Back to Journals » Drug Design, Development and Therapy » Volume 9
Nanoparticle formulation by Büchi B-90 Nano Spray Dryer for oral mucoadhesion
Authors Harsha S ![]() , E Aldhubiab B, Nair A, Abdulrahman Alhaider I, Attimarad M, Venugopala K, Srinivasan V, Gangadhar N, Asif A
, E Aldhubiab B, Nair A, Abdulrahman Alhaider I, Attimarad M, Venugopala K, Srinivasan V, Gangadhar N, Asif A
Received 22 April 2014
Accepted for publication 12 June 2014
Published 23 January 2015 Volume 2015:9 Pages 273—282
DOI https://doi.org/10.2147/DDDT.S66654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Sree N Harsha,1 Bander E Aldhubiab,1 Anroop B Nair,1 Ibrahim Abdulrahman Alhaider,1 Mahesh Attimarad,1 Katharigatta N Venugopala,1 Saminathan Srinivasan,2 Nagesh Gangadhar,2 Afzal Haq Asif3
1Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa, Saudi Arabia; 2Department of Pharmaceutics, East Point College of Pharmacy, Bangalore, India; 3Department of Pharmacy Practice, College of Clinical Pharmacy, King Faisal University, Al-Ahsa, Saudi Arabia
Abstract: Diabetes is considered one of the main threats to global public health in this era. It is increasing rapidly in every part of the world; the prevalence of the disease will grow to the point where 366 million people will be affected by 2030. The prevalence of diabetes mellitus (DM) in the Saudi population is high, and the majority of patients suffer from type 2 DM. Marketed oral antidiabetic drugs have indicated poor tolerability during chronic treatments, and this contributes to the moderately large proportion of type 2 DM patients that remain inadequately managed. Vildagliptin nanospheres were prepared with aminated gelatin using a spray-drying method; narrow particle-size distribution was seen at 445 nm. The angle of repose was found to be θ <33.5°. The nanospheres appeared to be spherical with a smooth surface. The drug content and percentage yield of the nanospheres were found to be 76.2%±4.6% and 83%±2%, respectively. The nanosphere-swell profile was found to be 165%±7%. The pure drug was 100% dissolved in 30 minutes, and the nanosphere formulation took 12 hours to dissolve (97.5%±2%), and followed a Korsmeyer–Peppas kinetic model with an R2 of 0.9838. The wash-off test of nanospheres found that they exhibited an excellent mucoadhesive property at 86.7% for 8 hours. The stability-study data showed no changes in the physicochemical properties of the nanospheres, and suggested that the nanospheres be stored below room temperature. The amount of vildagliptin retained was 1.6% within 3 hours, and in comparison with the gelatin vildagliptin nanoparticles formulation, the percentage that was retained was much higher (98.2% in 12 hours).
Keywords: nanospheres, vildagliptin, Büchi Nano Spray Dryer, diabetes
Introduction
Diabetes is considered one of the main threats to global public health in this era. It is increasing rapidly in every part of the world; the prevalence of the disease will grow to the point that 366 million people will be affected by 2030,1–3 and about 90% of these individuals have type 2 diabetes.4
Data from the International Diabetes Federation have revealed that diabetes devastatingly affects 16.8% of individuals in Saudi Arabia. The figures in its neighboring countries are also similar – in Oman, diabetes affects 13.4%; in Bahrain, 15.4% have diabetes; in the United Arab Emirates, 18.7% are afflicted with the disease; and in Kuwait, 14.6% are affected. Saudi Arabia has over 28 million people,6 and it is a fast-developing country. Roughly 25% of the population has been diagnosed with diabetes,7 and a rise between 40% and 50% by 2020 is expected.8
Some oral antidiabetic drugs display poor tolerability during chronic treatment, which contributes to a large proportion of type 2 diabetes mellitus (T2DM) patients that remain inadequately managed. The presently existing oral antidiabetic agents do not directly restore the complex secretory function of the islet cells, and they are also limited by side effects. Therefore, there has been much interest in recognizing newer agents that are capable of restoring the complex secretory dysfunction of patients with T2DM.9
Dipeptidyl peptidase (DPP)-4 inhibitors are the most common accompaniments to the therapeutic armamentarium offered for treating T2DM. The source of the therapeutic effects of DPP-4 inhibitors basically increases the intact circulating levels of the incretin hormones – glucose-dependent insulinotropic polypeptide and glucagon like peptide 1 – and they achieve their insulinotropic effects by adhering to the specific receptor. In addition, they have a short half-life because of rapid enzyme inactivation. With respect to the mode of action, vildagliptin is a DPP-4 inhibitor; a detailed study has been carried out in clinical studies, and considerable developments have been made toward understanding the molecular interaction between vildagliptin and the DPP-4 enzyme in biological activity.10,11
Gelatin is a denatured, biodegradable polymer obtained from collagen and the best material for clinical and medicinal use. It has been widely used as an excipient for drugs and controls the release for many formulations.12–14 The safety of gelatin has been confirmed through its extended clinical usage as a plasma expander.15 Mucoadhesion is a complex process, and many hypotheses clarify the mechanisms, including wetting theory, electric theory, diffusion theory, and adsorption theory. Although no hypothesis alone can make clear this process, it is generally accepted that the first step of mucoadhesion relates to the intimate contact between the mucoadhesive materials and the mucus surface. Consequently, mucoadhesive agents would interact with mucin on the mucus layer and adhere with secondary chemical bonds, such as van der Waals and hydrogen bonds. Based on this consideration, polymers with hydrophilic groups, such as OH, NH2, and COOH, were considered to be crucial for successful mucoadhesion.16,17 The literature suggests aminated gelatin demonstrates a sustained release with considerable gastric mucoadhesive properties.12,18
Disappointingly, one of the major therapeutic drawbacks of using vildagliptin is its rapid metabolism and short elimination half-life of 1.32–2.43 hours.19 Due to the shorter half-life of the therapeutic agent, it is recommended that patients receiving the medical treatment need to adhere meticulously to the interval between doses, and the drug should be taken at a dose of 50 mg twice a day.20 Mucoadhesive drug-delivery systems offer abundant advantages when compared to other conventional/controlled-release systems. These dosage forms adhere to the target site of the gastrointestinal tract (GIT), and they release the drug by maintaining the therapeutic concentration for prolonged periods of time. Therefore, the uptake and consequent bioavailability of the therapeutic agent may be enhanced. In addition, the rate of recurrence of the dosing reduces when patient compliance is improved.21–23
Even with the current exciting drug delivery, more discoveries are still required to progress further the effectiveness of existing drugs. An attempt has been made to develop a mucoadhesive nanoparticle of vildagliptin using a spray-drying method. Several attempts have been developed in formulating a mucoadhesive drug-delivery system, and an additional formulation for vildagliptin has been attempted by preparing nanoparticles. Numerous methods, such as direct polymerization, salting out, supercritical fluid technology, solvent evaporation, nanoprecipitations, and various polymerization techniques have been developed for formulating nanoparticles.24–26 On the other hand, these techniques typically engage the use of organic solvents, which may serve as a potential reservoir of particle contamination and cause toxicity.
Furthermore, these techniques are inappropriate for mass production, because they do not produce a high yield or a reproducible quantity. At present, spray-drying is used in pharmaceutical industries. It is an entrenched drying process that is conventionally used to exchange liquid solutions to a powder form. Spray-drying is widely used for the microencapsulation of many therapeutic agents, due to its reproducibility, consistency, and control of particle-size distribution and drug release.27 The Nano Spray Dryer B-90 from Büchi Labortechnik AG (Flawil, Switzerland) offers patented technology, such as the production of small particles from a minimum sample, and it provides high yields and reduces research and development costs.28 In addition, spray-drying is a continuous single process; it is a one-step process, is simple to scale up, and shows minor variations in terms of feed flow, as well as the concentration of polymer and the temperature needed to fabricate the preferred particle-size distribution.
To formulate nanospheres by spray-drying, gelatin may be used as an effectual option, given that it is a wall material due to its favorable film formation, natural biodegradable polymer properties, edibility, emulsification ability, and water-solubility.29
The aim of this research was to formulate mucoadhesive nanospheres for the delivery of vildagliptin and to evaluate the GIT drug distribution in an in vivo rat model. Several studies have established the potential of gelatin as an excellent bioadhesive, and its controlled-release properties have been used extensively to enhance the delivery of active ingredients to various mucous membranes.30,31 To the best of our knowledge, based on a literature survey, this is the first mucoadhesive drug-delivery system to be designed for the formulation of a vildagliptin–gelatin nanospheres via a Büchi (Büchi Labortechnik AG) spray-dryer.
Materials and methods
Vildagliptin was purchased from Biokemix, London, UK. Gelatin Bloom 250 was a gift sample provided from Shree Ram Industry. All of the other chemicals used were of analytical reagent grade.
Preparation of aminated gelatin
Aminated gelatin was prepared by reaction between gelatin and 1,2-ethylenediamine. An excess amount of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride was added to gelatin solution, which had been previously dissolved in phosphate buffer (pH 5.3). At 37°C, the resultant solution was allowed to react for 1 hour, and was dialyzed against purified water for 2 days.18
Formulation of the vildagliptin nanospheres
Gelatin-loaded vildagliptin nanospheres were prepared via a spray-drying technique (B-90; Büchi Labortechnik AG) with a nanospray-dryer. This spray-dryer has a pulsating casing in the spray nozzle to atomize the feed, and the particles were gathered by an electrostatic particle accumulator. A 4 μm spray nozzle was used; the flow rate was set to about 100–110 L/minute, and the relative spray rate was fixed to 100%. The inlet temperature and outlet temperature were established at 120°C and 27°C, respectively. A solution of gelatin (1 g of both vildagliptin and gelatin in 200 mL of water) was sprayed. The viscosity of the solution was 6.2 cP. The parameters had been previously optimized in our laboratory. The solution was filtered prior to the spray-drying process to avoid nozzle blockage. The dried particles were collected from the particle chamber using a powder scraper, and they were then kept in a desiccator at a temperature of 25°C for further analysis.31
Characterization of nanospheres
Scanning electron microscopy
The uncoated vildagliptin nanospheres were analyzed by scanning electron microscopy (JSM-6390LA analytical scanning microscope; JEOL, Tokyo, Japan) at 20 kV using different magnifications (between 1,000× and 95,000×). Different batches were examined after they had been platinum-sputtered (JEOL JFC-1600).32,33
Analysis of the particle size of nanospheres
The particle-size distribution of the nanospheres was studied by laser light-diffraction techniques.34 The nanospheres were diluted tenfold with deionized water using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).
Drug content
Drug content was determined according to our previous report.35 Briefly, the weighed quantity of gelatin vildagliptin nanoparticles (GVN) was dissolved in methanol. The sample solution was further diluted with a mobile phase, and 10 μL was injected into a liquid chromatography mass spectrometry (LC-MS) system to determine the concentration of vildagliptin. The reported chromatographic method36 was modified slightly and used for the examination of vildagliptin in formulation and tissue extracts. The LC-MS system (1200 series; Agilent Technologies, Santa Clara, CA, USA) consisted of a binary pump, a 6120 mass (MS) detector, an autoinjector, and a Chem data module. Chromatographic separation of the active ingredients was accomplished using a Zorbax Eclipse C18 (150×4.6 mm internal diameter, 5 μm) reverse-phase analytical column at room temperature (approximately 25°C). The mobile phase consisted of 40% mobile phase A (ammonium acetate [10 mM], pH 8 adjusted with ammonia) and 60% mobile phase B (acetonitrile–methanol 10:90), and it was filtered through a 0.45 μm nylon filter before use. LC-MS was performed isocratically at a flow rate of 0.5 mL/minute, and the elute was continuously monitored by the MS detector in a positive-ion single-ion monitory mode at (M++1) mass to charge ratio 304.3. The tryouts were piloted in triplicate. The drug loading was calculated as the ratio between the quantity of vildagliptin loaded and the theoretical amount of vildagliptin loaded for each preparation. The production yield was calculated from the quotient of the average weight of the dried nanospheres (N1) that were recovered from batches to the sum of the first dry weight of the preliminary materials (N2). The average value of the drug content was represented as the drug loading.35,37,38
|
|
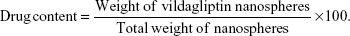
Production recovery
Nanosphere recovery was calculated as the relationship of the achieved weight of the nanospheres related to the entire amount of the drug and polymer initially used in the preparation process.32,37
|
|
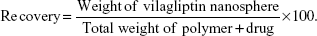
Determination of powder-flow properties
Powder flow is a prerequisite in pharmaceutical industries. The flowability of powder is carried out by a fixed-funnel method. The angle of repose is the angle between the horizontal surface and the slope of the pile of powder fall from a specific height. Briefly, a funnel with the end of its stem cut at right angles to the axis of symmetry is secured with its tip at a 2 cm height, H, above a graph sheet positioned on a flat, level surface. The nanospheres were cautiously shifted through the funnel until reaching the peak of the conical heap.39 The mean diameter, 2R, of H at the base of the powder cone was determined, and the tangent of the angle of repose was given by:
|
|
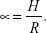
Interaction studies
Fourier-transform infrared spectroscopy (FTIR) spectra of GVN, vildagliptin, and aminated gelatin were analyzed independently after mixing with potassium bromide (1:10) and pressed to develop a thin suitable disk by applying pressure using a PerkinElmer model 883 spectroscope (range 400–4,000 cm−1).40
Measuring the swelling behavior of polymeric nanospheres
Measurement of the swelling of nanospheres was conducted in simulated gastric fluid. The sizes of the dried powder (GVN) and those after incubation in methanol for 15 minutes, 30 minutes, 1 hour, 3 hours, 6 hours, 10 hours, and 12 hours were calculated using a microscopic method. The percentage of swelling at the different time intervals was determined by calculating the difference between the diameter of nanospheres at time “t” (Vt) and the initial time (t =0 [V0]). This was calculated as follows41:
Swelling% = Vt − V0/V0 ×100. | (4) |
In vitro mucoadhesive wash-off method
The mucoadhesive properties of the nanospheres were determined with a slight modification of the method reported by Mankala et al.42 A small piece (2×2 cm) of rat stomach mucosa was mounted onto a glass slide that was 7.62×2.54 cm and secured with elastic bands. A known amount of nanospheres was placed on a mucosal wet surface; thereafter, the support was hung onto the arm of a US Pharmacopeia tablet-disintegrating apparatus (ZT 304 disintegration tester; Erweka, Heusenstamm, Germany). The disintegrator basket containing the mucosa was adjusted for regular up and down movement in a beaker containing phosphate buffer with a pH of 6.8 at 37°C. The drug content was estimated spectrophotometrically at different time intervals (1 hour, 3 hours, 5 hours, and 8 hours) to ascertain the remaining drug-adherence levels to the mucosa. The total amount of nanospheres that corresponded to the difference in the amount of drug was calculated. The adhered nanosphere amounts were estimated from the difference between the amount of the applied nanospheres and the nanoparticle-flow amount. The percentage of the nanospheres that adhered to the applied nanospheres was computed as the percentage of mucoadhesion.
Drug-release study of vildagliptin
In vitro drug release was carried out using a dialysis technique. Vildagliptin nanospheres were positioned in a dialysis bag and dialyzed against 250 mL of phosphate-buffered saline (PBS). The PBS was prepared to 250 mL with water. Then, 5 mL of the samples at 15 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, and 12 hours were withdrawn and replaced with a fresh sample of 5 mL of PBS. During the dissolution process, the temperature was maintained at 37°C±1°C, and the sink conditions were maintained throughout the course of the study. The drug content was analyzed by the LC-MS method, as described previously.35,43
In vitro mucoadhesiveness measurement
Male Sprague Dawley rats weighing 200±50 g were fasted for 24 hours before commencing the experiment. Water was provided ad libitum. Five groups of animals were employed, with each group containing 15 rats. The first group was orally administered an aqueous solution of vildagliptin. The second to fifth groups received vildagliptin nanospheres. The oral administration of the nanospheres was accomplished by suspending a 20 mg sample of the nanospheres in 1.0 mL of normal saline, and the nanoparticles were administered via a rubber tube under nonanesthetic conditions. Three rats were killed at each time point after 1 hour, 2 hours, 4 hours, 8 hours, 10 hours, and 12 hours of administration. The stomach and the entire length of the small intestine were isolated.41 The entire GIT was cut open to expose the inner mucosal surface. All nanospheres located in each part were collected by scratching the mucosa with a spatula. The mixture was mashed using a homogenizer to extract the vildagliptin, and the sample was kept for 24 hours. After centrifugation at 805 g for 20 minutes, the supernatant was determined spectrophotometrically.
LC-MS method
The respective GIT tissue samples were has revolutionized, and vildagliptin was extracted with double-distilled water five times. The collected extractions were diluted to 15 mL and then centrifuged at 12,000 rpm for 15 minutes. For protein precipitation, 500 μL of acetonitrile was added to 2 mL of the supernatant, and the mixture was vortexed for 1 minute and centrifuged at 8,000 rpm for 10 minutes. Then, the 10 μL supernatant was injected into the LC-MS system to determine the vildagliptin concentration, while adopting the method described in “Drug content”.
The stability of GVN
The formulation was kept in a glass bottle and stored for 12 months at 3°C–5°C, 15°C–25°C, and 37°C, respectively (Binder KMF 115; Tuttlingen, Germany). The surface examination and active pharmaceutical ingredient (API) content were evaluated periodically.35
Results and discussion
The Büchi Nano Spray Dryer B-90 has revolutionized today’s pharmaceutical industries, and it offers a variety of particle-engineering prospects. Additionally, it is capable of producing high levels of drug loading and efficient spray processes for the smallest amount at a high yield.31,44
The vildagliptin loading and the yield percentage were 76.2%±4.6% and 83%±2%, respectively. The high drug-encapsulation efficiency from 50% up to 100% is classical for spray-drying. The rationale for this is that the drug cannot separate into a continuous phase, as is the case with the water-in-oil emulsion method or solvent evaporation.45 However, the loss of the product occurred due to dehydrated particles sticking to the walls of the drying compartment, thus reducing the yield of the product collected, as shown in Figure 1. In contrast with conventional “particle engineering” methods, a spray-dryer provided maximum yields of fine powder.46–48
 | Figure 1 Dried particles adhere to the drying chamber. |
The particle-size distribution of vildagliptin nanospheres is shown in Figure 2. The prepared latex showed narrow particle-size distribution at 445 nm.49 Determining the particle-size distribution of the nanospheres is important, because the target site of the nanospheres that contains the therapeutic agent is dependent on the size distribution of the powder and its bioavailability. The drug plasma concentration will oscillate if the size deviation is broad.
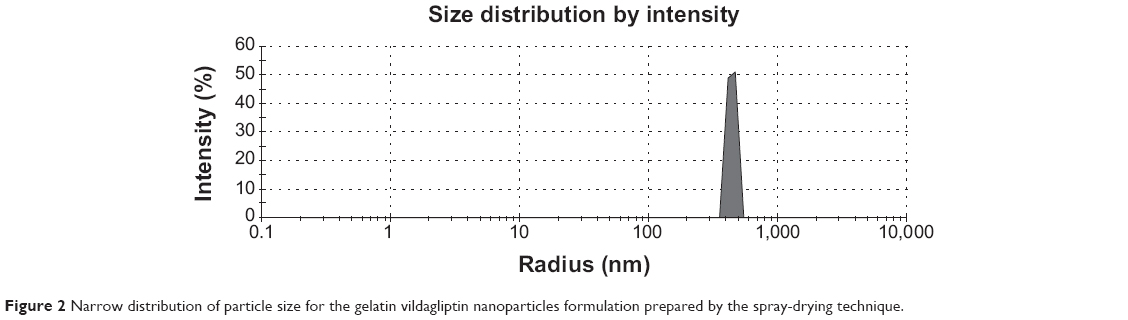 | Figure 2 Narrow distribution of particle size for the gelatin vildagliptin nanoparticles formulation prepared by the spray-drying technique. |
The flow behavior of the GVN-formulated nanospheres demonstrated good flowability, with θ <33.5°. The change in flowability upon the spray-drying of gelatin may be attributed to the relatively small and narrow particle-size distribution of the formulation,50 which was characterized by a mean size of 80.9 nm and 5.7 nm.
The surface morphology is a qualitative evaluation of a three-dimensional surface, and it was analyzed for the GVN formulation that was produced by the spray-drying technique. The nanospheres exhibited a spherical and smooth surface featuring dehydrated powder (Figure 3). Essentially, it was noted that the occurrence of nanospheres that were shriveled in shape could have been caused by a lack of surfactant. While the preparation was planned to reside in the GIT for a prolonged period of time, we did not use a surfactant or surface-active agent in our preparation, due to their possible toxic deposits upon ingestion into the GIT.51,52 Additionally, surface folding may also have been due to a loss of moisture in the drying chamber of the spray feed.31
 | Figure 3 Surface morphology of the gelatin vildagliptin nanoparticles preparation showing a smooth surface and surface-folding shape. |
Figure 4 illustrates the swelling profile of the nanospheres in stomach gastric fluid for GVN at different time intervals. The results indicated that all of the nanosphere formulations swelled gradually when immersed in stomach gastric fluid. This may have been due to the type of gelatin (type B) used in formulating the nanospheres. This type of gelatin is well known to be produced from an alkaline precursor, and would therefore be expected to swell in an acidic environment.53 The swelling profile of the GVN formulation was found to be 165%±7%.
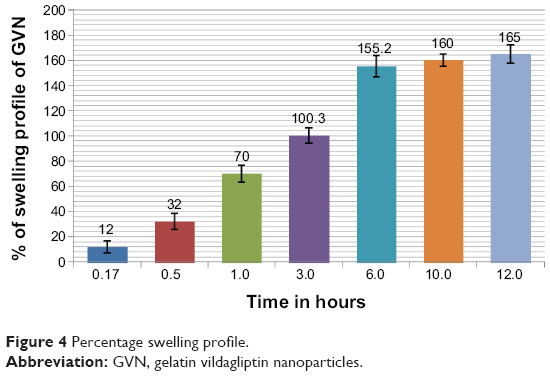 | Figure 4 Percentage swelling profile. |
In vitro release testing is an essential tool in analyzing drug-formulation performance for the majority of formulations. Figure 5 illustrates the in vitro release data for GVN and vildagliptin (API). The API was dissolved completely (100%) at 30 minutes (the first sampling point), at the same time API from the gelatin formulation was (70%). The in vitro profile showed a burst release for the GVN formulation, and it released the API alone (25%±5%) in 30 minutes. The data obtained were not surprising, because the literature54,55 reveals that the drug-release profile typically exhibits two phases. The initial “burst effect” occurs due to free drug binding at the surface of the nanospheres, and this will help to achieve the minimum therapeutic level, followed by second-phase release of drug up to 12 hours (97.5%±2%).56 The mechanism of drug transport/permeation through the nanospheres was considered by fitting the release profile to the five kinetic models (first-order, Higuchi, Korsmeyer–Peppas, Baker and Lonsdale, and Hixon and Crowell). The data were analyzed by fitting them to Sigma plot version 9, and the best-fit curve with an R2 of 0.9838 was acknowledged for the Korsmeyer–Peppas model. Other models that fit the release data were less significant. This may have been due to the diffusion and swelling mechanism of the nanoparticles.
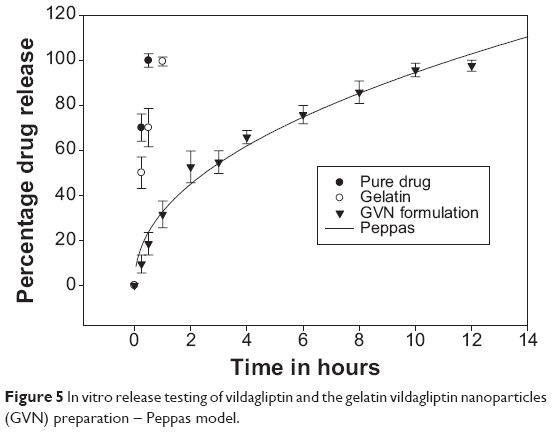 | Figure 5 In vitro release testing of vildagliptin and the gelatin vildagliptin nanoparticles (GVN) preparation – Peppas model. |
The FTIR spectra of vildagliptin alone, aminated gelatin, and formulation are shown in Figure 6. The FTIR spectrum of vildagliptin showed prominent peaks at 3,110–3,420 cm−1 (broad) and 3,265 cm−1, representing OH and N–H stretching vibrations. The prominent peaks at 2,250 cm−1 and 1,610 cm−1 were assigned to nitrile and amide C=O stretching bands, respectively. Aminated gelatin FTIR spectra showed peaks at 3,000–3,400 cm−1, representing O–H stretching vibration of COOH (intramolecular hydrogen bonding), other peaks at 3,220 cm−1 and 3,300 cm−1 were due to N–H stretching vibrations of the NH2 group, and absorption bands in the range of 1,550–1,640 cm−1 were due to amide C=O stretching and N-H–bending vibrations. In the case of the formulation (vildagliptin nanoparticles), overlapping of the characteristic peaks was observed, indicating the stable nature of the drug during spraying of the nanoparticles with amino gelatin.
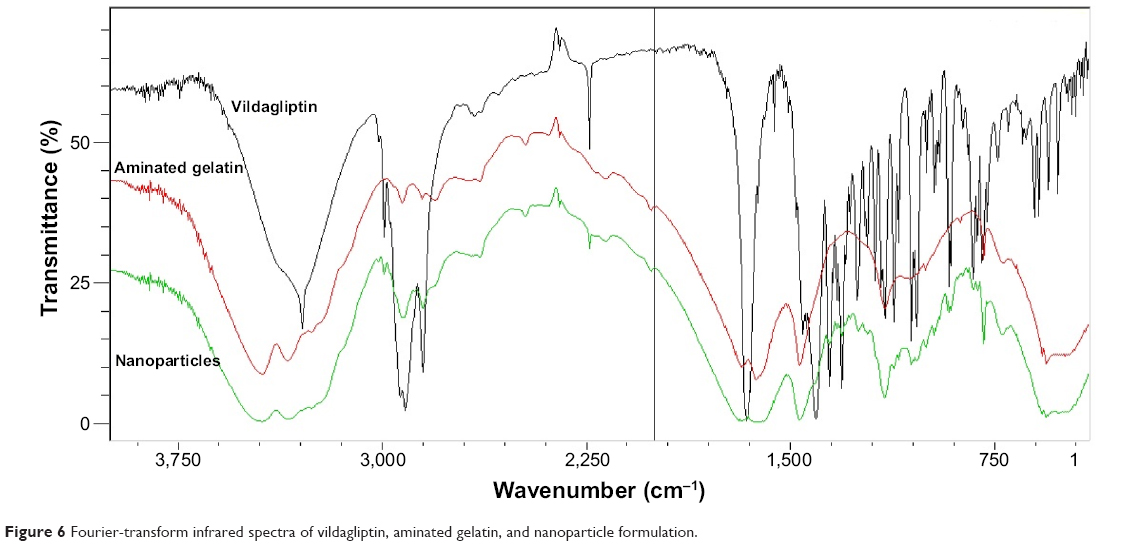 | Figure 6 Fourier-transform infrared spectra of vildagliptin, aminated gelatin, and nanoparticle formulation. |
From the wash-off test, the bioadhesion of the nanospheres was found to exhibit good mucoadhesive properties. The drug content from the PBS at different time intervals was found to be as follows: 1 hour, 14.5%; 3 hours, 38%; 5 hours, 67.3%; and 8 hours, 86.7%. The results confirmed that the nanospheres had excellent bioadhesive characteristics and could effectively adhere to the stomach mucosa. The results obtained are logical, because the mucosa is comprised of negatively charged mucin, and it will interact with a cationic charge of aminated gelatin (cationized gelatin with ethylenediamine).57 In addition, at lower pH conditions, a stronger electrostatic repulsion between the amino groups or nanospheres allows the nanosphere gel to swell and bind extensively.18
It is well known that the in vivo study of animals is a necessary and mandatory step for the development of any drug-delivery system. Vildagliptin was orally ingested by Sprague Dawley rats that had fasted for 24 hours; the vildagliptin dose was administered at 3 mg/kg body weight in the form of GVN or a vildagliptin solution (standard). The rats were killed at different time intervals to measure the mucoadhesiveness of the nanospheres in GIT. From Figure 7, it is evident that the remaining percentage of vildagliptin was significantly lower (5.3% within 4 hours). In comparison with the GVN formulation, the percentage remaining was much higher (42.4% in 4 hours). This may have been due to the aminated gelatin having a cationic charge, and due to the fact that it binds to the mucin of the GIT mucosa, which is negatively charged.57 This might highlight that the mucoadhesive/bioadhesive nanospheres we formulated had better mucoadhesive effects in the GIT, and they might reside longer in the stomach for more effective drug release among diabetics. Statistical analysis showed that the mucoadhesive strength of the GVN was significantly (P<0.05) superior to that of the pure vildagliptin solution.
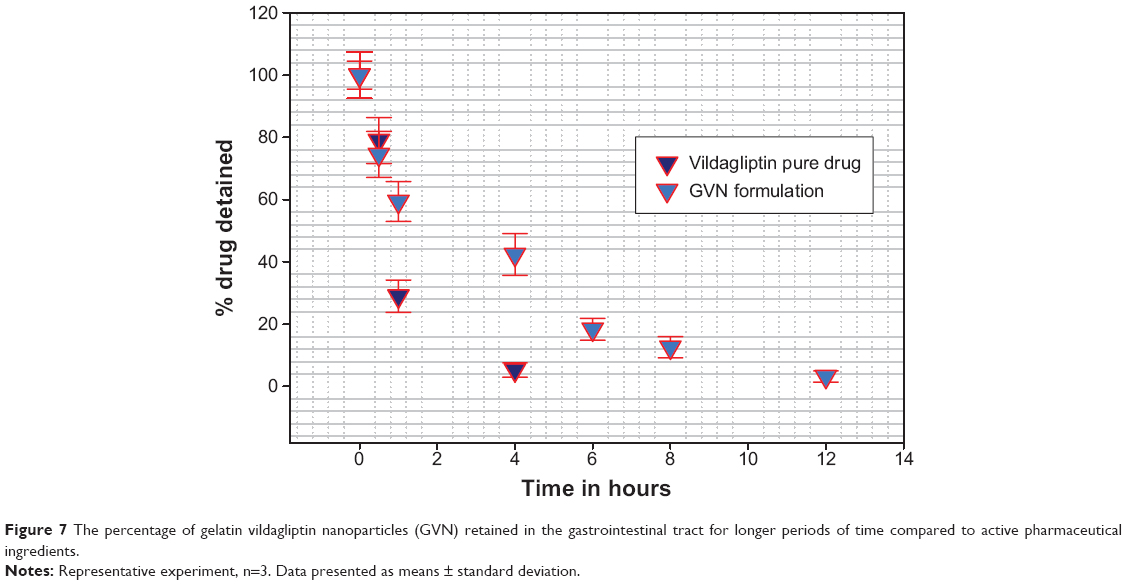 | Figure 7 The percentage of gelatin vildagliptin nanoparticles (GVN) retained in the gastrointestinal tract for longer periods of time compared to active pharmaceutical ingredients. |
Stability testing
This study examined the changes in the physicochemical properties of a GVN formulation during storage at 3°C–5°C/15°C–25°C for 12 months (Figure 8). The surface analysis and API content of GVN demonstrated no notable changes; still, at 37°C and relative humidity 75%, liquefaction was noted, and the particles were aggregated. This may have been due to the fact that the particles were stored at 37°C and at relative humidity 75%, so it is suggested that the particles can be stored at below room temperature. The vildagliptin-activity data was revealed, as solid circles had a linear regression line (solid) that overlapped. The lower 95% confidence line is also plotted. The approved definition of the shelf-life time is the x-axis coordinate for the intersection of the lower 95% confidence line with 90% drug activity (Guideline for Submitting Documentation for the Stability of Human Drugs and Biologics, US Food and Drug Administration, Department of Health and Human Services, 1987). This macrocomputes the shelf life analytically by solving the equation for the lower 95% confidence line and the 90% activity line – in our case (t90 =16.7 months label).
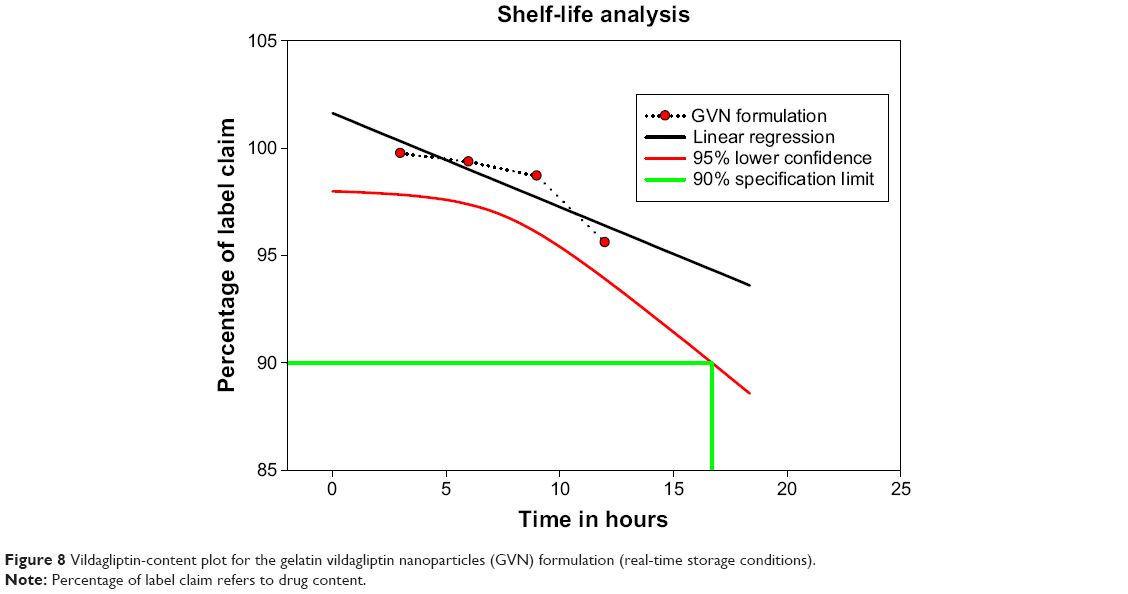 | Figure 8 Vildagliptin-content plot for the gelatin vildagliptin nanoparticles (GVN) formulation (real-time storage conditions). |
Conclusion
The oral drug-delivery route is one of the most favored routes of drug administration. The major problem with most drugs is that they often have a short half-life. The most important conclusion of our mucoadhesive drug-delivery route is that the nanospheres interacted with the mucosa of the GIT, and they were localized or trapped at the adhesive site by retaining nanospheres at the target site. Our research can be used as a benchmark for site-specific drug administration, and it can be further explored in vivo with other therapeutic agents, using slight modifications in the preparation method.
The mucoadhesive nanospheres prepared by the Büchi Nano Spray Dryer B-90 were considered suitable for oral administration by virtue of their favorable narrow particle-size distribution. Higher mucoadhesion results were due to aminated gelatin. The fact that the nanospheres were retained to a larger extent both in vitro and in vivo suggests that the formulated nanospheres could be used for oral administration, which in turn could eliminate frequent dosing intervals and prevent fluctuations of the therapeutic agent.
Acknowledgments
All of the authors thank the Deanship of Scientific Research, King Faisal University, Al-Ahsa, Saudi Arabia for their support (grant number 140131). We thank Journal Prep for editing our manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Sivaprasad S, Gupta B, Crosby-Nwaobi R, Evans J. Prevalence of diabetic retinopathy in various ethnic groups: a worldwide perspective. Survey of Ophthalmology. 2012;57(4):347–370. | ||
Dong HQ, Li M, Zhu F, Liu FL, Huang JB. Inhibitory potential of trilobatin from Lithocarpus polystachyus Rehd against α-glucosidase and α-amylase linked to type 2 diabetes. Food Chem. 2011;130(2):261–266. | ||
Kikuchi M, Haneda M, Koya D, et al. Efficacy and tolerability of vildagliptin as an add-on to glimepiride in Japanese patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2010;89(3):216–223. | ||
Boutayeb A, Boutayeb S. The burden of non communicable diseases in developing countries. Int J Equity Health. 2005;4(1):2. | ||
Farag YM, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrol Dial Transplant. 2011;26(1):28–35. | ||
Kheir N, Zaidan M, Younes H, Hajj ME, Wilbur K, Jewesson PJ. Pharmacy education and practice in 13 Middle Eastern countries. Am J Pharm Educ. 2009;72(6):133. | ||
Elhadd TA, Al-Amoudi AA, Alzahrani AS. Epidemiology, clinical and complications profile of diabetes in Saudi Arabia: a review. Am J Pharm Educ. 2007;27(4):241–250. | ||
[No authors listed]. Summary of the “Stakeholder opinions: type 2 diabetes in the UAE and Saudi Arabia growth opportunities in fragmented markets” report. Datamonitor. 2008;12:241–250. | ||
Kikuchi M, Abe N, Kato M, Terao S, Mimori N, Tachibana H. Vildagliptin dose-dependently improves glycemic control in Japanese patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2009;83(2):233–240. | ||
Ahrén B, Schweizer A, Dejager S, Villhauer EB, Dunning BE, Foley JE. Mechanisms of action of the dipeptidyl peptidase-4 inhibitor vildagliptin in humans. Diabetes Obes Metab. 2011;13(9):775–783. | ||
Nauck M, Schmidt W, Meier J. The incretin modulators – incretin mimetics (GLP-1 receptor agonists) and incretin enhancers (DPP-4 inhibitors). In: Mogensen C, editor. Pharmacotherapy of Diabetes: New Developments. New York: Springer; 2007:111–141. | ||
Morimoto K, Chono S, Kosai T, Seki T, Tabata Y. Design of cationic microspheres based on aminated gelatin for controlled release of peptide and protein drugs. Drug Deliv. 2008;15(2):113–117. | ||
Tabata Y, Ikada Y. Protein release from gelatin matrices. Adv Drug Deliv Rev. 1998;31(3):287–301. | ||
Hao Z, Wu H, Hao L, et al. Preparation, characterization, and release behavior of ceftiofur-loaded gelatin-based microspheres. J Appl Polym Sci. 2013;130(4):2369–2376. | ||
Cremers HF, Feijen J, Kwon G, et al. Albumin-heparin microspheres as carriers for cytostatic agents. J Control Release. 1990;11(1–3):167–179. | ||
Peppas NA, Buri PA. Surface, interfacial and molecular aspects of polymer bioadhesion on soft tissues. J Control Release. 1985;2:257–275. | ||
Wang J, Tabata Y, Bi D, Morimoto K. Evaluation of gastric mucoadhesive properties of aminated gelatin microspheres. J Control Release. 2001;73(2–3):223–231. | ||
Wang J, Tauchi Y, Deguchi Y, Morimoto K, Tabata Y, Ikada Y. Positively charged gelatin microspheres as gastric mucoadhesive drug delivery system for eradication of H. pylori. Drug Deliv. 2000;7(4): 237–243. | ||
He YL, Serra D, Wang Y, et al. Pharmacokinetics and pharmacodynamics of vildagliptin in patients with type 2 diabetes mellitus. Clin Pharmacokinet. 2007;46(7):577–588. | ||
Keating GM. Vildagliptin: a review of its use in type 2 diabetes mellitus. Drugs. 2010;70(16):2089–2112. | ||
Singh B, Chakkal SK, Ahuja N. Formulation and Optimization of controlled release mucoadhesive tablets of atenolol using response surface methodology. AAPS PharmSciTech. 2006;7(1):E3. | ||
Ponchel G, Irache J-M. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Adv Drug Deliv Rev. 1998;34(2–3):191–219. | ||
Bernkop-Schnürch A. Mucoadhesive systems in oral drug delivery. Drug Discov Today Technol. 2005;2(1):83–87. | ||
Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55(1):R1–R4. | ||
Bilati U, Allémann E, Doelker E. Sonication parameters for the preparation of biodegradable nanocapsules of controlled size by the double emulsion method. Pharm Dev Technol. 2003;8(1):1–9. | ||
Rao JP, Geckeler KE. Polymer nanoparticles: preparation techniques and size-control parameters. Prog Polym Sci. 2011;36(7):887–913. | ||
Mahajan HS, Tatiya BV, Nerkar PP. Ondansetron loaded pectin based microspheres for nasal administration: in vitro and in vivo studies. Powder Technol. 2012;221:168–176. | ||
Büchi. Nano Spray Dryer B-90: the “cost saver” for small samples and particles. Available from: http://www.buchi.com/sites/default/files/downloads/B-90_Product_Brochure_en_A.pdf. Accessed March 21, 2014. | ||
Su YL, Fu ZY, Zhang JY, et al. Microencapsulation of Radix salvia miltiorrhiza nanoparticles by spray-drying. Powder Technol. 2008;184(1):114–121. | ||
Gupta S, Samanta M, Raichur A. Dual-drug delivery system based on in situ gel-forming nanosuspension of forskolin to enhance antiglaucoma efficacy. AAPS PharmSciTech. 2010;11(1):322–335. | ||
Harsha S. Dual drug delivery system for targeting H. pylori in the stomach: preparation and in vitro characterization of amoxicillin-loaded Carbopol® nanospheres. Int J Nanomedicine. 2012;7:4787–4796. | ||
Harsha S, Chandramouli R, Rani S. Ofloxacin targeting to lungs by way of microspheres. Int J Pharm. 2009;380(1–2):127–132. | ||
Yeo JH, Lee KG, Lee YW, Kim SY. Simple preparation and characteristics of silk fibroin microsphere. Eur Polym J. 2003;39(6):1195–1199. | ||
Alexakis T, Boadi D, Quong D, et al. Microencapsulation of DNA within alginate microspheres and crosslinked chitosan membranes for in vivo application. Appl Biochem Biotechnol. 1995;50(1):93–106. | ||
Harsha NS, E. Aldhubiab B, Abdulrahman Al, Attimarad M, Nair A. Carbopol 934-P loaded with vildagliptin for diabetic delivery: in vitro and in vivo evaluation of nanoparticles. Curr Nanosci. 2013;9(5): 642–647. | ||
He YL, Sabo R, Riviere GJ, et al. Effect of the novel oral dipeptidyl peptidase IV inhibitor vildagliptin on the pharmacokinetics and pharmacodynamics of warfarin in healthy subjects. Curr Med Res Opin. 2007;23(5):1131–1138. | ||
Lu B, Zhang JQ, Yang H. Lung-targeting microspheres of carboplatin. Int J Pharm. 2003;265(1–2):1–11. | ||
Radha GV, Rani TS, Sarvani B. A review on proniosomal drug delivery system for targeted drug action. J Basic Clin Pharm. 2013;4(2): 42–48. | ||
Bayomi MA, al-Suwayeh SA, el-Helw AM, Mesnad AF. Preparation of casein-chitosan microspheres containing diltiazem hydrochloride by an aqueous coacervation technique. Pharm Acta Helv. 1998;73(4):187–192. | ||
Tayel SA, El-Nabarawi MA, Tadros MI, Abd-Elsalam WH. Positively charged polymeric nanoparticle reservoirs of terbinafine hydrochloride: preclinical implications for controlled drug delivery in the aqueous humor of rabbits. AAPS PharmSciTech. 2013;14(2):782–793. | ||
Dhaliwal S, Jain S, Singh HP, Tiwary AK. Mucoadhesive microspheres for gastroretentive delivery of acyclovir: in vitro and in vivo evaluation. AAPS J. 2004;10(2):322–330. | ||
Mankala SK, Korla AC, Gade S. Development and evaluation of aceclofenac-loaded mucoadhesive microcapsules. J Adv Pharm Technol Res. 2011;2(4):245–254. | ||
Harsha NS, Rani RH. Drug targeting to lungs by way of microspheres. Arch Pharm Res. 2006;29(7):598–604. | ||
Harsha S, Attimarad M, Khan T, et al. Design and formulation of mucoadhesive microspheres of sitagliptin. J Microencapsul. 2013;30(3):257–264. | ||
Cordin A, Nina S. Spray dried biodegradable polymers as target material for controlled drug delivery. Best@buchi. 2007;46. | ||
Atuah KN, Walter E, Merkle HP, Alpar HO. Encapsulation of plasmid DNA in PLGA-stearylamine microspheres: a comparison of solvent evaporation and spray-drying methods. J Microencapsul. 2003;20(3):387–399. | ||
Burrki K, Jeon I, Arpagaus C, Betz G. New insights into respirable protein powder preparation using a nano spray dryer. Int J Pharm. 2011;408(1–2):248–256. | ||
Schafroth N, Arpagaus C, Jadhav UY, Makne S, Douroumis D. Nano and microparticle engineering of water insoluble drugs using a novel spray-drying process. Colloids Surf B Biointerfaces. 2012;90:8–15. | ||
Paulal HC, Sombral FM, Abreu FO, de Paul RC. Lippia sidoides essential oil encapsulation by angico gum/chitosan nanoparticles. J Braz Chem Soc. 2010;21(12):2359–2366. | ||
Samy W, Elgindy N, El-Gowelli HM. Biopolymeric nifedipine powder for acceleration of wound healing. Int J Pharm. 2012;422(1–2):323–331. | ||
Coowanitwong I, Arya V, Kulvanich P, Hochhaus G. Slow release formulations of inhaled rifampin. AAPS J. 2008;10(2):342–348. | ||
Patil P, Joshi P, Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004;5(3):43–50. | ||
Adikwu MU. Biopolymers in Drug Delivery: Recent Advances and Challenges. Sharjah: Bentham Science; 2009. | ||
Pavanetto F, Genta I, Giunchedi P, Conti B. Evaluation of spray drying as a method for polylactide and polylactide-co-glycolide microsphere preparation. J Microencapsul. 1993;10(4):487–497. | ||
Fu YJ, Mi FL, Wong TB, Shyu SS. Characteristic and controlled release of anticancer drug loaded poly(D,L-lactide) microparticles prepared by spray drying technique. J Microencapsul. 2001;18(6):733–747. | ||
Jadhav NR, Tone JS, Irny PV, Nadaf SJ. Development and characterization of gelatin based nanoparticles for targeted delivery of zidovudine. Int J Pharm Investig. 2013;3(3):126–130. | ||
Van Der Walle C. Peptide and Protein Delivery. Amsterdam: Elsevier Science; 2011. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.