Back to Journals » Drug Design, Development and Therapy » Volume 9
Neuroprotective therapies for glaucoma
Authors Song W ![]() , Huang P, Zhang C
, Huang P, Zhang C
Received 8 January 2015
Accepted for publication 5 February 2015
Published 11 March 2015 Volume 2015:9 Pages 1469—1479
DOI https://doi.org/10.2147/DDDT.S80594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Wei Duan
Wei Song, Ping Huang, Chun Zhang
Department of Ophthalmology, Peking University Third Hospital, Beijing, People’s Republic of China
Abstract: Glaucoma is the second leading cause for blindness worldwide. It is mainly caused by glaucomatous optic neuropathy (GON) characterized by retinal ganglion cell loss, which leads to visual field defect and blindness. Up to now, the main purpose of antiglaucomatous therapies has been to lower intraocular pressure (IOP) through surgeries and medications. However, it has been found that progressive GON is still present in some patients with effective IOP decrease. Therefore, risk factors other than IOP elevation, like neurotrophin deprivation and excitotoxicity, contribute to progressive GON. Novel approaches of neuroprotection may be more effective for preserving the function of the optic nerve.
Keywords: glaucoma, glaucomatous optic neuropathy, retinal ganglion cells, neuroprotection
Introduction
Glaucoma is the second leading cause of blindness around the world, and is characterized by retinal ganglion cell (RGC) death, progressive glaucomatous optic neuropathy (GON), and glaucomatous visual field defect. Quigley and Broman estimated that the number of glaucoma patients worldwide would reach 79.6 million by 2020.1 Therefore, precautionary measures and therapeutic treatments of glaucoma are challenges for public health globally.
The pathological features of GON consist of cupping of optic nerve head, enlargement of vertical cup–disc ratio of >0.6, loss of neuroretinal rim, and notching with nerve fiber layer defect. These manifestations are mainly caused by RGC loss within the retina. Nowadays, it is commonly believed that pathological elevation of intraocular pressure (IOP) is a major risk factor for occurrence and development of GON (Figure 1). Therefore, the main purpose of current antiglaucomatous therapies, including topical application of ocular hypotensives, anterior chamber paracentesis, and antiglaucomatous surgeries is to lower the IOP.2
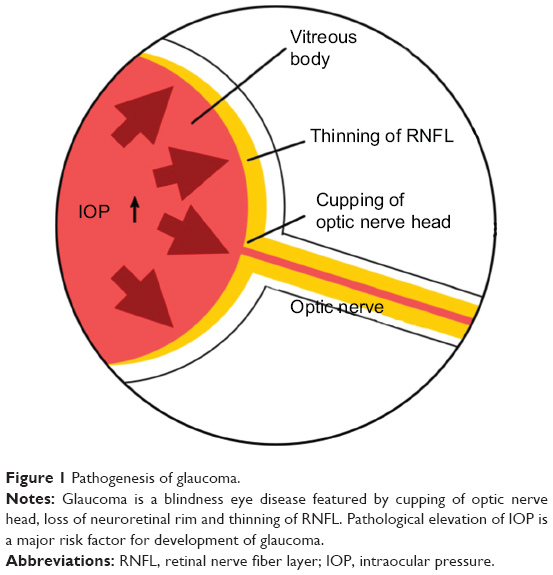 | Figure 1 Pathogenesis of glaucoma. |
However, the percentage of primary angle-closure glaucoma among glaucoma patients is not relatively low based on the epidemiological surveys completed in the past several years. A population-based glaucoma survey that was conducted in seven regions throughout Japan found that only 8% of the subjects were primary angle-closure glaucoma, while 49% were normal tension glaucoma (NTG), the IOP of which was in the normal range (10 mmHg <IOP <21 mmHg).3 Another population-based glaucoma surveys in Japan named the Tajimi study found that only 12% of the subjects were primary open-angle glaucoma (POAG), while 78% were NTG.4 The Singapore Indian Eye Study (SINDI) study performed in Singapore demonstrated that nearly half of the glaucoma patients were NTG.5 In the People’s Republic of China, one epidemiological study in a rural county in Handan (named the Handan Study) also reported that among POAG patients, 90% were NTG.6 In summary, we could easily conclude that more than half of glaucoma patients have IOPs in the normal range. Besides IOP, there may be unknown risk factors contributing to progressive GON and visual field defects. Thus, neuroprotective therapies as complements to conventional IOP-lowering treatments increase the chances of delaying RGC loss and structural/functional damage of the optic nerve in these patients.
Supplementation of neurotrophic factors
Neurotrophic factors refer to growth factors for neurons. They are essential for neuronal development, differentiation, and regeneration within the nervous system. Levi-Montalcini discovered the first neurotrophic factor, nerve-growth factor (NGF), in the 1950s.7 NGF belongs to the neurotrophin (NT) family of growth factors, which include NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5).8 In addition, novel neurotrophic factors, like GDNF (glial cell line-derived neurotrophic factor) and CNTF (ciliary neurotrophic factor), were discovered in the past few decades.9,10
BDNF is synthesized in the soma of neurons and transported to the postsynaptic sites by vesicles. At the postsynaptic side of synapses, BDNF molecules are secreted into the synaptic cleft, diffuse to the presynaptic side, and bind to the surface tropomyosin receptor kinase B (TrkB). The BDNF-TrkB complexes are next internalized through endocytosis, retrograde transported to the neuronal cell body, and regulate neuronal activities there (Figure 2).11 In 2000, Pease et al12 and Quigley et al13 reported for the first time that in experimental rats and monkeys of glaucoma, retrograde transport of BDNF from the superior colliculus to the optic nerve head was substantially obstructed by acute or chronic IOP elevation. Based on these experimental observations, they proposed the neurotrophic factor deprivation hypothesis for the pathogenesis of GON (Figure 2).12,13 Moreover, Johnson et al found that by 7 days of IOP elevation in an experimental rat model of glaucoma, the endogenous expression of BDNF and NT-4/5 in the optic nerve was completely diminished, in line with the RGC apoptosis.14 Sposato et al demonstrated that in experimental glaucomatous eyes of Sprague-Dawley (SD) rats, the expression levels of NGF and corresponding tropomyosin kinase receptor A (TrkA) within optic nerve were significantly decreased.15 In summary, shortage of neurotrophic factors in the optic nerve contributes to the occurrence and progression of GON caused by high IOP. Enhancement of neurotrophic support may delay RGC loss in glaucoma.
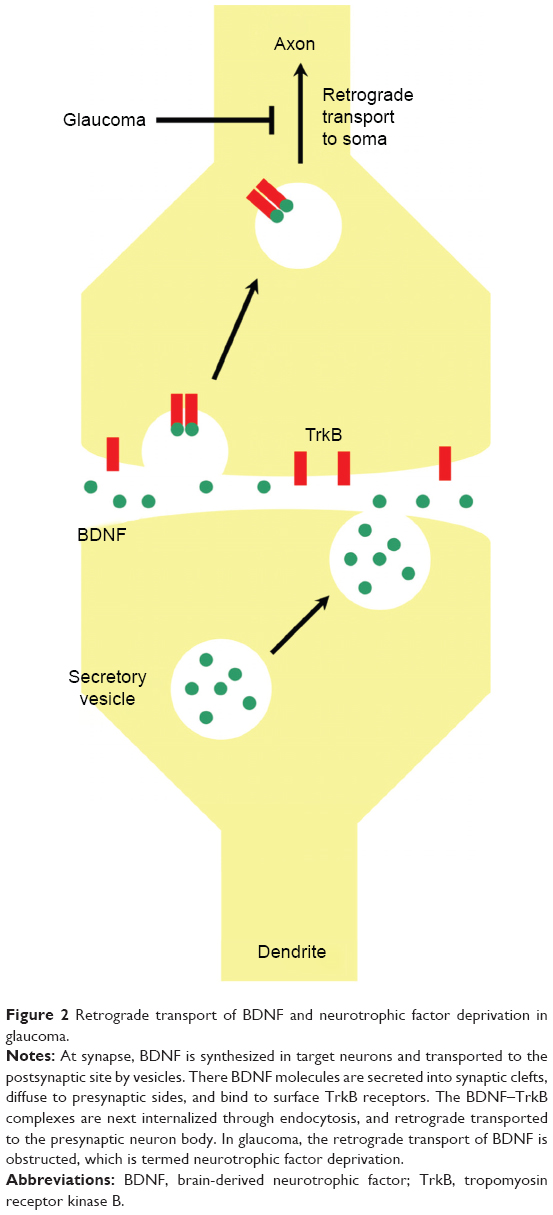 | Figure 2 Retrograde transport of BDNF and neurotrophic factor deprivation in glaucoma. |
Intravitreal injections of recombinant human BDNF in Wistar rats having chronic IOP elevation significantly enhanced RGC survival within the retina.16 However, intravitreal injections of BDNF simply are not able to deliver the protein continuously, and repeated administrations with time are required. Therefore, other novel approaches by using gene therapy or stem cells were developed later to overcome this disadvantage. Martin et al developed a modified adeno-associated viral (AAV) vector to overexpress BDNF in RGC in rats, followed by laser application to the trabecular meshwork to increase the IOP 2 weeks later. Results indicated that such pre-treatment significantly suppressed RGC axon loss in the optic nerve.17 Harper et al transplanted BDNF-MSCs (mesenchymal stem cells) intravitreally in a rat model of chronic glaucoma. Six weeks after receiving the transplantation, effective RGC preservation was obtained by counting the number of RGCs in optic nerve cross-sections. Further functional tests by computerized pupillometry and electroretinography showed significantly better function in BDNF-MSC-treated eyes.18 Since the effects of BDNF to enhance neuronal survival are mediated by its cognate TrkB receptor, Bai et al used a selective agonistic TrkB monoclonal antibody 1D7 instead of BDNF intravitreally in the experimental Wistar rats of ocular hypertension (acute/chronic), and showed delayed RGC death and maintenance of retinal nerve fiber layer.19 Brimonidine is a widely used ocular hypotensive by activation of α2 adrenergic receptor and decrease in aqueous humor production. Recent research found that intravitreal instead of topical administration of brimonidine significantly promoted RGC survival through upregulating endogenous expression of BDNF in RGCs.20 Although BDNF significantly reduced RGC loss and maintained optic nerve function in experimental glaucoma, long-term treatments with BDNF might limit its own neuroprotective actions, because it downregulated TrkB receptor with excessive application.21
In addition to BDNF, other neurotrophic factors were also applied in experimental glaucoma to determine their neuroprotective effects on the RGCs. NGF was the first discovered member of the NT family. Lambiase et al tested its neuroprotective effects by topical application in both experimental glaucoma and patients.22 In SD rats with chronic ocular hypertension, NGF has potential neuroprotective effects as indicated by reduced RGC loss. In three patients with effective IOP control, NGF eye drops could preserve the optic nerve function as indicated by improved visual field test results.22 Ji et al showed that intravitreal administration of CNTF was able to preserve RGCs from apoptosis for up to 4 weeks in SD rats that had ocular hypertension. Such neuroprotective effects were mediated by intracellular Janus kinase/signal transducer and activator of transcription 3 (JAK-STAT3) signaling pathway,23 which is required for the embryonic development of retina.24 By using modified AAV vector again, Pease et al overexpressed CNTF in the Wistar rats’ retinas with chronic ocular hypertension to examine its protective effects on RGCs. Exactly 15% less RGC axon loss was observed from the eyes receiving intravitreal injection of CNTF-AAV vectors.25 To evaluate the efficacy of another trophic factor GDNF, Jiang et al used GDNF-loaded biodegradable microspheres by intravitreal injection. The GDNF microsphere treatment displayed effective neuroprotective effects on RGCs in Brown Norway rats with chronic ocular hypertension, as indicated by increased number of RGC axons, thickness of the retinal inner plexiform layer (IPL), and level of glial fibrillary acidic protein expression.26 The advantages of microspheres used in this study included continuous delivery of the trophic factor and economically feasibility, because of the considerable expense of neurotrophic factors.
Blockage of excitotoxicity
Excitotoxicity is the pathological process in neurons resulting from overactivation of N-methyl-d-aspartic acid receptors (NMDARs) by the excitatory neurotransmitter glutamate. The extensive stimulation of NMDARs allows high level of Ca2+ to enter the cell, activating enzymes including phospholipases, endonucleases, and proteases such as calpain. These enzymes may lead to the damage of plasma membrane, cytoskeleton, and DNA, and other cell components (Figure 3).27 Dreyer et al showed a twofold elevation in the glutamate level in the vitreous body of glaucoma patients, indicating the possibility of an excitotoxic mechanism resulting in RGC death.28 Accordingly, NMDAR blockers and calcium channel blockers (CCBs) can be used to inhibit excitotoxicity in RGCs and to delay GON progression and visual field loss in glaucoma patients.
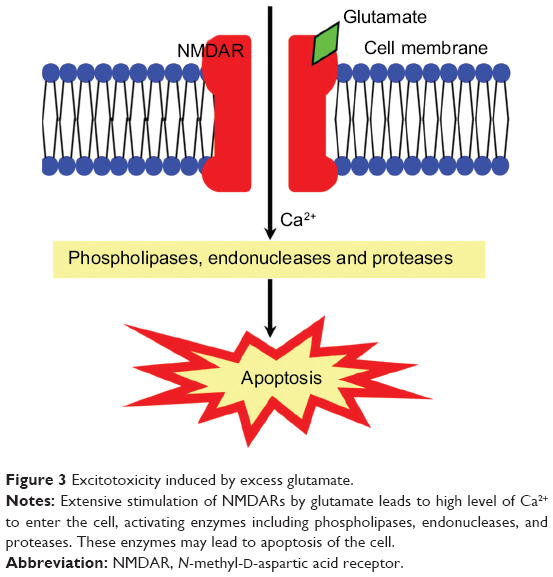 | Figure 3 Excitotoxicity induced by excess glutamate. |
MK801 is a non-competitive NMDAR antagonist. Intraperitoneal administration of MK801 in Wistar rats with experimentally introduced glaucoma significantly lowered the death rate of RGCs in vivo (from 14% to 3%).29 Systemic treatment with MK801 by intraperitoneal injections also reduced RGC loss in SD rats with acute glaucoma.30 Despite the effective neuroprotection of MK801 in experimental glaucoma, it is not suitable for long-term application due to its neurotoxic effects. Another NMDAR inhibitor, memantine, is commonly used to treat Alzheimer’s disease. In chronic ocular hypertensive animal models, long-term administration of memantine intraperitoneally for 3 months reduced RGC loss from 37% at the control level to 12% in the experimental group.31 Hare et al confirmed its efficacy in RGC structural and functional protection in macaque monkeys with chronic ocular hypertension.32,33 Although the outcomes in the experimental glaucoma were exciting, two registered randomized controlled trials (RCTs) (NCT00141882 and NCT00168350) in chronic glaucoma patients conducted by Allergan showed no potential beneficial effects.34 Therefore, well-designed clinical trials are required to further test its utility in glaucoma patients.
CCBs have been widely used in treating cardiac diseases such as hypertension and heart failure. In a rat model of acute IOP elevation by intraperitoneal injection of flunarizine, an L- and T-type Ca2+ channel blocker, significant protective effects were obtained, as revealed by less loss of RGCs and thinning of the inner retinal layer.35 Another study performed in rabbits with elevated IOP showed neuroprotective effects of flunarizine by topical application.36 Nilvadipine is a CCB used to treat hypertension and chronic major cerebral artery occlusion. The neuroprotective effects of nilvadipine on RGCs by intramuscular injection were also observed in rats with acute ocular hypertension.37 Pre-treatment with lomerizine, another novel, specific blocker for L- and T-type Ca2+ channels, also exhibited protective effects against glutamate-induced excitotoxicity in rat retinal cell cultures.38 Koseki et al reported that in a placebo-controlled study conducted in POAG patients, oral nilvadipine for 3 years slightly slowed visual field progression and maintained the optic disc rim.39 Moreover, one kind of β1-adrenoceptor antagonist, betaxolol, works as an L-type Ca2+ blocker in RGCs. Topical application of betaxolol in SD rats with acute ocular hypertension preserved the integrality of IPL and inner nuclear layer of retinas.40 Collignon-Brach reported that patients with ocular hypertension or chronic open-angle glaucoma received more benefits for their visual field after using betaxolol than timolol for 3 years, although timolol was more effective in lowering IOP.41
Inhibition of apoptosis
Apoptosis is the process of programmed cell death that occurs in multicellular organisms. The extrinsic pathway is mediated by Fas receptor or tumor necrosis factor receptor (TNFR), while the intrinsic pathway is initiated by efflux of cytochrome c from mitochondria. The initiation of these two apoptotic pathways leads to activation of downstream effectors caspases 3, 6, and 7, which proteolytically degrade a host of intracellular proteins to carry out the cell death program (Figure 4). The anti-apoptotic Bcl-2 proteins, including Bcl-2 and Bcl-xl, act by directly inhibiting mitochondrial apoptosis-induced channel formation.42 By using the terminal deoxynucleotidyl transferase dUTP nick end labeling method, Quigley et al confirmed that GON was partially caused by apoptosis of RGCs in experimental glaucoma;43 Kerrigan et al confirmed that this was also the case in glaucoma patients.44 Based on these observations, anti-apoptotic agents may preserve the optic nerve structurally and functionally in glaucoma.
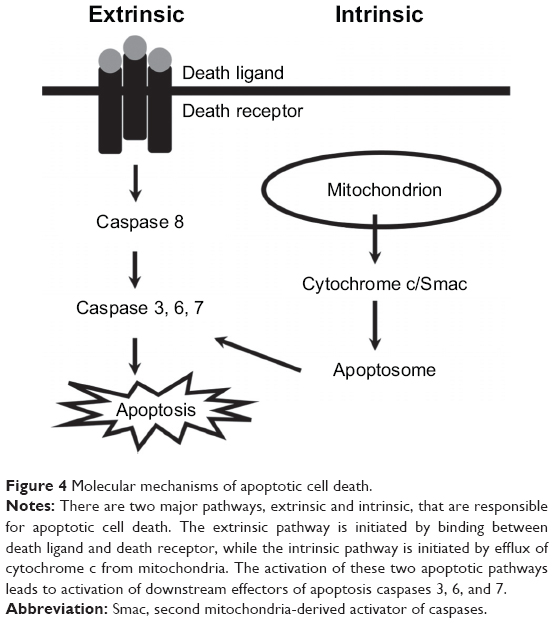 | Figure 4 Molecular mechanisms of apoptotic cell death. |
Agents to inhibit initiation of the apoptotic pathway have been developed. Since the extrinsic apoptotic pathway is mediated by activation of TNFR, the tumor necrosis factor alpha (TNF-α) inhibitor etanercept was applied intraperitoneally in a rat model of glaucoma. The axonal degeneration and RGC loss was effectively suppressed by etanercept, a result obtained by comparing the experimental treatment group to the control group without any management.45 Oral administration of calcineurin inhibitor FK506 for rats with acute increased IOP significantly decreased the release of cytochrome c from mitochondria and RGC apoptosis.46 McKinnon et al developed the baculoviral IAP repeat-containing protein-4 (BIRC4)-modified AAV vector to treat Brown Norway rats with chronic ocular hypertension. BIRC4 is a potent caspase 3 inhibitor. Intravitreal injection of the modified vectors significantly promoted optic nerve axon survival.47
Alternatively, medications that enhance the endogenous anti-apoptotic pathways also have neuroprotective effects on optic nerves. Brimonidine, an ocular hypotensive that activates α2 adrenergic receptor to decrease aqueous humor production, was able to elevate intracellular levels of anti-apoptotic proteins Bcl-2 and Bcl-xl in RGCs.48 Minocycline, a second-generation tetracycline, has been reported to enhance RGC survival in experimental glaucoma. One of the involved mechanisms was to increase gene Bcl-2 expression, as indicated by the observations obtained in experimental glaucoma rats with intraperitoneal injection.49
Suppression of oxidative stress
Oxidative stress occurs because of the imbalance between reactive oxygen species (ROS) production and clearance. ROS are chemically reactive molecules containing oxygen, including oxygen ions and peroxides, which are generated in normal metabolism of oxygen and are cleared by antioxidants. Excess ROS leads to damage of cellular components including DNA, fatty acids in lipids, and amino acids in proteins, by oxidation. Oxidative DNA damage was observed in the glaucoma patients, indicating its possible role in the pathogenesis of GON.50 The use of antioxidants to prevent and treat glaucoma is worth being explored.
Vitamin E and glutathione (GSH) are two important endogenous antioxidants responsible for ROS clearance. In experimental guinea pigs, a significant decrease in retinal GSH was induced by acute ocular hypertension. However, treatments with subcutaneous vitamin E reversed the decreased GSH level and prevented retinal injury induced by high IOP.51 Pre-treatments with subcutaneous vitamin E or lutein in Long-Evans rats with acute elevation of IOP were effective in inhibiting the production of malondialdehyde and decelerating GSH loss.52 Coenzyme Q10 (CoQ10), a component of the electron transport chain, is generated in aerobic cellular respiration. It also acts as an important endogenous antioxidant. Topical treatments with CoQ10 or vitamin E in established rat models of glaucoma showed their efficacy in preventing RGC apoptosis.53 Systemic administration of CoQ10 in DBA/2J mice preserved the axons in the optic nerve head through inhibiting oxidative stress, as revealed by suppressed superoxide dismutase-2 (SOD2) and heme oxygenase-1 protein expression.54 Alpha-lipoic acid and SOD are the other two essential antioxidants. Nebbioso et al found that Wistar rats that were fed with α-lipoic acid or SOD for 8 weeks were able to resist the damage to RGCs induced by acute ocular hypertension.55
Some traditional Chinese medicines (TCMs), used historically as tonics in the People’s Republic of China, show neuroprotective effects in glaucoma. It has been reported that oral administration of Ginkgo biloba extract EGb 761 for 5 months in a rat model of chronic glaucoma effectively suppressed high IOP-induced RGC loss.56 In NTG patients, oral administration of EGb 761 for 4 weeks significantly improved the visual field.57 Long-term treatment with G. biloba extract for 4 years in NTG subjects also significantly slowed the progression of visual field defects without affecting the IOP.58 One possible mechanism for its essential neuroprotective effects is based on the ability to clear ROS.59 However, one recent randomized and crossover clinical trial performed in newly diagnosed NTG patients in the People’s Republic of China indicated that oral administration of G. biloba extract for 4 weeks had no effects on mean defect or contrast sensitivity, which was extremely different from previous reports.60 Thus, further clinical investigations are required to prove its efficacy in the future. Lycium barbarum, another historic TCM widely used to treat ocular diseases, has also been shown by modern scientific methods to protect RGCs. Experimental evidence demonstrated that oral administration of aqueous extract of L. barbarum could promote RGC survival against elevated IOP in SD rats.61 Inhibition of oxidative stress mediated by polysaccharides extracted from L. barbarum may be involved in its neuroprotective effects.62 Since, in general, extracts of TCMs contains multiple active components, we are not able to infer the specific component that works as a protectant for RGCs. Therefore, a high degree of quality control and standardization for TCM extraction should be established to explore these promising neuroprotective agents.
Blocking neurotoxicity of nitric oxide
In the 1990s, Neufeld et al63 proposed the hypothesis that neurotoxicity caused by nitric oxide (NO) contributes to the progressive GON in glaucoma. Increased amounts of NO synthase-2 (NOS-2) were observed in the optic nerve head in POAG patients, indicating that the glaucomatous optic nerve head was exposed to a high level of NO. NO could be converted to peroxynitrite by ROS, which is harmful to RGCs.63,64
Based on these observations, Neufeld then investigated whether aminoguanidine, a specific inhibitor of NOS-2, was able to rescue RGCs in rats with ocular hypertension. Either oral administration or topical application of aminoguanidine in rat models with either chronic or acute IOP elevation significantly enhanced RGCs survival.65,66 Another NOS-2 inhibitor, SC-51, had similar protective effects on RGCs when used in a Brown Norway rat model with chronic high IOP.67 In another rat model, inducible NOS-2 was present in the inner retinal cells 5 days after acute IOP elevation, followed by 50% RGC loss. However, treatments with aminoguanidine or SC-51 for 2 weeks immediately after the IOP rise substantially prevented RGC loss.68 Therefore, inhibition of NOS-2 by application of its specific blockers is an effective neuroprotective approach for glaucoma treatment.
Interventions in microglia overactivation
Microglia are a type of glial cell that act as macrophages throughout the central nervous system. Microglial activation facilitates injured nerve rehabilitation by clearance of toxic components such as foreign materials, neurofibrillary tangles, DNA fragments, etc.69 Neufeld detected overactivated microglia present in the optic nerve head and lamina cribrosa in glaucomatous human eyes.65 Overactivation of microglia leads to excessive production of toxic materials surrounding RGCs, including TNF-α, NO, and ROS (Figure 5). Hence, interventions in microglia overactivation may be useful to enhance survival of RGCs in treating glaucoma.
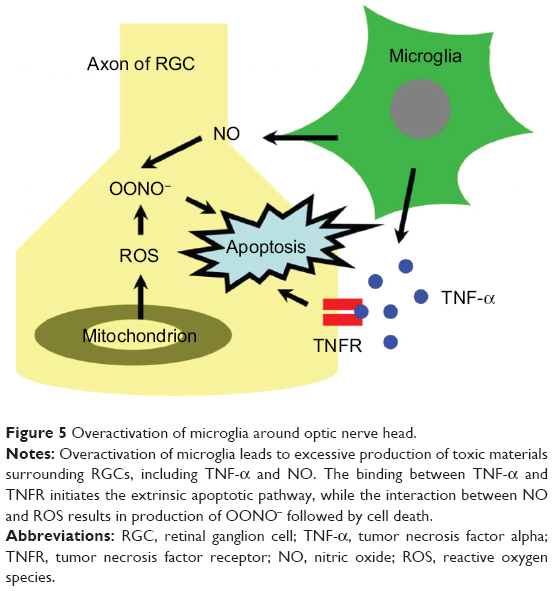 | Figure 5 Overactivation of microglia around optic nerve head. |
Minocycline, which acts as a microglia activation inhibitor, was found to be effective in maintaining RGC survival in experimental glaucoma. Intraperitoneal injections of minocycline in Wistar rats with elevated IOP significantly increased the survival rate of RGCs (84% compared with control group rate of 65%).70 Similarly, such treatments also enhanced RGC survival in DBA/2J mice with ocular hypertension.71 Although the neuroprotective effects of minocycline have only been tested in rodents, and further investigations are needed in primates and humans, minocycline’s safety has been proved in clinical practice; its ability to penetrate the blood–brain barrier suggests that it may be a promising application in prospective glaucoma treatments. Su et al demonstrated that intraperitoneal administration of rapamycin daily in a SD rat chronic ocular hypertension model protected RGCs for up to 6 weeks by suppressing the overactivation of microglia around the optic nerve head.72 Yang et al found a Chinese herb extract, triptolide, was able to improve RGC maintenance in DBA/2J mice by suppressing retinal microglia activation.73 Intravitreal injection of tetrandrine (the extract from the root of Stephania tetrandra), was also reported to have neuroprotective effects on RGCs in vivo,74 and the corresponding in vitro observations later indicated suppression of microglial activation was involved in producing its essential effects.75
Enhancement of protective autoimmunity
In nerve injuries, the immune system plays beneficial roles in limiting damage to the nervous system, termed protective autoimmunity.76 GON, like other nerve injuries, can be viewed as a primary neuronal degeneration by high IOP, followed by a secondary degeneration. Antihypertensive therapies are required to control IOP and to protect the optic nerve from primary neurodegeneration. Secondary degeneration mainly results from a continuous hostile environment, including the presence of ROS, shortage of neurotrophic factors, or overload of glutamate; the mechanisms of secondary degeneration could be obstructed by protective autoimmunity. Therefore, enhancement of protective autoimmunity by vaccination together with IOP-lowering treatments may prevent the progression of GON.77
Copolymer-1 (Cop-1; also named glatiramer acetate) was therefore applied to treat glaucoma, based on the purposes of protective autoimmunity. It acts as an antigenic copolymer that cross-reacts with a broad spectrum of self-reacting T cells. Application of Cop-1 in Lewis rats of chronic high IOP was beneficial to the survival rate of RGCs for 3 weeks.78 Vaccination with Cop-1 in SD rats of acute high IOP also provided protection to RGCs over the sites of secondary degeneration, without any autoimmune disorders.79 Ben Simon et al obtained similar results in Lewis rats with chronic ocular hypertension 1 week after vaccination with Cop-1.80 Later, Yang et al reported that vaccination with Cop-1 in Wistar rats with experimental glaucoma elevated the expression of NT-3 and its cognate receptor, TrkC, in RGCs, RNFL, and IPL. As a result, the RGC loss induced by chronic ocular hypertension was markedly decreased.81 Zhou et al demonstrated that vaccination with Cop-1 in SD rats of chronic glaucoma showed enhancement of endogenous BDNF expression in retinas, accompanied by suppression of RGC apoptosis.82 These observations may provide several possible underlying molecular mechanisms for the protective autoimmunity offered by Cop-1.
Outlook and conclusion
It is widely accepted that glaucoma is a multifactorial ocular disease. A variety of treatments other than conventional IOP-lowering therapies will be effective to delay progressive GON. In 2001, Wheeler et al pointed out four criteria to evaluate the therapeutic utility of newly developed neuroprotective approaches for glaucoma.83 However, most of the therapeutic methods discussed above were only proved in experimental glaucoma (Table 1). Although the effects of several newly developed medications have been tested in glaucoma patients, the number of enrolled subjects has never been large enough (Table 2). Brimonidine was the only effective agent, as proved by RCTs, which was supposed to be the potential neuroprotective medication in the future.84 Therefore, further clinical trials, especially RCTs, are necessary to confirm newly developed medications’ safety and utility.
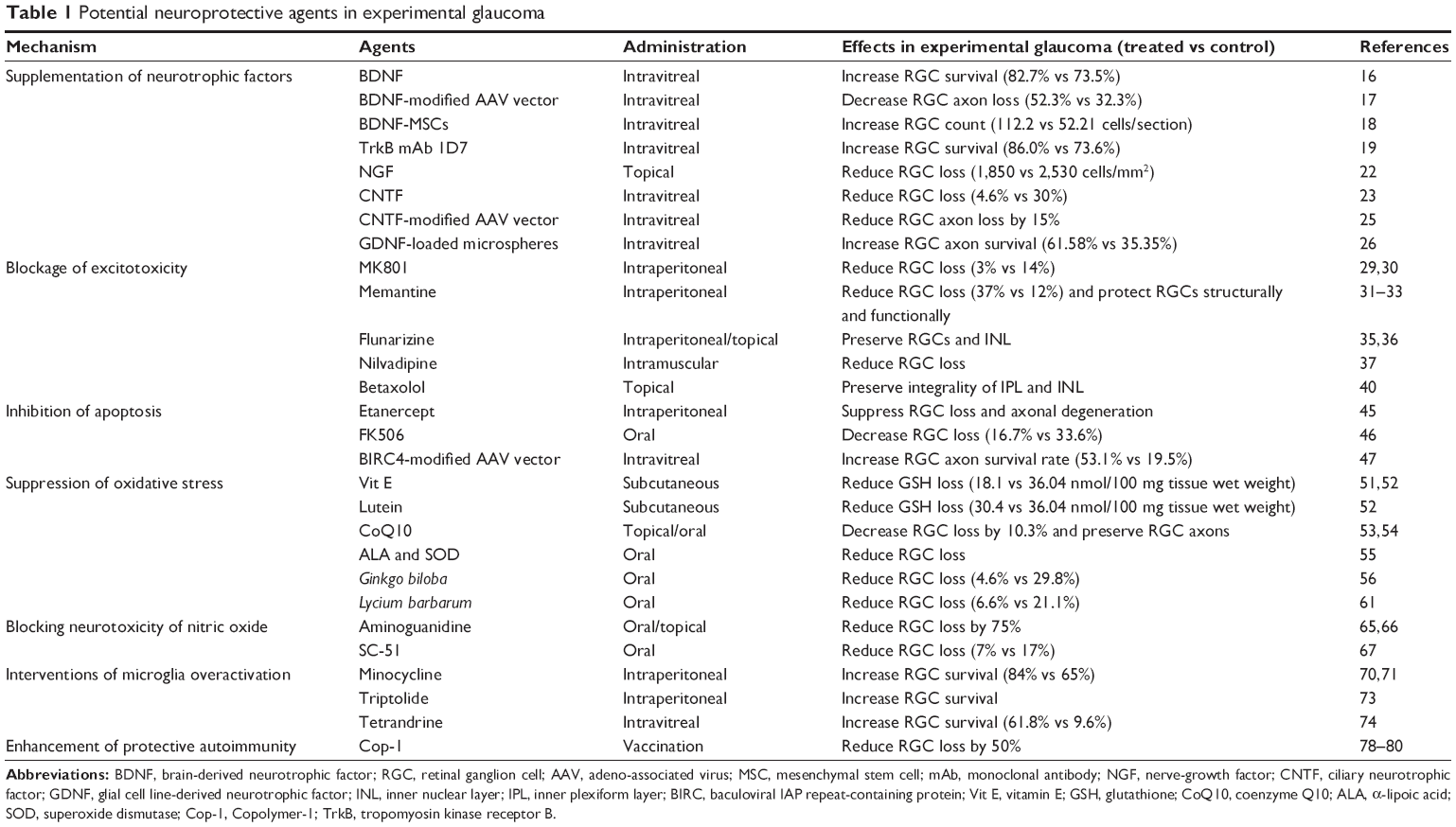 | Table 1 Potential neuroprotective agents in experimental glaucoma |
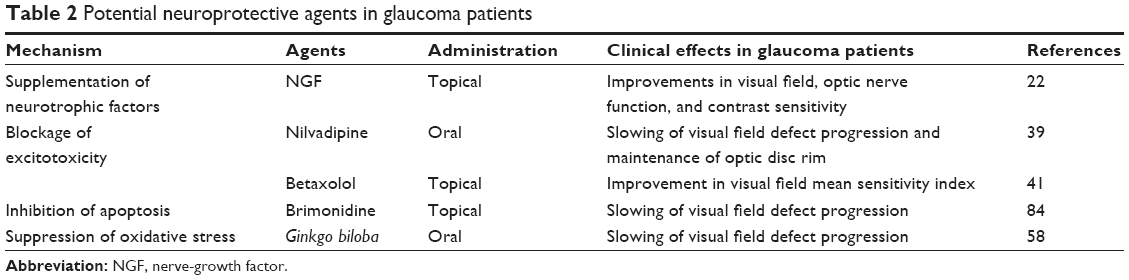 | Table 2 Potential neuroprotective agents in glaucoma patients |
At the same time, exploring other novel treatments based on the above-mentioned criteria may potentially increase the cure rate of glaucoma. For example, intravitreal injections of gene-modified stem cells are more effective to deliver target proteins continuously than simple intravitreal injections of the proteins themselves. Small interfering RNA is the essential agent to interfere with the expression of specific genes within cells. If the expression of NOS-2 or pro-apoptotic proteins could be blocked by specially designed small interfering RNA, less RGC loss, which leads to progressive GON, would occur. Despite so many exciting treatment options available for glaucoma, the challenges of neuroprotection drug discovery nowadays mainly arise from several issues, as indicated in Table 3.85 Thus, it will be worthwhile developing creative innovations that are aimed at solving these problems in both basic research and clinical trials in the future.
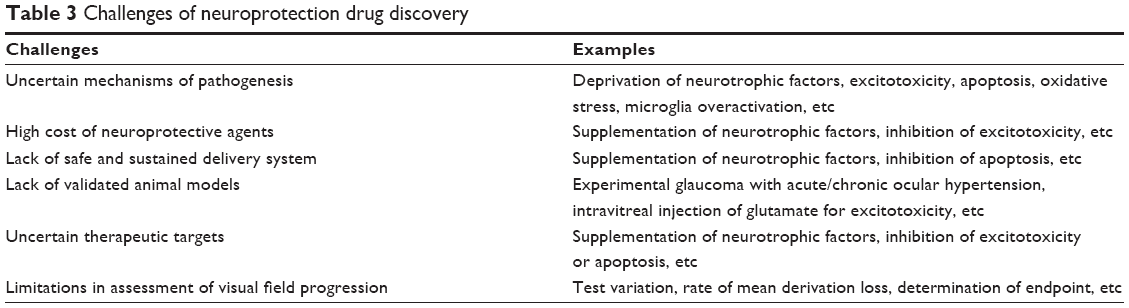 | Table 3 Challenges of neuroprotection drug discovery |
Acknowledgments
This review was supported by grants from Chinese State Natural and Science Foundation (grant number 81300756) and Beijing Natural Science Foundation (grant number 7132219). We thank Dr Yalong Dang, Ms Qiaojin Lin, and Mr Xiwan Jin for editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–267. | ||
Casson RJ, Chidlow G, Wood JP, Crowston JG, Goldberg I. Definition of glaucoma: clinical and experimental concepts. Clin Experiment Ophthalmol. 2012 ;40(4):341–349. | ||
Shiose Y, Kitazawa Y, Tsukahara S, et al. Epidemiology of glaucoma in Japan – a nationwide glaucoma survey. Jpn J Ophthalmol. 1991;35(2):133–155. | ||
Iwase A, Suzuki Y, Araie M, et al; Tajimi Study Group, Japan Glaucoma Society. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology. 2004;111(9):1641–1648. | ||
Narayanaswamy A, Baskaran M, Zheng Y, et al. The prevalence and types of glaucoma in an urban Indian population: the Singapore Indian Eye Study. Invest Ophthalmol Vis Sci. 2013;54(7):4621–4627. | ||
Liang YB, Friedman DS, Zhou Q, et al. Prevalence of primary open angle glaucoma in a rural adult Chinese population: the Handan eye study. Invest Ophthalmol Vis Sci. 2011;21:52(11):8250–8257. | ||
Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237(4819):1154–1162. | ||
Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. | ||
Adler R, Landa KB, Manthorpe M, Varon S. Cholinergic neuronotrophic factors: intraocular distribution of trophic activity for ciliary neurons. Science. 1979;204(4400):1434–1436. | ||
Henderson CE, Phillips HS, Pollock RA, et al. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266(5187):1062–1064. | ||
Poo MM. Neurotrophins as synaptic modulators. Nat Rev. 2001;2(1):24–32. | ||
Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41(3):764–774. | ||
Quigley HA, McKinnon SJ, Zack DJ, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000;41(11):3460–3466. | ||
Johnson EC, Deppmeier LM, Wentzien SK, Hsu I, Morrison JC. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000;41(2):431–442. | ||
Sposato V, Bucci MG, Coassin M, Russo MA, Lambiase A, Aloe L. Reduced NGF level and TrkA protein and TrkA gene expression in the optic nerve of rats with experimentally induced glaucoma. Neurosci Lett. 2008;446(1):20–24. | ||
Ko ML, Hu DN, Ritch R, Sharma SC, Chen CF. Patterns of retinal ganglion cell survival after brain-derived neurotrophic factor administration in hypertensive eyes of rats. Neurosci Lett. 2001;305(2):139–142. | ||
Martin KR, Quigley HA, Zack DJ, et al. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44(10):4357–4365. | ||
Harper MM, Grozdanic SD, Blits B, et al. Transplantation of BDNF-secreting mesenchymal stem cells provides neuroprotection in chronically hypertensive rat eyes. Invest Ophthalmol Vis Sci. 2011;52(7):4506–4515. | ||
Bai Y, Xu J, Brahimi F, Zhuo Y, Sarunic MV, Saragovi HU. An agonistic TrkB mAb causes sustained TrkB activation, delays RGC death, and protects the retinal structure in optic nerve axotomy and in glaucoma. Invest Ophthalmol Vis Sci. 2010;51(9):4722–4731. | ||
Gao H, Qiao X, Cantor LB, WuDunn D. Up-regulation of brain-derived neurotrophic factor expression by brimonidine in rat retinal ganglion cells. Arch Ophthalmol. 2002;120(6):797–803. | ||
Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8(6):1220–1230. | ||
Lambiase A, Aloe L, Centofanti M, et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc Natl Acad Sci U S A. Epub 2009 Aug 3. | ||
Ji JZ, Elyaman W, Yip HK, et al. CNTF promotes survival of retinal ganglion cells after induction of ocular hypertension in rats: the possible involvement of STAT3 pathway. Eur J Neurosci. 2004;19(2):265–272. | ||
Zhang SS, Wei JY, Li C, Barnstable CJ, Fu XY. Expression and activation of STAT proteins during mouse retina development. Exp Eye Res. 2003;76(4):421–431. | ||
Pease ME, Zack DJ, Berlinicke C, et al. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50(5):2194–2200. | ||
Jiang C, Moore MJ, Zhang X, Klassen H, Langer R, Young M. Intravitreal injections of GDNF-loaded biodegradable microspheres are neuroprotective in a rat model of glaucoma. Mol Vis. 2007;13:1783–1792. | ||
Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36(1):106–112. | ||
Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996;114(3):299–305. | ||
Chaudhary P, Ahmed F, Sharma SC. MK801-a neuroprotectant in rat hypertensive eyes. Brain Res. 1998;792(1):154–158. | ||
Nucci C, Tartaglione R, Rombolà L, Morrone LA, Fazzi E, Bagetta G. Neurochemical evidence to implicate elevated glutamate in the mechanisms of high intraocular pressure (IOP)-induced retinal ganglion cell death in rat. Neurotoxicology. 2005;26(5):935–941. | ||
WoldeMussie E, Yoles E, Schwartz M, Ruiz G, Wheeler LA. Neuroprotective effect of memantine in different retinal injury models in rats. J Glaucoma. 2002;11(6):474–480. | ||
Hare WA, WoldeMussie E, Lai RK, et al. Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, I: Functional measures. Invest Ophthalmol Vis Sci. 2004;45(8):2625–2639. | ||
Hare WA, WoldeMussie E, Weinreb RN, et al. Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, II: Structural measures. Invest Ophthalmol Vis Sci. 2004;45(8):2640–2651. | ||
Sena DF, Lindsley K. Neuroprotection for treatment of glaucoma in adults. Cochrane Database Syst Rev. 2013;2:CD006539. | ||
Takahashi K, Lam TT, Edward DP, Buchi ER, Tso MO. Protective effects of flunarizine on ischemic injury in the rat retina. Arch Ophthalmol. 1992;110(6):862–870. | ||
Osborne NN, Wood JP, Cupido A, Melena J, Chidlow G. Topical flunarizine reduces IOP and protects the retina against ischemia-excitotoxicity. Invest Ophthalmol Vis Sci. 2002;43(5):1456–1464. | ||
Uemura A, Mizota A. Retinal concentration and protective effect against retinal ischemia of nilvadipine in rats. Eur J Ophthalmol. 2008;18(1):87–93. | ||
Toriu N, Akaike A, Yasuyoshi H, et al. Lomerizine, a Ca2+ channel blocker, reduces glutamate-induced neurotoxicity and ischemia/reperfusion damage in rat retina. Exp Eye Res. 2000;70(4):475–484. | ||
Koseki N, Araie M, Tomidokoro A, et al. A placebo-controlled 3-year study of a calcium blocker on visual field and ocular circulation in glaucoma with low-normal pressure. Ophthalmology. 2008;115(11):2049–2057. | ||
Woo Cheon E, Hee Kim Y, Yun Cho Y, et al. Betaxolol, a beta1-adrenoceptor antagonist, protects a transient ischemic injury of the retina. Exp Eye Res. 2002;75(5):591–601. | ||
Collignon-Brach J. Longterm effect of topical beta-blockers on intraocular pressure and visual field sensitivity in ocular hypertension and chronic open-angle glaucoma. Surv Ophthalmol. 1994;38(Suppl):S149–S155. | ||
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. | ||
Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 1995;36(5):774–786. | ||
Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115(8):1031–1035. | ||
Roh M, Zhang Y, Murakami Y, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 2012;7(7):e40065. | ||
Huang W, Fileta JB, Dobberfuhl A, et al. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci U S A. 2005;102(34):12242–12247. | ||
McKinnon SJ, Lehman DM, Tahzib NG, et al. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol Ther. 2002;5(6):780–787. | ||
Tatton W, Chen D, Chalmers-Redman R, Wheeler L, Nixon R, Tatton N. Hypothesis for a common basis for neuroprotection in glaucoma and Alzheimer’s disease: anti-apoptosis by alpha-2-adrenergic receptor activation. Surv Ophthalmol. 2003;48(Suppl 1):S25–S37. | ||
Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch Clin Exp Ophthalmol. 2014;252(5):761–772. | ||
Izzotti A, Sacca SC, Cartiglia C, De Flora S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am J Med. 2003;114(8):638–646. | ||
Aydemir O, Naziroglu M, Celebi S, Yilmaz T, Kukner AS. Antioxidant effects of alpha-, gamma- and succinate-tocopherols in guinea pig retina during ischemia-reperfusion injury. Pathophysiology. 2004;11(3):167–171. | ||
Dilsiz N, Sahaboglu A, Yildiz MZ, Reichenbach A. Protective effects of various antioxidants during ischemia-reperfusion in the rat retina. Graefes Arch Clin Exp Ophthalmol. 2006;244(5):627–633. | ||
Nucci C, Tartaglione R, Cerulli A, et al. Retinal damage caused by high intraocular pressure-induced transient ischemia is prevented by coenzyme Q10 in rat. Int Rev Neurobiol. 2007;82:397–406. | ||
Lee D, Shim MS, Kim KY, et al. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2014;55(2):993–1005. | ||
Nebbioso M, Scarsella G, Tafani M, Pescosolido N. Mechanisms of ocular neuroprotection by antioxidant molecules in animal models. J Biol Regul Homeost Agents. 2013;27(1):197–209. | ||
Hirooka K, Tokuda M, Miyamoto O, Itano T, Baba T, Shiraga F. The Ginkgo biloba extract (EGb 761) provides a neuroprotective effect on retinal ganglion cells in a rat model of chronic glaucoma. Curr Eye Res. 2004;28(3):153–157. | ||
Quaranta L, Bettelli S, Uva MG, Semeraro F, Turano R, Gandolfo E. Effect of Ginkgo biloba extract on preexisting visual field damage in normal tension glaucoma. Ophthalmology. 2003;110(2):359–362; discussion. 362–354. | ||
Lee J, Sohn SW, Kee C. Effect of Ginkgo biloba extract on visual field progression in normal tension glaucoma. J Glaucoma. 2013;22(9):780–784. | ||
Droy-Lefaix MT, Cluzel J, Menerath JM, Bonhomme B, Doly M. Antioxidant effect of a Ginkgo biloba extract (EGb 761) on the retina. Int J Tissue React. 1995;17(3):93–100. | ||
Guo X, Kong X, Huang R, et al. Effect of Ginkgo biloba on visual field and contrast sensitivity in Chinese patients with normal tension glaucoma: a randomized, crossover clinical trial. Invest Ophthalmol Vis Sci. 2014;55(1):110–116. | ||
Chan HC, Chang RC, Koon-Ching Ip A, et al. Neuroprotective effects of Lycium barbarum Lynn on protecting retinal ganglion cells in an ocular hypertension model of glaucoma. Exp Neurol. 2007;203(1):269–273. | ||
Li H, Liang Y, Chiu K, et al. Lycium barbarum (wolfberry) reduces secondary degeneration and oxidative stress, and inhibits JNK pathway in retina after partial optic nerve transection. PLoS One. 2013;8(7):e68881. | ||
Neufeld AH, Hernandez MR, Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol. 1997;115(4):497–503. | ||
Liu B, Neufeld AH. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia. 2000;30(2):178–186. | ||
Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch Ophthalmol. 1999;117(8):1050–1056. | ||
Neufeld AH. Pharmacologic neuroprotection with an inhibitor of nitric oxide synthase for the treatment of glaucoma. Brain Res Bull. 2004;62(6):455–459. | ||
Neufeld AH, Das S, Vora S, et al. A prodrug of a selective inhibitor of inducible nitric oxide synthase is neuroprotective in the rat model of glaucoma. J Glaucoma. 2002;11(3):221–225. | ||
Neufeld AH, Kawai S, Das S, et al. Loss of retinal ganglion cells following retinal ischemia: the role of inducible nitric oxide synthase. Exp Eye Res. 2002;75(5):521–528. | ||
Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–139. | ||
Levkovitch-Verbin H, Kalev-Landoy M, Habot-Wilner Z, Melamed S. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol. 2006;124(4):520–526. | ||
Bosco A, Inman DM, Steele MR, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49(4):1437–1446. | ||
Su W, Li Z, Jia Y, Zhuo Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS One. 2014;9(6):e99719. | ||
Yang F, Wu L, Guo X, Wang D, Li Y. Improved retinal ganglion cell survival through retinal microglia suppression by a chinese herb extract, triptolide, in the DBA/2J mouse model of glaucoma. Ocul Immunol Inflamm. 2013;21(5):378–389. | ||
Li W, Yang C, Lu J, et al. Tetrandrine protects mouse retinal ganglion cells from ischemic injury. Drug Des Devel Ther. 2014;8:327–339. | ||
Dang Y, Xu Y, Wu W, et al. Tetrandrine suppresses lipopolysaccharide-induced microglial activation by inhibiting NF-kappaB and ERK signaling pathways in BV2 cells. PLoS One. 2014;9(8):e102522. | ||
Graber JJ, Dhib-Jalbut S. Protective autoimmunity in the nervous system. Pharmacol Ther. 2009;121(2):147–159. | ||
Schwartz M. Neurodegeneration and neuroprotection in glaucoma: development of a therapeutic neuroprotective vaccine: the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2003;44(4):1407–1411. | ||
Schori H, Kipnis J, Yoles E, et al. Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: implications for glaucoma. Proc Natl Acad Sci U S A. 2001;98(6):3398–3403. | ||
Bakalash S, Kessler A, Mizrahi T, Nussenblatt R, Schwartz M. Antigenic specificity of immunoprotective therapeutic vaccination for glaucoma. Invest Ophthalmol Vis Sci. 2003;44(8):3374–3381. | ||
Ben Simon GJ, Bakalash S, Aloni E, Rosner M. A rat model for acute rise in intraocular pressure: immune modulation as a therapeutic strategy. Am J Ophthalmol. 2006;141(6):1105–1111. | ||
Yang B, Shan L, Song W, Xiao Z, Shi L, Yuan H. Copolymer-1 immunization reduces damage in retinal ganglion cells under high intraocular pressure through altering the expression of retinal neurotrophins. J Ocul Pharmacol Ther. 2010;26(1):11–19. | ||
Zhou X, Xia XB, Xiong SQ. Neuro-protection of retinal stem cells transplantation combined with copolymer-1 immunization in a rat model of glaucoma. Mol Cell Neurosci. 2013;54:1–8. | ||
Wheeler LA, Gil DW, WoldeMussie E. Role of alpha-2 adrenergic receptors in neuroprotection and glaucoma. Surv Ophthalmol. 2001;45(Suppl 3):S290–S294; discussion S295–S296. | ||
Krupin T, Liebmann JM, Greenfield DS, Ritch R, Gardiner S; Low-Pressure Glaucoma Study Group. A randomized trial of brimonidine versus timolol in preserving visual function: results from the Low-Pressure Glaucoma Treatment Study. Am J Ophthalmol. 2011;151(4):671–681. | ||
Liu Y, Pang IH. Challenges in the development of glaucoma neuroprotection therapy. Cell Tissue Res. 2013;353(2):253–260. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.