Back to Journals » Drug Design, Development and Therapy » Volume 13
Neuroprotective effects of dexmedetomidine against isoflurane-induced neuronal injury via glutamate regulation in neonatal rats
Authors Wang X, Shan Y ![]() , Tang Z
, Tang Z ![]() , Gao L, Liu H
, Gao L, Liu H
Received 20 January 2018
Accepted for publication 9 October 2018
Published 21 December 2018 Volume 2019:13 Pages 153—160
DOI https://doi.org/10.2147/DDDT.S163197
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
Xue Wang,1,2,* Yangyang Shan,1,* Zhiyin Tang,1 Linlin Gao,3 Hongtao Liu1
1Department of Anesthesiology, Shengjing Hospital, China Medical University, Shenyang 110004, China; 2Department of Anesthesiology, Xiangyang Central Hospital, Affiliated Hospital of Hubei University of Arts and Sciences, Xiangyang 441000, China; 3Department of Medical Research, Shengjing Hospital, China Medical University, Shenyang 110004, China
*These authors contributed equally to this work
Background: Considerable evidences support the finding that the anesthesia reagent isoflurane increases neuronal cell death in young rats. Recent studies have shown that dexmedetomidine can reduce isoflurane-induced neuronal injury, but the mechanism remains unclear. We investigated whether isoflurane cause neurotoxicity to the central nervous system by regulating the N-methyl-D-aspartate receptor (NMDAR) and excitatory amino acid transporter1 (EAAT1) in young rats. Furthermore, we examined if dexmedetomidine could decrease isoflurane-induced neurotoxicity.
Methods: Neonatal rats (postnatal day 7, n=144) were randomly divided into four groups of 36 animals each: control (saline injection without isoflurane); isoflurane (2% for 4 h); isoflurane + single dose of dexmedetomidine (75 µg/kg, 20 min before the start of 2% isoflurane for 4 h); and isoflurane + dual doses of dexmedetomidine (25 µg/kg, 20 min before and 2 h after start of isoflurane at 2% for 4 h). Six neonates from each group were euthanatized at 2 h, 12 h, 24 h, 3 days, 7 days and 28 days post-anesthesia. Hippocampi were collected and processed for protein extraction. Expression levels of the NMDAR subunits NR2A and NR2B, EAAT1 and caspase-3 were measured by western blot analysis.
Results: Protein levels of NR2A, EAAT1 and caspase-3 were significantly increased in hippocampus of the isoflurane group from 2 h to 3 days, while NR2B levels were decreased. However, the -induced increase in NR2A, EAAT1 and caspase-3 and the decrease in NR2B in isoflurane-exposed rats were ameliorated in the rats treated with single or dual doses of dexmedetomidine.Isoflurane-induced neuronal damage in neonatal rats is due in part to the increase in NR2A and EAAT1 and the decrease in NR2B in the hippocampus.
Conclusion: Dexmedetomidine protects the brain against the use of isoflurane through the regulation of NR2A, NR2B and EAAT1. However, using the same amount of dexmedetomidine, the trend of protection is basically the same.
Keywords: dexmedetomidine, isoflurane, neurotoxicity, apoptosis, glutamate
Introduction
There are many newborns who need to receive anesthesia due to diagnosis or surgical needs each year.1 Generally, the most commonly used anesthetics, including isoflurane and ketamine, exert their effects by inhibiting the N-methyl-D-aspartate receptor (NMDAR) and/or potentiating the gamma amino acid type A receptor.1 The NMDAR is a heterotetramer with universal brain distribution. It is composed of NR1 and NR2 (2A, 2B, 2C, and 2D) subunits. NR2A and NR2B are the most dominant subunits in the brain.2–5 NMDARs have critical roles in synapse plasticity, long-term potentiation (LTP), and long-term depression (LTD). A persistent strengthening of synapses induces LTP to improve learning and memory,6–9 while a long-lasting decrease in synaptic strength causes LTD and results in learning and memory deficits.10 Meanwhile, LTD is needed for plasticity in the brain and prevents overexcitation of circuits. Studies have shown that prolonged or multiple exposures to NMDAR inhibitors such as isoflurane may affect cell proliferation, interrupt synaptic transmission, and induce apoptosis and neuronal injury in the central nerve system (CNS) during development in rodents.1,10–13 Liu et al11 found that a short exposure to isoflurane increased NR2B expression in adult mice and improved their cognitive functions. However, prolonged exposure to isoflurane decreased NR2B levels, increased the production of apoptotic proteins, and resulted in cognitive dysfunction. Frontal lobe and hippocampus demonstrated an age-dependent decrease in NMDAR and NR2B expression, along with delayed short-term memory, long-term memory impairment, and spatial cognitive dysfunction.7 Glutamate plays critical roles in synaptic plasticity and development by mediating the fast excitatory transmission. The extracellular glutamate concentration is kept low enough to avoid neurons from glutamate excitotoxicity.14 Excessive release of glutamate may lead to neuronal damage through receptor-mediated excitotoxicity, which plays an important role in many CNS diseases. Excitatory amino acid transporter 1 (EAAT1) is an important glutamate transporter in neurons and glial cells, which limits excessive increase of glutamate levels to reduce glutamate excitotoxicity.15
Dexmedetomidine is an agonist for α2-adrenoreceptors. It has been widely used in clinics and shows potent sedative, analgesic, and anxiolytic effects. Dexmedetomidine produces less stress responses and maintains a stable cardiovascular function. It has been shown that dexmedetomidine protects neuronal cells against isoflurane-induced hippocampal neuronal injury in neonatal mice.16,17 However, the mechanism underlying this protective effect has yet to be determined. In the present study, we investigated whether prolonged exposure to isoflurane generates neurotoxicity by changing the expression of NMDAR subunits and EAAT1. We further investigated if dexmedetomidine protects against isoflurane-induced neuronal injury and if glutamate neurotransmission could be influenced.
Materials and methods
Animals and grouping
Experimental animals were 144 male Sprague Dawley rats (specific pathogen free) at postnatal day 7 weighing 12–20 g obtained from the Experimental Animal Facility at Shengjing Hospital of China Medical University (Taichung, Taiwan). The Ohmeda 210 anesthesia machine and F-MCI sensor were purchased from Datex-Ohmeda (Datex-Ohmeda, Helsinki, Finland). Isoflurane was purchased from Abbott Laboratories (Chicago, IL, USA) and dexmedetomidine was purchased from Jiangsu Hengrui Medicine Co. Ltd. (Lianyungang, China; Lot 14101732).
All experimental procedures and protocols were approved by The Laboratory Animal Care Committee of China Medical University, Shenyang, China (2015PS223K) and were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health, USA. Rat pups were randomly divided into four groups (n=36 per group): control, isoflurane, isoflurane with a single dose of dexmedetomidine, and isoflurane with dual doses of dexmedetomidine. All pups initially stayed with their dams until postnatal day 21, when surviving rats were weaned and housed in a temperature- and light-controlled room (22°C; 12-hour light/dark cycle) with ad libitum access to food and water.
Experimental model
For all isoflurane groups, rats were placed in a customized anesthetic chamber (30×20×20 cm3) with a gas inlet and outlet. The gas inlet was connected to a compressed oxygen tank and a standard anesthesia vaporizer to direct a constant 1 L/min flow of oxygen containing 2% isoflurane through the chamber. The isoflurane levels were continuously monitored through the gas outlet by a gas monitor. The infusion time of isoflurane was 4 hours for all isoflurane groups. For the group receiving a single dose of dexmedetomidine, the drug was administered intraperitoneally (ip) at 75 μg/kg 20 minutes prior to isoflurane infusion. For the group receiving dual doses of dexmedetomidine, the drug was administered twice at 25 μg/kg (ip) 20 minutes before and 2 hours after beginning isoflurane infusion. Sham animals received single saline injections (ip) and were exposed to 30% O2 for 4 hours. During anesthesia, the oxygen concentration was continuously monitored and maintained at 30%. All rats were allowed to breathe spontaneously. After anesthesia, the rats were closely monitored until they regained consciousness, and then were returned to their dams in their home cages.
Tissue preparation
Rats were decapitated at 2 hours, 12 hours, 24 hours, 3 days, 7 days, and 28 days after the 4-hour anesthesia. Hippocampi were collected on ice and placed immediately into liquid nitrogen. The samples were removed from the liquid nitrogen 4 hours later and stored at −80°C until use.
Western blot assays
Lysis buffer (Beyotime, Shanghai, China) was added to hippocampal samples (10 μL per 1 mg tissue). Samples were sonicated and incubated at room temperature for 30 minutes. Lysates were centrifuged at 4°C, 12,000× g for 20 minutes, and the supernatants were collected. Bicinchoninic acid assay was used to measure total protein concentration. Equivalent quantities of protein (40 μg) were separated by 10% SDS-PAGE under reducing conditions and electrophoretically transferred to a polyvinylidene fluoride membrane. Membranes were blocked in 5% nonfat milk for 4 hours and washed three times. Membranes were then incubated overnight at 4°C with anti-GAPDH (1:5,000 dilution; Proteintech, Chicago, IL, USA), anti-NR2B rabbit monoclonal antibody (1:1,000 dilution; Abcam, Cambridge, UK), anti-EAAT1 rabbit monoclonal antibody (1:1,000 dilution; Cell Signaling Technology, Beverly, MA, USA) or anti-caspase-3 rabbit monoclonal antibody (1:1,000 dilution; Abcam). After three washes (10 minutes per wash), membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody at room temperature for 90 minutes. Membranes were then washed three times (10 minutes per wash) and labeled proteins were visualized by incubating with chemiluminescent substrates (Thermo Fisher Scientific, Waltham, MA, USA) for 5 minutes. X-ray films were developed in a C300 developing machine (Azure Biosystems, Dublin, CA, USA), and ImageJ was used to semi-quantify protein levels. Protein levels of NR2A, NR2B, EAAT1, and caspase-3 were normalized to their respective GAPDH levels.
Statistical analyses
Data were expressed as 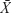 ±s. All statistical analyses were performed using SPSS (v19.0; IBM Corporation, Armonk, NY, USA). All continuous variables were tested for assumption of normality using the Shapiro–Wilk test. If the assumption of normality was met, one-way ANOVA followed by Tukey’s post hoc multiple comparison tests was used. If the assumption of normality was unmet, the Kruskal–Wallis H test was used. Significance was accepted at P<0.05.
±s. All statistical analyses were performed using SPSS (v19.0; IBM Corporation, Armonk, NY, USA). All continuous variables were tested for assumption of normality using the Shapiro–Wilk test. If the assumption of normality was met, one-way ANOVA followed by Tukey’s post hoc multiple comparison tests was used. If the assumption of normality was unmet, the Kruskal–Wallis H test was used. Significance was accepted at P<0.05.
Results
Caspase-3 levels
Compared with the control group, the levels of caspase-3 increased significantly at 2 hours, 12 hours, 24 hours, and 3 days after anesthesia in the isoflurane group (P<0.05, P<0.01, P<0.01, and P<0.01, respectively), but no significant difference was found between the control and isoflurane groups post-anesthesia 7 and 28 days after anesthesia. Compared with the isoflurane group, caspase-3 levels were significantly decreased in the isoflurane+ dexmedetomidine single-dose group at 12 hours, 24 hours, and 3 days (P<0.05, P<0.05, and P<0.05, respectively) after anesthesia. In the isoflurane+ dexmedetomidine dual-dose groups, caspase-3 levels were significantly decreased at 3 days (P<0.05) after anesthesia compared to isoflurane group (Figure 1).
 | Figure 1 (A) Dexmedetomidine reduces isoflurane-induced activation of caspase-3 (17 kD). (B) Caspase-3 protein expression level in each group at 2 h, 12 h, 24 h, 3 day, 7 day and 28 day after isoflurane anesthesia. Quantification of caspase-3 is normalized to GAPDH. |
NMDAR NR2A levels
Compared with the control group, the levels of NMDAR 2A increased significantly at 2 hours, 12 hours, 24 hours, and 3 days after anesthesia in the isoflurane group (P<0.05, P<0.01, P<0.01, and P<0.01, respectively), but no significant difference was found between the control and isoflurane groups post-anesthesia at 7 and 28 days after anesthesia. Compared with the isoflurane group, NMDAR 2A levels were significantly decreased in the isoflurane+ dexmedetomidine single-dose group at 2 hours, 12 hours, 24 hours, and 3 days (P<0.05, P<0.05, P<0.05, and P<0.01, respectively) after anesthesia. In the isoflurane+ dexmedetomidine dual-dose groups, NMDAR 2A levels were significantly decreased at 12 hours, 24 hours, and 3 days (P<0.05, P<0.05, and P<0.05, respectively) after anesthesia (Figure 2).
 | Figure 2 (A) Dexmedetomidine reduces isoflurane-induced activation of NMDAR 2A. (B) NMDAR 2A protein expression level in each group at 2 h, 12 h, 24 h, 3 day, 7 day and 28 day after isoflurane anesthesia. Quantification of NMDAR 2A is normalized to GAPDH. |
NMDAR NR2B levels
Compared with the control group, the levels of NMDAR 2B decreased significantly in the isoflurane group, but no significant difference was found between the control and isoflurane groups post-anesthesia 28 days after anesthesia. Compared with the isoflurane group, NMDAR 2B levels were significantly increased in the isoflurane+ dexmedetomidine single-dose group. In the isoflurane+ dexmedetomidine dual-dose groups, NMDAR 2A levels were significantly decreased at 12 hours and 3 days (P<0.05 and P<0.05, respectively) after anesthesia (Figure 3).
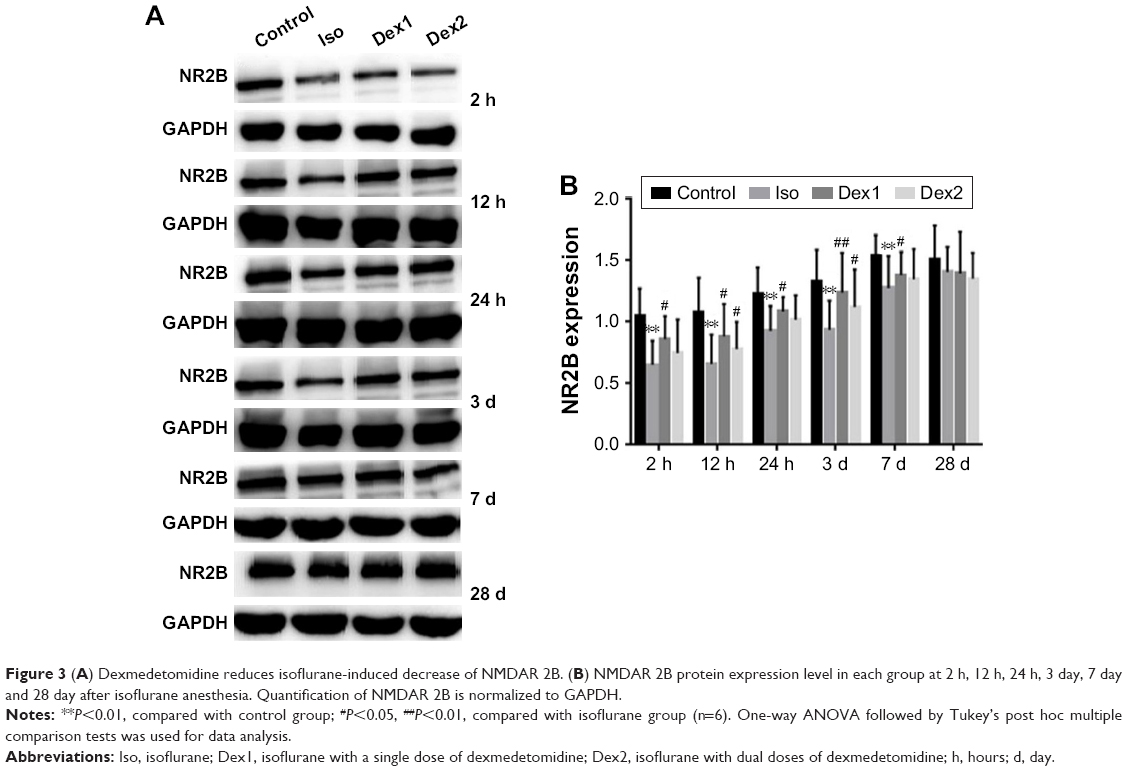 | Figure 3 (A) Dexmedetomidine reduces isoflurane-induced decrease of NMDAR 2B. (B) NMDAR 2B protein expression level in each group at 2 h, 12 h, 24 h, 3 day, 7 day and 28 day after isoflurane anesthesia. Quantification of NMDAR 2B is normalized to GAPDH. |
EAAT1 levels
Compared with the control group, the levels of EAAT1 increased significantly at 2, 12, and 24 hours after anesthesia in the isoflurane group (P<0.05, P<0.01, and P<0.01, respectively), but no significant difference was found between the control and isoflurane groups post-anesthesia at 3, 7, and 28 days after anesthesia. Compared with the isoflurane group, EAAT1 levels were significantly decreased in the isoflurane+ dexmedetomidine single-dose group at 2, 12, and 24 hours (P<0.05, P<0.05, and P<0.05, respectively) after anesthesia. In the isoflurane+ dexmedetomidine dual-dose groups, NMDAR 2A levels were significantly decreased at 2, 12, and 24 hours (P<0.05, P<0.05, and P<0.05, respectively) after anesthesia (Figure 4).
 | Figure 4 (A) Dexmedetomidine reduces isoflurane-induced activation of EAAT1. (B) EAAT1 protein expression level in each group at 2 h, 12 h, 24 h, 3 day, 7 day and 28 day after isoflurane anesthesia. Quantification of EAAT1 is normalized to GAPDH. |
Discussion
Our studies have demonstrated that 2% isoflurane for 4 hours increases hippocampal neuronal apoptosis in Sprague Dawley rat pups. The result is consistent with a previous study which reported that rat pups subjected to an initial 4% isoflurane and subsequently maintained at 1 minimum alveolar concentration (MAC) exhibited a marked increase in cell death in several brain areas, including hippocampus.18 Dexmedetomidine administration (both single and dual doses) protected against isoflurane neurotoxicity. Using different doses of dexmedetomidine, single or dual administration did not have significantly different effects. Furthermore, isoflurane decreased NR2B expression and increased NR2A, EAAT1, and caspase-3 expression while dexmedetomidine changed the expression of the subunits, suggesting that the glutamate neurotransmission could be influenced. Sanders et al showed that dexmedetomidine attenuates isoflurane-induced neuronal injury and neurocognitive impairment in the hippocampus and thalamus.19 The present study further demonstrates that glutamate systems involving NR2A, NR2B, and EAAT1 are involved in the toxicity of isoflurane and the neuroprotection provided by dexmedetomidine.
The caspase family plays essential roles in programmed cell death and caspase-3 is a central mediator. In the early phase of apoptosis, activated caspase-3 cleaves corresponding cytoplasmic nuclear substrates and leads to cell death.20,21 Li et al reported a significantly greater amount of apoptotic cells in the hippocampus of rat pups exposed to 2% isoflurane for 6 hours.22 In our expriment, Western blot analysis showed that the expression of caspase-3 in hippocampus of isoflurane group was significantly higher than that of other groups in 3 days, leading to an increase in apoptotic proteins and neurotoxicity. The levels of caspase-3 in the single-dose dexmedetomidine group were slightly but nonsignificantly lower than the dual-dose group, and both groups were lower than the isoflurane group. Our results agree with previous studies by Li et al10 in which neonatal rats were exposed to 0.75% isoflurane for 6 hours. They showed that there was no significantly different protective effect between single (75 μg/kg) and triple administrations (25 μg/kg per administration) of dexmedetomidine when all rats received same doses. Results from both our group and the Li group showed the toxicity of isoflurane and the neuroprotective effect of dexmedetomidine.
The duration of 3 months after pregnancy to 4 years after birth in humans is equivalent to 7–14 days after birth in rodents. During this period, synapses are rapidly increased. Thus, if exposed to NMDAR inhibitors such as isoflurane, ketamine, or nitrous oxide, brain development is inhibited to various degrees, leading to neuronal injury and long-term disability.23,24 Anesthetic exposure changes NMDAR subunits, affects calcium influx, induces cell signaling cascades, and generates LTP or LTD.25,26 The levels of the NR2B subunit in the hippocampus decrease with age. Increasing the NR2B levels in the hippocampi of old rats enhances excitatory postsynaptic potentials, strengthens memory, and improves synaptic transmission function.27 Our results showed that NR2A levels were increased in the isoflurane group while NR2B levels were decreased, which may affect the ability of learning and memory in adulthood. We will explore this issue in future experiments. Furthermore, in NR2A knock-out mice and NR2A regulatory signaling-deficient mice, activation of NR2B can generate LTP.28–30 NVP-AAM077, a NR2A specific inhibitor, is able to activate NMDARs and generate LTP.30,31 Compared to NR2A-dependent NMDARs, NR2B-dependent NMDARs carry a large amount of calcium ions which react preferentially with calmodulin kinase II, so they are more likely to induce LTP.32 Most neonatal rats have high levels of NR2B which begins to decline to adult levels 6–20 days after birth. On the other hand, the levels of NR2A subunits are low early after birth and begin to increase to adult levels 12 days after birth.33,34 Our data are consistent with results from Zhao et al35 showing that ketamine exposure in pregnant rats induces neuronal rats impairment, consistent with changes in NMDAR subunits. Thus, these results suggest that inhibition of NR2A and NR2B subunits is involved in isoflurane-induced neurotoxicity.
EAAT1 is predominantly expressed in astrocytes and is one of the glutamate transporters required to maintain glutamate levels in the extracellular space.36,37 Qu et al37 reported that isoflurane induced spatial learning and memory impairment and produced increases in glutamate and EAAT1 levels in the hippocampal CA1 region of old rats, suggesting that the increase in glutamate induced excitotoxicity, while the increase in EAAT1 protected against it and further improved isoflurane-induced spatial learning and memory dysfunction. In the present study, we also found that EAAT1 levels were significantly increased in the isofluran e group. Those changes suggest that the enhanced EAAT1 levels may be related to neuroprotective mechanisms in the CNS that remove excessive glutamate released by excitatory synapses, maintaining extracellular glutamate levels and protecting against excitotoxicity. The decreased EAAT1 levels in the dexmedetomidine groups may be due to the neuroprotective effects of dexmedetomidine, which leads to attenuation of the increase in EAAT1.
Taken together, the present study may provide guidance in the clinical use of drugs, suggesting that isoflurane anesthesia in pediatric patients may be combined with dexmedetomidine and other protective drugs to reduce brain damage. However, there are some limitations to our study. First, this study did not measure blood glucose, blood oxygen, or body temperature levels. Prolonged isoflurane anesthesia may cause hypoxia, hypercapnia, abnormal glucose levels, and hypothermia. All of these factors may lead to increased apoptosis, which can cause neurocognitive disorders. Second, NR2A and NR2B inhibitors were not used in the present studies, so we could not definitively determine whether apoptosis and the expression of certain subunits were directly related. Third, the plasma levels of dexmedetomidine were not measured and we could not determine the exact difference in the effective drug dosage in rats treated with high and low doses of dexmedetomidine.
Conclusion
Isoflurane caused an increase in apoptotic cells in neonatal rats, which was related to increased expression of NR2A and EAAT1 and decreased expression of NR2B. Isoflurane-induced neurotoxicity during critical developmental periods may be controlled by dexmedetomidine.
Acknowledgment
This work was made possible by grants from National Nature Science Foundation of China (No. 81271370 and No. 81501021).
Disclosure
The authors report no conflicts of interest in this work.
References
Sitdikova G, Zakharov A, Janackova S, et al. Isoflurane suppresses early cortical activity. Ann Clin Transl Neurol. 2014;1(1):15–26. | ||
Shipton OA, Paulsen O. GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci. 2014;369(1633):20130163. | ||
Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4(3):235–236. | ||
Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7(1):39–47. | ||
Mathur P, Graybeal C, Feyder M, Davis MI, Holmes A. Fear memory impairing effects of systemic treatment with the NMDA NR2B subunit antagonist, Ro 25-6981, in mice: attenuation with ageing. Pharmacol Biochem Behav. 2009;91(3):453–460. | ||
Sinner B, Friedrich O, Lindner R, Bundscherer A, Graf BM. Long-term NMDA receptor inhibition affects NMDA receptor expression and alters glutamatergic activity in developing rat hippocampal neurons. Toxicology. 2015;333:147–155. | ||
Brim BL, Haskell R, Awedikian R, et al. Memory in aged mice is rescued by enhanced expression of the GluN2B subunit of the NMDA receptor. Behav Brain Res. 2013;238:211–226. | ||
Zhao MG, Toyoda H, Lee YS, et al. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47(6):859–872. | ||
Bartlett TE, Bannister NJ, Collett VJ, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52(1):60–70. | ||
Li Y, Zeng M, Chen W, et al. Dexmedetomidine reduces isoflurane-induced neuroapoptosis partly by preserving PI3K/Akt pathway in the hippocampus of neonatal rats. PLoS One. 2014;9(4):e93639. | ||
Liu J, Wang P, Zhang X, Zhang W, Gu G. Effects of different concentration and duration time of isoflurane on acute and long-term neurocognitive function of young adult C57BL/6 mouse. Int J Clin Exp Pathol. 2014;7(9):5828–5836. | ||
Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283(5398):70–74. | ||
Huang L, Liu Y, Jin W, Ji X, Dong Z. Ketamine potentiates hippocampal neurodegeneration and persistent learning and memory impairment through the PKCγ-ERK signaling pathway in the developing brain. Brain Res. 2012;1476:164–171. | ||
Mayer ML, Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28(3):197–276. | ||
Kanai Y, Smith CP, Hediger MA. A new family of neurotransmitter transporters: the high-affinity glutamate transporters. FASEB J. 1993;7:l450–l459. | ||
Si Y, Zhang Y, Han L, et al. Dexmedetomidine acts via the JAK2/STAT3 pathway to attenuate isoflurane-induced neurocognitive deficits in senile mice. PLoS One. 2016;11(10):e0164763. | ||
Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol Scand. 2010;54(6):710–716. | ||
Lee BH, Chan JT, Kraeva E, Peterson K, Sall JW. Isoflurane exposure in newborn rats induces long-term cognitive dysfunction in males but not females. Neuropharmacology. 2014;83:9–17. | ||
Sanders RD, Xu J, Shu Y, et al. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110(5):1077–1085. | ||
Zhang G, Dong Y, Zhang B, et al. Isoflurane-induced caspase-3 activation is dependent on cytosolic calcium and can be attenuated by memantine. J Neurosci. 2008;28(17):4551–4560. | ||
Peng J, Drobish JK, Liang G, et al. Anesthetic preconditioning inhibits isoflurane-mediated apoptosis in the developing rat brain. Anesth Analg. 2014;119(4):939–946. | ||
Li X, Wei K, Hu R, et al. Upregulation of Cdh1 attenuates isoflurane-induced neuronal apoptosis and long-term cognitive impairments in developing rats. Front Cell Neurosci. 2017;11:368. | ||
Patel P, Sun L. Update on neonatal anesthetic neurotoxicity: insight into molecular mechanisms and relevance to humans. Anesthesiology. 2009;110(4):703–708. | ||
Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23(3):876–882. | ||
Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52(1):71–76. | ||
Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci. 2002;22(9):3628–3637. | ||
Brigman JL, Wright T, Talani G, et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30(13):4590–4600. | ||
Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor epsilon1 subunit. J Neurosci. 1998;18(17):6704–6712. | ||
Köhr G, Jensen V, Koester HJ, et al. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23(34):10791–10799. | ||
Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25(37):8386–8390. | ||
Berberich S, Punnakkal P, Jensen V, et al. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25(29):6907–6910. | ||
Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25(26):6037–6046. | ||
Laurie DJ, Bartke I, Schoepfer R, Naujoks K, Seeburg PH. Regional, developmental and interspecies expression of the four NMDAR2 subunits, examined using monoclonal antibodies. Brain Res Mol Brain Res. 1997;51(1–2):23–32. | ||
Wang YH, Bosy TZ, Yasuda RP, et al. Characterization of NMDA receptor subunit-specific antibodies: distribution of NR2A and NR2B receptor subunits in rat brain and ontogenic profile in the cerebellum. J Neurochem. 1995;65(1):176–183. | ||
Zhao T, Li Y, Wei W, Savage S, Zhou L, Ma D. Ketamine administered to pregnant rats in the second trimester causes long-lasting behavioral disorders in offspring. Neurobiol Dis. 2014;68:145–155. | ||
Hanson E, Armbruster M, Cantu D, et al. Astrocytic glutamate uptake is slow and does not limit neuronal NMDA receptor activation in the neonatal neocortex. Glia. 2015;63(10):1784–1796. | ||
Qu X, Xu C, Wang H, et al. Hippocampal glutamate level and glutamate aspartate transporter (GLAST) are up-regulated in senior rat associated with isoflurane-induced spatial learning/memory impairment. Neurochem Res. 2013;38(1):59–73. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.