Back to Journals » International Journal of Nanomedicine » Volume 12
Nanoparticle orientationally displayed antigen epitopes
improve neutralizing antibody level in a model of porcine circovirus type 2
Authors Ding P ![]() , Zhang T, Li Y, Teng M, Sun Y, Liu X, Chai S, Zhou E
, Zhang T, Li Y, Teng M, Sun Y, Liu X, Chai S, Zhou E ![]() , Jin Q, Zhang G
, Jin Q, Zhang G
Received 1 May 2017
Accepted for publication 26 June 2017
Published 24 July 2017 Volume 2017:12 Pages 5239—5254
DOI https://doi.org/10.2147/IJN.S140789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Peiyang Ding,1,2 Teng Zhang,2,3 Yafei Li,1,2 Man Teng,2 Yaning Sun,2 Xiao Liu,2,4 Shujun Chai,2 Enmin Zhou,1 Qianyue Jin,2,5 Gaiping Zhang1,2,4,5
1College of Veterinary Medicine, Northwest A&F University, Yangling, 2Henan Provincial Key Laboratory of Animal Immunology, Henan Academy of Agricultural Sciences, 3College of Life Sciences, Henan Agricultural University, 4College of Animal Science and Veterinary Medicine, Henan Agricultural University, Zhengzhou, 5Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou University, Yangzhou, People’s Republic of China
Abstract: Recent advancements in biotechnology have enabled the rapid identification and subsequent expression of pathogenic microbial major antigens that induce protective immune responses. However, subunit vaccines have not been successfully commercialized mainly due to the lack of sufficient levels of neutralizing antibodies (NAs). High levels of NA rely on the efficient recognition and cross-linking of multiple neutralizing epitopes with B-cell receptors (BCRs). Nanoparticles are able to display coupled antigenic arrays at high density and provide multiple binding molecular scenarios with BCRs. The high-resolution antigenic structure makes it possible to accurately display stable neutralizing epitopes. Therefore, the development of a nanovaccine that orientationally displays neutralizing epitopes is a feasible strategy. To address this hypothesis, the capsid (Cap) protein of porcine circovirus type 2 as model antigen was conjugated to gold nanoparticles (AuNPs) through direct reaction of the mercapto group of the unique cysteines with AuNPs, rendering Cap-AuNPs to have neutralizing epitopes on outer surface and an immunodominant epitope buried within the inner surface. In vitro studies showed that AuNPs promoted the phagocytosis of Cap protein and NA levels were significantly improved, meanwhile antibody levels against the immunodominant epitope was significantly reduced. In mouse studies, Cap-AuNP-immunized mice displayed a high production of interleukin (IL)-4, IL-10, and interferon-γ, suggesting that Cap-AuNPs can effectively activate CD4+ and CD8+ T cells and balance Th1 and Th2 cellular responses. This study presents a new vaccine design strategy based on antigen structure, where nanoparticles are coupled to antigens in well-ordered arrays and orientationally display neutralizing epitopes to enhance NA levels.
Keywords: gold nanoparticles, porcine circovirus type 2, neutralizing antibody, epitopes, structure, orientationally display
Introduction
Vaccines are a cost-effective and powerful weapon against human and animal infectious diseases.1 Although many diseases have been neutralized, potential vaccine-preventable diseases continue to threaten human and animal health.2,3 Considering the safety of vaccines, subunit vaccines have become increasingly popular. Improvements and innovation in heterologous protein expression technology have led to a sufficient source for candidate subunit antigens.4,5 Regrettably, most subunit vaccines induce lower levels of NA, which is the key factor in providing protective immunity that is accomplished via reducing or abolishing the biological activity of a living microorganism.6 Therefore, the major challenge in the development of subunit vaccines is identification of strategies that can enhance the levels of NA.
Over the past few decades, advances in immunological and high-throughput genome sequencing technology have enabled the identification of pathogen-associated protective antigens.7 However, some viruses, such as human immunodeficiency virus (HIV), influenza viruses, and porcine reproductive and respiratory syndrome virus, rapidly vary their surface antigens such that NA cannot effectively recognize new epitopes, leading to escape from the host’s anti-infective immune process.8–10 In addition, some pathogens reduce the level of NA level by generating antibodies against immunodominant epitopes, thus exacerbating diseases, as seen in Dengue virus, respiratory syncytial virus, porcine circovirus type 2 (PCV2), and Middle East respiratory syndrome coronavirus.11–14 Development of structural biology has increased the throughput of protein structure determination enabling the determination of the three-dimensional structure and structured electrostatic landscapes of most vaccine antigens and their epitopes.15,16 Based on the surface electrostatic landscapes of the antigen, stabilized and fully displayed neutralizing epitopes on the antigenic surface or the flexible scaffold allow for easy recognition by B cells, promoting proliferation and antibody affinity maturation, while removing or hiding the immunodominant epitopes.17
Subunit vaccine monomers are variedly and chaotically distributed within the vaccine system and thus cannot efficiently activate B cells.18 The orientation of antigen epitopes is critical for activation of B cells. Reliable affinity maturation of NA requires stable and full display of neutralizing epitopes rather than transient orientations through a state.17 Cross-linking of B-cell receptors (BCRs) is also critical for activation of B cells. An ideal immunogen should have multiple copies of epitopes, and bind to multiple BCRs, allowing for multimerization of signal transduction molecules involved in antigen recognition on the surface of B cells.19,20 Antigen processing would be better facilitated if antigens were nanoparticles with granular structure and repetitive surface organization, which would increase phagocytosis and the ability to activate the complement system and recruit other immune molecules.21
One reasonable strategy to improve NA level is based on antigenic epitope structure and to orientationally and repeatedly display neutralizing epitopes on the outer surface of nanoparticles by orientation coupling between antigen and nanoparticles. To verify this strategy, gold nanoparticles (AuNPs) as delivery vehicles have been developed and have shown several features that make them well suited for biomedical applications. Importantly, AuNPs can be readily conjugated with peptides through the gold-thiol (Au-S) bond by simply mixing AuNPs with peptides that contain cysteine, thus offering a highly flexible and customizable approach to provide a scaffold that can be used to fix proteins or peptides to form a certain spatial orientation.22,23
According to bioinformatics analysis, mainly based on antigen crystal structure and the corresponding pathogenic characteristics, we selected the capsid (Cap) protein of PCV2 as a model antigen. PCV2, classified in the family Circoviridae and one of the smallest DNA viruses, is widespread and has caused an estimated € 5.76 billion economic loss for the EU.24,25 The Cap protein can form virus-like particles (VLPs) dividing the structure of Cap protein into two faces, neutralizing epitopes on the outer surface, while the unique cysteine residue is on the inner face (Figure 1A and B).26 The decoy epitope, an immunodominant region consisting of residues 169–180 (Cap (169–180)), exposed on the surface of the Cap protein monomer but buried in virus particles or VLPs, allows PCV2 to evade humoral immunity by inducing low-level NA and high-level non-protection antibodies.13,27
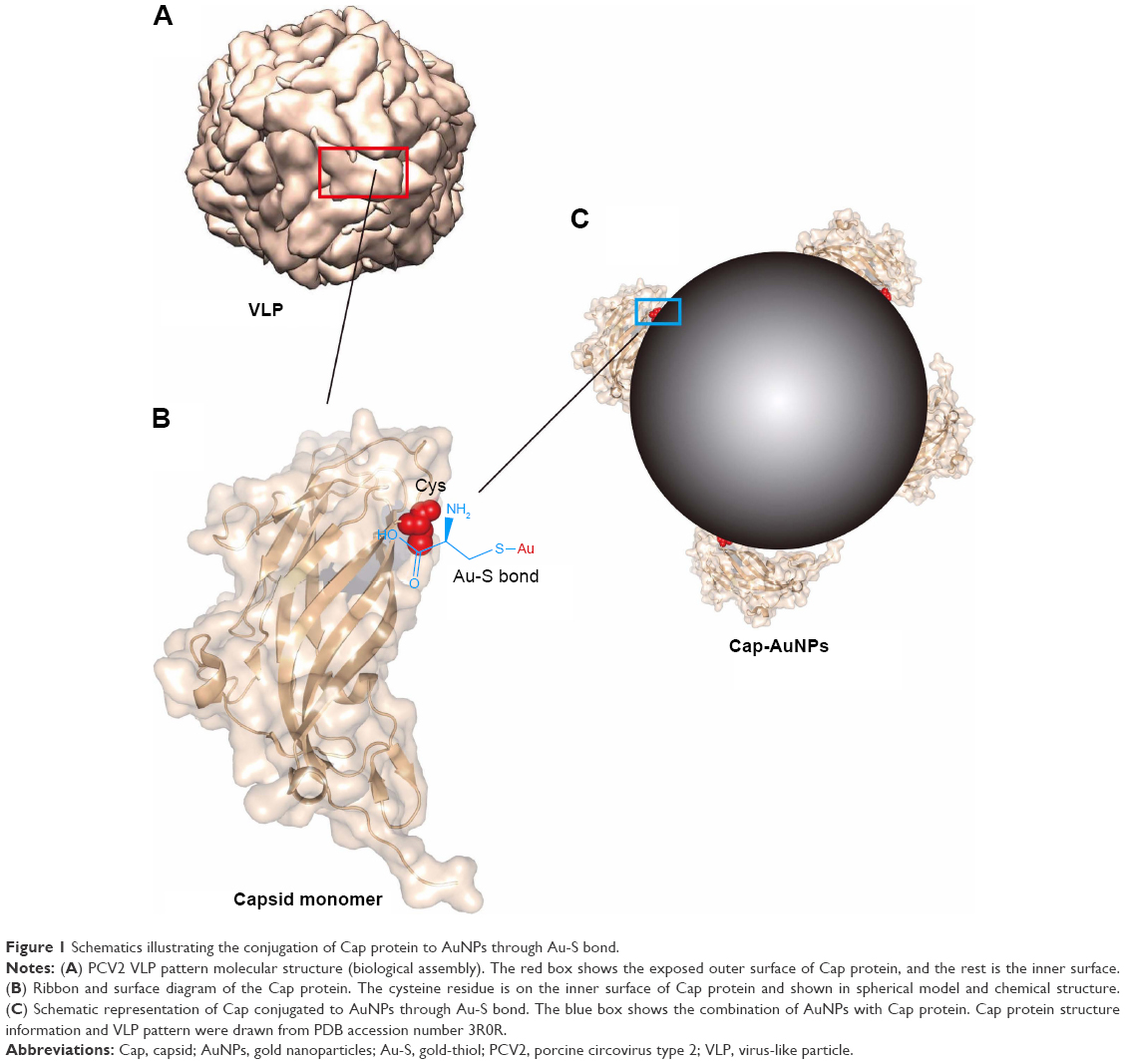 | Figure 1 Schematics illustrating the conjugation of Cap protein to AuNPs through Au-S bond. |
In the current study, we used AuNPs as an orientational-connect carrier to display Cap protein (Cap-conjugated AuNPs [Cap-AuNPs]), with neutralizing epitopes exposed on the outside surface of the nanoparticles (Figure 1C). The immune responses to Cap-AuNPs were evaluated in mice. To examine the mechanisms of the antibody production, the efficiency of cellular uptake of Cap-AuNPs into RAW264.7 macrophage cells and DC2.4 dendritic cells and subsequent cytokine secretion were measured. Our study lays the foundation for the development of a human and animal subunit vaccine based on antigen structural analysis that utilizes nanoparticles as a fixed-point connection carrier for antigens with reasonable conformation.
Materials and methods
Materials
Gold chloride trihydrate (HAuCl4·3H2O) and trisodium citrate dehydrate (Na3C6H5O7·2H2O) were purchased from Sigma-Aldrich (St Louis, MO, USA). Milli-Q water with a resistance of 18.2 M Ω was used in all experiments. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Fully cytosine-phosphate-guanine-oligodeoxynucleotide (CpG) 1826 with a sequence of 5′-TCCATGACGTTCCTGACGTT-3′ were synthesized and purified in Takara Company (Daliang, People’s Republic of China). PCV2 strain was stored in our lab. Porcine Kidney-15 cells, DC2.4 dendritic cells, and RAW264.7 macrophages were obtained from ATCC (Manassas, VA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), RPMI-1640, penicillin, and streptomycin were purchased from Hyclone (Logan, UT, USA). Fetal bovine serum (FBS), were purchased from GIBCO (Carlsbad, CA, USA). Carbopol® 971P NF polymer was purchased from Lubrizol (Cleveland, OH, USA). Freund’s adjuvant was purchased from Sigma-Aldrich. The oligopeptide 169-STIDYFQPNNKR was synthesized by GL Biochem (Shanghai, People’s Republic of China), a cysteine was added to the N-terminal end of the oligopeptide, and then conjugated to bovine serum albumin (BSA) using 4-(N-maleimidomethyl) cyclohexanecarboxylic acid N-hydroxysuccinimide ester according to the manufacturer’s instruction (Sigma-Aldrich).
Preparation and characterization of AuNPs
AuNPs were synthesized by the Turkevich’s method of reduction of HAuCl4·3H2O with Na3C6H5O7·2H2O.28 Briefly, 1 mL of 1% HAuCl4 (10 mg/mL) was added to 100 mL ddw and brought to boiling under vigorous stirring. Next, 8 mL of 1% Na3C6H5O7·2H2O (10 mg/mL) was added into the boiling solution. After 15 min of reaction, the solution color turned reddish and the reaction was stopped; then water was added to bring the nanoparticle solution to a volume of 100 mL. The shape, size, size distribution, and zeta potential of AuNPs obtained were characterized by transmission electron microscopic (TEM) (JEM-1400; Hitachi Ltd., Tokyo, Japan) and dynamic light scattering (DLS) (Malvern, Worcestershire, UK). AuNPs were monitored by scanning the surface plasmon peak using ultraviolet-visible spectroscopy (UV-Vis) (Thermo Fisher Scientific, Waltham, MA, USA).29
Preparation of recombinant Cap protein
The genomic DNA of PCV2 Cap protein without nuclear localization signal was codon optimized for Escherichia coli (E. coli) and cloned in pUC57 vector, which was synthesized by Sangon Company (Shanghai, People’s Republic of China) and stored in our lab. The coding sequence of Cap protein was PCR amplified. The PCR product and PET28a vector were doubly digested with BamHI and HindIII (NEB, Ipswich, MA, USA), treated with T4 DNA ligase (NEB), and then transformed into E. coli BL21 (DE3). For protein expression, bacteria were cultured in Luria-Bertani liquid medium (containing 0.1 mg/mL kanamycin sulfate) at 37°C with shaking until the optical density (OD) of the culture at 600 nm reached 0.6. Then, isopropyl-β-d-thiogalactoside (IPTG) was added to a final concentration of 0.1 mM to induce expression at 30°C for 10 h. The purification of Cap was performed by using a Ni-NTA His·Bind Resin (Novagen, Madison, WI, USA). Purity and bioactivity of Cap protein were determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting, respectively. The concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific). The endotoxin content of Cap proteins was detected by ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (GenScript, Piscataway, NJ, USA).
Preparation and characterization of Cap-AuNPs
Cap-AuNPs were prepared by mixing AuNPs (1 mL) with recombinant Cap protein (100 μL, 1 mg/mL) and stirred for 1 h at 4°C. To obtain washed Cap-AuNP formulation, excess free Cap protein was removed by repeated centrifugation (12,000 rpm for 60 min at 4°C) and resuspension of the pellet in PBS for a total of three wash cycles. The stability of the conjugate was assessed by adding 10% sodium chloride.30 The conjugates were confirmed by UV-Vis, DLS, and Fourier transform infrared spectroscopy (FTIR) (Thermo Fisher Scientific). The quantification of Cap-AuNPs was measured as follows: 1 mL Cap (20 μg/mL) was added into 1 mL AuNPs, stirred for 1 h at 4°C, and then AuNPs suspension was centrifuged down at 12,000 rpm for 60 min at 4°C. The supernatant was collected and the concentration of Cap was measured by Micro BCA Protein Assay Kit (Boster, Wuhan, People’s Republic of China).
Antigenic characterization of Cap-AuNPs
The spatial structure of Cap conjugated to AuNPs was measured by enzyme-linked immunosorbent assay (ELISA). Cap protein and Cap-AuNPs were serially diluted 2-fold by carbonate buffer (pH 9.6), then coated on 96-well microtiter plates, and incubated at 4°C overnight. After blocking with skim milk (2.5% in PBST), monoclonal antibodies (mAbs) (stored in our lab) 9F4 and 6A5 with neutralizing ability and anti-His tags mAb and 8A10 mAb with anti-Cap (169–180) epitopes were added to the wells and then incubated at 37°C for 1 h, respectively. After washing with PBST five times, goat anti-mouse IgG conjugated to HRP was added for 1 h at 37°C. The reaction was developed by using 3,3,5,5-tetramethylbenzidine (TMB) as the substrate. The OD value of each well was measured at 450 nm using an ELISA reader.
Uptake of Cap-AuNPs by DC2.4 cells and RAW264.7 cells
The uptake of Cap-AuNPs by antigen-presenting cells (APCs) was detected by indirect immunofluorescence (IFA). Briefly, RAW264.7 macrophages and DC2.4 dendritic cells were seeded on 96-well cell culture plates at 2×104 cells/well (100 μL) and allowed to grow overnight at 37°C in 5% CO2. Cap or Cap-AuNPs were diluted 10-fold by DMEM and 100 μL of Cap protein (1.2 μg/mL) or Cap-AuNPs (11.2 μg/mL) containing the same amount of Cap protein was added to incubate with the cells. After incubation for 1 h, the medium was removed, and the cells were fixed in precooled methanol for 10 min at room temperature, followed by three washes with PBS. After blocking with skim milk (5% in PBST), the cells were incubated with mouse anti-PCV2 serum, followed by a fluorescein isothiocyanate-labeled anti-mouse antibody and counterstained with 4′,6-diamidino-2-phenylindole before plates were examined by fluorescence microscopy.
Cytotoxicity assay
Cap-AuNP cytotoxicity was evaluated using the cell-counting kit 8 (CCK-8, Beyotime, Wuhan, People’s Republic of China).31 RAW264.7 and DC2.4 cells were seeded at 5×103 cells/well on 96-well cell culture plates, respectively. After cell adherence, different concentrations of Cap-AuNPs were added to incubate with the cells for 24 h. Then the medium was removed before adding 100 μL fresh medium containing 10% CCK-8 reagent. The culture plates were incubated for another 4 h. After incubation, the absorbance at 450 nm was measured using a microplate reader.
Cytokine production measurement
RAW264.7 cells were first primed with 50 ng/mL LPS for 4 h, which increased the expression of pro-interleukin (IL)-1β and pro-IL-18, the immature states of IL-1β and IL-18, in cells, respectively.32 After priming, the cells were seeded at 5×104 cells/well on 96-well cell culture plate. One hundred μL of AuNPs (10 μg/mL), Cap (1.2 μg/mL), or Cap-AuNPs (11.2 μg/mL) was added to incubate with the cells. Cap group and Cap-AuNPs group contain the same amount of Cap protein. Aluminum hydroxide was added as positive control. For IL-12 and tumor necrosis factor alpha (TNF-α) measurements, RAW264.7 cells were not primed with LPS. CpG was added as positive control.33 After incubation for 24 h, cytokine concentration of IL-1β, IL-12, IL-18, and TNF-α was measured by ELISA following the manufacturer’s protocols (R&D Systems, Minneapolis, MN, USA).
Animal experiments
Six-week-old female BALB/c mice (~20 g) were randomly allocated to 10 groups with 4 or 8 mice per group. Each mouse was subcutaneously immunized with 100 μL of the indicated vaccines: Cap alone, Cap-AuNPs alone, Cap mixed with CpG, Cap-AuNPs mixed with CpG, Cap mixed with Carbopol 971P, Cap-AuNPs mixed with Carbopol 971P, Cap mixed with Freund’s adjuvant, or Cap-AuNPs mixed with Freund’s adjuvant. The AuNPs were inoculated as negative controls. We also compared the immune response of lyophilized Cap-AuNPs with non-lyophilized Cap-AuNPs. Cap-AuNPs were lyophilized and stored for 3 months at 4°C. Before immunization, Cap-AuNP powder was resuspended in water and CpG was added. One hundred μL of each corresponding vaccine mixture contained 50 μg Cap, 25 μg CpG or 2.5 μg AuNPs. The final concentration of Carbopol 971P was 0.1%. Each group received an identical booster immunization 3 weeks after primary immunization. Sera were harvested at 0, 21, and 42 days post immunization (dpi).
Ethics statement
All BALB/c mice received humane care in compliance with the animal welfare guidelines of the Institutional Animal Care and Use Committee under the approval of Henan Academy of Agricultural Sciences (Approval number SYXK 2014-0007), and were purchased from Henan Experimental Animal Center and raised in isolation cages. All efforts were made to alleviate and minimize animal suffering.
Detection of anti-Cap, anti-Cap (169–180), and anti-PCV2 antibodies
Antibody titers were measured by ELISA. Briefly, Cap protein or BSA-Cap (169–180) was diluted to 5 μL/mL by carbonate buffer (pH 9.6), then coated on 96-well microtiter plates, and incubated at 4°C overnight. For detection of antibodies specific to PCV2, a commercial PCV2 antibody test kit (BioChek, Reeuwijk, Holland) was used. The ELISA microtiter plates were coated with purified and inactivated PCV2. After blocking with skim milk (2.5% in PBST), appropriate dilutions of serum samples were added to the wells and the plates were then incubated at 37°C for 1 h. After five washes with PBST, HRP-conjugated goat anti-mouse IgG and IgG subtype were added for 1 h at 37°C. The reaction was developed using TMB. The OD value of each well was measured at 450 nm using an ELISA microplate reader.
Virus neutralization test
The titers of PCV2 neutralizing antibody (NA) in sera were determined by the immunoperoxidase monolayer assay (IPMA) as previously described with minor modifications.34 Briefly, sera samples were heat-inactivated at 56°C for 30 min and serially diluted at 2-fold, then mixed with an equal volume of 200 TCID50 of PCV2 on 96-well culture plates with DMEM containing 2% FBS, and incubated for 1 h at 37°C. After incubation, the mixtures were added to 96-well plates containing 20% confluent PK15 cells. After incubation for 72 h, the cells were fixed for 30 min with cold ethanol at −20°C. After blocking with 5% skimmed milk for 1 h at 37°C, the cells were incubated with guinea pig anti-PCV2 polyclonal antibody, followed by HRP-conjugated goat anti-guinea pig IgG. The color cells were developed with 3-amino-9-ethylcarbazole and hydrogen peroxide in 0.05 M acetate buffer (pH 5.0). The neutralization titer was calculated as the reciprocal of the highest serum dilution that was able to completely block PCV2 infection in PK15 cells.
Lymphocyte proliferation assay
At 35 and 49 dpi, the lymphocytes were isolated from the spleens of 3 mice from each group using mouse lymphocyte separation medium (Solarbio, Beijing, People’s Republic of China) and resuspended at 5×105 cells/mL in RPMI-1640 complete medium containing 10% FBS. The splenocytes were seeded into 96-well plates at 100 μL per well and stimulated with 100 μL complete medium containing Cap protein (20 μg/mL) or 100 μL complete medium alone. After incubation for 48 h, the proliferative responses were determined using CCK-8. The stimulation index (SI) was calculated using the following formula: SI = (the mean of OD450 nm values of Cap-stimulated wells)/(the mean of OD450 nm values of complete medium-treated wells).
Cellular immune responses
To analyze the cellular immune responses, lymphocytes were prepared and stimulated as already described. After incubation for 60 h, the culture supernatants were collected to determine the amounts of IL-4, IL-10, and interferon (IFN)-γ using ELISA kits (R&D Systems) as already described.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA). All data are expressed as mean ± SEM. Statistical significance was determined by unpaired t-test when only two groups were compared or by one-way analysis of variance (ANOVA) when more than two groups were compared. Comparison of antibody titers, SI, and cytokine levels between groups was performed with two-way ANOVA. Statistical significance was determined at P<0.05 (*), P<0.01 (**), or P<0.001 (***).
Results
Characterization of Cap-AuNPs
The Cap protein was expressed in E. coli BL21 (DE3) and purified by affinity chromatography on a Ni2+-NTA column. SDS-PAGE and western blotting results showed that the Cap protein was successfully expressed and purified with an apparent molecular mass of approximately 26 kDa (Figure 2A). The Cap protein reacted with anti-PCV2 polyclonal antibody, indicating that the recombinant protein retained the biological activity of the wild-type protein. The endotoxin content of Cap proteins was 0.2 EU/mg.
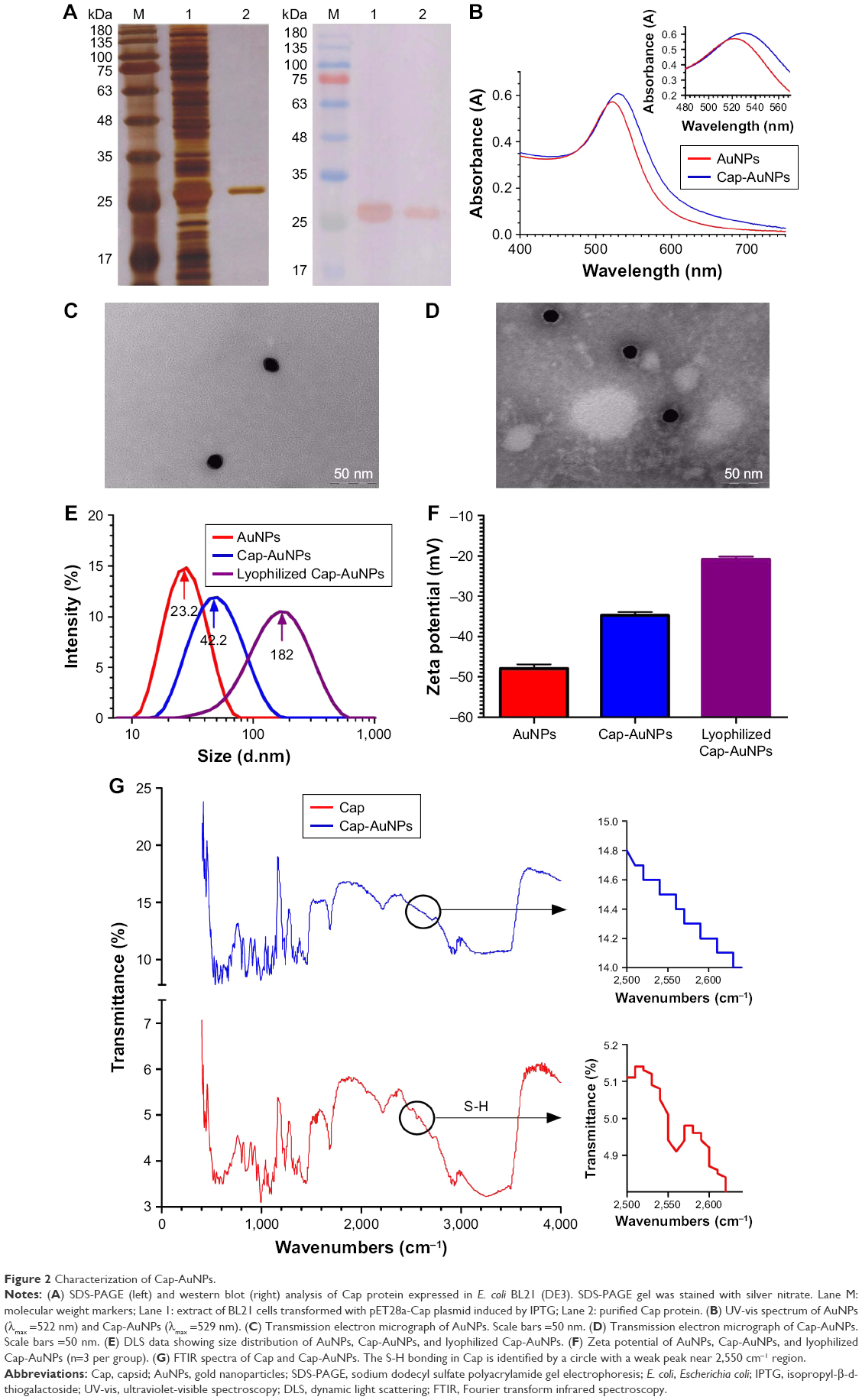 | Figure 2 Characterization of Cap-AuNPs. |
AuNPs were synthesized by the Turkevich’s method. Original AuNPs had an absorption maximum of 522 nm (Figure 2B). The mean diameter of AuNPs was 23.2 nm (~23 nm) (Figure 2C and E). After conjugation of Cap protein, the mean diameter increased to 42.2 nm (~42 nm) (Figure 2D and E), the absorbance wavelength shifted to 529 nm, and the zeta potential had a positive shift from −47.9 mV to −34.7 mV (Figure 2F), indicating stable conjugation between Cap protein and AuNPs. Cap-AuNPs aggregated when they were lyophilized and resuspended, with particle size increasing to 182 nm and zeta potential increasing to −20.8 mV (Figure 2E and F). The FTIR spectrum (Figure 2G) showed a weak peak near 2,550 cm−1 region, which virtually confirmed the presence of thiol (S-H) molecules in the sole cysteine molecule of Cap protein; however, the peak disappeared when AuNPs were coated with Cap protein (Figure 2G) indicating that S-H was changed to a new bond. This coincides with that reported by Aryal et al, who discovered the absence of S-H representative peak using FTIR spectrometer and the constitution of new Au-S bond when conjugating cysteine with AuNPs. The thiol molecules of cysteine could directly bind to AuNPs as the covalent Au-S bond is strongly favored. In this study, AuNPs were exploited as the support structure and conjugated with Cap through the Au-S bond.35
9F4 and 6A5 are mAbs with neutralizing ability that specifically recognize neutralizing epitopes, which are located on the outer surface of the Cap protein. However, a His tag located at the N-terminus of Cap protein replaced the nuclear localization sequence of Cap protein, which is located on the inner face of Cap protein. Furthermore, 8A10 can recognize the decoy epitope Cap (169–180), which was buried when forming VLPs. ELISA results showed that Cap-AuNPs were more easily recognized by 9F4 and 6A5 than Cap; on the contrary, Cap was more easily recognized by anti-His-tag mAb and 8A10 than Cap-AuNPs (Figure 3). These results indicate that the epitopes on the inner face of Cap protein were hidden, but the neutralizing epitopes located on the outer surface were exposed when Cap protein was conjugated to AuNPs via the Au-S bond.
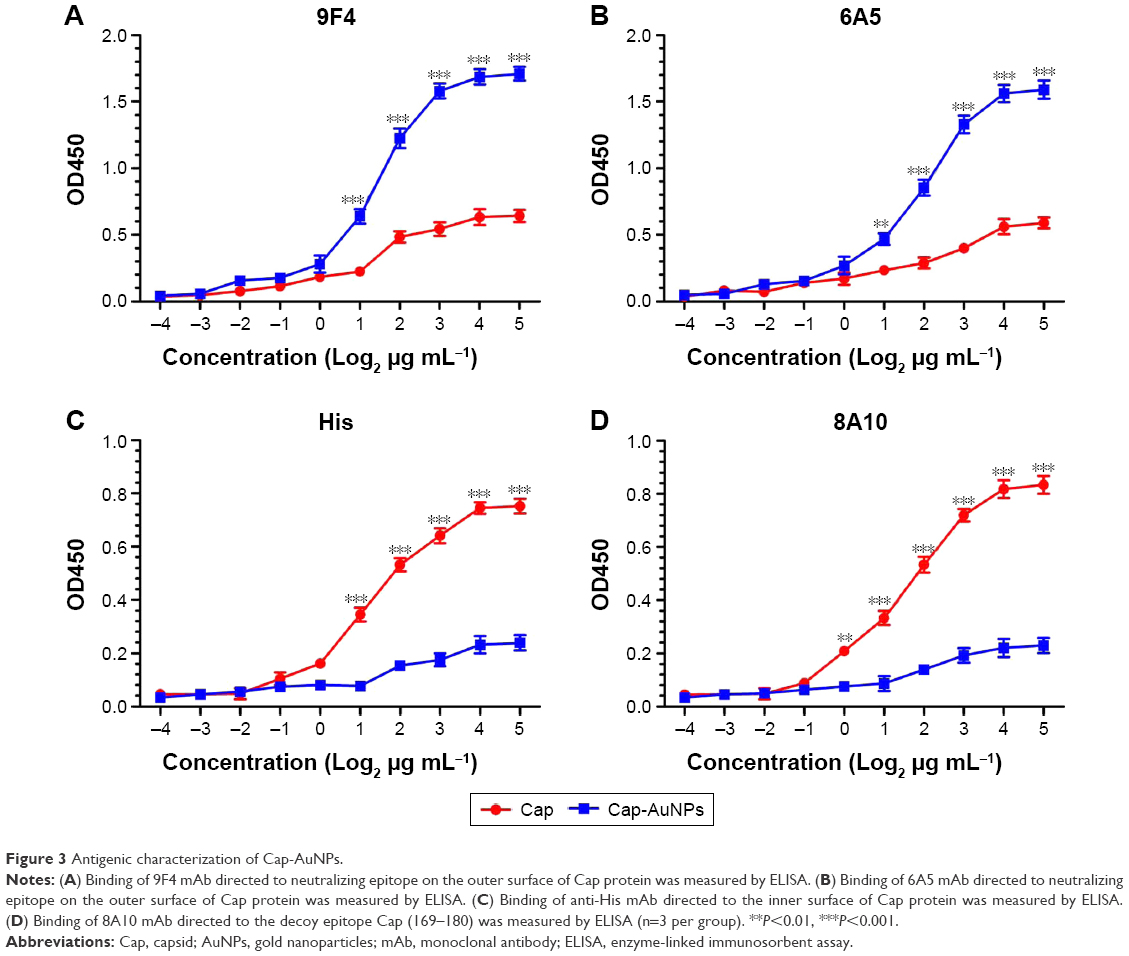 | Figure 3 Antigenic characterization of Cap-AuNPs. |
The amount of Cap protein conjugated to AuNPs was estimated by Micro BCA protein assay method; 1 mL of AuNPs (50 μg) could conjugate about 6.06 μg of Cap protein.
The formula used to calculate the number of Cap protein molecules was as follows:
|
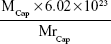
The formula used to calculate the number of AuNP molecules was as follows:36
|
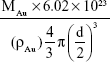
where MCap is the mass of Cap protein conjugated to AuNPs, which is 6.06 μg; MrCap is the relative molecular mass of Cap protein, which is 25.76 kDa; MAu is 50 μg in 1 mL solution, ρAu is the density of gold, which is 19.3 g/mL; and d is the diameter of the AuNPs, which is 23 nm. The number of AuNPs was calculated under the assumption that all gold chloride trihydrate reacted completely and that all resulting nanoparticles were of uniform size. All parameters use international systems of unit symbol. After calculation, the number of Cap protein molecule was 1.42×1014 and the total number of AuNPs in the 1 mL solution was found to be 3.96×1011. Based on this, we approximated that 358 molecules of Cap protein were conjugated on each AuNP, which is approximately six times the amount of Cap protein on PCV2 (60).26
Interestingly, the surface area of Cap-AuNP conjugates was about 6-fold to PCV2, whose diameter is about 17 nm, while the mean diameter of Cap-AuNPs is 42 nm. This indicated that Cap protein was closely distributed on the surface of AuNPs, with likely no voids. We hypothesized that the AuNPs reasonably represent the spatial location of Cap protein, forming an artificial VLP similar to PCV2.
Uptake of Cap protein by APCs and cytokine secretion
Uptake by APCs is the initial step for an antigen to arouse an immune response.37 We evaluated the uptake level of Cap protein using RAW264.7 cells and DC2.4 cells, which are often used as a model for primary macrophages and DCs, respectively. Additionally, antigen internalization, cytotoxicity, and cytokine release are size dependent. After incubation for 1 h, the Cap protein uptake by APCs was detected by IFA, and the fluorescence intensity corresponded to the number of Cap protein within APCs. As seen in Figure 4A, compared to the Cap protein group, the Cap-AuNP group had higher fluorescence intensity. More Cap protein was internalized when Cap protein was adsorbed on AuNPs (Figure 4B). These data suggested that the 23 nm diameter AuNPs improved the amount of Cap protein internalized into the APCs, and the Cap protein distributed in the cytoplasm, not the nucleus.
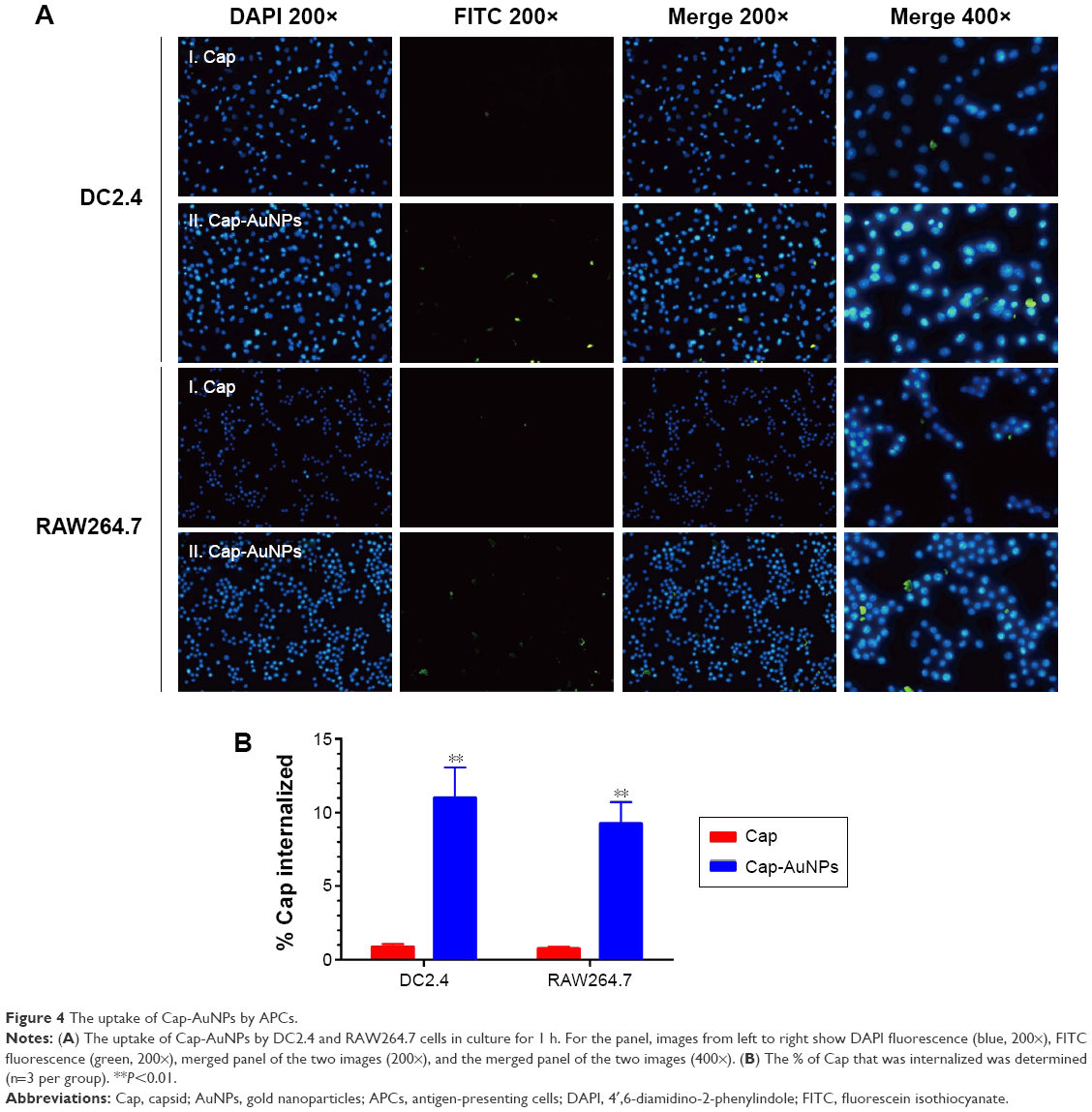 | Figure 4 The uptake of Cap-AuNPs by APCs. |
CCK-8 assays showed the Cap-AuNPs had no apparent cytotoxic effect on DC2.4 cells even at AuNP concentrations up to 100 μg/mL (Figure 5A), but had a significant cytotoxic effect on RAW264.7 cells at the concentration of 100 μg/mL (Figure 5B). This in vitro data suggested that 23 nm diameter AuNPs were a safe carrier.
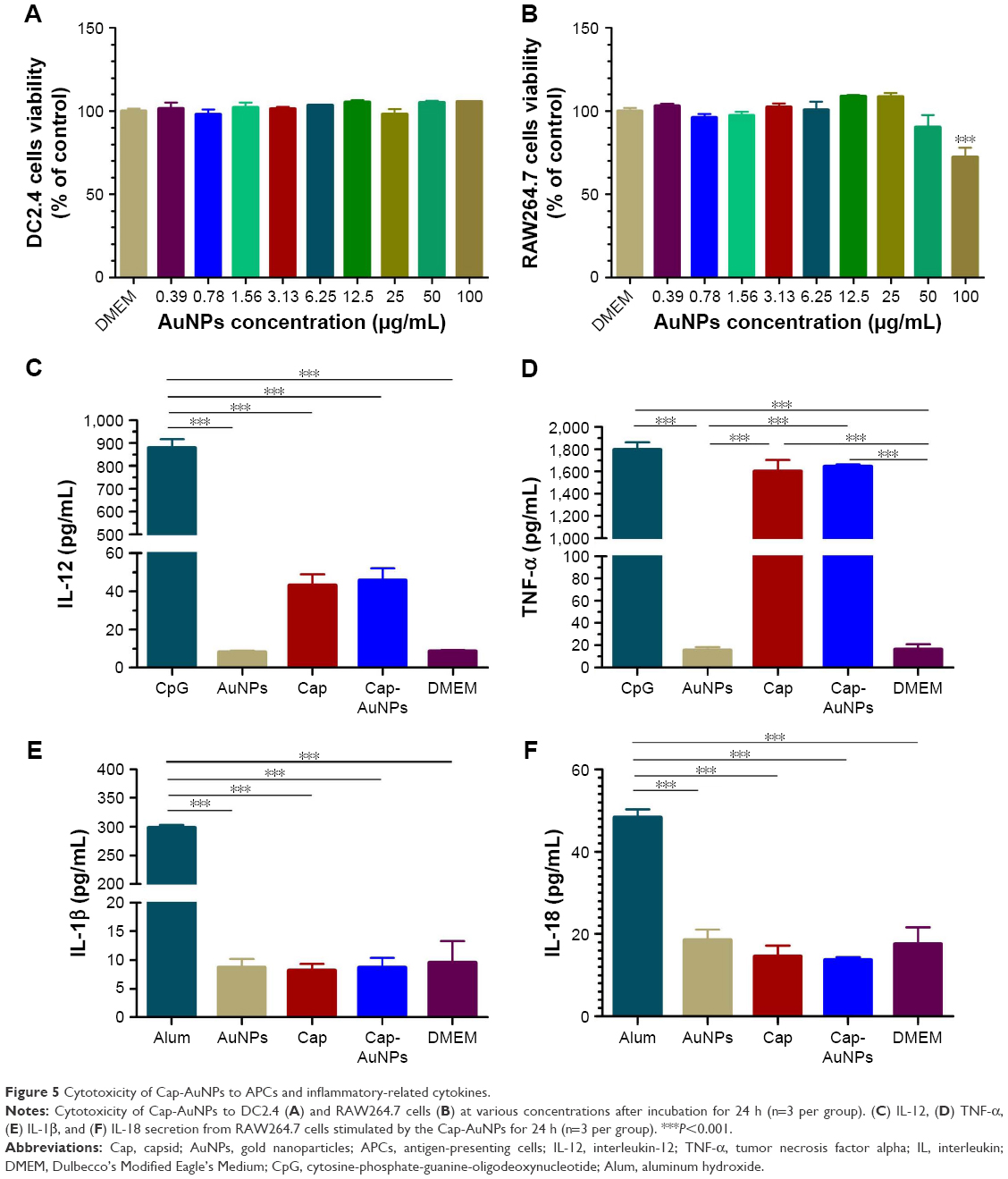 | Figure 5 Cytotoxicity of Cap-AuNPs to APCs and inflammatory-related cytokines. |
We next investigated the immunostimulatory effect of the Cap-AuNP conjugates and detected four types of cytokines: IL-12 (Figure 5C), TNF-α (Figure 5D), IL-1β (Figure 5E), and IL-18 (Figure 5F). IL-12 and TNF-α play pivotal roles in eliciting innate immune responses and regulation of adaptive immune responses, mainly produced by T cells and APCs.38,39 Although Cap could induce low-concentration IL-12 and high-concentration TNF-α, AuNPs had no influence on the release of IL-12 and TNF-α. Inflammasomes are known to be activated on exposure to nanoparticles due to lysosomal damage. We focused on IL-1β and IL-18, which promote inflammatory responses and are tightly regulated by inflammasomes. The concentration of IL-1β and IL-18 were low indicating no inflammasomes were produced after uptake of AuNPs by RAW264.7 cells.
These experiments showed that AuNPs can promote antigen phagocytosis and thus may help enhance the level of NAs. However, AuNPs did not selectively enhance antibodies against certain epitopes. Therefore, enhancing phagocytosis by APCs is not the underlying reason for AuNPs in enhancing NA levels.
Detection of antibodies levels
The antibody titers to Cap protein and PCV2 were detected by ELISA, while the NA titers were detected using IPMA. Vaccines are typically co-administered with adjuvants. In order to explore whether AuNPs are capable of enhancing NA levels by orientationally displaying epitopes in different types of adjuvants, we used three different types of adjuvants. CpG, a widely used soluble Th1-type adjuvant, is the ligand of TLR9 that recognizes pathogen-associated molecular patterns on foreign pathogens. Carbopol, a water-like substance, lightly cross-linked polymer of acrylic acid, has become a widely used adjuvant. Freund’s adjuvant, an oil-like substance, is the most commonly used adjuvant in animal experiments.40 We used these three representative adjuvants in the current study. As shown in Figure 6, AuNPs significantly increased the Cap-specific antibody levels in the no-adjuvant and CpG groups, while there was almost no effect in the Carbopol and Freund’s adjuvant groups (Figure 6A). We used the purified inactivated virus as fixative to detect PCV2-specific antibody levels. These antibodies bind to the outer surface of the virus. AuNPs significantly improved the PCV2-specific antibody levels with or without adjuvants, indicating that the addition of AuNPs resulted in more antibodies directed against the viral surface (Figure 6B).
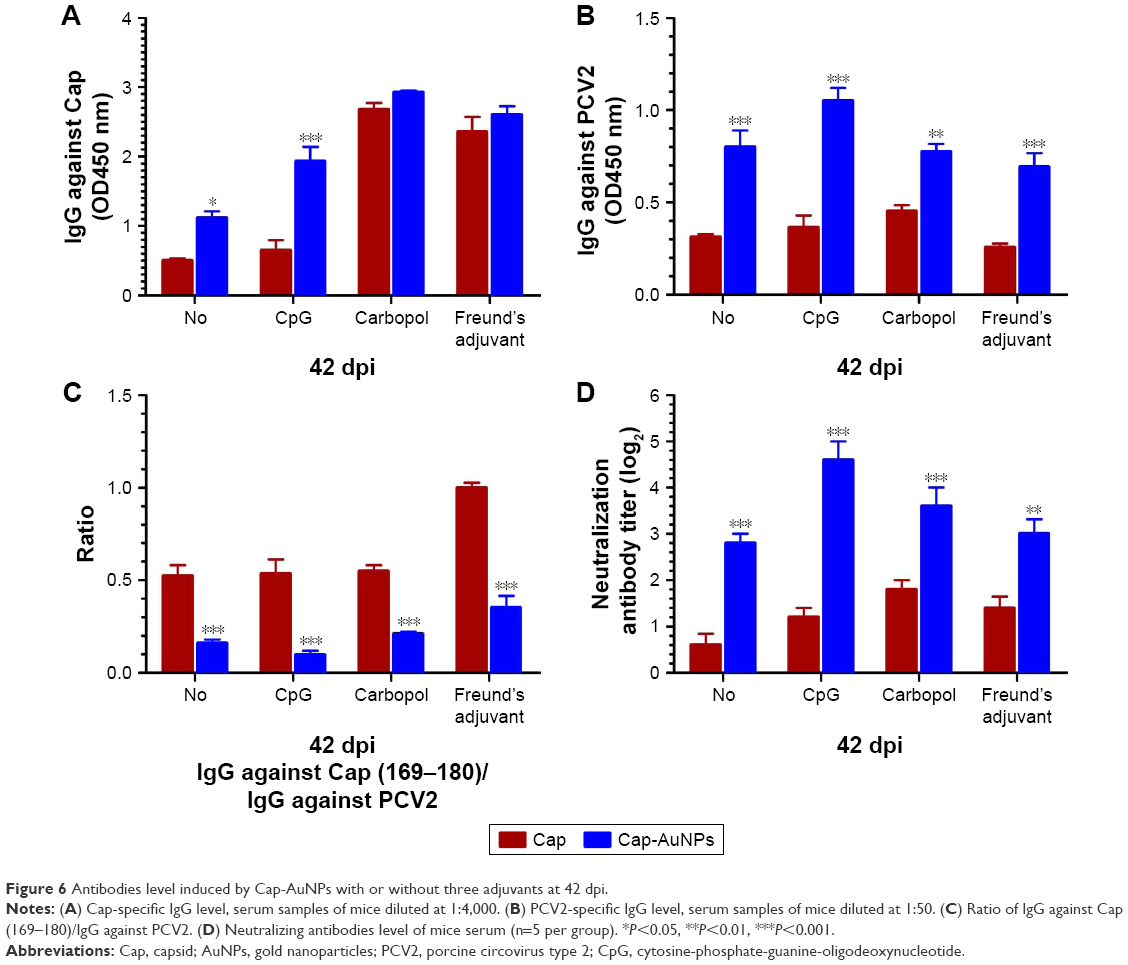 | Figure 6 Antibodies level induced by Cap-AuNPs with or without three adjuvants at 42 dpi. |
Antibodies against PCV2 were part of the antibody against Cap protein. The candidate vaccine used in animal experiments contained large amounts of uncoupled Cap protein (99.4%). Carbopol and Freund’s adjuvant are highly efficient adjuvants and can significantly increase IgG levels against Cap. Therefore, there was no significant difference in the levels of IgG between the Cap and Cap-AuNP groups when using Carbopol and Freund’s adjuvant (Figure 6A). High levels of antibody against PCV2 require epitopes on the surface of PCV2 to be fully recognized by APCs. Cap protein that binds to the surface of AuNPs can be efficiently recognized, swallowed, processed, and presented by APCs and plays a central role in activating immune response. Therefore, there was significant difference in the level of IgG against PCV2 between the Cap group and the Cap-AuNPs group when using Carbopol and Freund’s adjuvant (Figure 6B). Our previous experiments (data not shown) have confirmed that CpG weakly promoted the levels of anti-Cap and PCV2 IgG. Moreover, CpG had a synergistic effect with AuNPs and markedly enlarged immune effect. However, AuNPs played a key role rather than CpG. Similar results have been found by Tao et al.36 Thus, when CpG was used as adjuvant, there was a significant difference between the Cap and Cap-AuNPs groups for IgG against Cap and PCV2 (Figure 6A and B).
High levels of anti-Cap (169–180) antibodies is the result of host recognition of Cap protein monomer or fragments produced during virus replication and is associated with PCV2-SD, diverting the humoral response away from protective epitopes. The Cap (169–180) epitope located on the surface of Cap protein, however, due to its location and orientation, is not accessible on the surface of the PCV2. Cap protein is attached to AuNPs via an Au-S bond, thus masking some epitopes. Considering that AuNPs promoted the presentation of Cap protein and enhanced the antibody to all epitopes, the ratio of IgG against Cap (169–180)/IgG against PCV2 was more meaningful than IgG against Cap (169–180) level. After adding AuNPs, the ratio of IgG against Cap (169–180)/IgG against PCV2 was significantly reduced, indicating the Cap (169–180) epitope was buried (Figure 6C).
High levels of PCV2-specific antibodies and low levels of Cap (169–180)-specific antibody may help improve NA titers.27 NA titer was significantly improved after AuNPs were added (Figure 6D). On comparing the significant changes of Cap-specific antibodies with PCV2-specific antibodies and NA in the Carbopol and Freund’s adjuvant groups, we found that AuNPs significantly enhanced NA, rather than Cap-specific antibodies. These data indicated that AuNPs enhanced NAs mainly due to reasonable display of neutralizing epitopes and masking of the immunodominant epitope.
Effect of lyophilization on the immune response
As shown in Figure 7A, the Cap-specific antibody levels in non-lyophilized group was higher than in the lyophilized group (P=0.15), which may, in part, be due to the inactivation of a portion of Cap protein during lyophilization process. Interestingly, PCV2-specific and NA levels in lyophilized group were markedly higher than in non-lyophilized group (Figure 7B and D). The ratio of IgG against Cap (169–180)/IgG against PCV2 in lyophilized group was obviously lower than in non-lyophilized group (Figure 7C). These data suggested that the slight reduction of Cap-specific antibody did not affect NA and the stability of Cap-AuNPs conjugation was likely enhanced following lyophilization. Cap protein spatial orientation is more stable, thus offering an advantage in transportation and long-term preservation.41 Furthermore, these data confirmed that AuNPs promoted NA mainly due to the reasonable display of neutralizing epitopes.
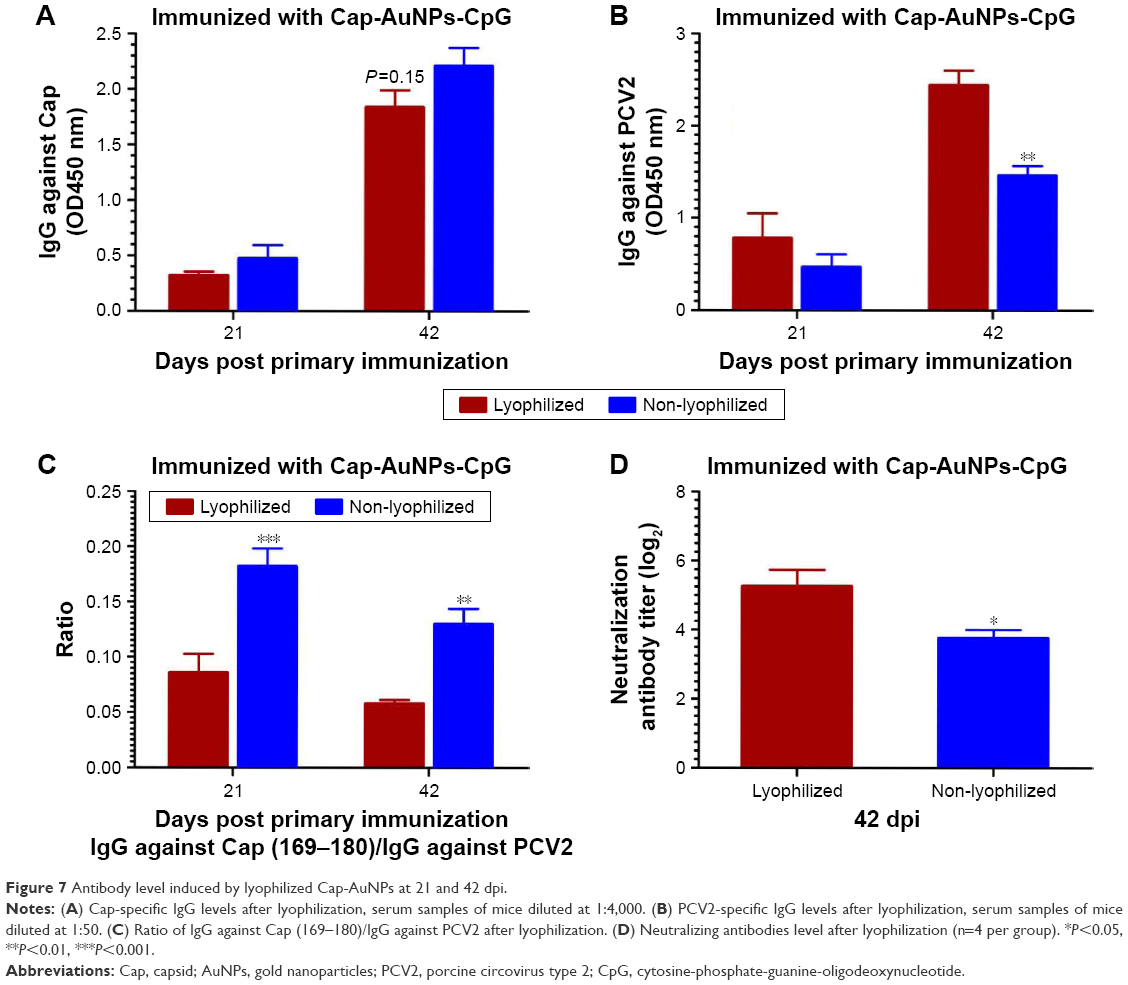 | Figure 7 Antibody level induced by lyophilized Cap-AuNPs at 21 and 42 dpi. |
The cellular immune effect of AuNPs
To evaluate the impact of Cap-AuNPs on T-cell proliferation in mice, Cap-specific lymphocyte proliferative responses were measured at 35 and 49 dpi (Figure 8A). The Cap-AuNP group induced significantly higher SI than the Cap or AuNP group.
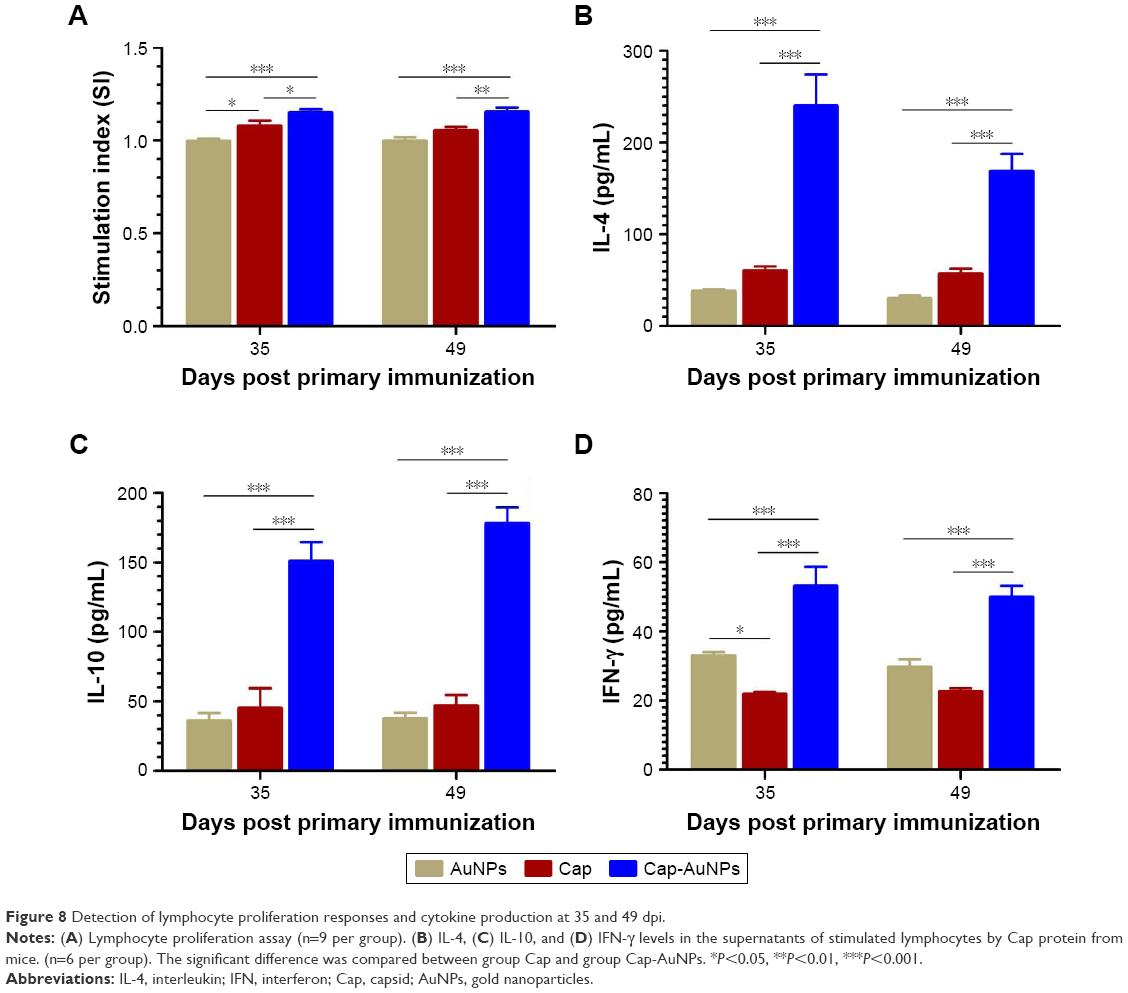 | Figure 8 Detection of lymphocyte proliferation responses and cytokine production at 35 and 49 dpi. |
The cytokine in spleen lymphocytes supernatant was measured by ELISA after the cells were stimulated with Cap protein. IL-4 and IL-10 are the archetypal Th2 cellular immune response cytokines that improve the production of IgG1. As shown in Figure 8B and C, Cap-AuNPs induced significantly high levels of IL-4 and IL-10 production than Cap or AuNPs. IFN-γ, Th1-biased cytokine, can promote the proliferation and differentiation of T cells, stimulate the secretion of IgG2a and IgG2b, activate macrophages, and enhance the cytotoxic effect of NK cells, conferring protection against viral infection. As shown in Figure 8D, Cap-AuNPs resulted in markedly higher level of IFN-γ than either the Cap or AuNPs. Interestingly, AuNPs induced a higher level of IFN-γ than Cap. We speculate that the increase in IFN-γ secretion may be due to the initial interaction of AuNPs with NK cells after AuNPs were injected into mice.42 In addition, it is likely that Cap protein mainly induced Th2 cell immunity and inhibited the secretion of Th1-based cytokines.43
Discussion
The mechanism that AuNPs improved the NA level of Cap-based PCV2 subunit vaccine can be attributed to two aspects: first, although Cap protein bound to AuNPs at a very low percentage (0.6%) of total immunized protein, this part of binding protein was efficiently taken up by APCs and played a key role in activating the immune response. Each AuNP was capable of displaying 6-fold the amount of Cap protein than PCV2, resulting in a higher degree of BCR cross-linking. The second and more important point, AuNPs directionally presented multiple neutralizing epitopes; thus, B cells simultaneously recognized more neutralizing epitopes, resulting in rapid proliferation of B-cell clones, which specifically target the neutralizing epitopes. Meanwhile AuNPs shielded the immunodominant epitope Cap (169–180), effectively reducing the antibody against this epitope, which plays a negative role in the production of NA.
Nanoparticles offer new horizons for subunit vaccine design.44 The most important mechanisms of nanoparticles in improving subunit vaccine effectiveness are by increasing the amount of antigen taken by APCs and multiplying the displayed subunit antigens in well-ordered arrays.18,19 Nanoparticles provide a customizable method for ligation of antigens by linking specific sites such as thiol groups to the gold particle. Furthermore, phosphorylation sites on proteins have a high affinity for aluminum hydroxide nanoparticles and tag polypeptides on antigens or domains on fusion-expressing antigens can be linked to nanoparticles, for example, poly-histidine tags can bind to various divalent metal cations and chitosan-binding domain can bind to chitosan nanoparticles. Nonetheless, only the antigens on the surface of the nanoparticles exhibit a certain orientation, and exposing the neutralizing epitope to the outside of the nanoparticles is sufficient for recognition by B cells.
High-resolution structures provide an opportunity to understand the structural basis for immunogenicity and immunodominance, improve vaccine efficacy, and broaden the range of vaccine-preventable diseases.45 According to the structural information of antigens, complete exposure of conserved or hidden neutralizing epitopes is the basic principle of structural vaccinology.46 Self-assembling protein nanoparticles have been used to achieve targeted display of antigens.18 Kanekiyo et al used a structure-based design strategy to generate a genetic fusion encoding hemagglutinin (HA) of influenza virus linked to ferritin. Interestingly, ferritin forms a nearly spherical particle composed of 24 subunits. HA was exposed on the outer surface of ferritin, mimicked its spatial position on the influenza H1N1 virus, and elicited broad NA against influenza virus.47 The fusion of the amino-terminus and carboxyl-terminus of the target antigen with the protein nanoparticles confines the spatial conformation of the antigen and thus it may be unfavorable to randomly select a connection point and the orientationally display of the antigen.48,49 Furthermore, a protein carrier is immunogenic, which is disadvantageous for the development of similar vaccines. These are limited to widely used structure-based nanoparticles in directional display strategy.
To the best of our knowledge, this is the first example of orientationally linking antigens to nanoparticles, to display neutralizing epitopes. The basic principle of designing such nanovaccines is to efficiently orient and repetitively display the neutralizing epitopes. First, identifying neutralizing epitopes depends on structural biology technology. Second, based on the antigenic structure, designing a site or domain which can adsorbe or conjugate to nanoparticles on the opposite surface of a neutralizing epitope is ideal. If possible, naturally occurring sites can be utilized, such as the unique thiol groups in this study. This takes into account that most neutralizing epitopes are formed from residues that are discontinuous in the primary amino acid sequence or buried by other residues; thus, there may be a need to redesign artificial scaffolds and display the neutralizing epitope on the surface of scaffold.46,49 Third, to synthesize nanoparticles that can efficiently bind to antigen or neutralizing epitope artificial scaffolds at a certain point, nanoparticles may need to be chemically modified. Antigen or artificial scaffolds can combine with biomaterials and then form nanoparticles. The introduction of structural-based antigen design methods combined with nanoparticles will contribute to the development of subunit vaccines.
Conclusion
It is possible to improve the level of NAs by directly orienting antigens to nanoparticles and fully exposing neutralizing epitopes based on the analysis of the antigen structure. In the current study, we selected the Cap protein of PCV2 as a model antigen, which has immunodominant epitopes and neutralizing epitopes. We conjugated Cap to AuNPs by the Au–thiol interaction, which was confirmed by FTIR, resulting in the immunodominant epitopes being obscured while fully exposing the neutralizing epitopes on the outer surface. AuNPs promoted phagocytosis of Cap protein by APCs without inducing inflammatory cytokine secretion, enhanced humoral and cellular immunity, and balanced Th1 and Th2 type cellular immunity. The NAs were significantly improved, while antibody against the immunodominant epitope was significantly reduced. Overall, this study demonstrates that the strategy of nanoparticles orientationally displaying antigen epitopes appears to be feasible for vaccine development.
Acknowledgments
The authors are grateful to Professor Ruiqin Li and Jianguo Li (Henan University of Chinese Medicine) for technical assistance with TEM analysis. The authors thank Yongxiao Chai for the assistance in preparation of the animal experiment. This work was funded by grants from the National Key Research and Development Program of China (2017YFD0501103, 2016YFD0500701, 2016YFD0500709), China Agriculture Research System (CARS-36), and Program of Henan Finance (No 201776-21).
Disclosure
The authors report no conflicts of interest in this work.
References
Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12(6):509–517. | ||
Goulder PJ, Lewin SR, Leitman EM. Paediatric HIV infection: the potential for cure. Nat Rev Immunol. 2016;16(4):259–271. | ||
Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nature Rev Immunol. 2014;14(5):315–328. | ||
van Oers MM, Pijlman GP, Vlak JM. Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J Gen Virol. 2015;96(Pt 1):6–23. | ||
Fahad S, Khan FA, Pandupuspitasari NS, et al. Recent developments in therapeutic protein expression technologies in plants. Biotechnol Lett. 2015;37(2):265–279. | ||
Bobbala S, Hook S. Is there an optimal formulation and delivery strategy for subunit vaccines? Pharm Res. 2016;33(9):2078–2097. | ||
Rappuoli R, Bottomley MJ, D’Oro U, Finco O, De Gregorio E. Reverse vaccinology 2.0: human immunology instructs vaccine antigen design. J Exp Med. 2016;213(4):469–481. | ||
Cicala C, Nawaz F, Jelicic K, Arthos J, Fauci AS. HIV-1 gp120: a target for therapeutics and vaccine design. Curr Drug Targets. 2016;17(1):122–135. | ||
Impagliazzo A, Milder F, Kuipers H, et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science. 2015;349(6254):1301–1306. | ||
Ostrowski M, Galeota JA, Jar AM, Platt KB, Osorio FA, Lopez OJ. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol. 2002;76(9):4241–4250. | ||
Smith SA, Nivarthi UK, de Alwis R, et al. Dengue virus prM-specific human monoclonal antibodies with virus replication-enhancing properties recognize a single immunodominant antigenic site. J Virol. 2015;90(2):780–789. | ||
Mok H, Lee S, Wright DW, Crowe JE Jr. Enhancement of the CD8+ T cell response to a subdominant epitope of respiratory syncytial virus by deletion of an immunodominant epitope. Vaccine. 2008;26(37):4775–4782. | ||
Trible BR, Suddith AW, Kerrigan MA, Cino-Ozuna AG, Hesse RA, Rowland RR. Recognition of the different structural forms of the capsid protein determines the outcome following infection with porcine circovirus type 2. J Virol. 2012;86(24):13508–13514. | ||
Du L, Tai W, Yang Y, et al. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat Commun. 2016;7:13473. | ||
Dormitzer PR, Grandi G, Rappuoli R. Structural vaccinology starts to deliver. Nat Rev Microbiol. 2012;10(12):807–813. | ||
Thomas S, Luxon BA. Vaccines based on structure-based design provide protection against infectious diseases. Expert Rev Vaccines. 2013;12(11):1301–1311. | ||
Dormitzer PR, Ulmer JB, Rappuoli R. Structure-based antigen design: a strategy for next generation vaccines. Trends Biotechnol. 2008;26(12):659–667. | ||
López-Sagaseta J, Malito E, Rappuoli R, Bottomley MJ. Self-assembling protein nanoparticles in the design of vaccines. Comput Struct Biotechnol J. 2015;14:58–68. eCollection 2016. | ||
Smith DM, Simon JK, Baker JR Jr. Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13(8):592–605. | ||
Yang J, Reth M. Receptor dissociation and B-cell activation. Curr Top Microbiol Immunol. 2016;393:27–43. | ||
Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–796. | ||
Salazar-González JA, González-Ortega O, Rosales-Mendoza S. Gold nanoparticles and vaccine development. Expert Rev Vaccines. 2015;14(9):1197–1211. | ||
Tsai DH, DelRio FW, Keene AM, et al. Adsorption and conformation of serum albumin protein on gold nanoparticles investigated using dimensional measurements and in situ spectroscopic methods. Langmuir. 2011;27(6):2464–2477. | ||
Segalés J. Best practice and future challenges for vaccination against porcine circovirus type 2. Expert Rev Vaccines. 2015;14(3):473–487. | ||
Burch D. Ten years of PCV2 in the UK. Pig Progress. 2009;25(6):21. | ||
Khayat R, Brunn N, Speir JA, et al. The 2.3-angstrom structure of porcine circovirus 2. J Virol. 2011;85(15):7856–7862. | ||
Trible BR, Rowland RR. Genetic variation of porcine circovirus type 2 (PCV2) and its relevance to vaccination, pathogenesis and diagnosis. Virus Res. 2012;164(1–2):68–77. | ||
Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A. Turkevich method for gold nanoparticle synthesis revisited. J Phys Chem B. 2006;110(32):15700–15707. | ||
Haiss W, Thanh NT, Aveyard J, Fernig DG. Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal Chem. 2007;79(11):4215–4221. | ||
Lou S, Ye JY, Li KQ, Wu A. A gold nanoparticle-based immunochromatographic assay: the influence of nanoparticulate size. Analyst. 2012;137(5):1174–1181. | ||
Niikura K, Matsunaga T, Suzuki T, et al. Gold nanoparticles as a vaccine platform: influence of size and shape on immunological responses in vitro and in vivo. ACS Nano. 2013;7(5):3926–3938. | ||
Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16(1):35–50. | ||
Tao Y, Zhang Y, Ju E, Ren H, Ren J. Gold nanocluster-based vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. Nanoscale. 2015;7(29):12419–12426. | ||
Fort M, Olvera A, Sibila M, Segalés J, Mateu E. Detection of neutralizing antibodies in postweaning multisystemic wasting syndrome (PMWS)-affected and non-PMWS-affected pigs. Vet Microbiol. 2007;125(3–4):244–255. | ||
Aryal S, Remant BKC, Dharmaraj N, Bhattarai N, Kim CH, Kim HY. Spectroscopic identification of S-Au interaction in cysteine capped gold nanoparticles. Spectrochim Acta A Mol Biomol Spectrosc. 2006;63(1):160–163. | ||
Tao W, Ziemer KS, Gill HS. Gold nanoparticle-M2e conjugate coformulated with CpG induces protective immunity against influenza A virus. Nanomedicine (Lond). 2014;9(2):237–251. | ||
Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220(Pt A):141–148. | ||
Novelli F, Casanova JL. The role of IL-12, IL-23 and IFN-gamma in immunity to viruses. Cytokine Growth Factor Rev. 2004;15(5):367–377. | ||
Wajant H, Scheurich P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol. 2001;33(1):19–32. | ||
Cranage MP, Fraser CA, Cope A, et al. Antibody responses after intravaginal immunisation with trimeric HIV-1 CN54 clade C gp140 in Carbopol gel are augmented by systemic priming or boosting with an adjuvanted formulation. Vaccine. 2011;29(7):1421–1430. | ||
Lévy R, Thanh NT, Doty RC, et al. Rational and combinatorial design of peptide capping ligands for gold nanoparticles. J Am Chem Soc. 2004;126(32):10076–10084. | ||
Liptrott NJ, Kendall E, Nieves DJ, et al. Partial mitigation of gold nanoparticle interactions with human lymphocytes by surface functionalization with a ‘mixed matrix’. Nanomedicine (Lond). 2014;9(16):2467–2479. | ||
Du Q, Huang Y, Wang T, et al. Porcine circovirus type 2 activates PI3K/Akt and p38 MAPK pathways to promote interleukin-10 production in macrophages via Cap interaction of gC1qR. Oncotarget. 2016;7(14):17492–17507. | ||
Zhao L, Seth A, Wibowo N, et al. Nanoparticle vaccines. Vaccine. 2014;32(3):327–337. | ||
Donnarumma D, Faleri A, Costantino P, Rappuoli R, Norais N. The role of structural proteomics in vaccine development: recent advances and future prospects. Expert Rev Proteomics. 2016;13(1):55–68. | ||
Correia BE, Bates JT, Loomis RJ, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507(7491):201–206. | ||
Kanekiyo M, Wei CJ, Yassine HM, et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499(7456):102–106. | ||
Burton DR. Scaffolding to build a rational vaccine design strategy. Proc Natl Acad Sci U S A. 2010;107(42):17859–17860. | ||
Ofek G, Guenaga FJ, Schief WR, et al. Elicitation of structure-specific antibodies by epitope scaffolds. Proc Natl Acad Sci U S A. 2010;107(42):17880–17887. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.