Back to Journals » International Journal of Nanomedicine » Volume 14
NLRP3 inflammasome-activating arginine-based liposomes promote antigen presentations in dendritic cells
Authors Li T, Zehner M, He J, Próchnicki T, Horvath G, Latz E, Burgdorf S, Takeoka S
Received 21 January 2019
Accepted for publication 13 March 2019
Published 13 May 2019 Volume 2019:14 Pages 3503—3516
DOI https://doi.org/10.2147/IJN.S202379
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Tianshu Li,1 Matthias Zehner,2 Jieyan He,3 Tomasz Próchnicki,4 Gabor Horvath,4 Eicke Latz,4 Sven Burgdorf,2 Shinji Takeoka1,3
1Institute for Advanced Research of Biosystem Dynamics, Waseda Research Institute for Science and Engineering, Waseda University, Tokyo, Japan; 2Molecular Immunology and Cell Biology, Life and Medical Sciences Institute, University of Bonn, Bonn, Germany; 3Department of Life Science and Medical Bioscience, Graduate School of Advanced Science and Engineering, Waseda University, Tokyo, Japan; 4Institute of Innate Immunity, Biomedical Center, University Hospitals, University of Bonn, Bonn, Germany
Purpose: The NLRP3 inflammasome activation has been proposed as a common mechanism for some adjuvants to boost the immune system, and cationic liposomes were reported to potentially activate the NLRP3 inflammasome. Herein, we questioned whether the NLRP3 inflammasome-activating cationic liposomes could promote antigen presentation and be applied as an immune adjuvant. In addition, we aimed to investigate the structure effect of lipid on triggering these immune responses.
Materials and methods: A series of structurally similar lipids, consisting of arginine (Arg) head group and varied lengths of alkyl chains or spacers in between were used to prepare cationic liposomes. Lipopolysaccharide-primed human or murine macrophages or phorbol 12-myristate 13-acetate-primed THP-1 cells were treated with these liposomes, and interleukin (IL)-1β secretion was measured to quantify the NLRP3 inflammasome activation. Lysosome rupture was examined in THP-1 cells by the fluorescence loss of acridine orange, a lysosome dye. Further, chicken ovalbumin (OVA) was loaded on the liposome surface and applied to murine bone marrow-derived dendritic cells (BMDCs), which activate OT-I and OT-II lymphocytes upon major histocompatibility complex (MHC) class I- and class II-mediated antigen presentation, respectively. OT-I and OT-II cell division and IL-2 secretion were measured to evaluate the antigen presentation efficiency. The expressions of MHC molecules and co-stimulatory molecules ie, CD80, CD86, and CD40 on BMDCs were investigated by flow cytometry.
Results: All the liposomes showed size distributions of 80–200 nm and zeta potentials of around 50 mV. A3C14 liposomes, consisting of Arg-C3-Glu2C14 lipids induced the most potent lysosome rupture and NLRP3 inflammasome activation. OVA-A3C14 also exhibited the most potent MHC class I- and class II-mediated antigen presentation in BMDCs without interfering MHC and co-stimulatory molecules.
Conclusion: The hydrophobic moieties of arginine-based liposomes are crucial in stimulating innate immune cells. A3C14 liposomes were non-immunogenic but strongly activated innate immune cells and promoted antigen presentation, and therefore can be applied as immune adjuvants.
Keywords: arginine, cationic liposomes, NLRP3 inflammasome activation, antigen presentation, lysosome rupture
Introduction
Cationic liposomes are widely used for protein delivery and gene transfection due to their electrostatic interaction with negatively charged macromolecules. To date, many attempts on modifying the structures of cationic lipids have been accumulated to enhance their drug delivery efficiency and meanwhile hurdle the toxicity.1–3 However, recent findings have shown that some cationic liposomes could cause the NLRP3 inflammasome activation in macrophages,4–6 revealing a potential of these liposomes being applied as immune stimuli.
The NOD-like receptors (NLRs) are intracellular sensors of invading microbes and “danger” molecules that are derived from the hosts such as ATP. The NLRP3 inflammasome is a multiprotein assembly of NLR-family protein NLRP3, pro-caspase-1, and an apoptosis-associated speck-like protein containing a CARD (ASC) adaptor, which is formed upon activation. It is crucial for innate immunity to trigger the release of pro-inflammatory cytokines such as interleukin (IL)-1β, and two sequential signals are needed for its secretion: one is the activation of toll-like receptor (TLR) such as TLR4, which accelerates the production of pro-IL-1β, and the other is the NLRP3 inflammasome activation, which activates caspase-1 to process pro-IL-1β into its matured form IL-1β. Hitherto knowledge regarding the molecular mechanism of the NLRP3 inflammasome activation proposes the involvement of reactive oxygen species (ROS) and/or lysosomal components released into cytosol upon phagolysosome rupture.7 Charged nanoparticles, in particular, cationic compositions, might induce the NLRP3 inflammasome activation through phagolysosome membrane destabilization following the phagoendocytosis,6 well-known example of which includes conventional adjuvant alum, QuilA, and chitosan particles.8,9 The released IL-1β recruits other immune cells to the site so that the immune responses can be amplified. Accordingly, the NLRP3 inflammasome activation is critical for these adjuvants to elicit immunological effects.
Upon the activation of the innate immune system, adaptive immune responses can be triggered through efficient antigen presentation by antigen presenting cells (APCs) such as dendritic cells (DCs) to activate T cells. There are mainly two pathways regarding the antigen presentation: major histocompatibility complex (MHC) class I molecules-mediated antigen presentation, which is recognized by the T cell receptor (TCR) on immature CD8+ T cell surface and promotes the maturation of CD8+ T cells (cytotoxic T cells, CTLs); and MHC class II molecules-mediated antigen presentation, which activates CD4+ T cells (T helper cells, Th). The activation of T cells results in the cell division as well as the secretion of IL-2, which further drives the proliferation and differentiation of T cells. In addition to TCR signaling, binding of co-stimulatory molecules is also synchronized to fully activate T cells. For example, CD80/CD86 recognizes CD28, and CD40 recognizes CD40-L on T cells. Deficient expression or recognition of co-stimulatory molecules on APCs will otherwise compromise the T cell activation.
Herein, we report an investigation of a series of structurally similar arginine-based cationic liposomes on the NLRP3 inflammasome activation in macrophages, as well as their potential application as antigen carriers and adjuvants in terms of antigen delivery to bone marrow-derived dendritic cells (BMDCs). MHC class I- and class II-mediated antigen presentations were studied by examining the secretion of IL-2 and proliferations of OT-I (OVA257–264 recognizing CD8+) and OT-II (OVA323–339 recognizing CD4+) T cells, respectively. Lysosome rupture induced by cationic liposomes was studied by flow cytometry, and the expressions of MHC and co-stimulatory molecules on BMDCs were evaluated after treatment with naked liposomes.
Material and methods
Reagents, antibodies, and mice
The following reagents were purchased: phorbol 12-myristate 13-acetate (PMA) and chicken ovalbumin (OVA) from Sigma–Aldrich; OVA conjugated with Alexa Fluor® 555 (OVA555) from Thermo Fisher Scientific; acridine orange (AO) from Dojindo Laboratories; adenosine 5′-triphosphate disodium salt hydrate (ATP) from TCI; lipopolysaccharide (LPS) Escherichia coli (E. coli) O111:B4 from InvivoGen; NLRP3 inhibitor CP-456773 (also known as CRID3) from Pfizer Inc; Caspase-1 inhibitor VX765 from Selleck Chemicals LLC; FITC-conjugated anti-mouse CD86 mAb (clone PO3) from Serotec; PE-conjugated anti-mouse MHC-I (H-2Db) mAb (clone 28-14-8), FITC-conjugated anti-mouse MHC-I (H-2Kb) mAb (clone AF6-88.5.5.3), eFluor® 450-conjugated anti-mouse CD80 mAb (clone 16-10A1), PE-conjugated anti-mouse CD40 mAb (clone 1C10) and PerCP/Cy5.5-conjugated anti-mouse CD11c mAb (clone N418) from eBioscience; and PerCP/Cy5.5-conjugated MHC-II (I-A/I-E) anti-mouse mAb (clone AF6-120.1) from BioLegend. Arginine-containing cationic lipids were synthesized according to the previous research.1–3 The cationic lipids were named according to the chemical structure, where Arg indicates the lipids with an arginine head group; the number in the middle indicates the carbon number of the spacers, and the last number indicates the carbon number of the alkyl chains (Figure 1).
 | Figure 1 Chemical structures of the arginine-containing cationic lipids. n indicates the carbon number of the alkyl chains, and x indicates the carbon number of the spacers between the hydrophilic and hydrophobic parts. |
C57BL/6, OT-II, and OT-I/RAG2-KO mice (female, 8–12 weeks old) were bred under specific pathogen-free conditions, and the experiments were carried out at the University of Bonn in compliance with institutional guidelines and the German law for Welfare of Laboratory Animals.
Cell culture
Human peripheral blood mononuclear cells were isolated from the buffy coat of healthy donors at University Hospital Bonn after written consent was given in accordance with the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board at University of Bonn. Positive selection of monocytes by CD14 MicroBeads (Miltenyi Biotec) was performed, followed by differentiation into macrophages using GM-CSF.6 These cells were cultured in RPMI1640 medium (Gibco®, Life Technologies) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin (PS) at 37°C under 5% CO2 atmosphere. Human macrophages (2.5×105/mL) were primed with 1 ng/mL LPS for 2 hrs before being treated with liposomes.
Immortalized murine macrophages were cultured in DMEM medium (Gibco®, Life Technologies) supplemented with 10% FCS and 1% PS at 37°C under 5% CO2 atmosphere. These cells (1.0×105/well) were seeded in 96-well plate and primed with 100 ng/mL LPS for 2 hrs before use.
Human leukemic THP-1 monocytes obtained from JCRB Cell Bank, Osaka, Japan were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum and 1% PS at 37°C under 5% CO2 atmosphere. THP-1 cells (4×105/mL) were primed with 100 nM PMA for 18 hrs before use. Then, the cells became adherent and the excess PMA was washed off thrice using pre-warmed Dulbecco’s Phosphate-Buffered Saline .
BMDCs were generated according to the reported protocol.10 Briefly, bone marrow of mice (C57BL/6) was isolated by flushing the hind limbs with PBS. Extracted cells were re-suspended and filtered through a 40 μm cell strainer. After centrifugation at 300×g for 5 mins, cells were re-suspended in BMDC culture medium, which was made from Iscove’s Modified Dulbecco’s Medium, 10% FCS, 2.5% GM-CSF-containing J558L cell culture supernatant, 50 μM beta-mercaptoethanol, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. Cells were cultured in 10 cm petri dishes at 37°C under 5% CO2 atmosphere for 3 days. Then, all the cells were harvested, centrifuged, re-suspended in fresh BMDC culture medium, and cultured in 10 cm petri dishes for 7–8 days before use.
OT-I and OT-II cells were collected from the spleen of OT-I/RAG2-KO and OT-II mice, respectively. Briefly, the spleen was homogenized in PBS and filtered through a metal mesh. After centrifugation at 1,500 rpm for 10 mins at 4°C, cells were resuspended in 2 mL of red blood cell lysis buffer and incubated for 5 mins at room temperature (rt). Then, 8 mL of PBS was added, and cells were filtered through 100 μm mesh and centrifuged at 1,500 rpm for 10 mins at 4°C. The cells were resuspended in T cell culture medium (RPMI medium containing 10% FCS and 1% PS) before use.
Preparation of cationic liposomes and loading with antigens
The cationic liposomes were prepared by hydration and bath-type sonication as previously reported.6 The lipid powder Arg-C0-Glu2C14, Arg-C3-Glu2C14, Arg-C5-Glu2C14, Arg-C7-Glu2C14, Arg-C0-Glu2C16, or Arg-C0-Glu2C18 was employed to fabricate A0C14, A3C14, A5C14, A7C14, A0C16, or A0C18 liposomes in 20 mM HEPES buffer, respectively. The liposome dispersions were stored at 4°C and homogenized by a vortex mixer for 10 s or re-suspended using a bath-type sonicator for 2–5 mins before use.
Antigen (ie, OVA)-loaded liposomes were freshly made by mixing soluble OVA and liposomes in HEPES buffer and standing at rt for 5 mins. Then, the resulting complex as well as the same volume of HEPES buffer (used for control groups) was diluted in culture medium and added to cells instantly.
Characterization of arginine-containing cationic liposomes
A dynamic laser scattering (DLS) spectrophotometer (N4 PLUS Submicro Particle Size Analyzer, Beckman Coulter) and a Zetasizer Nano S90 (Malvern Instruments) were employed to measure the particle diameter and zeta potential, respectively. Repeated measurements (n=3) were performed by using a dilution of cationic liposome dispersion containing 5 μg/mL lipids in 1 mL of HEPES buffer.
The NLRP3 inflammasome activation
Primed human or murine macrophages or THP-1 cells were exposed to a 100 μM (final concentration, fc) liposome-containing culture medium at 37°C for 18 hrs incubation and the supernatants were collected for ELISA tests (DuoSet®, R&D Systems) as previously reported.6 1 mM ATP was added 1 hr before the cytokine detection as a positive control. For inhibition assays, NLRP3 inhibitor CP-456773 (fc: 5 μM) or casp-1 inhibitor VX765 (fc: 25 μM) was added to primed cells 2 hrs prior to the addition of liposomes. Silica (fc: 400 μg/mL) was used as a positive control and kept with cells for 20 hrs. The secretion of IL-1ß was detected by homogenous time-resolved fluorescence and the inhibition rate was calculated according to equation (1):
Inhibition rate = (1- [IL-1ß]inhibitor + liposomes / [IL-1ß]liposomes) × 100% (1)
where [IL-1ß]liposomes indicates the concentration of IL-1ß released to the supernatant after treatment with liposomes, and [IL-1ß]inhibitor + liposomes indicates the concentration of IL-1ß released to the supernatant after treatment with each inhibitor and liposomes.
Lysosome rupture assay
AO was used to label and examine the rupture of lysosomes.11 Briefly, primed THP-1 cells were treated with 5 μg/mL AO for 15 mins at 37°C, followed by washing and overnight incubation with 100 μM liposomes. The cell population with AO fluorescence loss was gated and recorded using flow cytometry (FACSAria II Cell Sorter, BD Biosciences). The signals from AO-labeled and non-labeled THP-1 cells without exposure to liposomes were gated to reflect intact lysosomes and total lysosome loss, respectively.
Antigen uptake in BMDCs
BMDCs (1.6×105/well) were seeded in a 48-well cell culture plate for 2 hrs adhesion. Then, the culture medium was exchanged to that containing OVA555 (fc: 2.5 μg/mL)-loaded cationic liposomes (fc: 25 μM) and incubated with BMDCs for 2 hrs. After washing off twice with PBS, the cells were detached from the plate by Trypsin-EDTA, and the amount of internalized OVA was measured by the fluorescence intensity of Alexa Fluor® 555 using flow cytometry.
T cell activation and proliferation
BMDCs (7.5×104 cells/well) were seeded in a 96-well cell culture plate for 2 hrs adhesion prior to medium exchange with that containing OVA (fc: 25 μg/mL)-loaded cationic liposomes (fc: 25 μM). Likewise, free OVA without liposomal association and naked liposomes were added to the BMDCs culture plate as controls. The OVA was allowed to be internalized by BMDCs for 2 hrs. To label the splenocytes of mice, 1 μM carboxyfluorescein succinimidyl ester (CFSE) was used for 15 mins incubation at 37°C. The labeling was stopped with ice-cold PBS and washed off thrice with PBS by centrifugation at 1,500 rpm for 10 mins at 4°C. Then, BDMCs were washed twice with PBS and CF-labeled splenocytes from OT-I mice (1.25×105/well) or CF-labeled splenocytes from OT-II mice (1.6×105/well) were added into the BMDC culture plate. The supernatants were partially collected after overnight incubation for ELISA assay (ie, IL-2). After 48 hrs incubation, the cells were collected for flow cytometric analysis. OT-I cells and OT-II cells were gated by using anti-CD8 or anti-CD4 mAb, respectively. The T cell proliferation was evaluated by CF fluorescence intensity, and the average number of cell divisions (ie, division index, D.I.) was calculated using the formula (2):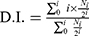
where i is the generation number (0 indicates the undivided cell population), and Ni is the cell number of generation i.
Expression of activation markers on BMDCs
BMDCs (4.5×105 cells/well) were seeded in a 24-well cell culture plate for 2 hrs adhesion. Then, 50 μM (fc) of each type of liposomes was added to the culture plate and kept with cells for 2 hrs. After washing off the liposomes twice with PBS, fresh culture medium was added for overnight incubation. Finally, the cells were detached from the culture plate and stained with two cocktails of activation markers at 4°C for 10 mins separately. One cocktail contained FITC-conjugated anti-CD86 mAb, PE-conjugated anti-MHC-I (H-2Db) mAb, and PerCP/Cy5.5-conjugated anti-MHC-II (I-A/I-E) mAb; and the other contained FITC-conjugated anti-MHC-I (H-2Kb), eFluor® 450-conjugated anti-CD80 mAb and PE-conjugated anti-CD40 mAb. After labeling, the cells were washed with PBS thrice and analyzed by flow cytometry.
Statistic analysis
Student’s t-tests were applied to examine the significant differences of A3C14 or A5C14 liposomes with the controls or other arginine-containing cationic liposomes independently in Figures 2, 3, 5 and 6 using Microsoft Excel. A P-value of less than 0.05 was considered statistically significant.
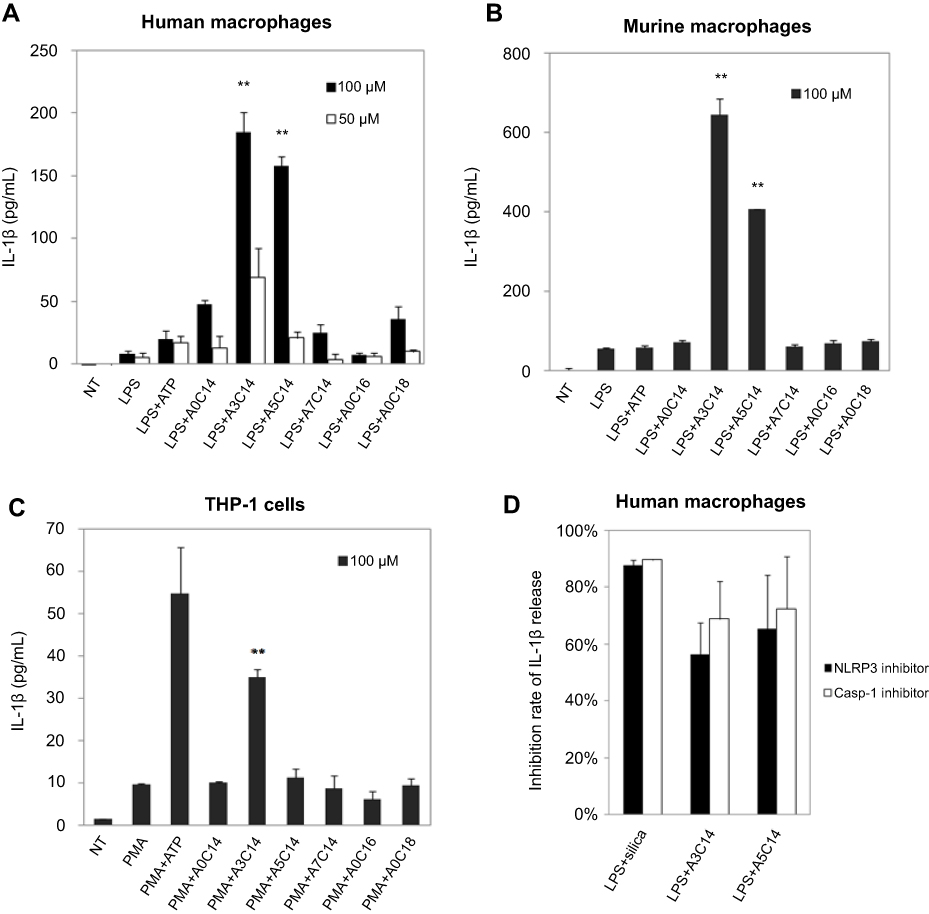 | Figure 2 NLRP3 inflammasome activation of macrophages and monocytes induced by arginine-containing cationic liposomes. LPS (1 ng/mL)-primed human macrophages (A), LPS (100 ng/mL)-primed immortalized murine macrophages (B), and PMA (100 nM)-primed THP-1 cells (C) were incubated with 100 μM or 50 μM cationic liposomes for 18 hrs. The NLRP3 inflammasome activation was evaluated by the release of IL-1ß using human or murine IL-1ß ELISA kit. Data show the mean + SD of three replicates; **P<0.01. (D) NLRP3 inhibitor CP-456773 and caspase-1 (casp-1) inhibitor VX765 suppressed NLRP3 inflammasome activation induced by A3C14 and A5C14 liposomes. 5 μM NLRP3 inhibitor CP-456773 (also known as CRID3) or 25 μM casp-1 inhibitor VX765 was added to the LPS-primed human macrophages prior to the addition of 100 μM liposomes. The release of IL-1β was detected by HTRF and the inhibition rate was calculated to indicate the suppression level of IL-1β by each inhibitor. Data show the mean + SE of two independent experiments (ie, macrophages were collected from two different donors and the measurements were performed at different days) with two replicates, respectively. |
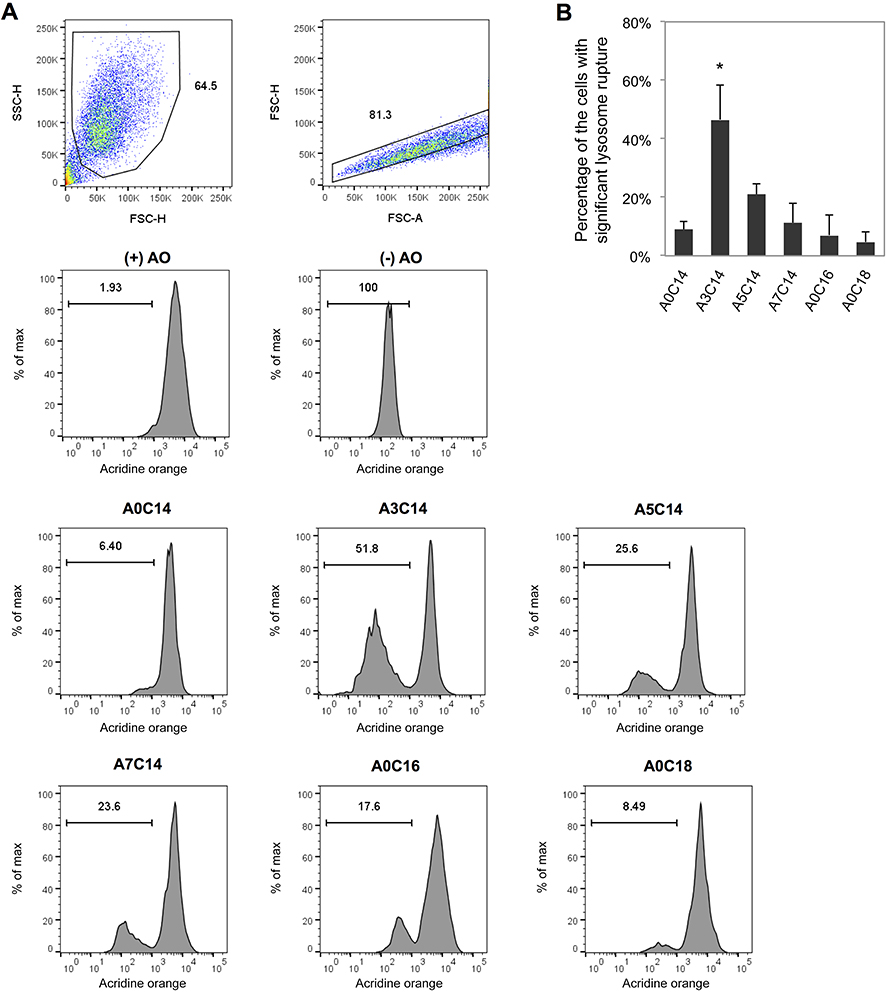 | Figure 3 Arginine-containing cationic liposomes induced the lysosome rupture in PMA-primed THP-1 cells. Acridine orange (AO) was incubated with cells for 15 mins prior to overnight exposure to cationic liposomes. (A) Gating strategies of THP-1 cells using flow cytometry. Cells were first gated by forward scatter (FSC)-height (H) vs side scatter (SSC)-H, and then gated by FSC-area (A) vs FSC-H. The fluorescence intensities of AO were recorded to evaluate the percentage of the cells with significant lysosome rupture (B). Data show the mean + SD of three independent experiments; *P<0.05. |
Results
Preparation and characterization of arginine-containing cationic liposomes
As listed in Table 1, the sizes of the cationic liposomes could be regulated to nanometer-magnitude, ranging from 80 to 200 nm by sonication. The zeta potentials, which reflect the surface electronic property of nanoparticles are basically determined by the head group of the composing lipids facing the aqueous environment. Because the head groups of these lipids are the same arginine group, similar zeta potentials were obtained, ranging from 43 to 60 mV, which are thought to be highly cationic to provide sufficient dispersion stability. To evaluate the homogeneity of the size of liposomes, polydispersity index (PDI) was employed to investigate the mono-dispersed state of the liposomes, the values of which varied from 0.15 to 0.22. In the DLS analysis, a single sharp peak was also observed for all the liposomes (
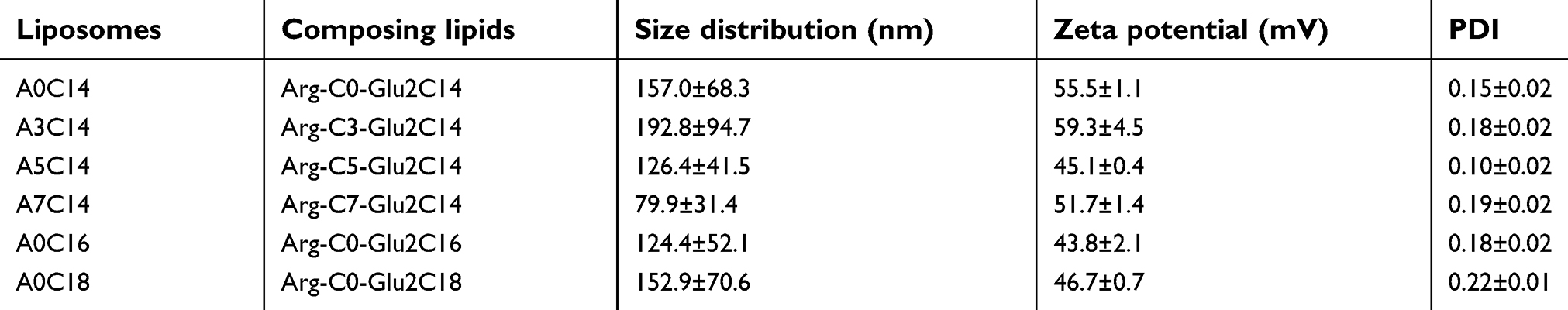 | Table 1 Formulation and characterization of cationic liposomes. Data show the mean ± SD (n=3) |
The NLRP3 inflammasome activation
As shown in Figure 2, A3C14 and A5C14 liposomes induced a significantly stronger release of IL-1β than all the other tested liposomes in both human and murine macrophages; while in THP-1 cells, A3C14 liposomes were the most potent to trigger IL-1β release. Decreasing the liposome concentration from 100 μM to 50 μM resulted in remarkably decreased secretion of IL-1β (Figure 2A). Thus, the NLRP3 inflammasome activation by arginine-containing cationic liposomes was concentration-dependent. However, none of these liposomes could trigger IL-1β release in unprimed immune cells (data not shown). Although the human macrophages from different donors may exhibit different sensitivities to liposomes, A3C14 liposomes were always the most potent to activate the NLRP3 inflammasome, followed by A5C14 liposomes; and the inhibitors of NLRP3 and casp-1 led to 55–75% decrease of IL-1β release in A3C14 and A5C14 liposome-treated cells (Figures 2D and
Lysosome rupture
The cellular lysosomes were labeled with AO, and the loss of AO fluorescence was acquired by flow cytometry to evaluate lysosome rupture (Figure 3A). As shown in Figure 3B, A3C14 liposomes which triggered the most potent NLRP3 inflammasome activation, induced the most potent lysosome rupture in primed THP-1 cells; ie, approximately 45% of the cell population exhibited significant lysosomal damage. In comparison, A5C14 liposomes caused moderate (approximately 20%) and other liposomes performed relatively weak (5–12%) potency of lysosome rupture.
Antigen uptake in BMDCs
Assuming that the isoelectric point (pI) value of OVA is around 4.6–4.9,12 OVA shows a negative charge at neutral pH, allowing the loading on the surface of cationic liposomes through electrostatic association. In comparison to naked liposomes, all the OVA/liposome complexes exhibited size increases of approximately 50 nm in diameter and zeta potential decreases to 20–30 mV, indicating the fact of OVA loading and a similar association pattern of OVA on these cationic liposomes (
As shown in Figure 4, the OVA uptake in BMDCs was increased when they were loaded to A3C14, A5C14, and A7C14 cationic liposomes. By contrast, A0C18, A0C16 or A0C14 liposomes did not increase the cellular uptake of associated OVA.
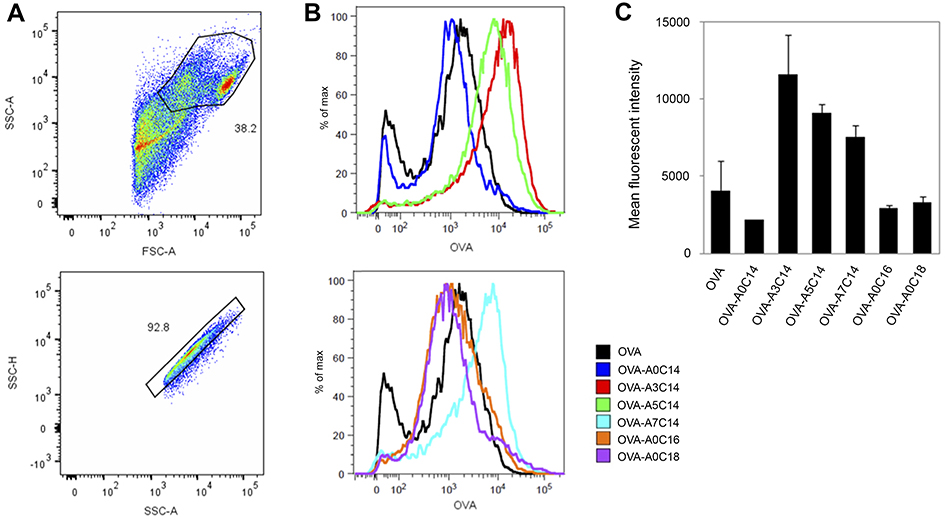 | Figure 4 Antigen delivery efficiency of arginine-containing cationic liposomes in BMDCs. Alexa Fluor 555-conjugated OVA (fc: 2.5 μg/mL) was freely dispersed or loaded on cationic liposomes (fc: 25 μM), and incubated with BMDCs for 2 hrs. The extracellular OVA-liposomes were washed off, and the intracellular OVA was measured by flow cytometry. (A) Cells were first gated by FSC-A vs SSC-A, and then gated by SSC-A vs SSC-H. (B) Representative histograms and (C) mean fluorescent intensities of intracellular OVA. Data show the mean + SD of two independent experiments with two replicates, respectively.Abbreviations: BMDCs, bone marrow-derived dendritic cells; OVA, ovalbumin; FSC, forward scatter; SSC, side scatter; A, area; H, height. |
Antigen presentation and activation of T cells
Antigen presented by APCs will consequently stimulate T cells that they encounter to release IL-2. Thus, MHC-I and MHC-II mediated antigen presentations can be evaluated by the secretion of IL-2 from OT-I and OT-II cells, respectively. J558L-GM-CSF derived BMDCs have been established and used for the study of cross-presentation,20 therefore we applied these cells to study the antigen presenting pathways of the liposomes. As shown in Figure 5, liposomes alone did not lead to T cell activation. However, OVA-associated A3C14 liposomes remarkably enhanced IL-2 secretion in OT-I cells, whereas other OVA-liposomes exhibited medium or slight enhancement in comparison to that of free OVA (Figure 5A). Similarly, the promotion of MHC-II mediated antigen presentation was also observed in OVA-A3C14 liposomes (Figure 5B).
 | Figure 5 Activation of OT-I and OT-II cells through MHC-I and MHC-II mediated antigen presentation pathways, respectively. BMDCs were incubated with OVA (fc: 25 μg/mL)-loaded cationic liposomes (fc: 25 μM) or the same concentration of soluble OVA, or the same concentration of liposomes for 2 hrs, and then extracellular OVA or liposomes were washed off with DPBS. CFSE was used to label splenocytes from OT-I mice (A, C, E) or OT-II mice (B, D, F) and then labeled cells were added into the BMDC culture plate. The supernatant was partially collected after overnight incubation to detect the secretion of IL-2 using an ELISA kit (A, B); and the cells were collected after 48 hrs incubation to analyze the OT-I cell and OT-II cell division using flow cytometry (C, D, E, F, see gating strategies of OT-I and OT-II cells in |
Furthermore, T cell proliferation was investigated to evaluate the liposomal effect on antigen presentation. CF-labeled OT-I and OT-II cells were gated by anti-CD8 or anti-CD4 mAb, respectively (
Influence of liposomes on the expression of MHC and co-stimulatory molecules
The expression of co-stimulatory molecules such as CD80, CD86, and CD40 on the surface of APCs is necessary to efficiently activate T lymphocytes during antigen presentation by MHC molecules. To investigate the influence of liposomes, the expression levels of these markers were examined using flow cytometry (see gating strategies in
As shown in Figure 6A, A3C14 liposomes did not interfere with any of the tested markers on BMDCs, including MHC-I and MHC-II molecules, CD80, CD86, and CD40 co-stimulatory molecules. However, treating with other liposomes might have influenced the expression of these markers (Figure 6B). A resemblance of A5C14, A7C14, A0C16, and A0C18 liposomes was observed in the down-regulation all of the tested activation markers except MHC-II molecules; on the contrary, A0C16 liposomes amplified the expression of MHC-II molecules. For A0C14 liposomes, MHC-II molecules and CD40 have been up-regulated, whereas the expressions of CD80 and CD86 were suppressed.
 | Figure 6 Flow cytometry of activation markers on BMDCs. BMDCs were incubated with each type of the liposomes (fc: 50 μM) for 2 hrs and then washed off. After further incubation overnight in the fresh culture medium, the cells were labeled with fluorescent antibodies against MHC-I (H-2Db), MHC-I (H-2Kb), MHC-II (I-A/I-E), CD80, CD86, and CD40 followed by flow cytometry (A, see gating strategies in |
Discussion
Arginine-containing cationic liposomes were prepared using a hydration of the arginine-type cationic lipid powder followed by a bath-type sonication method as previously reported.6 The advantage of this method is the easy-handling and manipulation of the size of liposomes by adjusting the time of sonication. Herein, all the arginine-containing lipids could be hydrated and sonicated for the same time. The resulting liposomes showed similar zeta potentials but various sizes, suggesting the influences of spacers and hydrophobic tails of the composing lipids in their molecular assembling properties. As with lysine-containing cationic lipids in our previous report,6 increasing the length of hydrophobic tails of arginine-containing lipids from 14 carbons to 16 or 18 carbons or inserting a spacer (ie, 3, 5, or 7 carbons) between the head group and the hydrophobic part, could increase the stability of the liposomes in view of their constant size distributions after storage at 4°C for one month. Otherwise, aggregation and sedimentation of the liposomes (ie, A0C14) could be observed. Besides the surface electrostatic repulsion among the cationic liposomes, a more stable liposome in aqueous solution was thought to be obtained by increasing the hydrophobicity of the bilayer, that was strengthened enough to overcome the tendency of fusion with each other.
Cationic liposomes can be readily internalized into the cells and applied as protein or gene delivery systems.1,3,13,14 To induce the NLRP3 inflammasome activation, cellular internalization of the arginine-containing cationic liposomes was indispensable (
Classically, MHC-I molecules process the endogenous antigens distributed in the cytosol, whereas the presentation of exogenous antigens is restricted to MHC-II mediated pathway, which undergoes endocytosis/phagocytosis and loads antigens derived from lysosomes. However, cross-presentation of the endocytosed antigens by MHC-I molecules also occurs, leading to the activation of CD8+ T cells.16,17 Recent evidence has indicated that the endocytosis mechanisms of antigens greatly influence the antigen presentation pathway.18 For example, soluble antigens uptaken by the mannose receptor (MR) tend to be routed toward the early endosomal compartment, where a mildly acidic environment facilitates the MHC class I-restricted antigen presentation. In contrast, pinocytosis or scavenger receptor (SR)-mediated endocytosis directs the antigens toward the lysosomal compartment, where MHC-II-restricted antigen presentation takes place. Since OVA can target MR and SR,19,20 it can be presented by both MHC-I and MHC-II molecules, and similar presenting capacity through these two routings was obtained under our experimental conditions (Figure 5). Interestingly, although A3C14 liposomes enhanced the total uptake of loaded OVA and meanwhile facilitated both MHC-I and MHC-II mediated antigen presentation, more enhanced T cell activation and proliferation via MHC-I was observed than that of MHC-II mediated antigen presentation. Obviously, A3C14 liposomes altered the intracellular routing of OVA so that they contributed to the increased ratio of cross-presentation. OVA association on the surface of cationic liposomes was achieved through electrostatic interaction at a neutral pH (7.4, HEPES buffer), and this force could be diminished at lower pHs and eliminated when the pH drops to the pI of OVA (4.6–4.9).12 Thus, the unloading of OVA would gradually occur as the acidification of endosome compartments continues, and fully expose the cationic liposomes at lysosomal pH. It is reasonable that A3C14 liposomes were the most capable of lysosome rupture, thereby the release of carried antigens into cytosol consequently endowed the antigen presentation by MHC class I molecules.
Similar discussion has also been proposed by Welsby et al, that adjuvant saponin QS-21 directly activated human monocyte-derived DCs and promoted a pro-inflammatory transcriptional program through lysosomal destabilization, which contributed to its adjuvanticity to antigens for both cellular and humoral immune responses.21 Vaccine adjuvants such as alum which are widely used to boost the immune response of antigens were also reported to trigger the NLRP3 inflammasome activation.9,22 Although the intrinsic relevance between NLRP3 inflammasome activation and antigen presentation is still unclear, we found that A3C14 and A5C14 liposomes, which remarkably activated NLRP3 inflammasome in macrophages, were also capable to promote antigen presentation in BMDCs in the same order of potency. Herein, we propose that NLRP3 inflammasome activation can be utilized as an indicator in the preliminary screening of similar particulate adjuvants to estimate their immune activities and adjuvanticities.
To fulfill the T cell activation, docking with the co-stimulatory molecules on APCs is crucial to strengthen and prolong the cell–cell interaction so as to efficiently and specifically present the antigens to T cells. Deficient expression or recognition of these co-stimulatory molecules would otherwise lead to a compromised or failed T cell activation. In contrast to A3C14 liposomes, which did not affect the expressions of MHC and co-stimulatory molecules on BMDCs, A5C14, and A7C14 liposomes decreased the expressions of CD80, CD86, and CD40 (Figure 6). Accordingly, the T cell activation was not enhanced as much as that of A3C14 liposomes (Figure 5) although comparable antigen delivery efficiencies were obtained (Figure 4). However, these liposomes could still facilitate antigen presentation in comparison to free OVA through both MHC-I and MHC-II pathways, indicating the importance of the amount of antigens that have been internalized and processed by the APCs. On the other hand, A0C16 liposomes moderately increased MHC-II mediated antigen presentation despite their mediocre antigen uptake and negative impact on CD80, CD86, and CD40 expressions. This phenomenon can be explained by the upregulation of MHC-II molecules on BMDCs after exposure to A0C16 liposomes. Surprisingly, OVA-A0C18 liposomes also moderately enhanced T cell activation although the amount of antigen uptake by APCs was similar to that of free OVA and the expressions of MHC and co-stimulatory molecules were partially suppressed. There is a possibility that the interaction of liposomes with intracellular organelles, for example, lysosome destabilization, facilitated the antigen processing in BMDCs as discussed above.21 It should be noted that although some naked arginine-containing cationic liposomes have shielded some activation markers on BMDCs, OVA-loaded liposomes significantly increased T cell activation and proliferation as final outcomes, indicating their predominant contribution to antigen presentation (See the summary of Figures in
In consistent with lysine-based cationic liposomes, the size (80–200 nm) and zeta potential (40–60 mV) of arginine-based cationic liposomes were not the key factors that determine their reactivity with the immune cells; instead, the chemical structures of the hydrophobic tails greatly contributed to the NLRP3 inflammasome activation and the promotion of antigen presentation by APCs for T cell activation. However, in comparison to lysine-based liposomes, A3C14 showed more improved antigen delivery efficacy to BMDCs than L3C14 bearing the same spacer and hydrophobic moiety in terms of T cell activation (approximately ten-fold in IL-2 secretion and two-fold in D.I. of those of L3C14 in both OT-I and OT-II cells,
Unlike conventional vaccine adjuvants such as alum, which is toxic and has a long-lasting cellular exposure,23 A3C14 liposomes are easy to be metabolized owing to the hydrolytic properties of the composing lipids, and they did not show significant cytotoxicity in vitro when the concentration was less than 100 μM. Another advantage of A3C14 liposomes is the high potential to activate CTLs through MHC-I mediated antigen presentation, of which conventional adjuvants often lacks in anticancer treatment.24,25 Therefore, in the future, in vivo investigations are expected to validate the adjuvanticity and to study other immune effects that may be elicited by A3C14 liposomes.
Conclusion
The structure of hydrophobic moieties of cationic lipids is crucial for liposome-immune cell interaction. Slight variation of the alkyl chains could lead to different biological outcomes of the liposomes or the loaded cargos, eg, NLRP3 inflammasome activation and antigen presentation. Among all the tested arginine-based cationic liposomes, A3C14, bearing propyl spacers (C3) between the arginine head group and ditetradecyl tails (C14) exhibited the strongest NLRP3 inflammasome activation and promotion of both MHC class I- and class II-mediated antigen presentations without interfering the MHC and co-stimulatory molecules. The potent cellular uptake and inducement of lysosome rupture are proposed as a common mechanism for arginine-based cationic liposomes in the inflammasome activation and enhanced antigen presentation.
Acknowledgments
This work was partly supported by the EUIJ Waseda program ‘Research Competition for Science and Engineering’ and the JSPS Core-to-Core program, A. Advanced Research Networks for international collaboration, and Waseda University Grant for Special Research Projects (2017B-229, 2018B-214). EL received funding from the ERC InflammAct and the SFB1123, TRR83, TRR57, SPP1923 and GRK1923 provided by the Deutsche Forschungsgemeinschaft.
Disclosure
EL is a co-founder and consultant of IFM Therapeutics. ST reports a patent JP2007073266 issued to Waseda University. The authors report no other conflicts of interest in this work.
References
1. Obata Y, Suzuki D, Takeoka S. Evaluation of cationic assemblies constructed with amino acid based lipids for plasmid DNA delivery. Bioconjug Chem. 2008;19(5):1055–1063. doi:10.1021/bc700416u
2. Obata Y, Saito S, Takeda N, Takeoka S. Plasmid DNA-encapsulating liposomes: effect of a spacer between the cationic head group and hydrophobic moieties of the lipids on gene expression efficiency. Biochim Biophys Acta. 2009;1788(5):1148–1158. doi:10.1016/j.bbamem.2009.02.014
3. Sarker SR, Aoshima Y, Hokama R, Inoue T, Sou K, Takeoka S. Arginine-based cationic liposomes for efficient in vitro plasmid DNA delivery with low cytotoxicity. Int J Nanomedicine. 2013;8:1361–1375. doi:10.2147/IJN.S38903
4. Zhong Z, Zhai Y, Liang S, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi:10.1038/ncomms2608
5. Lonez C, Bessodes M, Scherman D, Vandenbranden M, Escriou V, Ruysschaert JM. Cationic lipid nanocarriers activate Toll-like receptor 2 and NLRP3 inflammasome pathways. Nanomedicine. 2014;10(4):775–782. doi:10.1016/j.nano.2013.12.003
6. Li T, He J, Horvath G, Prochnicki T, Latz E, Takeoka S. Lysine-containing cationic liposomes activate the NLRP3 inflammasome: effect of a spacer between the head group and the hydrophobic moieties of the lipids. Nanomedicine. 2017;4(2):279–288.
7. Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010;22(1):28–33. doi:10.1016/j.coi.2009.12.004
8. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–856. doi:10.1038/ni.1631
9. Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181(1):17–21.
10. Franken L, Kurts C, Burgdorf S. Monitoring the intracellular routing of internalized antigens by immunofluorescence microscopy. Methods Mol Biol. 2013;960:371–377. doi:10.1007/978-1-62703-218-6_27
11. Duewell P, Latz E. Assessment and quantification of crystal-induced lysosomal damage. Methods Mol Biol. 2013;1040:19–27. doi:10.1007/978-1-62703-523-1_3
12. Lapinska U, Saar KL, Yates EV, et al. Gradient-free determination of isoelectric points of proteins on chip. Phys Chem Chem Phys. 2017;19(34):23060–23067. doi:10.1039/c7cp01503h
13. Obata Y, Ciofani G, Raffa V, et al. Evaluation of cationic liposomes composed of an amino acid-based lipid for neuronal transfection. Nanomedicine. 2010;6(1):70–77. doi:10.1016/j.nano.2009.04.005
14. Sarker SR, Hokama R, Takeoka S. Intracellular delivery of universal proteins using a lysine headgroup containing cationic liposomes: deciphering the uptake mechanism. Mol Pharm. 2014;11(1):164–174. doi:10.1021/mp400363z
15. Chen Y, Li X, Boini KM, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 2015;1853(2):396–408. doi:10.1016/j.bbamcr.2014.11.012
16. Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86(4):353–362. doi:10.1038/icb.2008.3
17. den Brok MH, Bull C, Wassink M, et al. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat Commun. 2016;7:13324. doi:10.1038/ncomms13324
18. Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20(1):89–95. doi:10.1016/j.coi.2007.12.002
19. Kindberg GM, Magnusson S, Berg T, Smedsrod B. Receptor-mediated endocytosis of ovalbumin by two carbohydrate-specific receptors in rat liver cells. The intracellular transport of ovalbumin to lysosomes is faster in liver endothelial cells than in parenchymal cells. Biochem J. 1990;270(1):197–203.
20. Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316(5824):612–616. doi:10.1126/science.1137971
21. Welsby I, Detienne S, N’Kuli F, et al. Lysosome-dependent activation of human dendritic cells by the vaccine adjuvant QS-21. Front Immunol. 2016;7:663.
22. Kool M, Petrilli V, De Smedt T, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181(6):3755–3759.
23. Shaw CA, Tomljenovic L. Aluminum in the central nervous system (CNS): toxicity in humans and animals, vaccine adjuvants, and autoimmunity. Immunol Res. 2013;56(2–3):304–316. doi:10.1007/s12026-013-8403-1
24. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33(4):492–503. doi:10.1016/j.immuni.2010.10.002
25. Temizoz B, Kuroda E, Ishii KJ. Vaccine adjuvants as potential cancer immunotherapeutics. Int Immunol. 2016;28(7):329–338. doi:10.1093/intimm/dxw015
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.