroids in non-eosinophilic asthma: A prospective trial in real life. Clin Exp Allergy. 2018;48(5):525–535. | ||
Thomson NC. Novel approaches to the management of noneosinophilic asthma. Ther Adv Respir Dis. 2016;10(3):211–234. | ||
Chapman RW, Minnicozzi M, Celly CS, et al. A novel, orally active CXCR1/2 receptor antagonist, SCH527123, inhibits neutrophil recruitment, mucus production, and goblet cell hyperplasia in animal models of pulmonary inflammation. Journal of Pharmacology and Experimental Therapeutics. 2007;322(2):486–493. | ||
Gernez Y, Tirouvanziam R, Chanez P. Neutrophils in chronic inflammatory airway diseases: can we target them and how? Eur Respir J. 2010;35(3):467–469. | ||
Banerjee K, Biswas PS, Kim B, Lee S, Rouse BT, et al. CXCR2-/- mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J Immunol. 2004;172(2):1237–1245. | ||
Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M, et al. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proceedings of the National Academy of Sciences. 1996;93(13):6682–6686. | ||
Chaudhuri R, Livingston E, Mcmahon AD, et al. Effects of Smoking Cessation on Lung Function and Airway Inflammation in Smokers with Asthma. Am J Respir Crit Care Med. 2006;174(2):127–133. | ||
Maghni K, Lemière C, Ghezzo H, Yuquan W, Malo J-L. Airway inflammation after cessation of exposure to agents causing occupational asthma. Am J Respir Crit Care Med. 2004;169(3):367–372. | ||
Nair P, Gaga M, Zervas E, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy. 2012;42(7):1097–1103. | ||
O’Byrne PM, Metev H, Puu M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2016;4(10):797–806. | ||
Wenzel SE, Barnes PJ, Bleecker ER, et al. A Randomized, Double-blind, Placebo-controlled Study of Tumor Necrosis Factor-α Blockade in Severe Persistent Asthma. Am J Respir Crit Care Med. 2009;179(7):549–558. | ||
Erzurum SC. Inhibition of Tumor Necrosis Factor α for Refractory Asthma. N Engl J Med Overseas Ed. 2006;354(7):754–758. | ||
Chu DK, Al-Garawi A, Llop-Guevara A, et al. Therapeutic potential of anti-IL-6 therapies for granulocytic airway inflammation in asthma. Allergy, Asthma & Clinical Immunology. 2015;11(1):14. | ||
Peters MC, Mcgrath KW, Hawkins GA, et al. National Heart, Lung, and Blood Institute Severe Asthma Research Program. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med. 2016;4:574–584. | ||
Hilvering B, Hinks TSC, Stöger L, et al. Synergistic activation of pro-inflammatory type-2 CD8+ T lymphocytes by lipid mediators in severe eosinophilic asthma. Mucosal Immunol. Epub 2018 Jun 15. | ||
Busse WW, Holgate S, Kerwin E, et al. Randomized, Double-Blind, Placebo-controlled Study of Brodalumab, a Human Anti–IL-17 Receptor Monoclonal Antibody, in Moderate to Severe Asthma. Am J Respir Crit Care Med. 2013;188(11):1294–1302. | ||
Hernandez ML, Mills K, Almond M, et al. IL-1 receptor antagonist reduces endotoxin-induced airway inflammation in healthy volunteers. J Allergy Clin Immunol. 2015;135(2):379–385. | ||
Kim RY, Pinkerton JW, Essilfie AT, et al. Role for NLRP3 Inflammasome–mediated, IL-1β–Dependent Responses in Severe, Steroid-Resistant Asthma. Am J Respir Crit Care Med. 2017;196(3):283–297. | ||
Kim RY, Horvat JC, Pinkerton JW, et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase–mediated suppression of histone deacetylase 2. J Allergy Clin Immunol. 2017;139(2):519–532. | ||
Mercado N, Hakim A, Kobayashi Y, et al. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS One. 2012;7(7):e41582. | ||
Marks-Konczalik J, Costa M, Robertson J, et al. A post-hoc subgroup analysis of data from a six month clinical trial comparing the efficacy and safety of losmapimod in moderate-severe COPD patients with ≤2% and >2% blood eosinophils. Respir Med. 2015;109(7):860–869. | ||
Southworth T, Plumb J, Gupta V, et al. Anti-inflammatory potential of PI3Kδ and JAK inhibitors in asthma patients. Respir Res. 2016;17(1):124. | ||
Rabe KF, Calverley PMA, Martinez FJ, Fabbri LM. Effect of roflumilast in patients with severe COPD and a history of hospitalisation. Eur Respir J. 2017;50(1):1. | ||
Bardin P, Kanniess F, Gauvreau G, Bredenbröker D, Rabe KF. Roflumilast for asthma: Efficacy findings in mechanism of action studies. Pulm Pharmacol Ther. 2015;35 Suppl(Suppl):S4–S10. | ||
Chaudhuri R, Norris V, Kelly K, et al. Effects of a FLAP inhibitor, GSK2190915, in asthmatics with high sputum neutrophils. Pulm Pharmacol Ther. 2014;27(1):62–69. | ||
Gibson PG, Yang IA, Upham JW, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10095):659–668. | ||
Schögler A, Kopf BS, Edwards MR, et al. Novel antiviral properties of azithromycin in cystic fibrosis airway epithelial cells. Eur Respir J. 2015;45(2):428–439. | ||
Essilfie AT, Horvat JC, Kim RY, et al. Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax. 2015;70(5):458–467. | ||
Kobayashi Y, Wada H, Rossios C, et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-κB inhibition. J Pharmacol Exp Ther. 2013;345(1):76–84. | ||
Maneechotesuwan K, Kasetsinsombat K, Wamanuttajinda V, Wongkajornsilp A, Barnes PJ. Statins enhance the effects of corticosteroids on the balance between regulatory T cells and Th17 cells. Clin Exp Allergy. 2013;43(2):212–222. | ||
Morissette MC, Shen P, Thayaparan D, Stämpfli MR. Impacts of peroxisome proliferator-activated receptor- activation on cigarette smoke-induced exacerbated response to bacteria. Eur Respir J. 2015;45(1):191–200. | ||
Radovanovic D, Santus P, Blasi F, Mantero M. The evidence on tiotropium bromide in asthma: from the rationale to the bedside. Multidiscip Respir Med. 2017:12:12. | ||
Laxmanan B, Hogarth DK. Bronchial thermoplasty in asthma: current perspectives. J Asthma Allergy. 2015;8:39–49. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.