Back to Journals » Journal of Pain Research » Volume 9
Nicoboxil/nonivamide cream effectively and safely reduces acute nonspecific low back pain – a randomized, placebo-controlled trial
Authors Blahova Z, Holm JC, Weiser T, Richter E, Trampisch M, Akarachkova E
Received 28 July 2016
Accepted for publication 12 October 2016
Published 14 December 2016 Volume 2016:9 Pages 1221—1230
DOI https://doi.org/10.2147/JPR.S118329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Michael Schatman
Zuzana Blahova,1 Janina Claudia Holm,1 Thomas Weiser,2 Erika Richter,2 Matthias Trampisch,2 Elena Akarachkova3
1Boehringer Ingelheim RCV GmbH & Co KG, Vienna, Austria; 2Boehringer Ingelheim Pharma GmbH & Co KG, Ingelheim am Rhein, Germany; 3I.M. Sechenov First Moscow State Medical University, Moscow, Russian Federation
Background/objective: Low back pain affects many patients and has a high socioeconomic impact. Topical capsaicinoids have been used for decades to treat musculoskeletal pain. This study investigated the effects of the fixed dose combination (FDC) of nonivamide (a capsaicinoid) and nicoboxil (a nicotinic acid ester) cream in the treatment of acute nonspecific low back pain.
Materials and methods: This phase III randomized, double-blind, placebo-controlled, multinational, multi-center trial investigated efficacy, safety, and tolerability of topical nicoboxil 1.08%/nonivamide 0.17% (Finalgon® cream) in treatment of acute nonspecific low back pain with the endpoints: pain intensity (PI) difference between pre-dose baseline and 8 hours after first application and the end of treatment, mobility score, and efficacy score.
Results: Patients (n=138), 21–65 years of age, were treated for up to 4 days with FDC or placebo cream. Mean baseline PI was 6.8 on a 0–10 point numerical rating scale. After 8 hours, pain was more reduced with the FDC than with placebo (adjusted means: 2.824 vs. 0.975 points; p<0.0001). On the last treatment day, mean pain reduction by the FDC was stronger than with placebo (adjusted means: 5.132 vs. 2.174 points; p<0.0001). Mobility on Day 1 was in favor of the FDC when compared to placebo (odds ratio [95% confidence interval {CI}]: 7.200 [3.609, 14.363], p<0.0001). At the end of treatment, patients treated with the FDC rated efficacy significantly higher than placebo (odds ratio [95% CI]: 11.370 [5.342, 24.199], p<0.0001). Both treatments were tolerated well. No serious adverse events were reported.
Conclusion: Nicoboxil/nonivamide cream is an effective and safe treatment for acute nonspecific low back pain, adding a promising treatment option.
Keywords: Finalgon®, pain reduction, mobility score, efficacy
Introduction
Low back pain is a widespread ailment and the leading cause of activity limitation and work absence globally, causing more global disability than any other condition.1,2
Data from the European Health surveys show a wide variation in the prevalence of self-reported low back pain. It is estimated that 12%–30% of adults have low back pain at any time and the lifetime prevalence varies between 60% and 85%.3 One year prevalence is reported to be up to 82%.4 The results of a recent World Health Organization epidemiological study on the risk factors and disability associated with low back pain in adults aged ≥50 years from six developing countries have shown that the prevalence was highest in the Russian Federation (56%).5
Usually, acute low back pain is nonspecific, self-limiting, and with a high percentage of remission within 6 weeks.6 Nevertheless, low back pain accounts for high direct costs associated with clinical diagnostics and therapy as well as high indirect costs due to the inability to work.7
Medical intervention can reduce the risk of chronification and support the patients until the remission of pain, increasing their quality of life.
An analysis of national and international guidelines for the treatment of low back pain identified as common practice the need to inform the patient and the advice on staying as active as possible.8 Recommended medication consists of paracetamol as the first option and nonsteroidal anti-inflammatory drugs as the second option, with varying guideline recommendations on other treatment options such as opioids, muscle relaxants, antidepressants, benzodiazepines, or capsaicin.8
A recent study by Williams et al concluded that paracetamol did not improve low back pain.9
Clinical evidences of the efficacy of the treatments for acute nonspecific low back pain are scarce and inconsistent. In this context, topically applied, locally acting treatments could be an attractive alternative to the systemic medication, reducing the risk of side effects and drug interactions.
Transient receptor potential vanilloid receptor agonists (such as capsaicin or nonivamide) have been shown to reduce musculoskeletal pain.10 Topical nicoboxil 1.08%/nonivamide 0.17% cream (Finalgon® Cream, Boehringer Ingelheim) has been used for decades to treat musculoskeletal pain. This drug product combines a capsaicinoid (nonivamide) with a nicotinic acid ester (nicoboxil), inducing fast hyperemia of the skin and in the musculature below.11,12
A recently published study demonstrated that fixed dose combination (FDC) of nicoboxil 2.5%/nonivamide 0.4% ointment provided more pronounced pain relief compared to placebo and nicoboxil.13 The ointment differs from the cream in the doses of the active ingredients (higher in the ointment) and composition of the vehicle (ointment versus cream base). The vehicle plays a key role in appearance, feel, and successful application of topical drug, and potentially improves adherence, efficacy, tolerability, and safety outcomes.14 Therefore, this study examined the efficacy, tolerability, and safety of nicoboxil 1.08%/nonivamide 0.17% cream in the treatment of adults with acute nonspecific low back pain.
Methods
Study design
This was a prospective, double-blind, placebo-controlled, randomized, multinational, multi-center, parallel group trial to assess the efficacy and safety of multiple doses of 1.08% nicoboxil/0.17% nonivamide cream in the treatment of acute nonspecific low back pain. The trial was conducted in 6 investigational sites (5 of them recruiting) located in the Russian Federation and 3 investigational sites located in Ukraine.
Ethical considerations
Prior to the start of the study, the clinical trial protocol, the patient information, the informed consent form, and other locally required documents were reviewed and approved by the respective ethics committees (EC) and regulatory bodies of the respective countries. The study was designed as a phase III trial and approved as such by The Ethic Council of the Ministry of Healthcare of the Russian Federation.
The constitution of each of the EC met the requirements and definitions of International Conference on Harmonization of Good Clinical Practice (ICH-GCP) and of the respective country. The study was carried out in compliance with the Clinical Trial Protocol, the principles of the Declaration of Helsinki, the ICH-GCP, with applicable regulatory requirements, and Boehringer Ingelheim standard operating procedures. Prior to the enrolment, patients gave their written informed consent according to the local regulatory and legal requirements and the ICH-GCP.
Patient population
After providing informed consent, a total of 138 adult patients of either gender, aged 21–65 years with diagnosis of acute low back pain (ICD-10 code: M54.5) for >2 days and <21 days, with reported low back pain ≥5 on an 11-point (0–10) numerical rating scale (NRS) were screened by the investigators. All the enrolled patients were randomized and treated. Women of childbearing potential had to have a negative urine pregnancy test and had to be using a highly effective method of birth control.
The following exclusion criteria were defined: multilocular pain or panalgesia; history of more than 3 low back pain episodes in the previous 6 months; patients with low back pain due to neurological causes; neurogenic bladder and/or rectum dysfunction; acute low back pain due to vertebral collapse or neoplastic, inflammatory (ankylosing spondylitis), traumatic, or infective origins; any condition, disease, or concomitant treatment that in the judgment of the investigator affected the patient’s ability to participate in the clinical trial or that influenced the test methodology used; negative experience in the past with heat treatment for muscle complaints; history of treatment of back pain with centrally acting analgesics (eg, opioids) and muscle relaxants; surgery due to back pain or rehabilitation due to back pain in the previous 12 months; spinal injection back pain treatment within 6 months prior to enrolment; intake of antidepressant/antipsychotic medication within 4 weeks prior to enrolment; treatment of the recent low back pain period with oral analgesics for more than 4 consecutive days; locally applied medication to the back within 48 hours prior to enrolment; administration of other analgesics within 24 hours prior to enrolment (exception: acetylsalicylic acid up to 100 mg/daily for anti-platelet aggregation therapy); non-pharmacological low back pain treatment (physiotherapy, heat treatment [eg, hot water bottle, and heat patch], or massages) within 12 hours prior to enrolment ; participation in an investigational drug or device trial within 4 weeks prior to enrolment; hypersensitivity to nicoboxil, nonivamide, or paracetamol; known hypersensitivity to any other ingredient; skin lesions (eg, rash, dermatitis, bruising, and laceration) in the back region; drug dependence and/or alcohol abuse; severe hepatocellular insufficiency; patients who were pregnant or breast-feeding.
Treatments
A total of 138 patients were randomly allocated in a 1:1 ratio to treatment with nicoboxil/nonivamide cream (69 patients) or matching placebo cream (69 patients). A validated system using a pseudorandom generator was used to generate the randomization list, so that the resulting treatment was both reproducible and non-predictable. The patients were randomized in blocks to double-blind treatment to ensure that equal numbers of patients were allocated to each treatment group. A random block size of 4 was used. Access to the treatment codes was controlled and documented. Each patient received blinded treatment for the overall treatment period lasting up to 4 days. Each medication kit consisted of 2 tubes, each containing either nicoboxil/nonivamide cream or matching placebo cream. To guarantee the double-blind design of the study, all treatments were indistinguishable for the patient as well as for the investigator.
Except the absence of active ingredients, the placebo cream had the same composition as the nicoboxil/nonivamide cream, including the same amount of fragrance mixture. Thus, both creams were identical concerning look, feel, and smell.
The patients had to administer the cream up to 3 times a day for a maximum of 4 days. The first 2 doses were to be applied at baseline (time point 0 on Day 1) and after 4 hours on Day 1. After the second application on Day 1, the patients could administer study medication as needed but not earlier than 8 hours after the first application for up to a total of 4 days. The intervals between each application of study medication had to be at least 4 hours; a maximum of three applications in a 24-hour period was allowed. A 2-cm long cream line was applied to an area of ~20 cm×20 cm on the patient’s low back where he/she felt the pain to be most pronounced.
With regard to the “warming effect” of the nicoboxil/nonivamide, it was considered that the blindness will be hold in an acceptable way because the individual sensitivity to cutaneous application of capsaicinoids varies considerably from person to person, and that the lower back is generally less sensitive to thermal stimulus. This assumption was confirmed by the heat perception assessments at times relevant for the primary endpoint (0–8 hours), with 4%–48% of the FDC patients perceiving no heat at all, whereas 10%–45% of the patients in the placebo group perceiving at least mild heat.
Paracetamol tablets were allowed as rescue medication (1 to 2 tablets of 500 mg, up to a maximum of four times per day). The first dose of rescue medication was allowed after 8 hours following the first dose of study medication and after having the patient completed the pain intensity (PI) and heat score assessments at the 8-hour time point. The patients were instructed to record the dose, time, and reason of paracetamol intake in the patient diary as often as deemed convenient but at minimum at the end of each treatment day.
Efficacy endpoints
The patients were asked to rate the degree of their PI on an 11-point NRS ranging from 0 = “no pain” to 10 = “worst pain possible” in a diary.15 Time points of assessment were pre-dose (i.e., directly before the first application of study drug on Day 1) and 0.5, 1, 2, 3, 4, 6, and 8 hours after the first cream application on Day 1. The pre-dose PI score was used as baseline PI. Moreover, the patients assessed the average pain intensity (API) of their low back pain on the same rating scale in the diary at the end of each treatment day. Primary endpoint was pain intensity difference (PID) between baseline (pre-dose) and 8 hours after the first application (PID8h).13 Secondary endpoints were PID between baseline (pre-dose) and 4 hours after the first application (PID4h), average PID versus baseline (APID) on the last individual treatment day (APIDLID), and patient assessment of efficacy on the last individual treatment day on a 4-point verbal rating scale (VRS) with the categories: 0 = “poor,” 1 = “fair,” 2 = “good,” 3 = “very good.”15 PID, APID, and APIDLID were calculated and presented in tables and figures with negative values indicating pain reduction.
Other efficacy endpoints included daily APID, time to onset of pain relief (7-point VRS with categories 0 = “within 30 minutes,” 1 = “between 30 minutes and 1 hour,” 2 = “between 1 and 2 hours,” 3 = “between 2 and 4 hours,” 4 = “between 4 and 8 hours,” 5 = “after >8 hours,” 6 = “no effect”), daily mobility score (4-point VRS with categories 0 = “no or poor improvement,” 1 = “fair improvement,” 2 = “good improvement,” 3 = “very good improvement”), patient’s assessment of efficacy on each treatment day, final overall investigator’s assessment of efficacy, heat score (4-point VRS with categories 0 = “no heat perceived,” 1 = “mild heat perceived,” 2 = “moderate heat perceived,” 3 = “strong heat perceived”), number of applications performed during each treatment day, and number of patients taking rescue medication.
Safety endpoints
The evaluation of safety and tolerability was based on the following assessments: physical examination, vital signs, incidence of adverse events, patient’s assessment of tolerability on each treatment day, and final overall investigator’s assessment of tolerability.15 Adverse events (AEs) and serious adverse events (SAEs) were defined according to the International Conference for Harmonization Note for Guidance on Clinical Safety Data Management: Definitions and Standards for Expedited Reporting. Changes in vital signs (blood pressure and pulse rate) and physical examination results were recorded as AEs or SAEs if they were judged clinically relevant by the investigator. Investigators recorded the intensity of AEs as mild (signs or symptoms that were easily tolerated), moderate (enough discomfort to cause interference with usual activity), or severe (incapacitating or causing inability to work or to perform usual activities). Investigators recorded the relatedness as possibly related or as not related to the study treatment.
Tolerability was assessed by the patients at the end of each treatment day, before going to bed, in the patients diary using a 4-point VRS (0 = “poor,” 1 = “fair,” 2 = “good,” 3 = “very good”).
The final overall tolerability was assessed by the investigators at the end of the treatment period using the same 4-point VRS.
Sample size
In a study with a similar design (investigating 2.5% nicoboxil/0.4% nonivamide ointment; Gaubitz et al13), the absolute treatment difference for PID8h between nicoboxil/nonivamide and placebo was ~1.36 on a 0–10 NRS and the common standard deviation was ~1.75. For this study, the sample size per group was planned to be 66. This sample size had 90% power to detect a treatment difference between 1.08% nicoboxil/0.17% nonivamide cream and placebo of about 1 point on a 0–10 NRS, assuming the same common standard deviation and using a 0.05 two-sided significance level.
Statistical analysis
Definition of datasets
The “treated set” (TS) was defined as all randomized patients who used at least 1 dose of study medication. The “full analysis set” (FAS) included all patients of the treated set who provided any post-treatment data for the primary efficacy endpoint. The “per-protocol set” (PPS) was composed of all patients who were part of the FAS and complied with the protocol without any important protocol violations (IPVs). Efficacy endpoints were to be analyzed primarily with the FAS and safety endpoints with the TS.
Primary endpoint
The analysis for the primary endpoint PID8h used a restricted maximum likelihood-based repeated measures approach and included all available longitudinal PI observations at each post-baseline time point up to 8 hours. The statistical model was applied to the analysis of change from baseline in PI at 0.5, 1, 2, 3, 4, 6, 8 hours and included the fixed, categorical effects of country, treatment, time, and treatment-by-time interaction, the continuous covariate of baseline PI, and the residual error term. Within-patient errors were modeled by unstructured covariance. The Kenward–Roger approximation was used to estimate the denominator degrees of freedom. Differences between the treatment group effects with regard to the primary endpoint of PID8h were estimated by reference to the adjusted least square means and the corresponding 95% confidence intervals (CIs). Sensitivity analysis was performed adding the interaction term “gender-by-treatment” to the model. Furthermore, the primary model was evaluated on the PPS.
Secondary and other analyses
PID4h and APID on Days 1 to 4 were analyzed with the corresponding model used for the primary endpoint. APIDLID was analyzed by analysis of covariance, with treatment and country as fixed effects and baseline PI as a continuous covariate. Patient assessment of efficacy on the last individual treatment day was analyzed by a cumulative logit model fit to the underlying ordinal data with treatment and country as fixed effects and baseline PI as a continuous covariate. Odds ratios together with 95% CIs were used to quantify the effect of treatment and to compare the treatment groups. For the endpoint “time to onset of pain relief,” a Kaplan–Meier analysis was performed, and the log-rank test was used to determine the differences between the treatment groups. The time was censored at 9 hours post-dose and displayed as >8 hours. The mobility score on each treatment day was analyzed correspondingly to the assessment of efficacy on the last individual treatment day (LID), but adding the fixed factors day and treatment-by-day interaction. Safety endpoints were analyzed descriptively for the TS.
General considerations
Missing baseline PI values were replaced by the next available post-dose value if post-treatment data were available within the first hour. An assessment of efficacy was assigned the worst category “poor” if it was missing because of discontinuation due to lack of efficacy. No imputations or replacements were made for analyses utilizing a likelihood-based repeated measures model as well as for other missing data.
All statistical analyses were performed using SAS® version 9.4 (SAS Institute, Cary, NC, USA).
Results
Patients
In total, 138 patients were enrolled and randomized; 69 patients each were randomized to nicoboxil/nonivamide and placebo (full analysis set; Figure 1). All randomized patients were treated. Gender was almost balanced in the placebo group (men: 46.4%, female: 53.6%), whereas the nicoboxil/nonivamide group consisted of more women (71.0%) than men (29.0%). Age, weight, height, body mass index, baseline back pain, and duration of back pain were similar in the two treatment groups, with a trend toward more severe pain and longer duration of pain in the nicoboxil/nonivamide group (Table 1). Before patients received trial medication, the mean ± SD (standard deviation) for the intensity of their low back pain was 6.8±1.3 points on the 11-point NRS with mean ± SD duration of 6.8±3.8 days. Medical history was similar in the treatment groups (data not shown). All 138 patients completed the trial. Of the 138 randomized patients, 19 (13.8%) discontinued the trial medication prematurely; all of them were treated with placebo. Of those, 18 refused to continue applying the study medication, and 1 patient gave no specific reason. All patients in the nicoboxil/nonivamide arm completed the trial as planned.
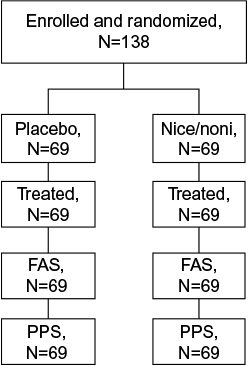 | Figure 1 Patient disposition Abbreviations: FAS, full analysis set; Nico, nicoboxil; Noni, nonivamide; PPS, per protocol set. |
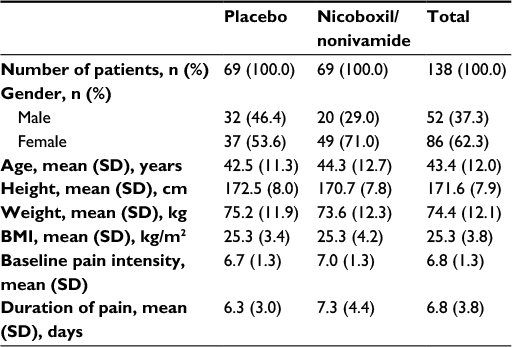 | Table 1 Patient demographics and baseline characteristics Abbreviations: BMI, body mass index; SD, standard deviation. |
Compliance
All patients complied with the treatment instructions.
Efficacy
Primary endpoint: PID8h
After 8 hours, pain was more reduced with the FDC (adjusted mean [95% CI]: 2.824 [2.384, 3.264] points) than with placebo (adjusted mean [95% CI]: 0.975 [0.546, 1.404] points), p<0.0001 (Figure 2). Thus, compared to placebo, the FDC reduced the PI 8 hours after the onset of treatment by additional 1.849 points.
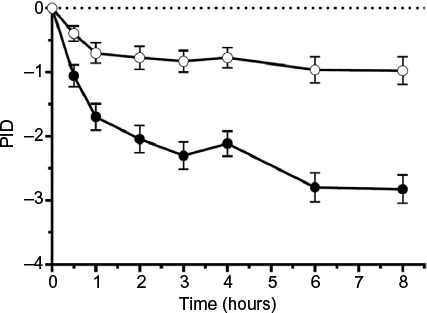 | Figure 2 Adjusted mean (±SEM) pain intensity differences up to 8 hours after the first application of placebo cream, or the combination of nicoboxil and nonivamide cream. Notes: Closed symbols: nicoboxil/nonivamide, open symbols: placebo. Treatment was applied at 0 and 4 hours. The statistical model included baseline pain intensity, country, time (0.5, 1, 2, 3, 4, 6, and 8 hours), treatment and treatment-by-time interaction. Number of patients was 69 for placebo and 69 for nicoboxil/nonivamide. Abbreviations: PID, pain intensity difference; SEM, standard error of the mean. |
Sensitivity analysis of the primary endpoint including gender-by-treatment interaction confirmed the results of the primary analysis (data not shown).
Secondary and other endpoints
PI was decreased more in the nicoboxil/nonivamide group 4 hours after the onset of treatment as observed by PID4h (adjusted mean [95% CI]=2.113 [1.723, 2.503]) than in the placebo group (0.772 [0.460, 1.084]; p<0.0001). The combination reduced PI by 1.341 points more compared to placebo at this early time point.
On the last individual treatment day, the FDC provided more pronounced average pain reduction compared to placebo: the adjusted mean (95% CI) was 5.132 (4.581, 5.683) points with the FDC versus 2.174 (1.635, 2.712) points with placebo, p<0.0001, as indicated by the higher decrease in APIDLID (Figure 3A).
 | Figure 3 (A) Adjusted mean (±SEM) average pain intensity difference on the last individual treatment day. Closed bar: nicoboxil/nonivamide (n=69), open bar: placebo (n=68). (B) Adjusted mean (±SEM) average pain intensity difference from Day 1 to Day 4. Closed symbols: nicoboxil/nonivamide, open symbols: placebo. Abbreviations: APIDLID, average pain intensity difference versus baseline on the last individual treatment day; APID, average pain intensity difference; BL, baseline; SEM, standard error of the mean. |
Average PI versus baseline continuously decreased from Days 1 to 4 in both treatment groups as indicated by the decrease in APID over time (Figure 3B). On all treatment days, the adjusted treatment differences for APID were in favor of nicoboxil/nonivamide compared to placebo (p<0.0001). Table 2 summarizes the results for the primary, secondary, and other pain-related endpoints per treatment group.
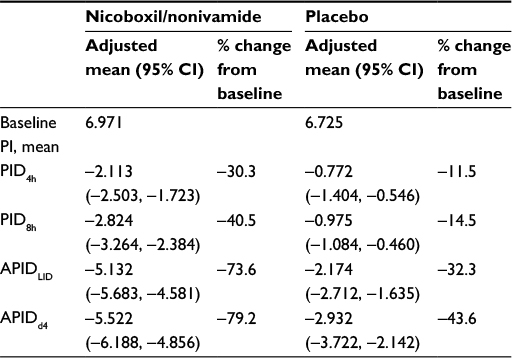 | Table 2 Comparison of various pain-related endpoints; % change from baseline Abbreviations: APIDd4, average pain intensity difference versus baseline on Day 4; APIDLID, average pain intensity difference versus baseline on the last individual treatment day; CI, confidence interval; PI, pain intensity; PID4h, pain intensity difference between baseline and 4 hours after the first application; PID8h, pain intensity difference between baseline and 8 hours after the first application. |
Time to onset of pain relief was earlier with the combination of nicoboxil and nonivamide than with placebo: 25 (36.2%) patients reported onset of pain relief within the first 30 minutes after the onset of treatment, compared to only 8 (11.6%) patients in the placebo group. The median category of time to onset of pain relief was lower for the FDC group (between 1 and 2 hours) compared to the placebo group, in which 48 (69.6%) patients indicated “no effect”( Table 3).
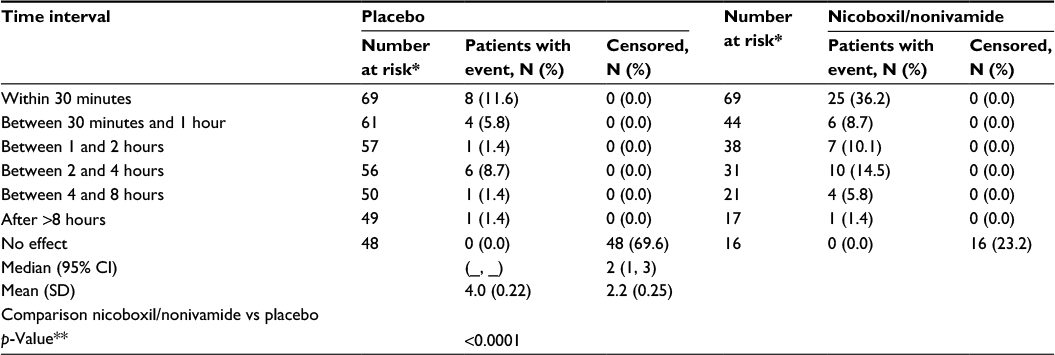 | Table 3 Time to onset of pain relief after the first application Notes: *Number of patients entering the respective time interval; **log-rank test. Abbreviations: CI, confidence interval; SD, standard deviation. |
In the nicoboxil/nonivamide group, the patients assessed the improvement of their mobility as better compared to placebo (p<0.0001) on all 4 treatment days. The odds ratios ranged from ~7 to 12 over Days 1–4 (Table 4). On the first treatment day only 12 (17.4%) of nicoboxil/nonivamide patients reported “no” or “poor” improvement of low back mobility, compared to 45 (65.2%) for placebo (Figure 4A).
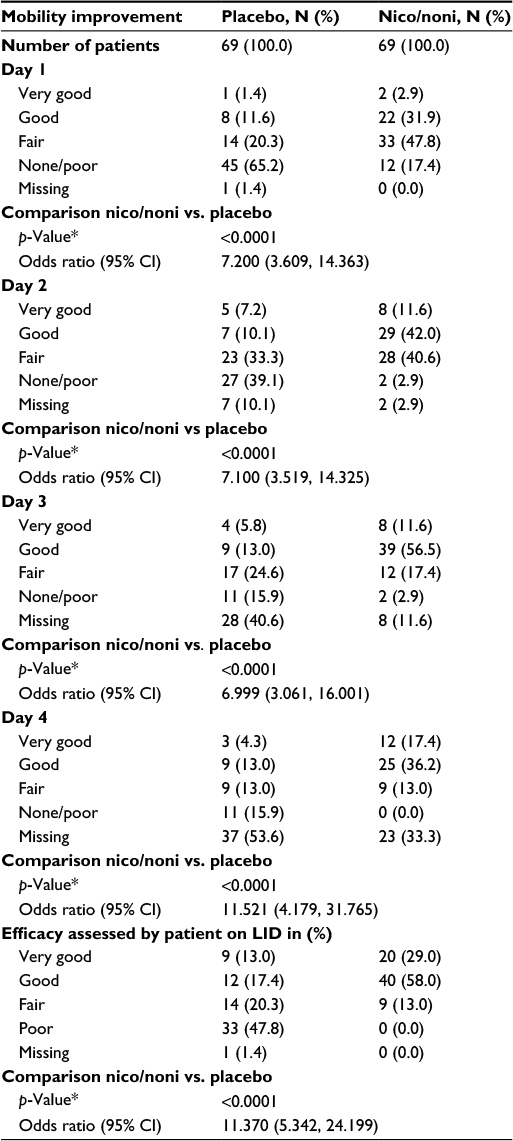 | Table 4 Patient assessment of mobility improvement on each treatment day and efficacy assessment on LID Note: *Logistic regression. Abbreviations: CI, confidence interval; LID, last individual treatment day; Nico, nicoboxil; Noni, nonivamide. |
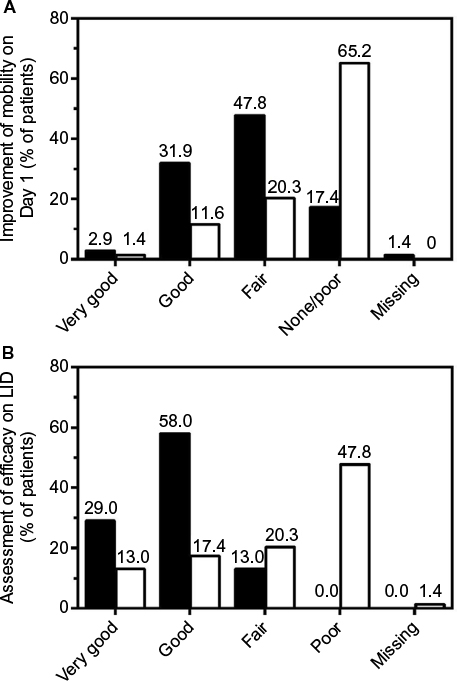 | Figure 4 (A) Improvement of mobility on Day 1 as reported by the patients. (B) Patient assessment of efficacy on the LID. Closed bars: nicoboxil/nonivamide, open bars: placebo. Abbreviation: LID, last individual treatment day. |
The patients in the FDC group assessed the efficacy on their last individual treatment day as better than the patients in the placebo group with an odds ratio (95% CI) of 11.370 (5.342, 24.199), p<0.0001. Efficacy was rated as “very good” or “good” by 60 (87.0%) patients treated with the FDC, compared to 21 (30.4%) patients treated with placebo (Figure 4B). The investigator assessment of efficacy confirmed this superiority of efficacy (data not shown). In the FDC group, 22 (31.9%) of patients took rescue medication, compared to 32 (46.4%) in the placebo arm. No rescue medication was taken within the first 8 hours after baseline.
Safety
A total of 9 (6.5%) patients experienced AEs during the study, 6 (8.7%) treated with the FDC and 3 (4.3%) treated with placebo. No serious AE was reported. AEs reported by 3 patients in the FDC group were assessed as drug-related, 1 case each of dysgeusia, application site pruritus, and urticaria (Table 5).
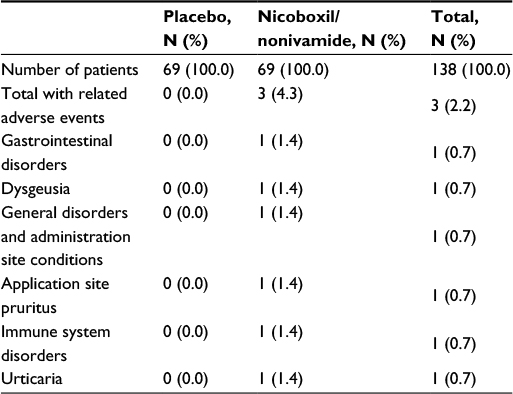 | Table 5 Number (%) of patients with drug-related adverse events |
At the end of treatment, the patients in the placebo group rated the tolerability of the study medication slightly better when compared to the group treated with the FDC (odds ratio [95% CI]: 0.465 [0.236, 0.916], p=0.0269). However, in both treatment arms, the majority of patients assessed the tolerability of study medication as “very good” or “good” (85.5% vs. 89.8% for the FDC and placebo, respectively; data not shown).
Discussion and conclusion
The burden put on society by low back pain and the need for clinically proven treatment interventions is high. The “Global Burden of Disease Study 2013” ranked low back pain to be the top cause of years lived with disability worldwide.1,2 Patients suffering from acute pain expect fast onset of action and pronounced reduction of PI from analgesic treatment.16,17
The results of a double-blind, randomized, parallel-group, placebo-controlled trial investigating the efficacy, tolerability, and safety of a topical FDC consisting of nicoboxil 1.08% and nonivamide 0.17% cream in the treatment of acute nonspecific low back pain are presented in this paper. A pronounced reduction of PI was achieved in this trial in the active treatment arm: PI compared to baseline was reduced fast by the nicoboxil/nonivamide cream combination, and increased further over the entire treatment period of up to 4 days. Comparison of the pain-related endpoints with the parameters defined by Moore et al18 showed that the FDC provided “moderate clinical benefit” (≥30% PI reduction) already at 4 hours and 8 hours post-treatment , and “significant clinical benefit” (≥50% PI reduction) at the last individual day, as well as after 4 days of treatment (Table 2).
According to the treatment guidelines for nonspecific low back pain, it is desirable to get patients back to their normal life as soon as possible; this includes work and everyday physical activity.8 Patients in this trial rated their improvement of low back pain mobility on each treatment day higher for the FDC, compared to placebo. Thus, it can be suggested that analgesic efficacy and improvement of low back mobility jointly contribute to well-being and functional recovery of the patients. This could explain the high percentage of patients assessing the efficacy of the combination as “very good” or “good.” Similar effects were observed in a randomized, placebo-, and active-controlled clinical trial investigating another topical FDC (2.5% nicoboxil/0.4% nonivamide ointment formulation) in the treatment of acute low back pain.13 Both nicoboxil/nonivamide ointment and cream combinations have been in clinical use for decades and their efficacy in the indication of acute nonspecific low back pain has now been confirmed in two state-of-art clinical trials.
Limitations of the presented study include no evaluation of durability of endpoints beyond several days for a more prolonged use of the nicoboxil/nonivamide cream.
What other treatment options for acute low back pain exist? In a review of national and international clinical low back pain guidelines, one of the common recommendations is that patients should stay as active as possible and gradually increase their activity levels (including returning to work even in the presence of back pain).8 Regarding pharmacological treatment options, the guidelines recommend paracetamol as first choice analgesic and nonsteroidal anti-inflammatory drugs (NSAIDs) in case paracetamol would not work. All of these drugs require systemic application.
In the meantime, some interesting relevant new studies and reviews have been published. Remarkably, a large clinical study on acute low back pain run in Australia did report no treatment effect for paracetamol in comparison to placebo.9 A meta-analysis of treatments for nonspecific low back pain found only small effects of NSAIDs (using their categories, nicoboxil/nonivamide would have been rated having “large treatment effects”).19 This was confirmed by another meta-analysis investigating the effects of over-the-counter medications for acute low back pain.20 The centrally acting muscle-relaxant cyclobenzaprine has been shown to be effective; however, up to 68% of the patients reported drowsiness as adverse event.21 NSAIDs (lornoxicam and diclofenac at prescription only doses) provided some acute low back pain relief.22 Besides the fact that systemic treatments have not provided a pronounced pain relief, one should bear in mind that agents such as NSAIDs or centrally acting muscle analgesics are prone to cause systemic adverse effects, which might limit their use. Such adverse effects are less likely to occur with topical treatments like the one investigated in this study.
Conclusion
In summary, this study demonstrates that nicoboxil/nonivamide cream is an effective, tolerable, and safe treatment option for acute nonspecific low back pain. Literature comparison of the data in acute low back pain suggests that the clinical efficacy of this topical treatment could be comparable or superior with that of systemic over-the-counter or prescription analgesics and muscle relaxants; however, this has to be further investigated in well-designed, controlled, parallel-group studies.18,19 Thus, the topical combination of nicoboxil/nonivamide adds a promising option for the treatment of acute nonspecific low back pain.
Acknowledgments
The skilled editorial assistance by Jessica Krysa is highly acknowledged.
This study was sponsored by Boehringer Ingelheim.
This research was presented as a poster at World Congress on Osteoporosis, Osteoarthritis and Musculoskeletal Diseases, Malaga, Spain, on April 14–17, 2016, and the abstract has been published in World Congress on Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (WCO-IOF-ESCEO 2016): Poster Abstracts, Osteoporosis International, April 2016, Volume 27, Supplement 1, pp. 79–548.
Disclosure
ZB, JCH, TW, ER, and MT are employees at Boehringer Ingelheim. EA was principal investigator in this study. She received honoraria for consultation from Boehringer Ingelheim. The authors report no other conflicts of interest in this work.
References
Hoy D, March L, Brooks P, et al. The global burden of low back pain: estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. 2014;73(6):968–974. | ||
Vos T. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386(9995):743–800. | ||
Eumusc.net [homepage on the Internet]. European musculoskeletal conditions surveillance and information network; musculoskeletal health status in Europe: report v 5.0. 2012. Available from: http://www.eumusc.net/myUploadData/files/Musculoskeletal%20Health%20in%20Europe%20Report%20v5.pdf. Accessed January 20, 2014. | ||
Hoy D, Brooks P, Blyth F, Buchbinder R. The epidemiology of low back pain. Best Pract Res Clin Rheumatol. 2010;24(6):769–781. | ||
Stewart Williams J, Ng N, Peltzer K, et al. Risk factors and disability associated with low back pain in older adults in low- and middle-income countries. results from the WHO study on global AGEing and adult health (SAGE). PLoS One. 2015;10(6):e0127880. | ||
Casser HR. Definition, Epidemiologie und sozioökonomische Bedeutung des nichtspezifischen Kreuzschmerz [Definition, epidemiologie and socialeconomic importance of nonspecific back pain]. Orthopaede. 2012;41:311–312.German. | ||
Leitlinien.de/nvl [homepage on the Internet]. Nationale VersorgungsLeitlinie (NVL) Kreuzschmerz [National disease management guideline low back pain]. Short version (english), 1st edition, version 5, January 2011, last amended: October 2015. Available from: http://www.leitlinien.de/mdb/downloads/nvl/kreuzschmerz/kreuzschmerz-1aufl-vers5-short.pdf. Accessed January 21, 2014.German. | ||
Koes BW, van Tulder M, Lin CW, Macedo LG, McAuley J, Maher C. An updated overview of clinical guidelines for the management of non-specific low back pain in primary care. Eur Spine J. 2010;19(12):2075–2094. | ||
Williams CM., Maher CG, Latimer J, McLachlan AJ, Hancock MJ, Day RO, Lin CW. Efficacy of paracetamol for acute low-back pain: a double-blind, randomised controlled trial. Lancet. 2014;384(9954):1586–1596. | ||
Chrubasik S, Weiser T, Beime B. Effectiveness and safety of topical capsaicin cream in the treatment of chronic soft tissue pain. Phytother Res. 2010;24(12):1877–1885. | ||
Stücker M, Reuther T, Hoffmann K, Aicher B, Altmeyer P. The effect of the base on the kinetics of action of the capsaicinoid nonivamide: evaluation with a hyperemic test. Skin Pharmacol Appl Skin Physiol. 1999;12(5):289–298. | ||
Warnecke JM, Wendt T, Winkler S, Schak M, Schiffer T, Kohl-Bareis M. Evaluation of changes in the haemoglobin of skin and muscle tissue of the calf, as induced by topical application of a nonivamide/nicoboxil cream. Can J Physiol Pharmacol. 2014;92(2):149–154. | ||
Gaubitz M, Schiffer T, Holm C, Richter E, Pisternick-Ruf W, Weiser T. Efficacy and safety of nicoboxil/nonivamide ointment for the treatment of acute pain in the low back - A randomized, controlled trial. Eur J Pain. 2015;20(2):263–273. | ||
Barry B. Transdermal drug delivery. In: Aulton ME, Editor. Pharmaceutics: The Science of Dosage Form Design. Churchill Livingston; 2002:499–533. | ||
ema.europa.eu [homepage on the Internet]. Guideline on the clinical development of medicinal products intended for the treatment of pain 2nd Draft from 17 December 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/12/WC500199242.pdf). Accessed September 12, 2016. | ||
Moore RA, Derry S, Straube S, Ireson-Paine J, Wiffen PJ. Faster, higher, stronger? Evidence for formulation and efficacy for ibuprofen in acute pain. Pain. 2014;155(1):14–21. | ||
Pageler L, Diener HC, Pfaffenrath V, Peil H, Aicher B. Clinical relevance of efficacy endpoints in OTC headache trials. Headache. 2009;49(5):646–654. | ||
Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta-analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain. 2011;152(5):982–989. | ||
Machado LA, Kamper SJ, Herbert RD, Maher CG, McAuley JH. Analgesic effects of treatments for non-specific low back pain: a meta-analysis of placebo-controlled randomized trials. Rheumatology (Oxford). 2009;48(10):520–527. | ||
Abdel Shaheed C, Maher CG, Williams KA, McLachlan AJ. Interventions available over the counter and advice for acute low back pain: systematic review and meta-analysis. J Pain. 2014;15(1):2–15. | ||
Weil AJ, Ruoff GE, Nalamachu S, Altman CA, Xie F, Taylor DR. Efficacy and tolerability of cyclobenzaprine extended release for acute muscle spasm: a pooled analysis. Postgrad Med. 2010;122(4):158–169. | ||
Herrmann WA, Geertsen MS. Efficacy and safety of lornoxicam compared with placebo and diclofenac in acute sciatica/lumbo-sciatica: an analysis from a randomised, double-blind, multicentre, parallel-group study. Int J Clin Pract. 2009;63(11):1613–1621. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.