Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Neuroprotection by triptolide against cerebral ischemia/reperfusion injury through the inhibition of NF-ΚB/PUMA signal in rats
Authors Zhang B, Song C, Feng B, Fang W
Received 7 February 2016
Accepted for publication 10 March 2016
Published 24 May 2016 Volume 2016:12 Pages 817—824
DOI https://doi.org/10.2147/TCRM.S106012
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor De Yun Wang
Bin Zhang,1 Cunfeng Song,1 Bo Feng,1 Weibing Fan2
1Department of Neurology, The Third Hospital of Liaocheng, Liaocheng, Shandong, 2Department of Neurology, The Third Hospital of Changsha, Changsha, Hunan, People’s Republic of China
Abstract: Triptolide, an active compound extracted from the Chinese herb thunder god vine (Tripterygium wilfordii Hook F.), has potent antitumor activity. Recently, triptolide was found to have protective effects against acute cerebral ischemia/reperfusion (I/R) injury through inhibition of cell apoptosis. However, the regulatory mechanism of the effect remains unclear. We hypothesize that the regulatory mechanisms of triptolide are mediated by nuclear factor κB (NF-κB) and p53-upregulated-modulator-of-apoptosis signal inhibition. To verify this hypothesis, we occluded the middle cerebral artery in male rats to establish focal cerebral I/R model. The rats received triptolide or vehicle at the onset of reperfusion following middle cerebral artery occlusion. At 24 hours after reperfusion, neurological deficits, infarct volume, and cell apoptosis were evaluated. The expression levels of NF-κBp65, PUMA, and caspase-3 were determined by Western blot. Real-time polymerase chain reaction was used to determine the levels of NF-κBp65 mRNA, PUMA mRNA, and caspase-3 mRNA. NF-κB activity was determined by electrophoretic mobility shift assay. Apoptotic cells were detected using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining. In I/R group, neurological deficit scores, cerebral infarct volume, expression of NF-κBp65, PUMA, caspase-3, NF-κB activity, and TUNEL-positive cells were found to be increased at 24 hours after I/R injury. The I/R/triptolide rats showed significantly better neurological deficit scores, decreased neural apoptosis, and reduced cerebral infarct volume. In addition, the expression of NF-κBp65, PUMA, caspase-3, and NF-κB activity was suppressed in the I/R/triptolide rats. These results indicate that the neuroprotective effects of triptolide during acute cerebral I/R injury are possibly related to the inhibition of apoptosis through suppression of NF-κB/PUMA signaling pathway.
Keywords: ischemia/reperfusion, apoptosis, triptolide, neuroprotection
Introduction
Ischemic stroke, which accounts for 85% of all stroke cases, is associated with high morbidity and mortality worldwide.1,2 Following cerebral ischemia/reperfusion (I/R) injury, cerebral microcirculatory damage and apoptotic death of neurons have been found to play a vital role in causing subsequent disability and mortality.3
Currently, the mechanisms of neuronal injury and death induced by cerebral I/R are not completely known. Since it was found that the activation of nuclear factor κB (NF-κB) induced by transient ischemia is prior to DNA fragmentation, accumulating evidences have demonstrated that NF-κB plays an important role in regulating transient ischemia-induced neuronal death.4–6
NF-κB regulates the expression of genes involved in a broad range of biological processes. In the central nervous system, NF-κB plays a dual role in neuronal survival following nerve injury. It participates in the regulation of apoptotic and inflammatory genes.7 A possible role of NF-κB genes in ischemia-induced activities was originally illustrated in clinical studies examining the expression of NF-κB family members in brain tissues of patients who died after stroke. Increased expression of RelA and p50 (NF-κB1) was detected in the ischemic and penumbra regions, implicating a regulatory role of these NF-κB subunit genes after ischemic stroke.8 In animal models of transient and permanent occlusion of the middle cerebral artery (MCA), the expression of NF-κB members p65 and p50 increased after cerebral ischemia.9 Several neuroprotective reagents showed an effect of decreasing NF-κB expression and/or activity, suggesting that NF-κB contributes to ischemic brain damage.10,11
PUMA is one of the most potent killers among the BCL-2 homology 3-only subgroup of BCL-2 family members. It drives apoptosis induced by p53-independent or -dependent signals, such as growth factor deprivation and exposure to glucocorticoids or phorbol ester.12,13 PUMA was previously shown to be upregulated in neurons after transient global cerebral ischemia, and inhibition of PUMA upregulation protected neurons from delayed ischemic death.14
It has been recently found that PUMA is a direct target of NF-κB and mediates TNF-α-induced apoptosis in vitro and in vivo.15 In colon cancer cells, PUMA was directly activated by p65 through the canonical NF-κB pathway following AKT inhibition.16 Kupchan et al17 have reported that astragalus saponin IV provided protection against I/R-induced renal injury by reducing apoptosis and inflammation through inhibition of NF-κB activity and PUMA expression. We therefore suggested that NF-κB/PUMA signal might be one of the causes to result in I/R injury. Agents that can prevent NF-κB/PUMA signal are believed to have therapeutic potentials toward brain I/R.
Triptolide is a diterpene triepoxide antibiotic compound that can be isolated from the extracts of the medicinal plant, Tripterygium wilfordii Hook F., which has been used for a number of years in traditional Chinese medicine.18 T. wilfordii Hook F. and triptolide have immunosuppressive and anti-inflammatory properties.19,20 Triptolide causes apoptosis by inducing the activation of caspases.21 Proliferation of rheumatoid synovial fibroblasts associated with rheumatoid arthritis has also been shown to decrease with triptolide treatment through a mechanism of increasing caspase-3 activity.22 Recently, it has been reported that triptolide may be a potential neuroprotective agent for I/R-induced injury in heart transplantation, I/R-induced lung injury, and I/R-induced hepatic injury.23–25 Fewer studies have reported that triptolide may be a potential neuroprotective agent for cerebral I/R injury.26–29 However, the mechanisms of possible protective effects of triptolide against cerebral I/R injury are not clear.
In the present study, we used a rat middle cerebral artery occlusion (MCAO) model to investigate the protective effects of triptolide on cerebral I/R injury and to determine the possible mechanism for this effect.
Materials and methods
Ethics statement
This study was approved by the research committee of the third hospital of Changsha, Hunan, People’s Republic of China, and all animal experiments were conducted in accordance with the guidelines of the Xiangya Hospital Animal Experimentation Committee. The Institutional Ethics Committee of Xiangya Hospital approved the animal study (20130830XY18).
Animals
Male Sprague Dawley rats weighing 280–300 g were purchased from Charles River Laboratories International, Inc. (Wilmington, MA, USA). Animals were housed in an environmentally controlled room at 22°C±2°C, with a 12/12-hour light/dark cycle and were allowed free access to food and water throughout the entire study.
Establishing MCAO model
Mice were anesthetized with chloral hydrate (400 mg/kg, intraperitoneal), and the core temperature (rectum) was maintained at 36.5°C–37.5°C using a homeothermic heating pad throughout the surgery. Cerebral focal ischemia was produced by intraluminal occlusion of the left MCA using a silicone rubber-coated nylon monofilament. The cerebral blood flow was reduced by >85% using a laser Doppler flowmeter (VMS™-LDF1; Moor Instruments, Axminster, UK). After 2 hours of ischemia, the occlusion was released for a 24-hour reperfusion period. The arterial blood pressure and heart rate were monitored through the left femoral artery using a monitoring system. Arterial blood samples for blood gas analysis were collected through the left femoral artery using an i-STAT®1 analyzer (Abbott Laboratories, Abbott Park, IL, USA) prior to (baseline) and 15 minutes after the onset of MCAO (ischemia) and reperfusion (reperfusion).
Experimental protocol
A total of 60 rats were randomly divided into four groups (n=15 in each group). 1) Sham group (sham), 2) I/R, 3) I/R + dimethyl sulfoxide [DMSO], and 4) I/R + triptolide. Triptolide (5 mg/kg; Sigma-Aldrich Co., St Louis, MO, USA, dissolved in pure DMSO) was intraperitoneally injected at the onset of reperfusion in rats belonging to the triptolide group; a corresponding volume of vehicle (pure DMSO) was administered to the rats in the I/R group. At the conclusion of the reperfusion period, the neurological deficit scores were evaluated. The rats were then sacrificed for the collection of tissue samples.
Infarct rate
The brains were removed and sliced into four coronal sections, which were 2 mm thick, 24 hours after MCAO. Sections were immediately stained with 0.5% 2,3,5-triphenyl tetrazolium chloride (TTC; Sigma-Aldrich Co.,) at 37°C for 30 minutes. The staining images were recorded by a digital camera and then quantified by using Motic Images Plus 2.0ML (Image, Xiamen, People’s Republic of China). The volume was calculated using the following equation:
|
|
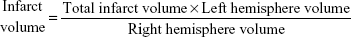
|
|
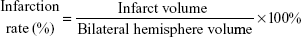
Assessment of neurological deficit scores
Neurological symptoms were assessed 24 hours after reperfusion using a neurological deficit score as previously described.16 The neurological evaluation parameters are described in Table 1.
 | Table 1 Neurological evaluation parameters |
Real-time polymerase chain reaction assay
Total RNA was isolated using TriReagent (Molecular Research Center Inc., Cincinnati, OH, USA), and 3 μg was primed with random hexamers and reverse transcribed using Superscript II (Thermo Fisher Scientific, Waltham, MA, USA) in a final volume of 50 μL. One microliter of this mixture was polymerase chain reaction amplified in a 10 μL reaction using AmpliTaq DNA polymerase (Thermo Fisher Scientific) with the addition of 5% DMSO. All of the oligonucleotide primers were designed using PerlPrimer software and synthesized commercially (Sangon Biotechnology, Shanghai, People’s Republic of China). NF-κBp65: 5′-GCG AGA GGA GCA CAG ATA CC-3′ and 5′-CTG ATA GCC TGC TCC AGG TC-3′; PUMA: 5′-CGACCTCAACGCACAGTACGA-3′ and 5′-AGGCACCTAATTGGGCTCCAT-3′. Activated caspase-3: 5′-AAGCTGAGCGAG TGTCAG-3′ and 5′-CAAAGTAGAAAAGGGCGAC-3′; GAPDH: 5′-GGTCTCCTCTGACA-3′ and 5′-AGCCAAATTCGTAC-3′. We performed the reactions according to standard protocols.
Western blot assay
The frozen tissues were placed in homogenate buffer and homogenized and then sonicated three times for 10 seconds at 4°C. The sonicated samples were subjected to centrifugation (10,000× g). Separate cytosol and nuclear protein lysates were prepared by using the Active Motif Nuclear Extract Kit (Active Motif Europe, Rixensart, Belgium), according to the manufacturer’s protocol. For routine quantitation of proteins, following the manufacturer’s protocol (Thermo Fisher Scientific). Equal amounts of protein samples (50 μg protein) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% Tris-glycine gels. After incubation in blocking buffer and being washed three times with Tris-buffered saline and Tween 20 (TBST) buffer (10 mM Tris-base, 100 mM NaCl, and 0.1% Tween 20; pH 7.5), blots were treated with an anti-NF-κBp65 polyclonal antibody (1:1,000), an anti-PUMA monoclonal antibody (mAb; 1:1,000), and an antiactivated caspase-3 pAb (1:250), or an anti-β-actin mAb (1:2,000) in TBST buffer overnight. Blots were subsequently washed with TBST and incubated with a secondary horseradish peroxidase-conjugated goat anti-mouse mAb for 1 hour. Blots were then washed, and the immunoreactive protein was detected using film exposed to enhanced chemiluminescence detection reagents. The bar graph depicts the ratios of semiquantitative results obtained by scanning reactive bands and quantifying the optical density using video densitometry.
Electrophoretic mobility shift assays
Nuclear protein extracts were prepared as described previously.30,31 Protein content of the recovered nuclear extracts was determined using the Bradford assay. The sequence of the NF-κB oligonucleotide probe was 5′-AGTTGAGGGACTTTCCCAGGC-3′. Electrophoretic mobility shift assay was performed as described previously.30,31
TUNEL staining
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was assessed by the In Situ Cell Death Detection Kit, POD (Hoffmann-La Roche Ltd., Basel, Switzerland). After 24-hour reperfusion, the sections were prepared, and the staining was performed according to the protocol provided by the manufacturer. All sections were counterstained with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific). In each case, ten fields in the infarcted cortex were randomly selected for apoptotic cell counting in a blinded manner using an Olympus IX51-reflected light fluorescence microscope (Olympus Corporation, Tokyo, Japan). The extent of apoptosis was calculated and expressed as a ratio of TUNEL-positive neurons versus total neurons.
Statistical analysis
Data are expressed as mean ± standard error of mean. Differences were evaluated by two-tailed Student’s t-test or analysis of variance followed by Tukey’s post hoc test with GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was set at P<0.05.
Results
Neurological deficit scores
The neurological deficit scores (total score per group, n=15) in I/R and I/R + DMSO groups were 22.2±2.8 and 19.6±2.4, respectively; however, in the triptolide-treated group, the neurological deficit score was only 8.4±1.2, which was significantly lower than that in the I/R or I/R + DMSO group, respectively (P<0.01, Figure 1A).
 | Figure 1 Protective effects of triptolide against cerebral I/R. |
Triptolide inhibits I/R-induced cerebral injury
Two hours of MCAO followed by a 24-hour reperfusion period induced an infarct volume of 41%±3.6% and 39%±3.2% in I/R and I/R + DMSO rats, respectively. Treatment with 5 mg/kg triptolide reduced the infarct volume to 19.4%±3.3%. The cerebral infarction rate in I/R and I/R + DMSO groups was significantly higher than that of triptolide-treated group (Figure 1B and C, P<0.01).
Triptolide reduces apoptotic cell death in the brain
After reperfusion for 24 hours, the number of apoptotic cells in the penumbral area was observed by TUNEL staining, which involves identification of DNA fragmentation. As can be seen from Figure 2, in the I/R and I/R + DMSO groups, the numbers of TUNEL-positive cells were significantly increased, as compared to sham rats. In the triptolide-treated group, the numbers of TUNEL-positive cells were significantly decreased, as compared to the I/R group. These results suggested that triptolide treatment effectively prevented expansion of apoptotic cell death in the I/R model. Data are expressed as mean ± SD vs control, *P<0.001; vs I/R, **P<0.01.
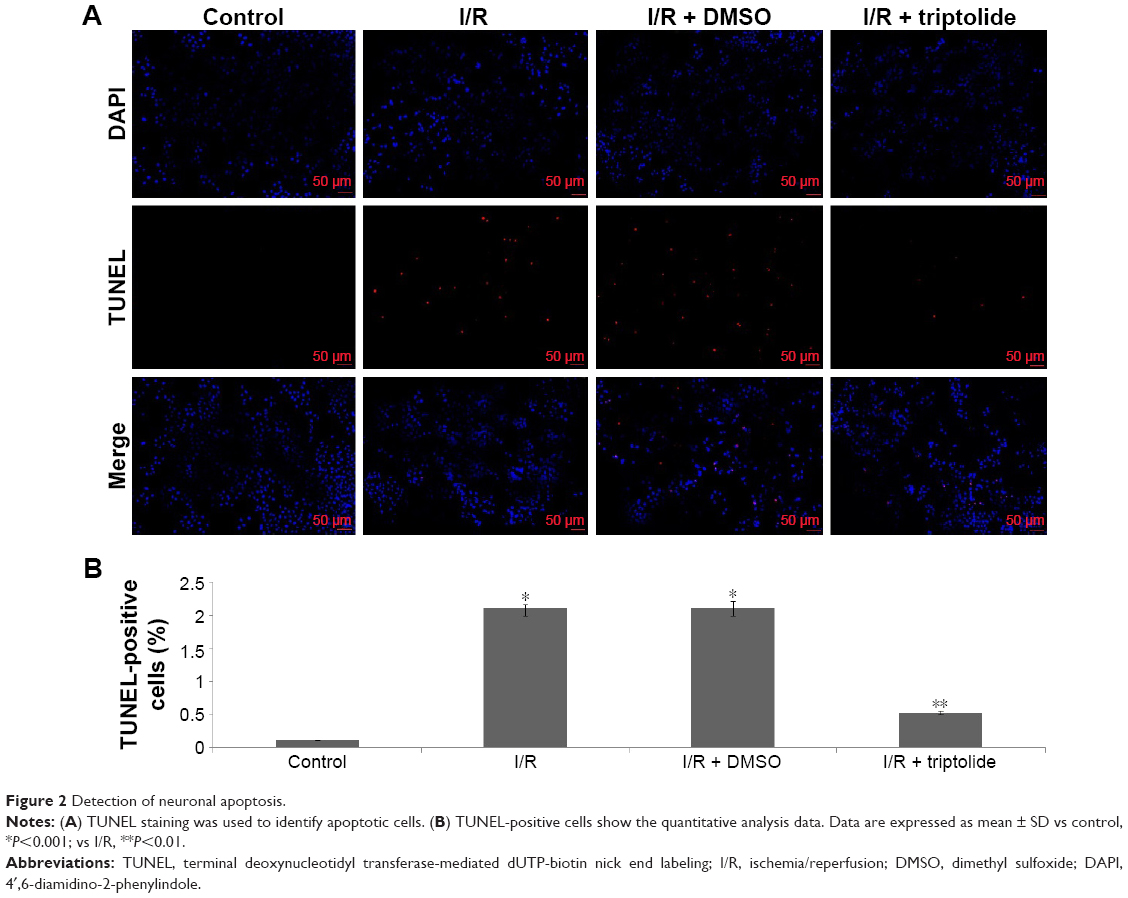 | Figure 2 Detection of neuronal apoptosis. |
NF-κBp65, PUMA, and activated-caspase-3 expression in rat brain tissue infarction side
NF-κBp65 expression from nuclear protein was detected by Western blot analysis. Total RNA was isolated from brain tissue infarction side using TriReagent. Real-time polymerase chain reaction was used to detect NF-κBp65 mRNA, PUMA mRNA, and activated-caspase-3 mRNA. Separate total cytosol and nuclear protein lysates were prepared by using the Active Motif Nuclear Extract Kit. Western blot was used to detect the expression of NF-κBp65, PUMA, and activated-caspase-3 protein. The results showed that higher levels of mRNA and protein expression of NF-κBp65, PUMA, and activated-caspase-3 were found in the I/R or I/R + DMSO rats (Figure 3A and B). After treatment with triptolide, mRNA and protein expression of NF-κBp65, PUMA, and activated-caspase-3 was inhibited (Figure 3A and B). In addition, tissues of I/R or I/R + DMSO rats exhibited a strong NF-κB activity as shown by electrophoretic mobility shift assay (Figure 3C); however, NF-κB activity was significantly decreased with triptolide treatment (Figure 3C).
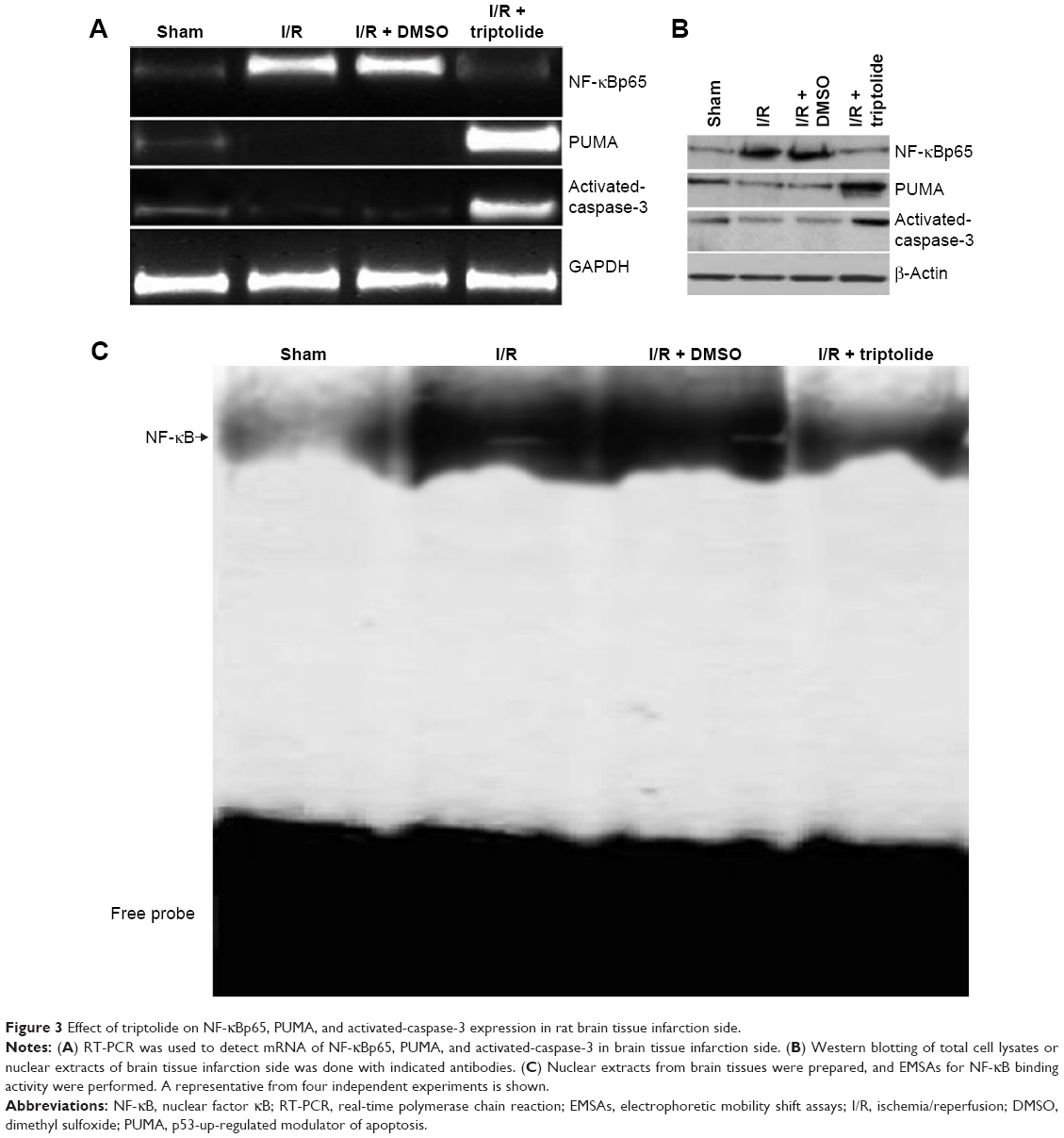 | Figure 3 Effect of triptolide on NF-κBp65, PUMA, and activated-caspase-3 expression in rat brain tissue infarction side. |
Discussion
In the present study, we provide the first evidence that NF-κB and PUMA were activated by I/R injury followed by increased cell apoptosis and brain injury. Treatment with triptolide improved neurological deficit scores and successfully reduced brain infarcted volume. In addition, triptolide treatment decreased neuronal apoptosis and inhibited NF-κB signaling activation during ischemic stroke. Furthermore, triptolide treatment inhibited PUMA signaling pathway.
NF-κB regulates the expression of multiple genes involved in inflammation, apoptotic cell death, and cell survival.32,33 Several studies have demonstrated that NF-κB is activated during development of brain injury, but the role of NF-κB in I/R brain damage is complex, and conflicting results have been obtained when exploring NF-κB inhibition as a potential neuroprotective therapy. NF-κB can promote cell survival through upregulation of the expression of antiapoptotic factors, including Bcl-xL and Bcl-2. Bcl-2 and Bcl-xL bind to Bax and Bak, thereby preventing Bax/Bak pore formation in the mitochondrial membrane.34,35 On the other hand, NF-κB can activate the proapoptotic machinery, eg, via upregulation of the tumor suppressor p53 and its downstream target PUMA.36–38 In our study, NF-κB expression and activity showed a significant increase in infarct volume in stroke models, suggesting that NF-κB plays a detrimental role in the response to cerebral ischemia. Triptolide at the onset of reperfusion significantly suppressed the NF-κB translocation and activity following MCAO. The protective effects of triptolide against cerebral I/R injury are therefore related to its inhibitory effects on the activation of NF-κB.
Recent study has shown that PUMA is under transcriptional control of NF-κB and consistent with NF-κB-dependent upregulation.15 In our study, activated NF-κB increased PUMA expression at 24 hours after MCAO. Inhibition of NF-κB activity by triptolide decreased PUMA expression at 24 hours after MCAO. NF-κB activity may promote apoptosis by PUMA upregulation, and reduced NF-κB activation may lead to lower expression of PUMA, by which cell apoptosis is inhibited. This study provides novel insights into the role of NF-κB during MCAO and sheds new light on the complex controversial results obtained in the past when studying inhibition of NF-κB as a potential therapeutic intervention in models of cerebral damage.
Conclusion
We conclude that the effects of triptolide could be brought about by the attenuation of NF-κB and PUMA cellular signaling processes. We suggest that triptolide could be further developed as a potential therapeutic agent for the treatment of cerebral I/R injury.
Study limitation
In the present study, although triptolide inhibited NF-κB activation and PUMA expression, we did not know whether knockdown of NF-κB or PUMA has the same results as triptolide. Further studies are needed to fully elucidate the biological activity of triptolide on cerebral I/R injury.
Acknowledgment
This study was supported by grants from the Hunan Nature Science Foundation (no 2015XM2293C).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Lu Y, Zhang J, Ma B, et al. Glycine attenuates cerebral ischemia/reperfusion injury by inhibiting neuronal apoptosis in mice. Neurochem Int. 2012;61(5):649–658. | ||
Zhao L, Liu X, Liang J, et al. Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res. 2013;1503(5):78–88. | ||
Wang JY, Wang P, Li SH, et al. Mdivi-1 prevents apoptosis induced by ischemia-reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species-activated mitochondrial pathway. J Stroke Cerebrovasc. 2014;23(6):1491–1499. | ||
Clemens JA, Stephenson DT, Dixon EP, et al. Global cerebral ischemia activates nuclear factor-kappa B prior to evidence of DNA fragmentation. Brain Res Mol Brain Res. 1997;48(2):187–196. | ||
Zhang W, Potrovita I, Tarabin V, et al. Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25(1):30–40. | ||
Li WL, Yu SP, Chen D, et al. The regulatory role of NF-κB in autophagy-like cell death after focal cerebral ischemia in mice. Neuroscience. 2013;244(6):16–30. | ||
Pizzi M, Spano P. Distinct roles of diverse nuclear factor-kappaB complexes in neuropathological mechanisms. Eur J Pharmacol. 2006; 545(1):22–28. | ||
Nurmi A, Lindsberg PJ, Koistinaho M, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35(4):987–991. | ||
Stephenson D, Yin T, Smalstig EB, et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20(3):592–603. | ||
Chen B, Liao WQ, Xu N, et al. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273(1):129–137. | ||
Garg P, Duncan RS, Kaja S, Koulen P. Intracellular mechanisms of N-acylethanolamine-mediated neuroprotection in a rat model of stroke. Neuroscience. 2010;166(1):252–262. | ||
Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27(Suppl 1):S71–S83. | ||
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7(3):673–682. | ||
Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40(2):618–625. | ||
Wang P, Qiu W, Dudgeon C, et al. PUMA is directly activated by NF-κB and contributes to TNF-α-induced apoptosis. Cell Death Differ. 2009;16(9):1192–1202. | ||
Sun J, Knickelbein K, He K, et al. Aurora kinase inhibition induces PUMA via NF-κB to kill colon cancer cells. Mol Cancer Ther. 2014;13(5):1298–1308. | ||
Kupchan SM, Court WA, Dailey RG Jr, Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc. 1972;94(20):7194–7195. | ||
Wen HL, Liang ZS, Zhang R, Yang K. Anti-inflammatory effects of triptolide improve left ventricular function in a rat model of diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:50. | ||
Xue M, Jiang ZZ, Liu JP, et al. Comparative study on the anti-inflammatory and immune suppressive effect of Wilforlide A. Fitoterapia. 2010;81(8):1109–1112. | ||
Ma JX, Sun YL, Wang YQ, Wu HY, Jin J, Yu XF. Triptolide induces apoptosis and inhibits the growth and angiogenesis of human pancreatic cancer cells by downregulating COX-2 and VEGF. Oncol Res. 2013;20(8):359–368. | ||
Liu C, Zhang Y, Kong X, et al. Triptolide prevents bone destruction in the collagen-induced arthritis model of rheumatoid arthritis by targeting RANKL/RANK/OPG signal pathway. Evid Based Complement Alternat Med. 2013;2013:626038. | ||
He JK, Gu ZL, Yu SD, Shen ZY. Triptolide on inhibition of reperfusion injury and prolongation of allograft survival in rat cardiac transplants. Chin J New Drugs Clin. 2001;20(6):423–426. | ||
He JK, Yu SD, Zhu HJ, Wu JC, Qin ZH. Triptolide inhibits NF-kappaB activation and reduces injury of donor lung induced by ischemia/reperfusion. Acta Pharmacol Sin. 2007;28(12):1919–1923. | ||
Wu C, Wang P, Rao J, et al. Triptolide alleviates hepatic ischemia/reperfusion injury by attenuating oxidative stress and inhibiting NF-κB activity in mice. J Surg Res. 2011;166(2):e205–e213. | ||
Li W, Yang Y, Hu Z, Ling S, Fang M. Neuroprotective effects of DAHP and Triptolide in focal cerebral ischemia via apoptosis inhibition and PI3K/Akt/mTOR pathway activation. Front Neuroanat. 2015;9(3):48. | ||
Jin XQ, Ye F, Zhang JJ, Zhao Y, Zhou XL. Triptolide attenuates cerebral ischemia and reperfusion injury in rats through the inhibition the nuclear factor kappa B signaling pathway. Neuropsychiatr Dis Treat. 2015;11(3):1395–1403. | ||
Bai S, Hu Z, Yang Y, et al. Anti-inflammatory and neuroprotective effects of triptolide via the NF-κB signaling pathway in a Rat MCAO model. Anat Rec. 2016;299(2):256–266. | ||
Yu H, Shi L, Zhao S, et al. Triptolide attenuates myocardial ischemia/reperfusion injuries in rats by inducing the activation of Nrf2/HO-1 defense pathway. Cardiovasc Toxicol. Epub 2015 Sep 2. | ||
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–476. | ||
Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86(7):2336–2340. | ||
Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11(5):1475–1489. | ||
Nichols TC. NF-kappaB and reperfusion injury. Drug News Perspect. 2004;17(2):99–104. | ||
Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2014;18(11):2195–2224. | ||
Bouchier-Hayes L, Lartigue L, Newmeyer DD. Mitochondria: pharmacological manipulation of cell death. J Clin Invest. 2015;115(10):2640–2647. | ||
Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15(6):691–699. | ||
Shishodia S, Aggarwal BB. Nuclear factor-kappaB activation: a question of life or death. J Biochem Mol Biol. 2002;35(1):28–40. | ||
Burstein E, Duckett CS. Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr Opin Cell Biol. 2013;15(6):732–737. | ||
Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22(56):8961–8982. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.