Back to Journals » Drug Design, Development and Therapy » Volume 20
Nanoparticle-Mediated PPAR Modulation of Macrophage Polarization in Radiation Enteritis: A Narrative Review
Authors Zhang JW ![]() , Weng L, Fu Y, Liu L, Chen J, Zhou JY
, Weng L, Fu Y, Liu L, Chen J, Zhou JY ![]() , Ma CY
, Ma CY
Received 12 January 2026
Accepted for publication 22 March 2026
Published 25 March 2026 Volume 2026:20 595650
DOI https://doi.org/10.2147/DDDT.S595650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Solomon Tadesse Zeleke
Jing-Wen Zhang,1,* Ling Weng,2,3,* Yi Fu,1 Lou Liu,1 Jie Chen,1 Ju-Ying Zhou,1 Chen-Ying Ma1
1Department of Radiation Oncology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, People’s Republic of China; 2Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, People’s Republic of China; 3School of Pharmacy, Nanjing University of Traditional Medicine, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chen-Ying Ma; Ju-ying Zhou, Department of Radiation Oncology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, 215006, People’s Republic of China, Tel +86 15695280357 ; +86 13962142066, Email [email protected]; [email protected]
Abstract: Radiation enteritis (RE) is a frequent and clinically important adverse effect of abdominal radiotherapy, characterized by acute inflammatory injury and longer-term fibrotic remodeling of the intestinal tract. Most patients receiving pelvic or abdominal irradiation experience measurable worsening of intestinal function. Macrophages orchestrate RE pathogenesis: tissue injury initially triggers a pro-inflammatory M1 phase that amplifies damage, followed by an M2 phase that promotes repair. Peroxisome proliferator-activated receptors (PPARs) have emerged as key regulators of macrophage polarization, with PPARγ playing a prominent role in promoting anti-inflammatory and tissue-restorative responses. This review explores the molecular mechanisms linking macrophage polarization and PPAR signaling in RE, reviews current PPAR-targeted therapies, and describes nanotechnology-based strategies for targeted intestinal delivery of PPAR modulators. We discuss how PPAR agonists modulate immune responses and how formulations engineered for localized intestinal activity may reduce systemic adverse effects. We also highlight progress in nanoparticle design, including carriers responsive to pH or reactive oxygen species and ligand-coated systems (e.g. hyaluronic acid or mannose) that target macrophages, thereby enhancing therapeutic delivery and cellular uptake. Finally, we summarize emerging omics-based approaches (single-cell transcriptomics, spatial profiling, and artificial-intelligence-driven design) that are accelerating the identification of novel therapeutic targets and informing the development of precision nanomedicines for RE.
Keywords: radiation enteritis, PPAR, macrophage, nanoparticle drug delivery, targeted nanotherapy
Introduction
Radiation enteritis (RE) is an inflammatory injury of the intestine secondary to abdominopelvic radiotherapy for malignancy. Acute gastrointestinal toxicity remains common after pelvic irradiation. In a prospective randomized double-blind placebo-controlled study of 234 patients receiving pelvic radiotherapy, grade 1 and grade 2 radiation-induced enteropathy occurred in 56%–64% and 22%–26% of patients, respectively.1 Chronic bowel injury also remains clinically relevant: in the updated Phase III NRG Oncology-RTOG 1203 trial, one year after radiotherapy, frequent diarrhea persisted in 15.1% of patients treated with conventional pelvic radiotherapy versus 5.8% of those treated with IMRT, and greater antidiarrheal use also remained more common in the conventional RT arm.2 In China, for example, >61% of patients with pelvic cancer receive therapeutic irradiation, with 75% experiencing acute intestinal injury and 5%–20% developing late effects.3 Complementing these clinical data, translational evidence has demonstrated chronic low-grade inflammation, mucus barrier disruption, and bacterial infiltration in irradiated intestinal mucosa up to two decades after pelvic radiotherapy, supporting a biological basis for persistent or late RE.4 Recent qualitative evidence further suggests that chronic bowel symptoms after pelvic radiotherapy remain under-recognized and insufficiently addressed in survivorship care, despite their substantial impact on quality of life.5 The condition can cause severe malabsorption, bleeding, and life-threatening complications, imposing a substantial burden on survivors’ quality of life.
RE results from radiation-induced DNA damage and epithelial cell death in the rapidly proliferating intestinal mucosa, accompanied by oxidative injury and maladaptive immune responses. Although advances in endoscopic and radiologic techniques facilitate diagnostic confirmation of RE, their value for preemptive therapeutic intervention remains limited. No approved curative therapies exist to prevent or reverse radiation injury; clinical management is largely palliative (eg., fluid support, dietary modification, antidiarrheals, corticosteroids), with surgery reserved for complications that arise. Therefore, there is an urgent need for molecularly targeted therapies that modulate the radiation response and accelerate mucosal healing. Macrophages are well-established mediators of intestinal homeostasis and inflammation. Tissue-resident and recruited macrophages span a functional spectrum that is often categorized into pro-inflammatory M1 and anti-inflammatory/wound-healing M2 phenotypes.6 This M1/M2 balance is a key determinant of whether tissue injury resolves or progresses to chronic fibrosis. After radiation injury, ionizing radiation skews macrophage polarization toward an M1 phenotype, triggering a pro-inflammatory cytokine cascade (eg., TNF-α, IL-1β, and IL-6) and increasing reactive oxygen species production, thereby exacerbating mucosal damage. In contrast, M2 macrophages secrete IL-10, TGF-β, and growth factors that support epithelial repair and dampen inflammation.7,8 Accordingly, suppressing M1 activity while promoting M2 polarization represents a promising therapeutic strategy.
Peroxisome proliferator-activated receptors (PPARs) are nuclear hormone receptors that regulate transcriptional programs involved in metabolism and inflammation.9 Among them, PPARγ is highly expressed in macrophages and plays a central role in macrophage polarization. PPARγ agonists attenuate inflammatory responses in colitis models, suggesting that targeting PPAR signaling in macrophages may ameliorate RE. In addition, nanotechnology-based drug delivery systems can improve tissue specificity. Nanoparticles (NPs) can be engineered to encapsulate PPAR-modulating agents, enhance their stability, and selectively target the inflamed gut or intestinal macrophages.6 In the following sections, we review the roles of macrophages in RE, PPAR signaling mechanisms in macrophages, current PPAR-targeted therapies for intestinal inflammation, and emerging nanodelivery approaches designed for precise intervention in radiation enteritis.
Macrophage Polarization in Radiation Enteritis
Macrophages are plastic innate immune cells that adopt distinct phenotypes in response to microenvironmental cues. They are broadly categorized as M1 (classically activated) and M2 (alternatively activated), each with characteristic markers and functions10 (Figure 1). M1 macrophages express surface markers such as CD86 and secrete pro-inflammatory mediators (TNF-α, IL-1β, and nitric oxide produced via iNOS). Although essential for host defense against pathogens and tumors, in sterile inflammation, they can cause collateral tissue damage. In contrast, M2 macrophages are characterized by receptors such as CD163 and CD200R, and by the production of anti-inflammatory cytokines (IL-10 and TGF-β), which promote wound healing and tissue remodeling. However, persistent or dysregulated M2 activity has been implicated in fibrosis and tumor progression.11
 |
Figure 1 Macrophage polarization. This diagram illustrates the two primary macrophage activation states. Classically activated M1 macrophages, stimulated by agents such as LPS or IFN-γ, express the surface marker CD86 and produce pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, which act to amplify tissue inflammation. Alternatively activated M2 macrophages, stimulated by IL-4 or IL-13, express markers like CD163 and CD200R and secrete anti-inflammatory mediators, such as IL-10 and TGF-β, which facilitate tissue repair. In RE, an initial M1-dominant response helps clear damaged cells but also exacerbates mucosal injury; a subsequent M2 response aids regeneration but can contribute to fibrosis if excessive or dysregulated. |
In RE, macrophages exhibit spatiotemporal dynamics that are pivotal in the transition from acute injury to chronic fibrosis. Under homeostatic conditions, intestinal macrophages primarily reside in the villi beneath the epithelium. Following irradiation, a distinct migratory pattern emerges: by day 3, macrophages begin relocating from villi to crypts; their numbers peak around regenerating crypts by day 6; and by day 30, they return to baseline villous localization.12 Functionally, M1-type macrophages predominate in the acute phase, exacerbating mucosal injury by releasing pro-inflammatory mediators (eg., IFN-γ, IL-6, IL-1β, and TNF-α), thereby amplifying the inflammatory cascade. Subsequently, the repair phase is dominated by M2-type macrophages, which coordinate restoration of the mucosal barrier, support antimicrobial defenses, and help prevent post-radiation infection.13 Macrophages are also major sources of enzymes involved in extracellular matrix turnover. Prolonged M1 activity can therefore promote pathological sequelae, including fibrosis and vascular sclerosis, before the onset of M2-dominated repair. This duality highlights the double-edged role of macrophages in RE: M1-mediated debris clearance is necessary but may sustain injurious inflammation, whereas M2-mediated healing can inadvertently promote fibrotic remodeling if unregulated.
Given these dynamics, modulating macrophage polarization is a rational therapeutic strategy for RE. Shifting macrophages from an M1- to an M2-like state may help interrupt the cycle of inflammation and tissue injury. Consistent with this idea, agents that modulate macrophage phenotype have shown efficacy in experimental colitis. Notably, PPARγ signaling has emerged as an endogenous switch that favors M2 polarization.14 The next section examines how PPAR receptors regulate these phenotypes.
PPAR Signaling in Macrophage Function
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors in the nuclear receptor family. They comprise three isoforms, PPARα, PPARβ/δ, and PPARγ, each encoded by a distinct gene and exhibiting unique tissue distributions. All PPARs share a modular structure, including an N-terminal ligand-independent activation function (AF-1), a highly conserved DNA-binding domain (DBD containing zinc fingers), a hinge region, and a C-terminal ligand-binding domain (LBD). In the canonical mechanism, ligand binding promotes heterodimerization of PPARs with retinoid X receptors (RXRs), enabling the complex to bind PPAR response elements (PPREs) in the promoters of target genes.15 This interaction activates transcriptional programs involved in lipid metabolism, glucose homeostasis, cell proliferation, and inflammation.16
PPAR function is regulated at multiple levels (Figure 2). Most prominently, ligand binding induces conformational changes that reshape interactions with co-activators and corepressors. In addition, PPAR activity is modulated by post-translational modifications (PTMs), including phosphorylation, SUMOylation, ubiquitination, and acetylation, with subtype-specific PTM patterns.
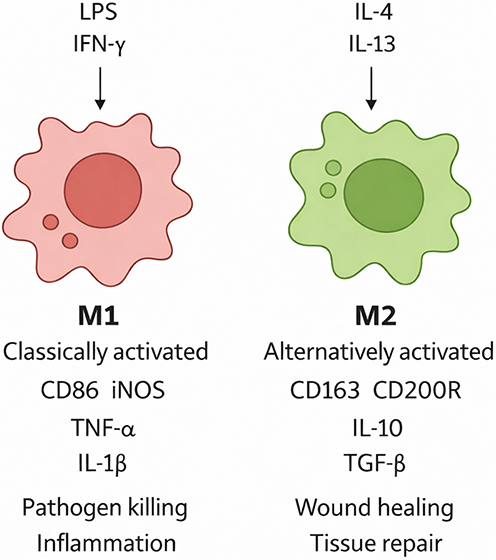 |
Figure 2 PPAR structure and mechanism. Schematic of PPAR isoforms (α, β/δ, γ) with their DNA-binding (DBD) and ligand-binding (LBD) domains, and illustration of two primary transcriptional regulatory modes. Upon ligand binding, PPAR–RXR heterodimers bind to PPREs and recruit co-activator complexes to initiate gene transcription (trans-activation). PPARs can also inhibit inflammatory gene expression by interfering with pro-inflammatory transcription factors, such as NF-κB/AP-1, by recruiting corepressor complexes to their target promoters (trans-repression). |
For PPARα, phosphorylation has been reported at serine residues 12, 21, 179, and 230. Phosphorylation at Ser12 and Ser21, mediated by mitogen-activated protein kinase (MAPK), enhances AF-1 activity, whereas phosphorylation at Ser179 and Ser230, mediated by protein kinase C (PKC), reduces NCoR recruitment and thereby increases PPARα activity. In contrast, SUMOylation of PPARα occurs at lysine residues 185 and 385, selectively recruiting NCoR to inhibit its transcriptional activity.17 Furthermore, the E3 ubiquitin ligase Mouse Double Minute 2 Homolog (MDM2) regulates PPARα stability by promoting its ubiquitination and degradation, whereas disruption of the MDM2–PPARα interaction stabilizes PPARα.18
PPARγ PTMs exhibit greater diversity and functional consequences. Phosphorylation of PPARγ plays a crucial role in regulating metabolic homeostasis. Phosphorylation of tyrosine residue 78 significantly suppresses the expression of pro-inflammatory genes in adipocytes and alleviates adipose tissue inflammation.19 Conversely, CDK5-mediated phosphorylation at serine 273 is linked to abnormal expression of genes like adiponectin and insulin resistance.20,21 PKCα-mediated phosphorylation at Thr166 impairs the induction of beige adipocytes within white adipose tissue (WAT).22 This modification has therefore emerged as a therapeutic target: certain PPARγ ligands with weak transcriptional activity can exert hypoglycemic effects by blocking phosphorylation at Thr166, thereby decoupling metabolic benefits from the classic thiazolidinedione (TZD) adverse effects of weight gain and fluid retention.23
Furthermore, PPARγ phosphorylation influences tumor cell proliferation and apoptosis. In the setting of DNA damage, agents such as carboplatin induce phosphorylation of PPARγ at Ser273, thereby enhancing DNA damage repair. In contrast, non-classical PPARγ agonist ligands (NALs) inhibit this process and can sensitize tumor cells to chemotherapy.21 Phosphorylation of PPARγ at Ser84 activates the PFKFB4 promoter, increasing expression of this key glycolytic enzyme and thereby promoting glycolysis and proliferation in hepatocellular carcinoma cells.24 This phosphorylation event also enhances proliferation in human HT1080 fibrosarcoma cells and increases resistance to 5-fluorouracil.25 Collectively, these findings suggest that inhibiting site-specific phosphorylation of PPARγ may be a viable strategy for anticancer drug development.
SUMOylation of PPARγ, which predominantly occurs at lysine residues within the AF-1 domain, is another key regulatory modification with diverse functional effects. One well-described anti-inflammatory mechanism involves SUMOylation at sites such as K177, which promotes stable association of corepressor complexes (eg., NCoR) at NF-κB response elements. This interaction potently transrepresses NF-κB–driven gene expression, reducing pro-inflammatory cytokine production and promoting a shift in macrophages toward an anti-inflammatory phenotype.26,27
However, studies suggest that the role of the K365 site is nuanced. Although it is implicated in this pathway, evidence indicates that it may not be a direct SUMOylation target but instead facilitates ligand-dependent SUMOylation within the AF-1 domain via the ligand-binding domain (LBD).28 PPARγ SUMOylation also serves as a pivotal hub linking lipid metabolism regulation to tumor suppression. Work by the Katafuchi group identified SUMOylation at the K107 site of PPARγ in white adipose tissue (WAT). This modification enhances insulin sensitivity without the weight gain commonly associated with TZDs.28 Furthermore, the antitumor effects of certain TZDs are mechanistically linked to SUMOylation at K107 and K395. This modification triggers a cascade of metabolic alterations, including upregulation of lipid synthesis and fatty acid β-oxidation genes, leading to NADPH depletion, increased mitochondrial reactive oxygen species (ROS), and disruption of cellular redox balance, collectively inhibiting lung cancer cell proliferation.29
The acetylation status of PPARγ, which is dynamically regulated by opposing acetyltransferase and deacetylase enzymes, profoundly influences its metabolic functions. PPARγ acetylation promotes the conversion of brown adipose tissue (BAT) to white adipose tissue (WAT) and accelerates the progression of age-related metabolic disorders.30 Conversely, PPARγ deacetylation reverses these effects, confirming the critical role of this modification.31 Moreover, targeted deacetylation at Lys268 and Lys293 enhances insulin sensitivity with efficacy comparable to thiazolidinediones (TZDs), while avoiding some of their typical side effects, thereby identifying a potential molecular target for the treatment of obesity and diabetes.32
Cellular PPARγ abundance is predominantly controlled by the ubiquitin–proteasome pathway. This process is mediated by E3 ubiquitin ligases, which can be functionally divided into two classes: pro-degradative and pro-synthetic, that engage distinct domains of PPARγ. Ultimately, both classes catalyze PPARγ ubiquitination, promoting proteasomal degradation and suppressing lipogenic activity.33,34
In contrast to PPARγ, the regulatory landscape of PPARδ/β is less well characterized. Epidermal growth factor (EGF) stimulates phosphorylation of tyrosine 108 in PPARδ, thereby recruiting heat shock protein 90 (HSP90) to prevent lysosomal degradation.35 SUMO-specific protease 2 (SENP2) promotes fatty acid oxidation (FAO)-associated gene expression by de-SUMOylating lysine 104 of PPARδ.36
PPARγ is the most extensively studied member of the PPAR family and is widely expressed in adipose tissue and the intestinal epithelium, as well as in macrophages and dendritic cells. In macrophages, PPARγ drives polarization toward an anti-inflammatory phenotype. Macrophages derived from PPARγ conditional knockout mice exhibit significantly reduced arginase activity, a canonical marker of M2 polarization.37 Mechanistically, ligand-activated PPARγ suppresses the production of pro-inflammatory cytokines by antagonizing key transcription factors. For example, PPARγ can directly interact with NF-κB and AP-1: upon ligand binding, PPARγ–RXR dimers recruit corepressor complexes to NF-κB/AP-1 target promoters, thereby mediating transrepression of inflammatory genes. In vitro, treatment of macrophages with PPARγ agonists inhibits LPS-induced TNF-α and IL-6 expression through this mechanism.38 Similarly, PPARγ activation suppresses the NLRP3 inflammasome by reducing ROS and thioredoxin-interacting protein (TXNIP), thereby further inhibiting IL-1β maturation.39 Endogenously, colonic cells synthesize PPARγ ligands; notably, 13-oxo-ODE (a metabolite of linoleic acid) binds PPARγ with high affinity in the gut epithelium and exerts anti-inflammatory effects.40 Diminished PPARγ activity has been observed in patients with inflammatory bowel disease (IBD), implicating this pathway in intestinal inflammation.
In contrast, PPARα and PPARβ/δ also exhibit anti-inflammatory properties that are relevant to intestinal macrophage function and RE pathophysiology. PPARα, which is highly expressed in the liver, heart, muscle, and immune cells, plays a crucial role in regulating intestinal macrophage phenotypes. Mechanistically, PPARα suppresses pro-inflammatory cytokine secretion and promotes IL-10 expression by inhibiting the NF-κB/AP-1 pathway. It also maintains gut microbiota homeostasis, preventing abnormal macrophage activation, ultimately suppressing intestinal inflammation and preserving mucosal immune balance. In PPARα-deficient mice, intestinal macrophages abnormally secrete elevated levels of IL-1β, IL-6, and TNF-α, leading to persistent intestinal mucosal inflammation.41 Moreover, PPARα’s classical function is to regulate lipid metabolism. Protein-protein interaction (PPI) analysis confirms direct interactions between PPARα and lipid metabolism-related genes (eg., ACOX1, CPT1A), ensuring energy supply for macrophages within the inflammatory microenvironment and indirectly supporting their function.9 For example, fibrates (PPARα agonists) increase IL-10 and decrease IL-17 in colitis models.42
Similarly, PPARβ/δ, which is broadly expressed, exerts anti-inflammatory effects by modulating the immune-inflammatory pathways directly. Activated PPARβ/δ binds directly to the κB element region of pro-inflammatory gene promoters (such as TNFα), competitively displacing the NF-κB p65 subunit bound there and thereby inhibiting transcription of pro-inflammatory genes. Furthermore, PPARβ/δ recruits the co-inhibitory factor BCL6 to further suppress NF-κB-mediated transcriptional activation.43 PPARβ/δ agonists can also directly alter the morphological and functional phenotype of human macrophages, inducing IL-4-mediated M2-type anti-inflammatory characteristics while reversing or inhibiting LPS/IFN-γ-induced M1-type pro-inflammatory polarization.43 Pan-PPAR and dual PPARα/γ agonists are under active investigation. Collectively, PPARs act as molecular rheostats, shifting macrophage gene expression from pro-inflammatory programs toward pro-resolution and repair. These observations provide a strong rationale for targeting PPAR signaling in RE to promote tissue-protective macrophage phenotypes.
PPAR-Targeted Therapeutic Strategies
Given the anti-inflammatory functions of PPARγ (and, to a lesser extent, PPARα and PPARδ), synthetic agonists have been investigated in models of colitis and other GI inflammation. Although the initial injury in RE arises from physical ionizing radiation, the downstream pathological cascade it triggers exhibits substantial histological and molecular similarities to ulcerative colitis (UC) and to chemically induced animal models. This includes depletion of intestinal epithelial stem cells, breakdown of the mucosal barrier due to decreased expressions of tight junction protein, secondary dysregulation of the immune microenvironment (including NF-κB pathway activation and pro-inflammatory cytokine storms), and gut microbiota dysbiosis.44,45 These intestinal diseases exhibit high consistency in the pathophysiological progression of mucosal damage and in treatment endpoints centered on ‘mucosal healing’. Consequently, chemically induced colitis (such as DSS and TNBS) and clinical UC patient cohorts are frequently utilized as valuable translational parallel alternative models. They are widely applied to investigate the pathogenic mechanisms of RE and to screen potential targeted therapeutic interventions.
The thiazolidinedione (TZD) class of drugs, including rosiglitazone and pioglitazone, comprises high-affinity PPARγ agonists originally developed for diabetes. In experimental colitis models (eg., dextran sodium sulfate or TNBS), TZDs consistently ameliorate mucosal damage, suppress pro-inflammatory cytokines, and improve barrier function.46 For example, in Clostridioides difficile–infected mice, pioglitazone alleviated colitis symptoms and downregulated TNF-α, IL-6, and other cytokine genes.46 In vitro, pioglitazone-treated macrophages exhibit reduced NF-κB activity.47 However, systemic TZDs are associated with significant adverse effects, including fluid retention, weight gain, bone loss, and increased cardiovascular risk.48 Pioglitazone has been shown to promote bladder cancer in rodents.49 Although no clear carcinogenic risk has been identified in human clinical studies, long-term monitoring is warranted. Meanwhile, the first-generation TZD rosiglitazone has been withdrawn from the market because of severe hepatotoxicity. Such adverse events constrain the clinical utility of TZDs for chronic GI conditions. An innovative strategy to overcome this limitation is site-specific delivery: for instance, AS002 is an oral PPARγ agonist engineered to act locally in the colon. In preclinical studies, AS002 showed negligible systemic absorption yet upregulated the expression of PPARγ-responsive genes in the colonic mucosa. In biopsies from patients with ulcerative colitis, AS002 potently induced adipophilin (a PPARγ target) and reduced TNF-α and IL-1β levels. In a DSS colitis mouse model, rectal AS002 ameliorated and even reversed inflammation, as evidenced by lower myeloperoxidase and cytokine levels.50 Together, these findings indicate that gut-restricted PPARγ agonism can provide therapeutic benefit while minimizing systemic exposure. Endogenous PPARγ ligands are primarily derived from host metabolism or the intestinal microbiota and generally exhibit high biocompatibility, low toxicity, and favorable safety profiles. 13-Oxo-ODE and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) function as endogenous PPARγ ligands,40 activating PPARγ-mediated anti-inflammatory pathways while potentially avoiding the systemic metabolic side effects associated with thiazolidinediones (TZDs).51 Butyrate, an endogenous PPARγ ligand produced by gut bacteria, mitigates intestinal inflammation by suppressing the dysbiotic expansion of Enterobacteriaceae, suggesting a potential therapeutic approach for inflammatory bowel diseases.52 Recent preclinical work has also identified natural PPARγ modulators. Ellagic acid activated the RXRβ–PPARγ pathway in a CT26 tumor-induced cachexia mouse model, supporting its relevance as a natural PPARγ modulator.53
PPARα agonists (fibrates) have also shown anti-inflammatory potential. Fenofibrate and WY-14643, for example, enhanced IL-10 production and expanded regulatory T cells in colitis models.54 In one mouse colitis study, fenofibrate improved mucosal healing. However, fibrates can elevate plasma homocysteine and creatinine levels, and their gut-specific effects are less well characterized.55 Endogenous PPARα agonists primarily include fatty acids and their derivatives, endocannabinoids, and specific arachidonic acid metabolites. Palmitoylethanolamide (PEA) significantly attenuates colitis symptoms in mice by activating PPARα, thereby inhibiting the expression of pro-inflammatory markers such as iNOS and COX-2.56 PEA occurs naturally in egg yolks and peanuts. Given its favorable safety profile, low incidence of side effects, and minimal risk of dependence or toxicity, PEA has been used clinically as a dietary supplement.57
Dual PPAR agonists and pan-PPAR modulators represent another therapeutic avenue, as coactivation of multiple PPARs may synergize anti-inflammatory and metabolic effects. For example, the PPARα/γ agonist saroglitazar has demonstrated antifibrotic properties in intestinal fibroblasts.58,59 The PPARα/δ agonist elafibranor (ELA) inhibits M1 polarization signals (TLR4, IFNγ) in the ileum while stimulating IL-10/STAT3 signaling to promote M2 polarization, thereby restoring intestinal barrier integrity in DSS-induced colitis mice.60 Conjugated linoleic acid (CLA), a natural PPAR ligand, ameliorates colitis by activating PPARγ and PPARδ.61 Punicic acid (PUA) is another natural dual agonist of PPARδ that simultaneously activates PPARγ. It inhibits the secretion of pro-inflammatory factors by macrophages while promoting intestinal mucosal repair.62
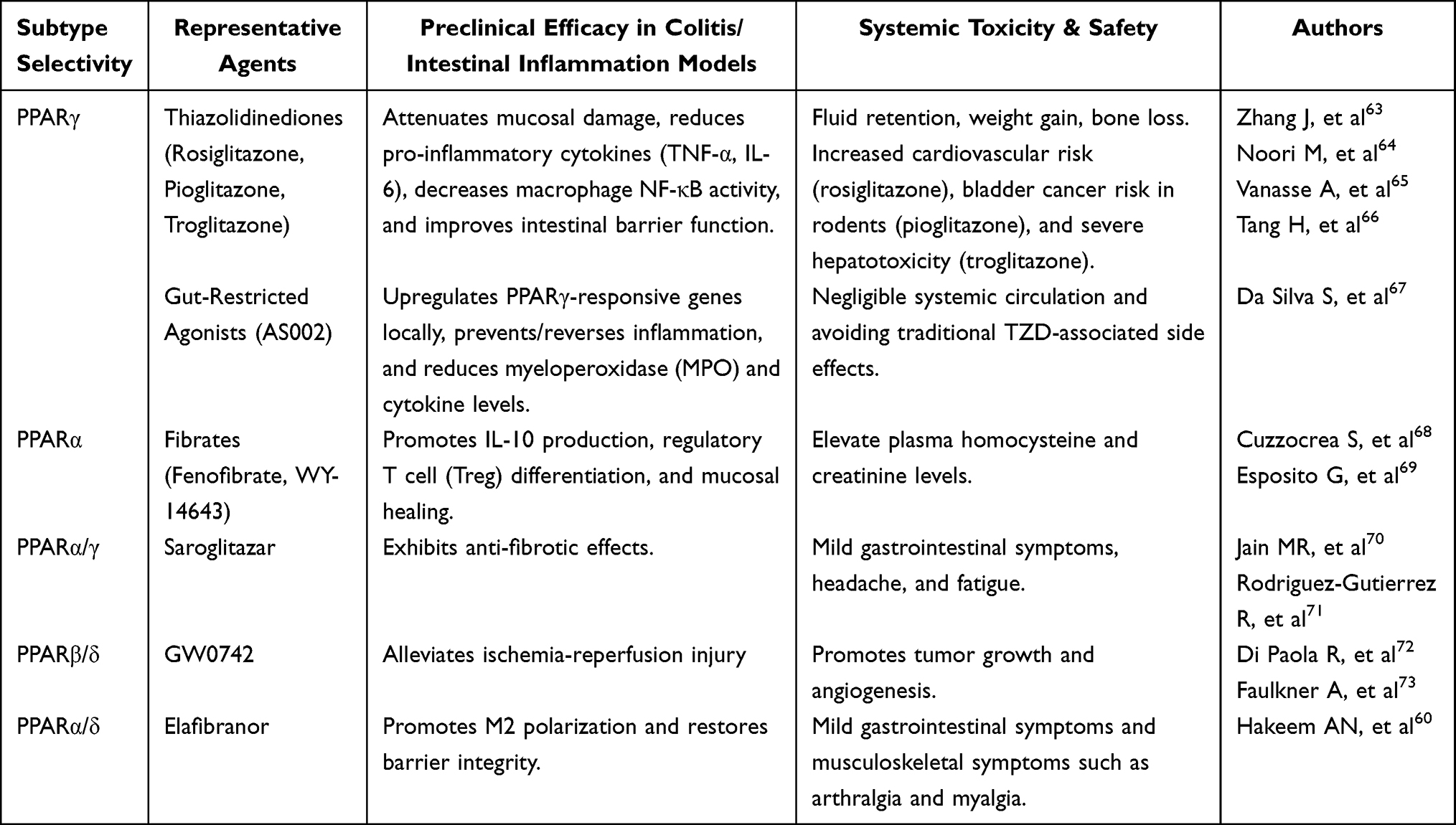 |
Table 1 Comparison of the Therapeutic Efficacy and Safety of Synthetic PPAR Receptor Agonists |
 |
Table 2 Comparison of the Therapeutic Efficacy and Safety of Endogenous PPAR Receptor Agonists |
In summary, both synthetic and endogenous PPAR agonists demonstrate therapeutic potential in experimental intestinal inflammation. As shown in Table 1, synthetic agonists, including TZDs, fibrates, gut-restricted PPARγ agonists, and dual PPAR modulators, can attenuate mucosal injury, suppress inflammatory signaling, and promote repair. However, their systemic adverse effects remain a major obstacle to translation.60,63–73 As shown in Table 2, endogenous and naturally derived agonists generally provide a safer and more intestine-compatible alternative, with favorable anti-inflammatory activity but variable potency and limited translational validation.62,69,74–78 Together, these findings suggest that PPAR-targeted therapy is promising for RE. Nevertheless, its clinical application will likely depend on strategies that deliver these agents specifically to the gut and macrophages while minimizing systemic toxicity. Nanoparticle-based platforms may provide such a solution through targeted and controlled drug delivery.
Nanoparticle Delivery Systems for RE
Nanotechnology offers a versatile platform for the precise delivery of therapeutics to diseased tissue. In the context of RE, ideal nanocarriers should (1) tolerate the harsh gastrointestinal milieu, (2) release their therapeutic cargo within inflamed intestinal mucosa, and (3) be selectively internalized by macrophages. Advances in materials science have produced diverse nanoparticle (NP) architectures capable of meeting these objectives.79 Nanoparticles can encapsulate small-molecule drugs, biologics, or nucleic acids, protecting them from degradation and enabling controlled release. They can also be conjugated with targeting ligands or engineered to release cargo in response to specific biological stimuli.
Lipid-based systems are widely used for their biocompatibility and their ability to fuse with cell membranes.80 Polymeric NPs enable tunable degradation rates and drug-release profiles.81 Inorganic nanocarriers, such as mesoporous silica, iron oxide, or gold NPs, provide robust scaffolds with versatile surface chemistry.82 Bio-derived carriers, including exosomes and cell membrane–coated NPs, exploit natural vesicles to evade the immune system. For example, intestinal macrophages can be targeted by coating NPs with macrophage cell membranes, leveraging homotypic interactions.83
Key advantages of nanocarriers in RE include improved drug stability in the GI tract and the potential for colon targeting. Oral nanoparticles can be engineered to withstand gastric acidity and enzymatic degradation. Once in the colon, inflammation can increase vascular permeability and promote local retention of nanomaterials. A recent review highlights that nanoparticle-based oral systems can enhance the bioavailability and sustained retention of therapeutics in the colon, thereby improving outcomes in ulcerative colitis.84 By confining the drug to the site of pathology, NPs diminish systemic side effects and permit lower dosages.
To preferentially engage macrophages, nanoparticle surfaces can be decorated with ligands that bind cognate receptors expressed on macrophages. Hyaluronic acid (HA) is one such ligand: it binds CD44, which is highly expressed on activated macrophages in inflamed tissue. A 2024 study engineered rapamycin-loaded liposomes incorporating HA and a ROS-cleavable linker. These ROS-responsive, HA-coated liposomes (Ra@TH) remained inert in healthy tissue but released rapamycin in response to elevated ROS in inflamed mucosa. In vitro and in mouse colitis models, Ra@TH preferentially delivered the drug to CD44+ cells, induced autophagy, and inhibited NF-κB activation.85 This strategy illustrates how coupling inflammatory cues (ROS) with cell-surface targeting (HA) can localize therapy to macrophages in the injured gut.
Zhao et al86 synthesized lactoferrin-modified liposomes (LF-lipo). Because activated colonic macrophages overexpress low-density lipoprotein receptor-related protein-1 (LRP-1), and lactoferrin binds this receptor with high affinity, these liposomes can be actively guided to their targets. Liu et al87 used PEGylated lipids conjugated to the S2P peptide to recognize the Stabilin-2 (STAB2) receptor on M1 macrophages, thereby achieving selective targeting. Huang et al88 developed an oral nanomedicine with a surface coating of low-molecular-weight heparin (LMWH). LMWH selectively binds integrin αM, which is abundant on macrophages within inflamed gut tissue. Both cell-based and in vivo experiments confirmed this selective binding and the associated therapeutic benefit.
Dextran and mannose are also widely used macrophage-targeting ligands. Macrophages readily internalize dextran-coated NPs via scavenger receptors. For example, Jain et al89 formulated dextran-based carriers encapsulating IL-10; these particles were efficiently taken up by M1 macrophages and reprogrammed them toward an M2 phenotype. Polyethylenimine NPs modified with mannose have likewise been shown to target monocytes/macrophages and promote M1-to-M2 polarization. These approaches could be adapted to deliver PPAR agonists; for instance, a mannose-decorated NP carrying a PPARγ ligand could preferentially modulate gut macrophages to induce an anti-inflammatory program.
Other targeting strategies include Fc fragments, peptides, or antibodies directed against macrophage markers. The nanoparticle drug delivery platform demonstrates unique advantages in pharmacokinetics and targeted distribution. Compared to conventional free drugs that are rapidly degraded, nanoparticle carriers effectively resist destruction by gastrointestinal digestive fluids and prolong retention time in damaged intestines. Following oral administration, Lip-LF primarily targets and accumulates in the small intestine—a region highly consistent with the key lesion site in radiation-induced intestinal injury (RIII). Cy5 fluorescence labeling and tracking further demonstrate that Lip-LF exhibits a significantly longer retention time in the gastrointestinal tract compared to free LF.90 Zhang et al91 developed a microalgae-nanoparticle integrated system (SP@ASXnano) that exhibits a significantly longer retention time in the gastrointestinal tract than standalone ASXnano, thereby substantially enhancing drug exposure duration at target sites. Importantly, nanocarriers can also passively target macrophages via phagocytosis; in inflamed tissue, these cells avidly internalize particulate material. Uptake efficiency is size-dependent, with diameters of 50–200 nm yielding maximal phagocytic sequestration.92 Specific small-molecule polymers can employ the Enhanced Permeability and Retention (EPR) effect to target inflamed tissues by penetrating the inflamed, hyperpermeable vascular walls. Therefore, by carefully optimizing NP properties, one can maximize macrophage specificity while minimizing uptake by other cells.
The radiation enteritis microenvironment exhibits mild acidosis (pH ≈ 6.5) and pronounced oxidative stress (elevated H2O2). Nanocarriers engineered with pH-sensitive polymers can exploit these cues: the polymers remain stable at physiological pH but selectively solubilize in the acidic, inflamed mucosa, enabling site-specific drug release.93 Similarly, ROS-sensitive linkers can cleave in the presence of elevated ROS, as demonstrated in the RA@TH system. Enzyme-responsive NPs can also be designed to respond to proteases or matrix metalloproteinases that are enriched in RE.
Zhang et al used oxybutylcyclodextrin (OxbCD) to assemble a nanoscale drug-delivery carrier. Markedly elevated H2O2 concentrations at sites of colonic inflammation trigger OxbCD hydrolysis, leading to carrier disruption and release of the encapsulated drug. The OxbCD nanomatrix may also mimic catalase activity, conferring H2O2-scavenging capacity. This can synergize with the loaded drug to reduce the major ROS species (•O2−, H2O2, and •OH), thereby producing anti-inflammatory and antioxidant effects.94 Thus, the nanoparticle remains structurally intact during systemic circulation or luminal transit and releases its therapeutic payload only upon encountering inflamed intestinal mucosa; such stimulus-responsive delivery increases local drug concentrations while minimizing systemic exposure.
Beyond the RA@TH (ROS-responsive, hyaluronic acid-coated liposomes loaded with rapamycin) paradigm, additional proof-of-concept systems support the utility of targeted, stimulus-responsive delivery. Polymeric nanoparticles surface-functionalized with cell-penetrating peptides have been used to selectively deliver curcumin to colonic macrophages in experimental colitis, resulting in marked attenuation of disease activity.95 Nanoparticles coated with anionic heparin can bind integrins and accumulate in inflamed colonic mucosa, serving as “Trojan horses” for small-molecule drugs. Lipid nanoparticles that mimic high-density lipoprotein (HDL) can deliver PPARα agonists to macrophages via scavenger receptors.96 Iron oxide or cerium oxide NPs have intrinsic antioxidant properties and can both neutralize ROS and shift macrophage polarization.96 Moreover, engineered cerium oxide nanoparticles exhibit remarkably selective radioprotective effects. Specifically, they provide substantial protection to normal tissues against radiation damage while offering no protection to tumor cells. Zamyatina et al97 and Xiang et al98 independently confirmed their highly selective radiosensitizing effects on EMT6/P cells (mouse adenocarcinoma) and on lung metastasis, respectively. Although many of these systems remain at the proof-of-concept stage, they underscore the feasibility of targeting macrophage PPAR pathways in GI inflammation.
Light-mediated therapies, including photothermal therapy (PTT), photodynamic therapy (PDT), and photobiomodulation (PBM), represent an emerging class of stimuli-responsive strategies for RE treatment. Characterized by non-invasiveness and precise temporal and spatial control, this approach has been widely applied in fields such as oncology, inflammation, and tissue repair. Common types include photodynamic therapy (PDT), photothermal therapy (PTT), and photobiomodulation (PBM). In recent years, the anti-inflammatory and tissue-repairing effects of light-mediated therapy in radiation-induced intestinal injury have garnered significant attention. Such strategies enable precise temporal and spatial activation of nanoparticle delivery systems, thereby enabling targeted regulation of macrophage polarization within the intestinal inflammatory microenvironment following radiotherapy. The site of radiation enteritis features a thick, viscous mucus barrier, making it difficult for conventional nanoparticles to reach macrophage surfaces. The localized thermal effect generated by Transient mild photothermia (TMP; ≈ 42°C, 10 min) reduces the viscoelasticity of the mucus layer while imparting a “nanomotor”-like mechanical motion to nanoparticles, significantly enhancing penetration efficiency.99
The photothermal effect triggers structural changes in the nanocarrier and controlled release of its payload. For example, PBN (Prussian Blue + NO donor SNP) exhibits strong near-infrared (808 nm) absorption. Upon NIR irradiation, it efficiently converts light energy into mild thermal energy (temperature rises to 49–51.3°C). This approach avoids the non-specific damage associated with conventional high-temperature PTT while simultaneously breaking the Fe-NO coordination bond within PBN. This controlled release of nitric oxide (NO) promotes intestinal epithelial cell proliferation and migration, directly repairing damaged intestinal mucosa. Additionally, the photothermal effect disrupts the electrostatic cross-linked network of the FU/CS hydrogel, accelerating drug release, increasing drug concentration at the site of inflammation, and enhancing anti-inflammatory effects.100 Photodynamic therapy further synergistically amplifies PPAR agonist-mediated anti-inflammatory and reparative signals. ROS generated by photodynamic processes modulate signaling pathways such as PI3K/AKT/mTOR, inducing macrophage autophagy and metabolic reprogramming to reshape inflammatory responses.101 He et al102 developed the orally administered prebiotic-modified nanoplatform CBF@LCP. This platform targets M1 macrophages overexpressing folate receptors at colonic inflammatory sites via folate-mediated delivery. Upon near-infrared light activation, it triggers synergistic photodynamic/photothermal therapy (PDT/PTT) to ablate M1 macrophages, promote polarization toward the M2 phenotype, simultaneously restore gut microbiota balance, and inhibit the PI3K/AKT signaling pathway.
Exosomes, as natural nanocarriers, enable specific targeting of intestinal macrophages without complex artificial modifications, thereby avoiding off-target effects associated with systemic distribution. Under inflammatory pathological conditions, compromised intestinal epithelial barriers and increased local vascular permeability allow exosomes—with their nanoscale dimensions (30–150 nm)—to passively accumulate at diseased mucosal sites via a mechanism analogous to the “Enhanced Permeability and Retention (EPR) effect”. Furthermore, the outer leaflet of the exosome lipid bilayer naturally enriches phosphatidylserine, which serves as a classic “eat-me” signal. This signal is efficiently recognized by scavenger receptors on the surface of locally resident intestinal macrophages, triggering endocytosis. Furthermore, extensively expressed transmembrane proteins (eg., CD9, CD63, CD81) and specific integrins on the exosome surface mediate specific adhesion to extracellular matrix components and cellular receptors within the inflammatory microenvironment.103,104 Fluorescent labeling has confirmed their efficient delivery to intestinal tissues via tail vein injection, and colocalization analysis demonstrates their active uptake by intestinal macrophages. Qian et al105 found that compared with exosomes prepared under normoxic conditions, hypoxia-pretreated adipose-derived mesenchymal stem cell-derived exosomes (HExos) strongly induced macrophage polarization from pro-inflammatory M1 to anti-inflammatory M2. This significantly reduced levels of pro-inflammatory factors such as TNF-α, IL-1β, and IL-6, increased secretion of anti-inflammatory factors such as IL-10, and simultaneously upregulated M2 markers (Arg1, CD206) and downregulated M1 markers (iNOS). Liu et al106 extracted endometrial regenerative cell-derived exosomes (ERC-Exos) and demonstrated that SIRT6 within these exosomes directly binds to the NF-κB p65 subunit, inhibiting the phosphorylation of IKKα/β and p65, blocking NF-κB inflammatory pathway activation. This induces macrophage polarization from pro-inflammatory M1 to anti-inflammatory M2 types. Both in vivo and in vitro experiments have demonstrated its therapeutic efficacy against intestinal inflammation. Yang’s team developed RSPO1-loaded small extracellular vesicles (evRSPO1) using an innovative approach. Unlike conventional vesicle cargo encapsulation, these engineered vesicles employ a surface-loading mode mediated by heparan sulfate proteoglycans (HSPGs).
Engineered extracellular vesicles (EVs) are emerging not merely as passive natural carriers, but as programmable nanoplatforms whose material architecture, surface display strategy, and loading method directly determine target engagement and regenerative efficacy. In the dual-ligand exosome system reported by Yang et al107 WNT ligands were actively loaded onto exosomes through donor-cell engineering using the Wntless (WLS) scaffold, whereas RSPO1 was co-displayed on the same vesicle to generate exoWNT3A/RSPO1, thereby enabling synergistic activation of WNT/β-catenin signaling and downstream crosstalk with the PPARα pathway. This dual-ligand configuration promoted organoid growth and accelerated tissue repair in acute and chronic injury models.107 In contrast, the orally deliverable evRSPO1 platform used a surface-loading mechanism mediated by heparan sulfate proteoglycans (HSPGs), allowing active RSPO1 protein to remain functionally exposed on the small extracellular vesicle surface; after oral administration, evRSPO1 activated WNT/β-catenin signaling in the intestinal crypt stem-cell niche and significantly accelerated regeneration in a radiation-induced intestinal injury model.108 Together, these studies indicate that the therapeutic performance of engineered EVs depends not only on cargo identity but also on whether bioactive ligands are incorporated by donor-cell engineering, surface tethering, or post-isolation modification, as these choices shape vesicle stability, receptor accessibility, tissue biodistribution, and signaling potency. In the context of radiation enteritis, such engineering strategies are especially attractive because they combine the intrinsic biocompatibility of natural vesicles with the potential to deliver instructive regenerative signals rather than anti-inflammatory drugs alone.
To better illustrate the engineering rationale and therapeutic workflow of these bio-derived nanovesicles, Figure 3 summarizes the major steps involved in donor-cell engineering, vesicle preparation and characterization, delivery, intestinal targeting, and downstream regenerative signaling. A comparative perspective is also informative. Compared with conventional synthetic nanoparticles, engineered EVs offer superior biological compatibility, membrane protein-mediated cellular recognition, and a lower risk of mucosal irritation, which may be advantageous in the irradiated intestine.
 |
Figure 3 Engineering workflow and therapeutic mechanism of extracellular vesicle-based regenerative nanoplatforms for radiation enteritis. Donor cells are first selected and genetically or biochemically engineered to enable ligand display or cargo loading onto extracellular vesicles (EVs). Two representative strategies are illustrated: (i) WLS-mediated active loading of WNT ligands into engineered exosomes, and (ii) HSPG-mediated surface loading of RSPO1 onto small extracellular vesicles. After isolation and characterization, the engineered vesicles can be administered either intravenously or orally. Intravenously delivered EVs may preferentially accumulate in injured intestinal tissue, whereas orally delivered vesicles can access the intestinal crypt niche after transit through the gastrointestinal tract. Following target engagement, these vesicles activate WNT/β-catenin signaling and induce downstream PPARα-associated reparative programs, thereby promoting intestinal stem cell proliferation, epithelial regeneration, barrier restoration, and the resolution of inflammation. Abbreviations: EV, extracellular vesicle; WLS, Wntless; HSPG, heparan sulfate proteoglycan; RSPO1, R-spondin1; PPARα, peroxisome proliferator-activated receptor alpha. |
A comparative perspective is also informative. Compared with conventional synthetic nanoparticles, engineered EVs offer superior biological compatibility, membrane protein-mediated cellular recognition, and a lower risk of mucosal irritation, which may be advantageous in the irradiated intestine. However, they also pose distinct manufacturing challenges, including batch-to-batch heterogeneity, lower production yields, limited control over ligand density, and the need for standardized potency assays. Mechanistically, exoWNT3A/RSPO1 represents a donor-cell-programmed, dual-ligand signaling vesicle optimized for pathway amplification, whereas evRSPO1 is an orally deliverable, surface-loaded regenerative vesicle optimized for intestinal access and stem-cell activation. These differences suggest that future RE-oriented EV design should explicitly match engineering strategy to therapeutic objective: intravenous systems may be preferable for systemic homing to irradiated bowel, while orally delivered vesicles may be more suitable for direct mucosal and crypt targeting.
From a translational standpoint, several issues deserve closer investigation. First, head-to-head comparisons between engineered EVs and synthetic nanocarriers should be performed in radiation-specific intestinal injury models to define differences in intestinal retention, macrophage uptake, epithelial repair, and systemic exposure. Second, manufacturing variables—including donor-cell source, ligand-display strategy, purification route, and storage stability—should be standardized, because these parameters are likely to influence EV potency and reproducibility. Third, quantitative pharmacokinetic and biodistribution studies are needed to determine whether orally delivered vesicles primarily act luminally, crypt-specifically, or after trans-epithelial transport, whereas intravenously delivered vesicles require assessment of spleen/liver sequestration versus injured-intestine homing. Finally, future studies should evaluate whether engineered EVs can be combined with PPAR agonists, antioxidants, or microbiota-modulating interventions to achieve synergistic mucosal repair in RE.
These recent studies indicate that engineered EVs are not merely passive carriers but biologically instructive nanoplatforms whose preparation strategies and surface architectures directly influence signaling potency and tissue repair. The therapeutic efficacy of engineered EVs depends on both vesicle construction and delivery route, which together determine target accessibility, signaling activation, and regenerative outcome.
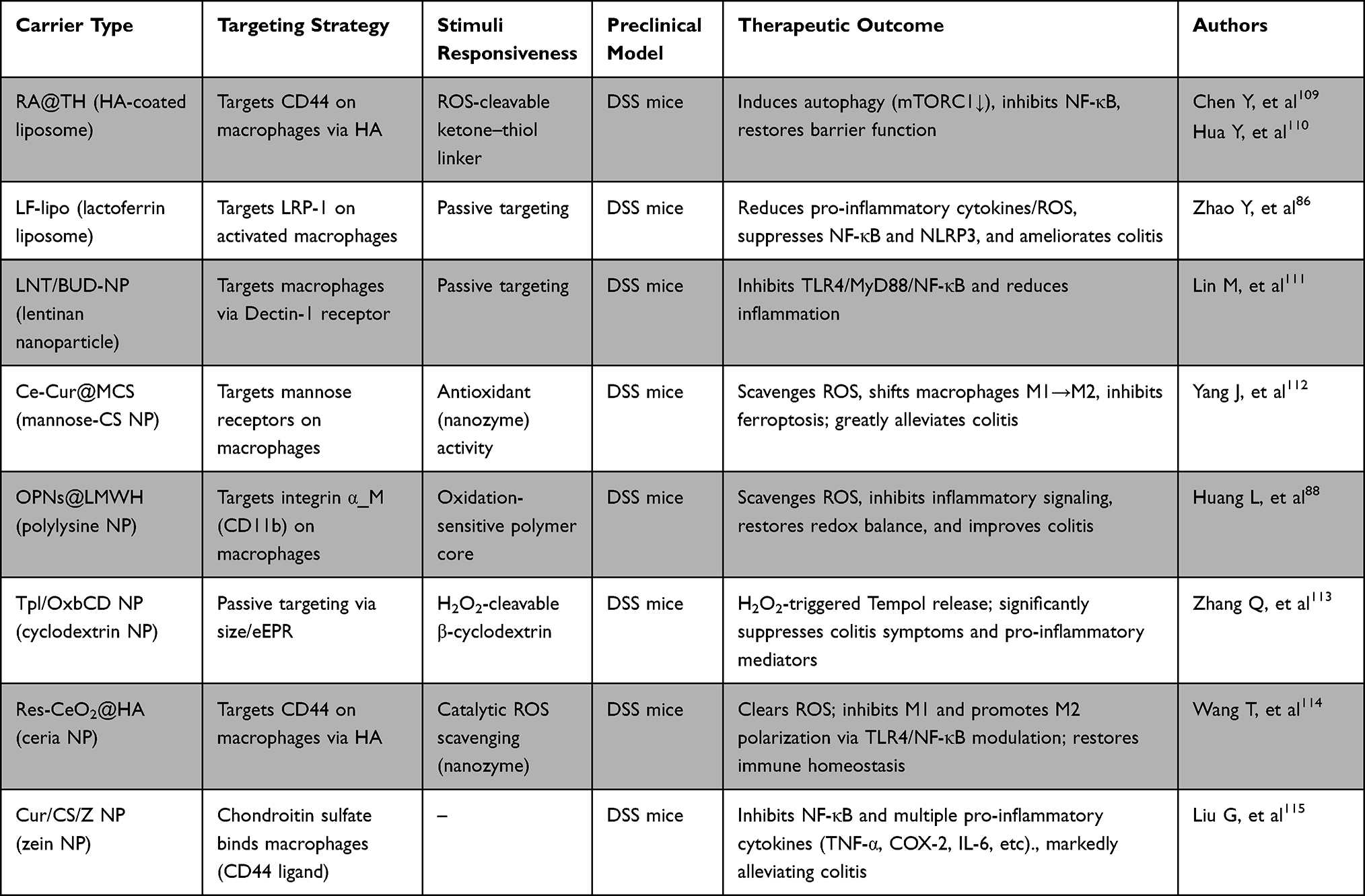 |
Table 3 Comparison of Nanoparticle Platforms for Intestinal Macrophage Targeting |
To facilitate comparison across these emerging strategies, Table 3 summarizes the major nanoparticle platforms developed for intestinal macrophage targeting, including their material composition, targeting ligands, stimulus-responsive features, representative payloads, and principal therapeutic advantages.86,88,109–115 Collectively, these platforms highlight several convergent design principles, namely enhanced accumulation in inflamed intestinal tissue, selective uptake by macrophages, and controlled drug release in response to the local inflammatory microenvironment.
To further evaluate the translational relevance of these platforms, recent studies have begun to clarify how nanoparticle composition and formulation influence intestinal biodistribution, local retention, and safety in irradiated or inflamed bowel. Recent evidence suggests that radiation-induced intestinal injury may enhance the local accumulation and retention of nanocarriers in the gut. For example, orally administered lactoferrin-loaded liposomal nanoparticles displayed markedly prolonged intestinal residence compared with free lactoferrin; at 120 min after administration, the liposomal formulation remained predominantly in the jejunum, whereas free lactoferrin had largely transited to the cecum and colon.90 Likewise, intravenously injected mesenchymal stem cell-derived extracellular vesicles (MSC-EVs) preferentially accumulated in irradiated intestines in a dose-dependent manner, with significantly stronger fluorescence signals detected in the intestines of mice exposed to 8–15 Gy abdominal irradiation than in non-irradiated controls.116 These findings indicate that epithelial injury, mucus alteration, and increased permeability after irradiation may create a microenvironment that facilitates nanoparticle adhesion and retention. However, despite encouraging biodistribution data, no nanoparticle-based therapy has yet entered clinical trials specifically for inflammatory bowel disease (IBD) or radiation enteritis (RE), underscoring the persistent translational gap between preclinical proof-of-concept studies and clinical application.117 In addition, safety profiles differ substantially across material classes. Lipid-based formulations generally exhibit favorable gastrointestinal biocompatibility; repeated oral administration of lactoferrin-loaded liposomes did not induce histopathological abnormalities in major organs in mice.90 In contrast, certain inorganic nanoparticles, particularly metal- or metal oxide-based systems, may provoke oxidative stress, inflammatory responses, and uncertain long-term tissue accumulation, which are of particular concern in the context of radiation-damaged mucosa.94 Collectively, these observations highlight that nanoparticle design for RE should consider not only therapeutic payload delivery but also formulation-dependent pharmacokinetics, intestinal biodistribution, biodegradability, and platform-specific safety.
Despite their promise, nanoparticles face several obstacles. In the bloodstream, proteins can adsorb onto NPs, potentially masking targeting ligands and reducing targeting efficiency. Repeated dosing may also induce anti-PEG antibodies, leading to accelerated blood clearance.118 After oral administration, NPs encounter two principal physiological barriers: the viscous mucus layer and rapid gastrointestinal transit.
To address these challenges, investigators are developing (i) mucoadhesive surface coatings that transiently anchor carriers to the epithelium and (ii) nanoparticle-encapsulating microparticles with pH-insensitive shells that dissolve specifically in the colonic lumen, thereby releasing nanoparticles at the intended site of action (Figure 4). Notably, recent clinical trials in IBD indicate that nano-medicine approaches are advancing toward clinical reality.
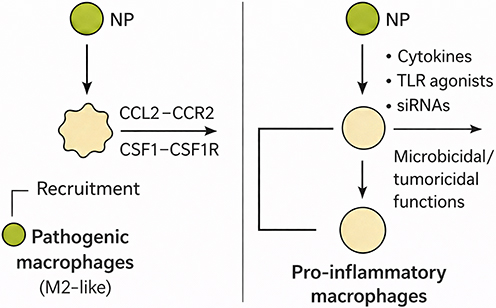 |
Figure 4 Nanoparticle strategies to modulate macrophages. Schematic illustration of nanoparticle (NP)-mediated modulation of macrophage polarization and function in inflammatory contexts. The diagram depicts two distinct pathways by which NPs influence macrophage behavior. On the left, NPs facilitate the recruitment of pathogenic, M2-like macrophages through the CCL2-CCR2 or CSF-1-CSF1R signaling axes, promoting their migration to sites of inflammation or pathology. On the right, NPs loaded with cytokines, TLR agonists, or siRNAs activate pro-inflammatory macrophages, enhancing their microbicidal and tumoricidal functions. This dual mechanism underscores the versatility of NPs in reprogramming macrophage phenotypes, potentially via PPAR pathways, to mitigate radiation-induced intestinal damage by balancing pro- and anti-inflammatory responses. |
Research Frontiers and Emerging Technologies
While engineered nanoparticle delivery systems have significantly advanced targeted modulation of PPAR pathways in macrophage polarization for the management of radiation enteritis (RE), integrating research frontiers such as single-cell omics and microbiomics promises to unravel intricate cellular and microbial dynamics, thereby facilitating the development of next-generation precision therapies.
Single-cell omics analyzes biomolecular information, including genomics, transcriptomics, proteomics, and epigenomics, at the resolution of individual cells. It overcomes key limitations of traditional bulk omics by resolving cellular heterogeneity and delineating the molecular features and functional states of single cells within complex biological systems. In studies of radiation-induced intestinal injury, single-cell omics has provided additional evidence that PPARs modulate radiation enteritis pathology by acting on macrophages. Motevasseli et al119 analyzed macrophage gene-expression matrices using pySCENIC (a Python-based tool for scRNA-seq analysis) and identified PPAR as a key transcription factor governing macrophage phenotypic transitions. Lu et al120 used scRNA-seq to show that Ly6c⁺ monocytes give rise to pro-inflammatory and resident macrophages in the context of radiation-induced intestinal injury (RII). They further elucidated a positive feedback loop between macrophages and endothelial cells that exacerbates the inflammatory response, suggesting that macrophages are key targets for mitigating early radiation-induced inflammation. Zhang et al113 applied gene co-expression network analysis (hdWGCNA) and showed that M1 macrophages stimulate fibroblasts via PPAR, ECM, and interferon-γ signaling pathways, thereby driving excessive ECM deposition. Single-cell omics also enables more precise assessments of therapeutic efficacy by identifying genes highly expressed in inflammatory macrophages and tracking changes in their expression after treatment.
Mennillo et al121 used scRNA-seq and CITE-seq to compare the expression of TIMP1, SOD2, TYMP, C15orf48, and CD63 in MNPs from healthy controls (HC), patients with ulcerative colitis (UC), and patients with treated UC. This analysis supported the efficacy of vedolizumab (VDZ) in controlling intestinal inflammation.
In radiation enteritis, the intestinal microbiota undergoes dysbiosis: taxonomic diversity contracts, and community structure shifts toward opportunistic lineages. Integrated 16S rRNA profiling, shotgun metagenomics, transcriptomics, and metabolomics indicate that pathobionts—particularly Actinobacteria and Proteobacteria—expand at the expense of protective commensals, exemplified by a marked decline in Bacteroides.122,123 Nepelska et al124 reported that most Actinobacteria strains elicit moderate or no PPARγ activation and may even indirectly reduce PPARγ levels. Conversely, certain Bacteroidetes strains activate PPARγ through ERK1/2-mediated phosphorylation cascades. Thus, the radiation-enteritis–associated perturbation of the gut microbiome converges with PPAR-dependent regulatory circuits. This congruence implies that dysbiosis partly fuels disease pathogenesis by suppressing PPARγ signalling. Conversely, pharmacological or endogenous activation of PPAR isoforms, exemplified by oleoylethanolamine (OEA), a high-affinity PPAR-α agonist, restructures the microbial community toward a probiotic-rich profile, thereby reinforcing the epithelial barrier and attenuating mucosal inflammation. This reciprocal regulation between gut microbiota and PPARs provides a microbiological basis for developing PPAR-targeted nanomedicines.125
In summary, nanocarriers enable precise localization of anti-inflammatory drugs to intestinal macrophages. By directing PPAR agonists to the right cells at the right time, these systems aim to maximize therapeutic benefit while minimizing adverse effects. The next generation of RE therapies will likely integrate immunology, lipid signaling, and nanotechnology to advance truly personalized medicine.
Conclusion
Radiation enteritis remains a significant unmet clinical need, and targeted therapies are still lacking. As reviewed above, macrophages play a central role in RE pathogenesis, and PPAR signaling is a key regulator of macrophage function. Activating PPARγ (and, to some extent, PPARα/δ) can shift macrophages away from pro-inflammatory programs and promote mucosal repair. Preclinical evidence consistently supports the efficacy of PPAR agonists in suppressing gastrointestinal inflammation. However, clinical translation will depend on minimizing systemic off-target toxicity and achieving site-restricted delivery to diseased mucosa.
Nano-medicine offers tools to address these challenges. Advances in nanoparticle engineering have enabled the development of carriers that target the injured gut and deliver PPAR modulators to macrophages. Recent studies illustrate this concept: ROS-sensitive, HA-targeted rapamycin-loaded liposomes and mannose-directed IL-10 nanoparticles both repolarize macrophages at sites of inflammation.
Analogous carriers can be loaded with PPAR agonists, mRNA, or gene editors; a single nanoparticle that both inhibits NF-κB and activates PPARγ could elicit a synergistic anti-inflammatory response.
However, the clinical translation of nanomedicines is hindered by the complexity of biological systems, unpredictable in vivo pharmacokinetics, and reliance on traditional, iterative development workflows. Advances in artificial intelligence (AI) are catalyzing a paradigm shift in nano-medicine research and development, moving from empirically guided (“experience-driven”) to computationally powered (“data-driven”) methodologies. By integrating multi-omics datasets, coupling CRISPR-based screening with machine learning, and analyzing single-cell data from inflammatory niches, AI can predict nanocarrier binding affinities for specific cellular markers. This capability streamlines target identification and supports the rational design of targeted formulations.126
AI also accelerates nanoparticle design and synthesis. Through in silico simulation of intracellular nanocarrier trafficking, AI models can predict stimulus-dependent release kinetics, facilitating the development of smart, stimuli-responsive delivery systems.127 In addition, by analyzing preclinical data, AI tools can forecast nanocarrier biodistribution, targeting efficiency, and potential toxicities. This addresses the persistent challenge of unpredictable nano-drug pharmacokinetics and may substantially reduce the time and cost of in vivo experimentation.128
Looking ahead, several areas warrant attention. First, additional in vivo studies in RE models are needed to evaluate these approaches. Most available data come from chemically induced colitis or infection models; radiation-specific studies are required to determine whether macrophage-targeting nanotherapies can prevent long-term fibrosis. Second, the safety and integrity of nanocarriers must be established. Immune clearance, off-target uptake, and the potential immunogenicity of novel materials require careful assessment. Third, clinical translation would benefit from biomarkers that identify patients most likely to respond to PPAR-targeted interventions. Finally, integrating nano-medicine with existing therapies may provide multipronged protection.
In conclusion, targeting macrophage PPAR signaling via precision nanodelivery holds substantial promise for improving outcomes in radiation enteritis. By leveraging expanding knowledge of inflammatory biology and nanotechnology, the field can move toward therapies that restore irradiated intestinal tissue while minimizing systemic side effects. Cross-disciplinary research will be essential to translate these strategies from the bench to the bedside.
Abbreviations
AF-1, activation function-1; AI, artificial intelligence; AP-1, activator protein-1; BAT, brown adipose tissue; C15orf48, chromosome 15 open reading frame 48; CD, cluster of differentiation; CDK, cyclin-dependent kinase; CITE-seq, cellular indexing of transcriptomes and epitopes by sequencing; CLA, conjugated linoleic acid; CRISPR, clustered regularly interspaced short palindromic repeats; DBD, DNA-binding domain; DNA, deoxyribonucleic acid; DSS, dextran sodium sulfate; EGF, epidermal growth factor; ELA, elafibranor; ERK1/2, extracellular signal-regulated kinase 1/2; EPR, enhanced permeability and retention; EVs, extracellular vesicles; FAO, fatty acid oxidation; GI, Gastrointestinal; Gy, gray; HA, hyaluronic acid; HC, healthy controls; HDL, high-density-lipoprotein; hdWGCNA, high-dimensional weighted gene co-expression network analysis; HSP90, heat shock protein 90; IBD, inflammatory bowel disease; IL, interleukin; iNOS, inducible nitric oxide synthase; LBD, ligand-binding domain; LF-lipo, lactoferrin-modified liposomes; LPS, lipopolysaccharide; LRP-1, low-density lipoprotein receptor-related protein-1; LMWH, low-molecular-weight heparin; MAPK, mitogen-activated protein kinase; MDM2, mouse double minute 2 homolog; MNPs, mononuclear phagocytes; MPO, myeloperoxidase; NALs, non-classical PPARγ agonist ligands; NcoR, nuclear corepressor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NOD-like receptor family pyrin domain-containing 3; NPs, nanoparticles;; OEA, oleoylethanolamine; OxbCD, oxybutylcyclodextrin; PEG, polyethylene glycol; PKC, protein kinase C; PEA, palmitoylethanolamide; PTMs, post-translational modifications; PPAR, peroxisome proliferator-activated receptor; PPREs, peroxisome proliferator-activated receptor response elements; Ra@TH, rapamycin-loaded hyaluronic acid; RE, radiation enteritis; ROS, reactive oxygen species; RT, radiotherapy; RXRs, retinoid X receptors; SENP2, SUMO-specific protease 2; scRNA-seq, single-cell RNA sequencing; SOD2, superoxide dismutase 2; STAB2, stabilin-2; SUMOylation, small ubiquitin-like modifier modification; TGF-β, transforming growth factor-β; TIMP1, tissue inhibitor of metalloproteinases 1; TNBS-2,4,6, trinitrobenzenesulfonic acid; TNF-α, tumor necrosis factor-α; TZD, thiazolidinedione; TXNIP, thioredoxin-interacting protein; TYMP, thymidine phosphorylase; UC, ulcerative colitis; VDZ, vedolizumab; WAT, white adipose tissue; 5-FU, 5-fluorouracil; 13-oxo-ODE, 13-oxo-octadecadienoic acid; 15d-PGJ2, 15-Deoxy-Δ12,14-Prostaglandin J2; 16S rRNA, 16S ribosomal RNA.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81602792); General Project of Jiangsu Provincial Health Commission (M2024004); Jiangsu Provincial Basic Science (Natural Science) Research General Project (23KJB310023); Jiangsu Provincial Maternal and Child Health Research Project (F202210); Jiangsu Provincial Key Medical Discipline (ZDXK202235); State Key Laboratory of Radiation Medicine and Radiation Protection Funding Project (GZK1202101); Youth Characteristic Technology Project of Clinical Diagnosis and Treatment Technology Innovation Project of the First Affiliated Hospital of Soochow University (2100201).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Jo YY, Kim YJ, Lee SH, et al. Prevention of radiotherapy-induced enteropathy by probiotics (prep): double-blind randomized placebo-controlled trial. Curr Oncol. 2024;31(10):5889–20. doi:10.3390/curroncol31100438
2. Yeung AR, Deshmukh S, Klopp AH, et al. Intensity-modulated radiation therapy reduces patient-reported chronic toxicity compared with conventional pelvic radiation therapy: updated results of a Phase III Trial. J Clin Oncol. 2022;40(27):3115–3119. doi:10.1200/JCO.21.02831
3. Yang XF, Zheng MY, An LY, et al. Quality evaluation of guidelines for the diagnosis and treatment of radiation enteritis. Radiat Oncol. 2023;18(1):14. doi:10.1186/s13014-023-02204-9
4. Devarakonda S, Thorsell A, Hedenström P, et al. Low-grade intestinal inflammation two decades after pelvic radiotherapy. EBioMedicine. 2023;94:104691. doi:10.1016/j.ebiom.2023.104691
5. Biran A, Dobson C, Rees CJ, et al. Healthcare provision for cancer survivors with chronic bowel symptoms post pelvic radiotherapy. “… and then you’re kind of cast adrift”: a qualitative study. Eur J Oncol Nurs. 2025;76:102895. doi:10.1016/j.ejon.2025.102895
6. Garrido-Trigo A, Corraliza AM, Veny M, et al. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat Commun. 2023;14(1):4506. doi:10.1038/s41467-023-40156-6
7. Hegarty LM, Jones GR, Biram A, et al. Tissue resident colonic macrophages persist through acute inflammation and adapt to aid tissue repair. Mucosal Immunol. 2026;19(1):1624–1635. doi:10.1016/j.mucimm.2025.11.007
8. Moyano A, Ferressini Gerpe NM, De Matteo E, et al. M1 macrophage polarization prevails in epstein-barr virus-infected children in an immunoregulatory environment. J Virol. 2022;96(1):e0143421. doi:10.1128/JVI.01434-21
9. Toobian D, Ghosh P, Katkar GD. Parsing the Role of PPARs in Macrophage Processes. Front Immunol. 2021;12:783780. doi:10.3389/fimmu.2021.783780
10. Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–386. doi:10.1016/j.immuni.2010.08.012
11. Trzebanski S, Kim JS, Larossi N, et al. Classical monocyte ontogeny dictates their functions and fates as tissue macrophages. Immunity. 2024;57(6):1225–1242.e6. doi:10.1016/j.immuni.2024.04.019
12. Moraitis I, Taelman J, Arozamena B, et al. Mucosal macrophages govern intestinal regeneration in response to injury. Gastroenterology. 2025;169(1):119–135.e26. doi:10.1053/j.gastro.2025.01.252
13. Jiang S, Yao S, Lu C. Lactobacillus reuteri FN041 alleviates radiation enteritis by regulating m2 macrophages polarization based on gut microbiota and its metabolites. Food Sci Nutr. 2025;13(9):e70871. doi:10.1002/fsn3.70871
14. Peng K, Deng N, Meng Y, et al. Alpha-Momorcharin Inhibits Pro-inflammatory Cytokine Expression by M1 Macrophages but Not Anti-Inflammatory Cytokine Expression by M2 Macrophages. J Inflamm Res. 2022;15:4853–4872. doi:10.2147/JIR.S372306
15. Chandra V, Huang P, Hamuro Y, et al. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature. 2008;456(7220):350–356. doi:10.1038/nature07413
16. Madsen MS, Broekema MF, Madsen MR, et al. PPARγ lipodystrophy mutants reveal intermolecular interactions required for enhancer activation. Nat Commun. 2022;13(1):7090. doi:10.1038/s41467-022-34766-9
17. Pourcet B, Pineda-Torra I, Derudas B, et al. SUMOylation of Human Peroxisome Proliferator-activated Receptor α Inhibits Its Trans-activity through the Recruitment of the Nuclear Corepressor NCoR. J Biol Chem. 2010;285(9):5983–5992. doi:10.1074/jbc.M109.078311
18. Qian X, Zhou Q, Ouyang Y, et al. Transferrin promotes fatty acid oxidation and liver tumor growth through PHD2-mediated PPARα hydroxylation in an iron-dependent manner. Proc Natl Acad Sci U S A. 2025;122(5):e2412473122. doi:10.1073/pnas.2412473122
19. Choi S, Jung JE, Yang YR, et al. Novel phosphorylation of PPARγ ameliorates obesity-induced adipose tissue inflammation and improves insulin sensitivity. Cell. Signalling. 2015;27(12):2488–2495. doi:10.1016/j.cellsig.2015.09.009
20. Dias MMG, Batista FAH, Tittanegro TH, et al. PPARγ S273 Phosphorylation Modifies the Dynamics of Coregulator Proteins Recruitment. Front Endocrinol. 2020;11:561256. doi:10.3389/fendo.2020.561256
21. Cazzaniga G, Capelli D, Montanari R, et al. Enhancing the activity of γ-hydroxy lactone derivatives as innovative peroxisome proliferator-activated receptor γ non-agonists inhibiting cyclin-dependent kinase 5-mediated phosphorylation. Eur J Med Chem. 2025;292:117657. doi:10.1016/j.ejmech.2025.117657
22. Yang N, Wang Y, Tian Q, et al. Blockage of PPARγ T166 phosphorylation enhances the inducibility of beige adipocytes and improves metabolic dysfunctions. Cell Death Differ. 2023;30(3):766–778. doi:10.1038/s41418-022-01077-x
23. Acton JJ, Black RM, Jones AB, et al. Benzoyl 2-methyl indoles as selective PPARγ modulators. Bioorg& Med Chem Lett. 2005;15(2):357–362. doi:10.1016/j.bmcl.2004.10.068
24. Shu Y, Lu Y, Pang X, et al. Phosphorylation of PPARγ at Ser84 promotes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget. 2016;7(47):76984–76994. doi:10.18632/oncotarget.12764
25. Jiao X, Tian L, Zhang Z, et al. Pparγ1 facilitates ErbB2-mammary adenocarcinoma in mice. Cancers. 2021;13(9):2171. doi:10.3390/cancers13092171
26. Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437(7059):759–763. doi:10.1038/nature03988
27. Xu T, Liu R, Lu X, et al. Lycium barbarum polysaccharides alleviate LPS-induced inflammatory responses through PPARγ/MAPK/NF-κB pathway in bovine mammary epithelial cells. J Anim Sci. 2022;100(1):skab345. doi:10.1093/jas/skab345
28. Diezko R, Suske G. Ligand Binding Reduces SUMOylation of the Peroxisome Proliferator-activated Receptor γ (PPARγ) Activation Function 1 (AF1) Domain. PLoS One. 2013;8(6):e66947. doi:10.1371/journal.pone.0066947
29. Niu X, Han P, Liu J, et al. Correction: regulation of PPARγ/CPT-1 expression ameliorates cochlear hair cell injury by regulating cellular lipid metabolism and oxidative stress. Mol Genet Genomics. 2023;298(4):977–978. doi:10.1007/s00438-023-02017-1
30. He Y, Zhang R, Yu L, et al. PPARγ acetylation in adipocytes exacerbates bat whitening and worsens age-associated metabolic dysfunction. Cells. 2023;12(10):1424. doi:10.3390/cells12101424
31. Qiang L, Wang L, Kon N, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of pparγ. Cell. 2012;150(3):620–632. doi:10.1016/j.cell.2012.06.027
32. Jiang X, Ye X, Guo W, et al. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J Mol Endocrinol. 2014;53(2):191–200. doi:10.1530/JME-14-0066
33. Lee KW, Kwak SH, Koo YD, et al. F-box only protein 9 is anE3ubiquitin ligase of PPARγ. Exp Mol Med. 2016;48(5):e234. doi:10.1038/emm.2016.31
34. Lee JM, Choi SS, Lee YH, et al. TheE3ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Exp Mol Med. 2018;50(10):1–11.
35. Gou Q, Zhang W, Xu Y, et al. EGFR/PPARδ/HSP90 pathway mediates cancer cell metabolism and chemoresistance. J Cell Biochem. 2021;122(3–4):394–402. doi:10.1002/jcb.29868
36. Kim PC, Lee JS, Chung SS, et al. Role of SUMO-Specific protease 2 in leptin-induced fatty acid metabolism in white adipocytes. Diabetes Metab J. 2023;47(3):382–393. doi:10.4093/dmj.2022.0156
37. Xu J, Lv H. PSTPIP2 alleviates obesity associated adipose tissue inflammation and insulin resistance in diabetes mice through promoting M2 macrophage polarization via activation of PPARγ. J Diabetes Complications. 2023;37(6):108479. doi:10.1016/j.jdiacomp.2023.108479
38. Zhou J, Ni W, Ling Y, et al. Human neural stem cell secretome inhibits lipopolysaccharide-induced neuroinflammation through modulating microglia polarization by activating peroxisome proliferator-activated receptor gamma. Stem Cells Dev. 2022;31(13–14):369–382. doi:10.1089/scd.2022.0081
39. Yang CC, Wu CH, Lin TC, et al. Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics. 2021;11(5):2424–2441. doi:10.7150/thno.46873
40. Shin M, Byun Y. Stereoselective syntheses of polyunsaturated fatty acids, 13-(S)-HODE and 15-(S)-HETE. J Org Chem. 2024;89(16):11293–11303. doi:10.1021/acs.joc.4c00983
41. Wang J, Xue X, Zhao X, et al. Forsythiaside A alleviates acute lung injury by inhibiting inflammation and epithelial barrier damages in lung and colon through PPAR-γ/RXR-α complex. J Adv Res. 2024;60:183–200. doi:10.1016/j.jare.2023.08.006
42. Walrath T, Malizia RA, Zhu X, et al. IFN-γ and IL-17A regulate intestinal crypt production of CXCL10 in the healthy and inflamed colon. Am J Physiol Gastrointest Liver Physiol. 2020;318(3):G479–G489. doi:10.1152/ajpgi.00208.2019
43. Villarroel-Vicente C, García A, Zibar K, et al. Synthesis of a new 2-prenylated quinoline as potential drug for metabolic syndrome with pan-PPAR activity and anti-inflammatory effects. Bioorg Med Chem Lett. 2024;106:129770. doi:10.1016/j.bmcl.2024.129770
44. Gao H, Sun M, Li A, et al. Microbiota-derived IPA alleviates intestinal mucosal inflammation through upregulating Th1/Th17 cell apoptosis in inflammatory bowel disease. Gut Microbes. 2025;17(1):2467235. doi:10.1080/19490976.2025.2467235
45. Elmassry MM, Sugihara K, Chankhamjon P, et al. A meta-analysis of the gut microbiome in inflammatory bowel disease patients identifies disease-associated small molecules. Cell Host Microbe. 2025;33(2):218–234.e12. doi:10.1016/j.chom.2025.01.002
46. Lai YH, Wu TC, Tsai BY, et al. Peroxisome proliferator-activated receptor-γ as the gatekeeper of tight junction in Clostridioides difficile infection. Front Microbiol. 2022;13:986457. doi:10.3389/fmicb.2022.986457
47. El-Shoura EAM, Sharkawi SMZ, Abdelzaher LA, et al. Reno-protective effect of fenofibrate and febuxostat against vancomycin-induced acute renal injury in rats: targeting PPARγ/NF-κB/COX-II and AMPK/Nrf2/HO-1 signaling pathways. Immunopharmacol Immunotoxicol. 2024;46(4):509–520. doi:10.1080/08923973.2024.2373216
48. Hong JH, Han KA, Hwang YC, et al. Efficacy and safety of high-dose pioglitazone as add-on therapy in patients with type 2 diabetes mellitus inadequately controlled with dapagliflozin and metformin: double-blind, randomized, placebo-controlled trial. Diabetes Metab J. 2025;28:1.
49. Suzuki S, Arnold LL, Pennington KL, et al. Effects of pioglitazone, a peroxisome proliferator–activated receptor gamma agonist, on the urine and urothelium of the rat. Toxicol Sci. 2010;113(2):349–357. doi:10.1093/toxsci/kfp256
50. Di Y, Li H, Yang J, et al. PPARγ/NF-κB axis contributes to cold-induced resolution of experimental colitis and preservation of intestinal barrier. Biochim Biophys Acta Mol Basis Dis. 2024;1870(7):167326. doi:10.1016/j.bbadis.2024.167326
51. Kim W, Jang JH, Zhong X, et al. 15-Deoxy-Δ12,14-Prostaglandin J2 Promotes Resolution of Experimentally Induced Colitis. Front Immunol. 2021;12:615803. doi:10.3389/fimmu.2021.615803
52. Xiao T, Kang J, Zhao C, Zhu R, Sun M, Wang Y. Butyrate ameliorates ulcerative colitis through targeting STING-dependent ER stress signaling and limiting CD4+ TRM T cells accumulation. J Inflamm. 2025;22(1):50. doi:10.1186/s12950-025-00475-5
53. Park WY, Kim B, Song G, et al. Ellagic acid alleviates abnormal fat reduction by activating the rxrβ-pparγ pathways in a CT26 tumour-induced cachexia mouse model. J Cachexia, Sarcopenia Muscle. 2026;17(1):e70176. doi:10.1002/jcsm.70176
54. Aldossary KM, Abdallah MS, Kamal N, et al. Therapeutic modulation of il-6/stat-3 and nitric oxide by fenofibrate in patients with ulcerative colitis: a randomized controlled pilot study. Pharmacotherapy. 2025;45(12):840–851. doi:10.1002/phar.70088
55. Li X, Sun W, Lu J, et al. Effects of fenofibrate therapy on renal function in primary gout patients. Rheumatology. 2021;60(11):5020–5027. doi:10.1093/rheumatology/keab231
56. Wang N, Kong R, Han W, et al. Honokiol alleviates ulcerative colitis by targeting PPAR-γ-TLR4-NF-κB signaling and suppressing gasdermin-D-mediated pyroptosis in vivo and in vitro. Int Immunopharmacol. 2022;111:109058. doi:10.1016/j.intimp.2022.109058
57. Valenza M, Facchinetti R, Steardo L, et al. Palmitoylethanolamide and White Matter Lesions: evidence for Therapeutic Implications. Biomolecules. 2022;12(9):1191. doi:10.3390/biom12091191
58. Francis MR, El-Sheakh AR, Suddek GM. Saroglitazar, a dual PPAR-α/γ agonist, alleviates LPS-induced hepatic and renal injury in rats. Int Immunopharmacol. 2023;115:109688. doi:10.1016/j.intimp.2023.109688
59. Di Paola R, Esposito E, Mazzon E, et al. GW0742, a selective PPAR-β/δ agonist, contributes to the resolution of inflammation after gut ischemia/reperfusion injury. J Leukocyte Biology. 2010;88(2):291–301. doi:10.1189/jlb.0110053
60. Hakeem AN, Kamal MM, Tawfiq RA, et al. Elafibranor modulates ileal macrophage polarization to restore intestinal integrity in NASH: potential crosstalk between ileal IL-10/STAT3 and hepatic TLR4/NF-κB axes. Biomed Pharmacother. 2023;157:114050. doi:10.1016/j.biopha.2022.114050
61. Pompili S, Vetuschi A, Latella G, et al. PPAR-Gamma orchestrates EMT, AGE, and cellular senescence pathways in colonic epithelium and restrains the progression of IBDs. Int J Mol Sci. 2023;24(10):8952. doi:10.3390/ijms24108952
62. Bassaganya-Riera J, DiGuardo M, Climent M, et al. Activation of PPARγ and δ by dietary punicic acid ameliorates intestinal inflammation in mice. Br J Nutr. 2011;106(6):878–886. doi:10.1017/S0007114511001188
63. Zhang J, Zhang Y, Xiao F, et al. The peroxisome proliferator-activated receptor γ agonist pioglitazone prevents NF-κB activation in cisplatin nephrotoxicity through the reduction of p65 acetylation via the AMPK-SIRT1/p300 pathway. Biochem Pharmacol. 2016;101:100–111. doi:10.1016/j.bcp.2015.11.027
64. Noori M, Azimirad M, Ghorbaninejad M, et al. PPAR-γ agonist mitigates intestinal barrier dysfunction and inflammation induced by Clostridioides difficile SlpA in vitro. Sci Rep. 2024;14(1):32087. doi:10.1038/s41598-024-83815-4
65. Vanasse A, Carpentier AC, Courteau J, et al. Stroke and cardiovascular morbidity and mortality associated with rosiglitazone use in elderly diabetic patients. Diab Vasc Dis Res. 2009;6(2):87–93. doi:10.1177/1479164109336047
66. Tang H, Shi W, Fu S, et al. Pioglitazone and bladder cancer risk: a systematic review and meta-analysis. Cancer Med. 2018;7(4):1070–1080. doi:10.1002/cam4.1354
67. Da Silva S, Åv K, Mohlin S, et al. A Novel Topical PPARγ Agonist Induces PPARγ Activity in Ulcerative Colitis Mucosa and Prevents and Reverses Inflammation in Induced Colitis Models. Inflammatory Bowel Diseases. 2018;24(4):792–805. doi:10.1093/ibd/izx079
68. Cuzzocrea S, Di Paola R, Mazzon E, et al. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors alpha (PPAR-α) in the development of inflammatory bowel disease in mice. Lab Investigation. 2004;84(12):1643–1654. doi:10.1038/labinvest.3700185
69. Esposito G, Capoccia E, Turco F, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut. 2014;63(8):1300–1312. doi:10.1136/gutjnl-2013-305005
70. Jain MR, Giri SR, Trivedi C, et al. Saroglitazar, a novel PPAR α/γ agonist with predominant PPAR α activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacology Research & Perspectives. 2015;3(3):e00136. doi:10.1002/prp2.136
71. Rodriguez-Gutierrez R, González JG, Parmar D, et al. Saroglitazar is noninferior to fenofibrate in reducing triglyceride levels in hypertriglyceridemic patients in a randomized clinical trial. J Lipid Res. 2022;63(7):100233. doi:10.1016/j.jlr.2022.100233
72. Di Paola R, Crisafulli C, Mazzon E, et al. GW0742, a high-affinity PPAR -beta/delta agonist, inhibits acute lung injury in mice. Shock. 2010;33(4):426–435. doi:10.1097/SHK.0b013e3181b8f2fb
73. Faulkner A, Lynam E, Purcell R, et al. Context-dependent regulation of endothelial cell metabolism: differential effects of the PPARβ/δ agonist GW0742 and VEGF-A. Sci Rep. 2020;10(1):7849. doi:10.1038/s41598-020-63900-0
74. Meerts IATM, Verspeek-Rip CM, Buskens CAF, et al. Toxicological evaluation of pomegranate seed oil. Food Chem Toxicol. 2009;47(6):1085–1092. doi:10.1016/j.fct.2009.01.031
75. Byndloss MX, Olsan EE, Rivera-Chávez F, et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357(6351):570–575. doi:10.1126/science.aam9949
76. Takagi T, Naito Y, Mizushima K, et al. 15-Deoxy-Δ12,14-prostaglandin J2 ameliorates dextran sulfate sodium-induced colitis in mice through heme oxygenase-1 induction. Arch Biochem Biophys. 2019;677:108183. doi:10.1016/j.abb.2019.108183
77. Altmann R, Hausmann M, Spöttl T, et al. 13-Oxo-ODE is an endogenous ligand for PPARγ in human colonic epithelial cells. Biochem Pharmacol. 2007;74(4):612–622. doi:10.1016/j.bcp.2007.05.027
78. Petrosino S, Iuvone T, Di Marzo V. N-palmitoyl-ethanolamine: biochemistry and new therapeutic opportunities. Biochimie. 2010;92(6):724–727. doi:10.1016/j.biochi.2010.01.006
79. Geng Y, Zou H, Li Z, et al. Recent advances in nanomaterial-driven strategies for diagnosis and therapy of vascular anomalies. J Nanobiotechnology. 2024;22(1):120. doi:10.1186/s12951-024-02370-2
80. Priya S, Desai VM, Singhvi G. Surface Modification of Lipid-Based Nanocarriers: a Potential Approach to Enhance Targeted Drug Delivery. ACS Omega. 2022;8(1):74–86. doi:10.1021/acsomega.2c05976
81. Lagarrigue P, Moncalvo F, Cellesi F. Non-spherical Polymeric Nanocarriers for Therapeutics: the Effect of Shape on Biological Systems and Drug Delivery Properties. Pharmaceutics. 2022;15(1):32. doi:10.3390/pharmaceutics15010032
82. Tran HV, Ngo NM, Medhi R, et al. Multifunctional Iron Oxide Magnetic Nanoparticles for Biomedical Applications: a Review. Materials. 2022;15(2):503. doi:10.3390/ma15020503
83. Sun T, Kwong CHT, Gao C, et al. Amelioration of ulcerative colitis via inflammatory regulation by macrophage-biomimetic nano-medicine. Theranostics. 2020;10(22):10106–10119. doi:10.7150/thno.48448
84. Zhang M, Merlin D. Nanoparticle-Based Oral Drug Delivery Systems Targeting the Colon for Treatment of Ulcerative Colitis. Inflamm Bowel Dis. 2018;24(7):1401–1415. doi:10.1093/ibd/izy123
85. Zhao Y, Yin W, Yang Z, et al. Nanotechnology-enabled M2 macrophage polarization and ferroptosis inhibition for targeted inflammatory bowel disease treatment. J Control Release. 2024;367:339–353. doi:10.1016/j.jconrel.2024.01.051
86. Zhao Y, Yang Y, Zhang J, et al. Lactoferrin-mediated macrophage targeting delivery and patchouli alcohol-based therapeutic strategy for inflammatory bowel diseases. Acta Pharm Sin B. 2020;10(10):1966–1976. doi:10.1016/j.apsb.2020.07.019
87. Liu B, Zhu L, Lei L, et al. Lesional Macrophage‐Targeted Nano-medicine Regulating Cholesterol Homeostasis for the Treatment of Atherosclerosis. Adv Mater. 2025;2025:1.
88. Huang L, Hu W, Huang LQ, et al. “Two-birds-one-stone” oral nanotherapeutic designed to target intestinal integrins and regulate redox homeostasis for UC treatment. Sci Adv. 2024;10(30):eado7438. doi:10.1126/sciadv.ado7438
89. Jain S, Tran TH, Amiji M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials. 2015;61:162–177. doi:10.1016/j.biomaterials.2015.05.028
90. Lin Y, Ding R, Zhang Y, et al. Lactoferrin-Loaded Liposomal Nanoparticles: enhanced Intestinal Stability and Bioactivity for Mitigating Radiation-Induced Intestinal Injury. Foods. 2025;14(19):3410. doi:10.3390/foods14193410
91. Zhang D, He J, Cui J, et al. Oral Microalgae-Nano Integrated System against Radiation-Induced Injury. ACS Nano. 2023;17(11):10560–10576. doi:10.1021/acsnano.3c01502
92. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: the Phagocyte Problem. Nano Today. 2015;10(4):487–510. doi:10.1016/j.nantod.2015.06.006
93. Ding N, Luo G, Li H, et al. A Cyclodextrin‐Based pH‐Responsive MicroRNA Delivery Platform Targeting Polarization of M1 to M2 Macrophages for Sepsis Therapy. Adv Healthc Mater. 2023;12(27). doi:10.1002/adhm.202301243.
94. Zhou M, Lu L, Xu X, et al. EUK nanozyme-loaded PL&GA coacervate droplets attenuate ulcerative colitis through restoring gut homeostasis and restricting intestinal cell ferroptosis. Mater Today Bio. 2025;35:102466. doi:10.1016/j.mtbio.2025.102466
95. Han X, Luo R, Qi S, et al. “Dual sensitive supramolecular curcumin nanoparticles” in “advanced yeast particles” mediate macrophage reprogramming, ROS scavenging and inflammation resolution for ulcerative colitis treatment. J Nanobiotechnol. 2023;21(1):321. doi:10.1186/s12951-023-01976-2
96. Kim J, Hong G, Mazaleuskaya L, et al. Ultrasmall Antioxidant Cerium Oxide Nanoparticles for Regulation of Acute Inflammation. ACS Appl Mater Interfaces. 2021;13(51):60852–60864. doi:10.1021/acsami.1c16126
97. Zamyatina EA, Goryacheva OA, Popov AL, et al. Novel Pyrroloquinoline Quinone-Modified Cerium Oxide Nanoparticles and Their Selective Cytotoxicity Under X-Ray Irradiation. Antioxidants. 2024;13(12):1445. doi:10.3390/antiox13121445
98. Xiang H, Li X, Wei H, et al. Biomimetic Engineering of Hybrid Radiosensitizers to Boost Radiotherapy against Cancer Metastasis. Adv Mater. 2025;37(34):e2501077. doi:10.1002/adma.202501077
99. Ma Y, Gou S, Zhu Z, et al. Transient Mild Photothermia Improves Therapeutic Performance of Oral Nano-medicines with Enhanced Accumulation in the Colitis Mucosa. Adv Mater. 2024;36(14):2309516. doi:10.1002/adma.202309516
100. Liang X, Yu X, Liu Y, et al. NIR-controlled photothermal intelligent nanozyme synergizes catalysis and gas therapy for multidimensional treatment of inflammatory bowel disease. J Nanobiotechnol. 2025;23(1):800. doi:10.1186/s12951-025-03878-x
101. Liu S, Kong X, Fang Y, et al. A dual-sensitive nanoparticle-mediated deepening synergistic therapy strategy involving DNA damage and ICD stimuli to treat triple-negative breast cancer. Biomater Sci. 2023;11(18):6325–6341. doi:10.1039/D3BM00781B
102. He N, Jiang H, Dai T, et al. Prebiotic-Engineered Oral Nanoplatform against Ulcerative Colitis via Photodynamic Remodeling of Gut Microbiota and Macrophage Polarization. Biomater Res. 2025;29:0283. doi:10.34133/bmr.0283
103. Tang D, Cao F, Yan C, et al. Extracellular Vesicle/Macrophage Axis: potential Targets for Inflammatory Disease Intervention. Front Immunol. 2022;13:705472. doi:10.3389/fimmu.2022.705472
104. Ma F, Zhang S, Akanyibah FA, et al. Exosome-mediated macrophage regulation for inflammatory bowel disease repair: a potential target of gut inflammation. Am J Transl Res. 2023;15(12):6970–6987.
105. Qian W, Huang L, Xu Y, et al. Hypoxic ASCs-derived Exosomes Attenuate Colitis by Regulating Macrophage Polarization via miR-216a-5p/HMGB1 Axis. Inflammatory Bowel Diseases. 2023;29(4):602–619. doi:10.1093/ibd/izac225
106. Liu X, Liu T, lu SC, et al. Endometrial regenerative cell exosome-derived Sirtuin 6 alleviates ulcerative colitis by remodeling macrophage polarization. Colloids Surf B. 2026;257:115196. doi:10.1016/j.colsurfb.2025.115196
107. Yang L, Wang S, Qiao Z, et al. Dual-ligand engineered exosome regulates WNT signaling activation to promote liver repair and regeneration. Nat Commun. 2025;16(1):9019. doi:10.1038/s41467-025-64069-8
108. Yang L, Wang X, Wei X, et al. Oral Delivery of R‐spondin1‐Loaded Small Extracellular Vesicles Activates WNT Signalling Pathway to Accelerate Intestinal Injury Repair and Reverse Ageing. J Extracell Vesicles. 2026;15(1):e70226. doi:10.1002/jev2.70226
109. Chen Y, Feng J, Chen Y, et al. ROS-responsive nano-medicine for navigating autophagy to enhance targeted therapy of inflammatory bowel disease. Int J Pharm. 2024;659:124117. doi:10.1016/j.ijpharm.2024.124117
110. Hua Y, Qin M, Lu S, et al. Hyaluronic acid-functionalized MOFs for combined sunitinib and siRNA therapy in renal cell carcinoma. Int J Biol Macromol. 2024;283:137317. doi:10.1016/j.ijbiomac.2024.137317
111. Lin M, Dong L, Chen Q, et al. Lentinan-Based Oral Nanoparticle Loaded Budesonide With Macrophage-Targeting Ability for Treatment of Ulcerative Colitis. Front Bioeng Biotechnol. 2021;9:702173. doi:10.3389/fbioe.2021.702173
112. Yang J, Bai Y, Shen S, et al. An oral nano-antioxidant for targeted treatment of inflammatory bowel disease by regulating macrophage polarization and inhibiting ferroptosis of intestinal cells. Chem Eng J. 2023;465:142940. doi:10.1016/j.cej.2023.142940
113. Zhang Q, Tao H, Lin Y, et al. A superoxide dismutase/catalase mimetic nano-medicine for targeted therapy of inflammatory bowel disease. Biomaterials. 2016;105:206–221. doi:10.1016/j.biomaterials.2016.08.010
114. Wang T, Lin X, Li W, et al. Resveratrol-embedded hollow cerium oxide nano-medicine targeted treat inflammatory bowel disease through ROS clearance, intestinal mucosal immune homeostasis recovery and gut microbiota modulation. Mater Today Bio. 2026;36:102765. doi:10.1016/j.mtbio.2026.102765
115. Liu G, Feng S, Sun Y, et al. Colonic macrophage-targeted curcumin nanoparticles alleviate DSS-induced colitis in mice through the NF-kappa B pathway. Food Bioscience. 2021;41:101089. doi:10.1016/j.fbio.2021.101089
116. He N, Dong M, Sun Y, et al. Mesenchymal stem cell-derived extracellular vesicles targeting irradiated intestine exert therapeutic effects. Theranostics. 2024;14(14):5492–5511. doi:10.7150/thno.97623
117. Tan AXX, Ong BYC, Dinesh T, et al. Nanomedicine Strategies in the Management of Inflammatory Bowel Disease and Colorectal Cancer. Int J Mol Sci. 2025;26(13):6465. doi:10.3390/ijms26136465
118. Kojima C, Yao J, Nakajima K, et al. Attenuated polyethylene glycol immunogenicity and overcoming accelerated blood clearance of a fully PEGylated dendrimer. Int J Pharm. 2024;659:124193. doi:10.1016/j.ijpharm.2024.124193
119. Motevasseli M, Darvishi M, Khoshnevisan A, et al. Distinct tumor-TAM interactions in IDH-stratified glioma microenvironments unveiled by single-cell and spatial transcriptomics. Acta Neuropathol Commun. 2024;12(1):133. doi:10.1186/s40478-024-01837-5
120. Lu H, Yan H, Li X, et al. Single-cell map of dynamic cellular microenvironment of radiation-induced intestinal injury. Commun Biol. 2023;6(1):1248. doi:10.1038/s42003-023-05645-w
121. Mennillo E, Kim YJ, Lee G, et al. Single-cell and spatial multi-omics highlight effects of anti-integrin therapy across cellular compartments in ulcerative colitis. Nat Commun. 2024;15(1):1493. doi:10.1038/s41467-024-45665-6
122. Manichanh C, Varela E, Martinez C, et al. The Gut Microbiota Predispose to the Pathophysiology of Acute Postradiotherapy Diarrhea. Am J Gastroenterol. 2008;103(7):1754–1761. doi:10.1111/j.1572-0241.2008.01868.x
123. Shen Z, Wang J, Huang Q, et al. Genetic modification to induce CXCR2 overexpression in mesenchymal stem cells enhances treatment benefits in radiation-induced oral mucositis. Cell Death Dis. 2018;9(2):229. doi:10.1038/s41419-018-0310-x
124. Nepelska M, De Wouters T, Jacouton E, et al. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci Rep. 2017;7(1):43199. doi:10.1038/srep43199
125. Li JK, Veeraperumal S, Aweya JJ, et al. Fucoidan modulates gut microbiota and immunity in Peyer’s patches against inflammatory bowel disease. Carbohydr Polym. 2024;342:122421. doi:10.1016/j.carbpol.2024.122421
126. Das KP, C J. Nanoparticles and convergence of artificial intelligence for targeted drug delivery for cancer therapy: current progress and challenges. Front Med Technol. 2023;4:1067144. doi:10.3389/fmedt.2022.1067144
127. Sun Z, Huang J, Fishelson Z, Wang C, Zhang S. Cell-Penetrating Peptide-Based Delivery of Macromolecular Drugs: development, Strategies, and Progress. Biomedicines. 2023;11(7):1971. doi:10.3390/biomedicines11071971
128. Hamilton S, Kingston BR. Applying artificial intelligence and computational modeling to nanomedicine. Curr Opin Biotechnol. 2024;85:103043. doi:10.1016/j.copbio.2023.103043
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.