Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanometer-scale siRNA carriers incorporating peptidomimetic oligomers: physical characterization and biological activity
Authors Utku Konca Y, Kirshenbaum K, Zuckermann RN
Received 12 November 2013
Accepted for publication 23 January 2014
Published 10 May 2014 Volume 2014:9(1) Pages 2271—2285
DOI https://doi.org/10.2147/IJN.S57449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Yeliz Utku Konca,1 Kent Kirshenbaum,2 Ronald N Zuckermann3
1Department of Chemistry, Koc University, Istanbul, Turkey; 2Department of Chemistry, New York University, New York, NY, 3Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
Abstract: Synthetic short interfering RNA (siRNA) oligonucleotides can trigger the RNA interference pathway and lead to selective gene silencing. Despite considerable enthusiasm and investment, formidable challenges remain that may deter translating this breakthrough discovery into clinical applications. In particular, the development of efficient, nontoxic, nonimmunogenic methods for delivering siRNA in vivo has proven to be exceptionally challenging. Thorough analysis of the relationship between the structure and function of siRNA carrier systems, both in isolation and in complex with RNA, will facilitate the design of efficient nonviral siRNA delivery vehicles. In this study, we explore the relationship between the physicochemical characteristics and the biological activity of "lipitoid" compounds as potent siRNA delivery vehicles. Lipitoids are cationic peptidomimetic oligomers incorporating a peptoid and a phospholipid moiety. Lipitoids can associate with siRNA oligonucleotides and self-assemble into spherical lipitoid-based nanoparticles (LNPs), with dimensions that are dependent upon the medium and the stoichiometric ratio between the cationic monomers of the lipitoid and anionic siRNA oligonucleotides. The morphology, gene silencing efficiency, and cytotoxicity of the siRNA-loaded LNPs are similarly sensitive to the stoichiometry of the complexes. The medium in which the LNPs are formed affects the assembled cargo particles' characteristics such as particle size, transfection efficiency, and stability. Formation of the LNPs in the biological, serum-free medium OptiMEM resulted in LNPs an order of magnitude larger than LNPs formed in water, and were twice as efficient in siRNA transfection compared to LNPs formed in water. Inhibitor studies were conducted to elucidate the efficiency of lysosomal escape and the uptake mechanism of the siRNA-loaded LNPs. Our results suggest that these lipitoid-based, siRNA-loaded spherical LNPs are internalized through a lipid raft-dependent and dynamin-mediated pathway, circumventing endosomal and lysosomal encapsulation. The lipitoid-siRNA nanospheres proved to be suitable platforms for investigating the critical parameters determining the efficiency of transfection agents, revealing the necessity for conducting characterization studies in biological media. The investigation of the LNP internalization pathway points to an alternative uptake route that bypasses the lysosome, explaining the surprisingly high efficiency of LNPs and suggesting that the uptake mechanism should be probed rather than assumed for the next generation of rationally designed transfection agents.
Keywords: lipitoid, siRNA delivery, therapeutic oligonucleotides, peptoid
Introduction
Short interfering ribonucleic acid (siRNA) can exert specific gene silencing effects in mammalian cells1,2 and thus have the potential to act as therapeutics for disease states that would otherwise be difficult to target via existing drugs.3 Early siRNA-based therapeutic regimens are currently in clinical trials, targeting more than a dozen diseases to date.4–6 Due to shortcomings regarding the biological stability and membrane permeability of naked siRNA,7 the routine clinical application of this promising therapeutic modality will rely upon the development of biocompatible molecular carrier systems that can efficiently deliver siRNAs to the target site.4,8,9 To date, a growing number of candidate molecules have been evaluated for their capability to facilitate intracellular nonviral uptake of siRNA.6,10–17 An ongoing challenge is to attain improved understanding of the relationship between the chemical composition, the overall structure, and the biological function of these delivery molecules.18 Developing a robust structure–activity relationship for carrier molecules may enable rational design and lead to the discovery of guidelines for improving the potency, stability, specificity, and safety of siRNA delivery vehicles.
In this study, we investigate a molecular siRNA delivery system termed “lipitoid”, which is composed of a cationic peptoid oligomer and a hydrophobic lipid moiety.19–21 A broad class of functionalized lipid materials shows promise as carriers for therapeutic siRNA. Not surprisingly, molecules similar to lipitoids, termed “lipidoids”, have subsequently been investigated and shown to be effective not only in cell culture but also in higher organisms.10,18,22–24 Ongoing studies in the rational design of these siRNA delivery vehicles have proposed designing smaller sized nanoparticle carriers that can penetrate the cells and support a mechanism of action entailing endocytic encapsulation and lysosomal escape,6,18,25 suggesting improved efficiency in delivery vehicles that can escape this pathway.25 Our molecule, containing a peptoid backbone, features many desirable properties of peptidomimetic oligomers, including facile solid-phase synthesis,26 access to exceptional side chain chemical diversity,27 and resistance to proteolytic degradation.28 Thus, peptoids constitute a modular oligomeric vehicle for siRNA delivery and provide an attractive platform to systematically investigate how alterations of particle formation parameters such as charge ratio, zeta potential, and morphology may influence transfection efficiency. Our earlier published studies have shown that this system is highly effective for oligonucleotide delivery, with efficiency comparable to, and in some cases superior to, commercial reagents such as Oligofectamine™, particularly for hard-to-transfect primary cell types.21,29 Here, our focus centers on investigating the origins of the functional potency. We show that this well-defined species, containing a nonamer hydrophilic peptoid backbone and two C12 lipid tails, associates with siRNA to form nanoparticle assemblies. In this study, we further explore the molecular and supramolecular structure–function relationships of this lipitoid-based nanoparticle (LNP) by probing the impact of complex formation conditions on the supramolecular morphology and the resulting effect on biological function. Our results reveal striking differences between both the physical properties and the biological function of LNPs formed in water and in biological media. The smallest LNPs were not necessarily the best performing ones, which may pose difficulties with intravenous injection but may ultimately improve the silencing efficiency of the LNPs by increasing the circulating half-life of these molecules in the bloodstream.8
The high silencing efficiency of the siRNA loaded LNPs may be due to the role of a different internalization pathway than commonly hypothesized for these molecules. Our results suggest a lipid raft/caveolae dependent internalization route, which bypasses the lysosome, and thus eliminates the need for a lysosomal escape step. Owing to their alternative internalization mechanism, these LNPs may further be exploited in the design of therapeutic biomaterials capable of intracellular targeting.
Materials and methods
Materials
The lipitoid agent consists of a single high performance liquid chromatography-purified compound, DMPE-(NaeNmpeNmpe)3, and was prepared as described earlier19 (dimirystoyl phosphatidyl-ethanolamine [DMPE]; N-(2-aminoethyl)glycine [Nae]; N-[2-(4′-methoxyphenyl)ethyl] glycine [Nmpe]). The peptoid portion of the lipitoid molecule was synthesized on Rink amide resin, using an automated robotic synthesizer at room temperature. The lipid was subsequently manually conjugated to the oligomer at 60°C. Following cleavage of the product from the solid support, the lipitoid was purified by high performance liquid chromatography and characterized by liquid chromatography-mass spectrometry. Green fluorescent protein (GFP)-22 siRNA (sense strand sequence GCAAGCUGACCCUGAAGUUCAU, molecular weight 17,001.9) and AllStars negative control siRNA (proprietary sequence, catalog number 1027280, molecular weight 14,857.1) were obtained from Qiagen (Venlo, the Netherlands). NM2C5 human breast cancer cells and NM2C5-GFP cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). U2OS human osteosarcoma cells and U2OS-GFP cell lines were a kind gift of the Pam Silver Lab (Harvard Medical School, Boston, MA, USA). Allophycocyanin (APC) conjugated annexin V was purchased from BD Biosciences (San Jose, CA, USA), propidium iodide (PI) was purchased from Roche (Basel, Switzerland), and all inhibitors were purchased from Sigma-Aldrich (St Louis, MO, USA). Cell culture reagents, including OptiMEM, Oligofectamine, Lipofectamine and RNAiMax were obtained from Life Technologies (Carlsbad, CA, USA). OptiMEM is an optimized modification of the serum-free Modified Eagle’s Medium.
Formation of lipitoid-siRNA complexes
Samples of lipitoid and siRNA complexes were prepared by combining siRNA (20 μM in Qiagen siRNA Suspension Buffer) and 500 μM lipitoid solution prepared in distilled water. The solutions were warmed to room temperature prior to complex formation. As a sample calculation, for generation of LNPs at 3/1 (positive/negative) charge ratio, 5 μL 20 μM siRNA was diluted in 95 μL OptiMEM, and in a separate tube, 7.5 μL 500 μM lipitoid was diluted in 92.5 μL OptiMEM. The two solutions were mixed by gentle aspiration, resulting in a complex formation solution composed of 500 nM siRNA and 18.75 μM lipitoid in a total volume of 200 μL in OptiMEM. The amounts of siRNA were kept fixed, and the amount of lipitoid was adjusted as required for the other charge ratios used. Following incubation for 10 minutes at room temperature with occasional agitation, the resulting LNP complexes were used immediately for transfection, scanning electron microscopy (SEM) analysis, dynamic light scattering (DLS) imaging, zeta potential measurements, or atomic force microscopy (AFM) imaging.
Transfection experiments
U2OS and NM2C5 cells were counted and plated 24 hours prior to transfection at a concentration that would afford 70%–80% cell density at the time of the experiment. Cells were typically plated in 6-well plates in 2 mL growth medium. On the day of transfection, the cells were washed twice with 1 mL phosphate buffered saline (PBS) and once with 1 mL OptiMEM. The cells were then incubated in 1.8 mL OptiMEM at 37°C while the complexes were being prepared. As soon as the complexes of lipitoid and siRNA were formed as described, 200 μL of the LNP-containing solution was gently added onto cells being incubated in 1.8 mL OptiMEM, resulting in final concentrations of 50 nM siRNA and 1.875 μM lipitoid (for LNPs formed at 3/1 charge ratio). The cells were incubated in the presence of LNPs at 37°C under 5% carbon dioxide for 20 minutes before 500 μL of 50% fetal bovine serum (FBS) was added to each well to achieve the normal growth conditions of 10% serum. The cells were then incubated for 48 hours prior to analysis by fluorescence assisted cell sorting (FACS). Negative controls consisted of nontreated cells, cells treated only with lipitoid, and cells treated only with siRNA (at 50 nM transfection concentration). Positive controls were performed with Oligofectamine, Lipofectamine®, and RNAiMAX™. Transfections with the commercial transfection reagents were carried out as instructed in the manufacturers’ protocols, with siRNA concentrations in the 20–200 nM range.
Preparing SEM samples
Silica wafers (5 × 7 mm) were purchased from Ted Pella (Redding, CA, USA) and were plasma treated in a plasma cleaner for 3 minutes to remove organic contaminants. Solutions containing the formed complexes were transferred onto plasma-cleaned silica wafers. A volume of 10 μL complex solution was sufficient to evenly cover the surface of the wafer. The solution was incubated on the silica surface for 5 minutes at room temperature. Generally, the complexes were incubated for 5 minutes under ambient conditions in solution while complexes were being formed, then incubated for an additional 5 minutes on the wafer, which provided a total complex formation time consistent with transfection and DLS experiments (10 minutes). The excess solution was then removed by tapping the side of the wafer onto a clean laboratory cleaning tissue. The wafer was twice rinsed with double distilled water to remove remaining salts that could interfere with SEM analysis. The sample was then dried further at room temperature and atmospheric pressure to complete dryness. The sample was analyzed under a Gemini Ultra-55 analytical scanning electron microscope (Zeiss, Oberkochen, Germany) at 1 kV beam voltage using either an in-lens or SE2 detector (Zeiss).
FACS analysis
Growth medium was aspirated from the cell culture plates, and cells were washed once with 1 mL PBS before being trypsinized until they detached from the growing plate. The detached cells were collected in a 15 mL tube and pelleted by centrifugation at 1,000 rpm for 5 minutes. The trypsin solution was removed, and cells were washed twice in 2 mL PBS. Finally, the cells were counted and resuspended in PBS at a concentration of 106 cells/mL. The prepared samples were analyzed for fluorescence intensity by FACS. FACS data were collected on a BD FACS ARIA2 flow cytometer (BD Biosciences) equipped with 488 nm and 633 nm lasers and suitable band pass filters. GFP fluorescence was detected by utilizing the 488 nm-laser with a 510/30 bp filter, allowing monitoring of emission detection at 515–545 nm.
Cellular viability tests
PI staining was performed in order to determine cell viability and to exclude apoptosis-inducing mechanisms. In addition, annexin V was used to quantify apoptosis in tandem. APC-conjugated annexin V was applied to treated cells as described,30 and apoptosis and cell death was quantified by FACS through quantification of APC and PI, respectively. Lipitoid-siRNA complexes were formed at varying charge ratios in OptiMEM, using GFP-targeting siRNA (or nontargeting control siRNA). Silencing results for the targeted GFP expression were analyzed in GFP-expressing U2OS cells 48 hours posttransfection by FACS. All results are averages of at least three experiments, and silencing values are normalized with respect to nontreated cells as a control. Cytotoxicity was assessed by PI staining and analyzed by FACS.
Inhibitor studies
U2OS-GFP cells were transfected with a 3/1 charge ratio of lipitoid:GFP siRNA complex, while being incubated with various internalization inhibitors. The complexes were formed for 10 minutes, and cells were preincubated with the inhibitors as was described in literature.31,32 Briefly, cells were incubated with 0.1% sodium azide (NaN3) and 50 mM 2-D-deoxyglucose (2DDG) for 1 hour, or with 50 μM chloroquine for 30 minutes, or with 5 mM methyl-b-cyclodextrin (mbcd) for 15 minutes, or with 10 μg/mL chlorpromazine for 15 minutes, or with 200 μM genistein for 1 hour, or with 80 μM dynasore for 1 hour. The cells were then coincubated with the complexes and inhibitors for 20 minutes. The medium containing the LNPs and inhibitors was removed, and following a PBS wash, fresh OptiMEM and FBS was added, yielding a final regular growth medium containing 10% serum. GFP expression by cells was quantified by FACS in 48 hours. All experiments were performed three times with the exception of incubation on ice.
AFM imaging
Complexes were prepared as described above and deposited on a piece of freshly cleaved mica. The solution was incubated for 5 minutes on the mica surface for complete attachment. Excess solution was removed with a gentle stream of nitrogen gas and the sample was imaged immediately in AC mode by an Asylum MFP-3D atomic force microscope (Asylum Research, Santa Barbara, CA, USA), equipped with a Multi-75Al tip.
DLS imaging and zeta potential measurements
Complexes were formed as described, placed into a Malvern Zetasizer nanoseries DTS 1061 capillary cell (Malvern Instruments, Malvern, UK), and analyzed with a Malvern Zetasizer Nano instrument at 25°C. A Polybead Sampler Kit III (Polysciences, Warrington, PA, USA) was used for standardization in DLS measurements.
Results and discussion
Biological activity
Lipitoid-siRNA stoichiometry
The overall net charge of cationic carrier systems for oligonucleotides is one of the critical parameters determining the efficiency of transfection.33 In order to investigate the influence of charge on the lipitoid-siRNA complexes, samples were prepared at various charge ratios by varying the amount of lipitoid and keeping the amount of siRNA constant. The primary amine groups on the Nae sidechains and the secondary amine on the linker between the lipid and the peptoid portion of the lipitoid contribute to the net positive charge of the lipitoid, yielding three overall positive charges per lipitoid molecule (Figure 1).
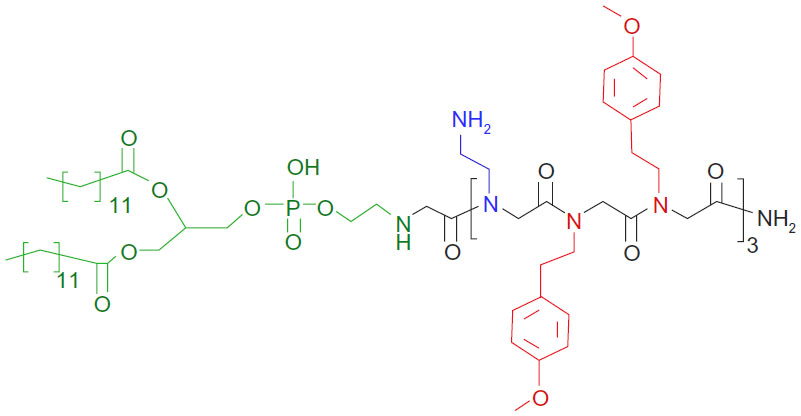 | Figure 1 Chemical structure of the lipitoid. |
Charge ratios of the formed LNPs were calculated by evaluating the ratio of the net positive charges on the lipitoid and the negative charges on the siRNA phosphodiester backbone (see the Materials and methods section for a sample calculation). LNPs were formed between the lipitoid and siRNA oligos targeting the GFP gene. Transfection experiments were carried out in two different human carcinoma cell lines, U2OS and NM2C5 expressing GFP, in order to rule out cell type specific response. Silencing efficiency was determined by quantifying the downregulation of GFP through FACS.
Lipitoid-siRNA particles formed between 0.5/1 and 3/1 charge ratio in OptiMEM exhibited the most potent silencing efficiency (>90% of GFP) in U2OS cells (Figure 2). The ANOVA test was significant for silencing, with P<0.001. The significantly different values resulting from Bonferroni corrected t-tests are marked on the graph for % GFP expression results (Figure 2). Previously, we had confirmed that this level of silencing efficiency is significantly greater than that of Oligofectamine,21 with enhanced downregulation potency as evaluated by quantifying targeted mRNA levels and the targeted protein level of endogenous, physiologically relevant genes. Since previous cytotoxicity tests with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (the MTT assay) showed no significant cytotoxicity, and there was no observable morphology change of the tested cells,21 toxicity was further probed by PI staining and later tested for early signs of apoptosis using annexin V. The lowest cytotoxicity was observed for complexes formed at charge ratios between 0.5/1 and 3/1 (Figure 2). Cytotoxicity increased significantly at charge ratios above 3/1, with a 5/1 charge ratio evoking significant toxicity to treated cells and a 10/1 charge ratio leading to the death of most cells. For cytotoxicity, ANOVA tests and Bonferroni corrected t-tests indicated that the 5/1 LNPs were significantly more toxic than all other LNPs except the 10/1 LNPs, and the 10/1 LNPs were significantly more toxic than all of the other tested LNPs. Annexin V assay was performed to confirm that the cell death was not the outcome of induced apoptosis. The apoptosis rate for all the tested samples, including the nontreated control cells, remained at 1%–2%, suggesting that none of the tested charge ratios contributed to a significant increase in apoptosis of the cells (data not shown). The 3/1 charge ratio represented the best performing charge ratio, and similar results were observed in NM2C5 cells (Figure S1), as toxicity was not evident for LNPs formed at or below a charge ratio of 3/1. Positive controls conducted with Oligofectamine, Lipofectamine, and RNAiMAX indicated that the 3/1 charge ratio LNPs were comparable to, and in some cases better than, commercial transfection reagents (Figure S2). Further experiments were conducted by forming the lipitoid-siRNA LNPs at 3/1 charge ratio unless stated otherwise.
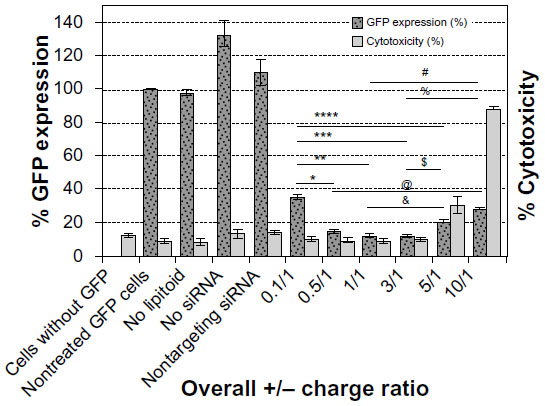 | Figure 2 The effect of charge ratio on silencing efficiency and cytotoxicity. |
Structural characteristics of the complexes
Lipitoid-siRNA nanoparticle morphology
In order to ascertain the nanometer-scale morphology of the active siRNA/lipitoid assemblies of 3/1 charge ratio, these complexes were analyzed by DLS, AFM, and SEM. Lipitoid-siRNA assemblies were initially formed in water as described in Materials and methods. SEM imaging of these assemblies revealed disk-shaped nanoparticles, with radii in the range of 20–80 nm (Figure 3A). SEM analysis of such soft matter may show slightly larger sizes, presumably due to slight flattening of the nanospheres on the silica surface as a result of water removal. To confirm the nanoparticle shapes and sizes in aqueous medium, the assemblies were also analyzed by DLS in water. The average hydrodynamic radius as determined by DLS was observed to be 34±2 nm. AFM imaging confirmed that the complexes were indeed spherical (Figure 3B and D). Together, these results confirm that the lipitoid interacts with siRNA in water to assemble into spherical LNPs of about 34 nm in radius in water.
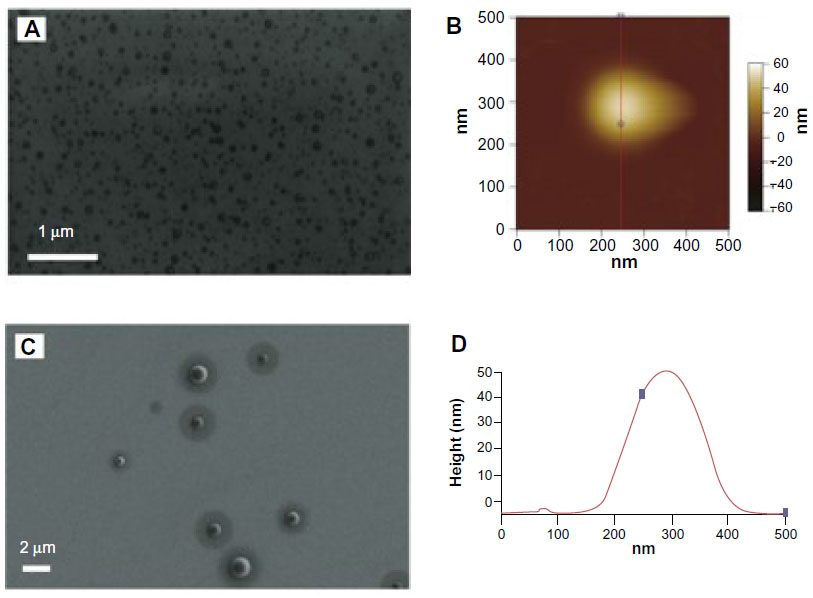 | Figure 3 Lipitoid and siRNA self-assemble to form spherical LNPs in water. |
Biological studies, however, are seldom conducted in pure water. In order to evaluate a more biologically relevant condition, lipitoid-siRNA complexes were formed in OptiMEM, a serum-free biological medium that is routinely used in cell culture experiments. SEM radii of the LNPs formed in OptiMEM were significantly larger, averaging 320±11 nm (Figures 3C and 4A). Similarly, spheres formed in the biological buffer PBS yielded complexes with an average hydrodynamic radius of 289±12 nm (Figure 5E). These order-of-magnitude enlargements of the LNP sizes revealed that the lipitoid-siRNA complexes respond very differently to biological media in comparison with water. The characterization of complexes formed in growth medium with supplemental serum was not carried out due to the complexity of the serum supplemented medium and the impossibility of probing the size of complexes in the presence of large serum proteins.
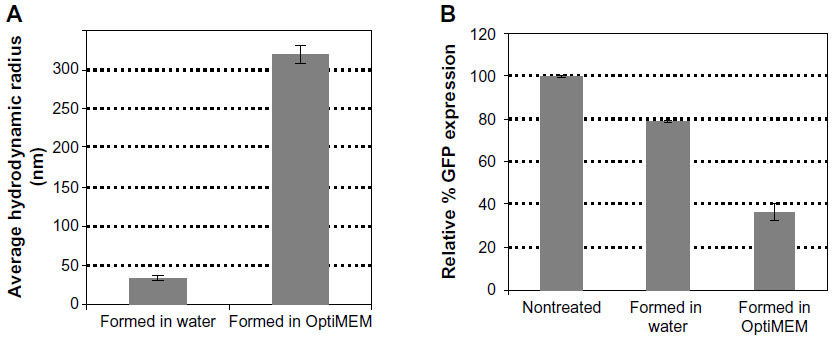 | Figure 4 siRNA loaded LNP sizes depend on the medium in which they are formed. |
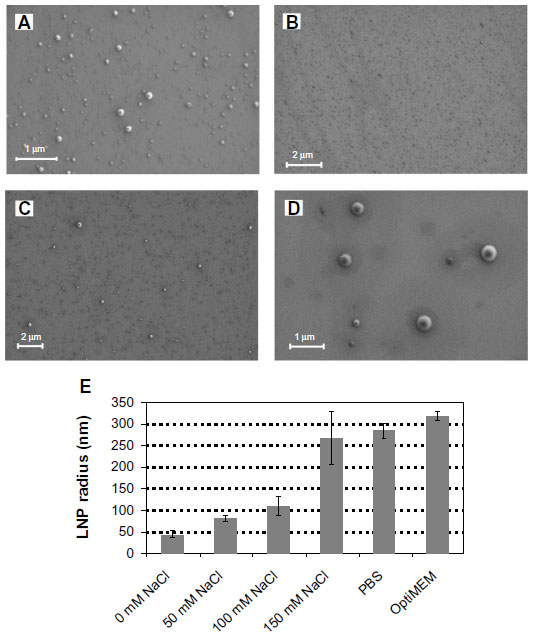 | Figure 5 Lipitoid-siRNA nanoparticle sizes increase with increasing monovalent salt concentration. |
Owing to their order-of-magnitude smaller sizes, the LNPs formed in water were expected to be more efficient in gene silencing. Surprisingly, biological results showed that LNPs formed in water were not as efficient as the LNPs formed in OptiMEM, seen from the GFP expression of 79±0.2 with the LNPs formed in water, as compared to 36±6 with the LNPs formed in OptiMEM. (Figure 4B). In order to study the effect of transferring the LNPs that were formed in water into a biological medium, the LNPs were formed in water as described in the Materials and methods and were then added to cell cultures growing in OptiMEM, following supplementation with 10% serum after transfection. The complexes grew in size when transferred into OptiMEM. However, these complexes were less stable and began to deteriorate within an hour of formation, as monitored by SEM, losing their spherical morphology (Figure S3). Gene silencing experiments conducted with these unstable complexes revealed that the silencing efficiency of the complexes formed in water was 66% lower than the silencing efficiency of the complexes formed in OptiMEM (data not shown). Thus, the LNPs formed in the biological medium OptiMEM were the largest in size and were also the best performing complexes as evaluated in cell culture. To further test the effect of serum on the biological activity of the LNPs, the best performing LNPs were used for transfection. LNPs formed at 3/1 charge ratio with a final concentration of 50 nM siRNA were prepared in OptiMEM and then used to transfect siRNA, either into cells incubated in the serum-free OptiMEM or into cells incubated in serum-supplemented OptiMEM. Silencing activity was 50% with the transfection in serum-supplemented medium, as opposed to the 80% silencing in serum-free medium (Figure S4). This result reinforces the potency of LNPs, demonstrating that they can still generate a significant silencing effect even in the presence of serum.
Complex size depends on ionic strength
The origins of the LNP size enlargement in OptiMEM relative to water were investigated by probing the system in varying concentrations of salt. The concentrations of NaCl during complex formation were varied, at 0 mM, 50 mM, 100 mM, and 150 mM (equivalent to the physiological monovalent salt concentration). Increasing the monovalent salt concentration resulted in the formation of LNPs with increasing spherical size. At 0 mM NaCl concentration the sizes of the nanospheres were homogeneous and averaged r=47±6 nm; at 50 mM NaCl the LNPs grew to r=85±3 nm; at 100 mM NaCl the average was r=110±20 nm; at 150 mM NaCl most of the spheres were between 150 nm and 300 nm in diameter, with an average of r=269±62 nm (Figure 5). As expected from previous results concerning the effect of ionic strength,34 within the tested concentration range of 0–150 mM, ionic strength is the main determinant of the complex sphere size – increasing salt concentration results in larger spheres. The major factor contributing to the size increase of the LNPs when formed in OptiMEM as opposed to in water is thus identified to be the monovalent salt concentration of the solution in which the LNPs are formed. The LNP sizes in 150 mM NaCl (r=269±62 nm) are almost identical to the LNP sizes in PBS (r=289±12 nm) and slightly smaller than the LNP sizes in OptiMEM (r=320±11 nm) (Figure 5E). The slightly larger LNP size in OptiMEM may be attributed to protein binding or to the other numerous variables in OptiMEM (such as divalent cations, amino acids, etc). The decreased biological activity in the presence of serum may be due to formation of large aggregates, which were impossible to characterize by DLS, SEM, or AFM. This aggregation of the LNPs may be due to protein binding, whereby the resulting conglomerate is too large to stick onto and stay on mica or silica surfaces, larger than the limits of detection of DLS, and too large to be taken up into the cells in vitro.
The effect of charge ratio on complex size and morphology
To investigate the effect of charge ratio on the structure of lipitoid-siRNA LNPs, complexes of lipitoid and siRNA were formed at varying charge ratios of cationic lipitoid and anionic siRNA as described above. The sizes and morphologies of the LNPs varied significantly (Figure 6A–G). Sizes of the spherical LNPs increased with increasing charge ratio (Figure 6G and Table S1); however, at the elevated charge ratio of 10/1, the spherical nature of the complexes was observed to be distorted (Figure 6F), and DLS readings were outside of the detectable range. These high charge ratio LNPs were also shown to be extremely toxic and less efficient in silencing (Figure 2). Despite the differences in size and morphology, the zeta potential of all complexes formed in OptiMEM was similar, in the range of −12 to −6 mV (Table S1). This phenomenon of slightly negative zeta potential in biological medium was also recently observed for other cationic cell penetrating systems.35 For comparison, the zeta potential of complexes formed in water was +18.5 mV. Both in water and in OptiMEM, the zeta potential of the LNPs was within the −20 to +20 mV range, which is considered mostly neutral, and the differences within this range are not considered significant. OptiMEM is abundant in physiological salts. Since zeta potential is a relative measure of the charges of the particles within the solution with respect to the surrounding environment, the slightly negative zeta potential in OptiMEM may suggest that OptiMEM is a more negatively charged medium than the LNPs. In support of this analysis, the zeta potential of OptiMEM alone was measured to be −30 mV. These observations illuminate the structure–function relationship between the complexes and their silencing effect. The charge ratio between the lipitoid and the siRNA is a crucial parameter in the formation of lipitoid-siRNA LNPs, affecting not only the size but also the morphology of the formed LNPs. Surprisingly, the charge ratio of LNPs does not seem to dramatically alter the zeta potential of the particles. The 3/1 charge ratio LNPs provided optimal conditions for assembly between the lipitoid molecules and the siRNA oligos, producing spheres of a homogeneous size in the submicrometer scale, with LNP radii of about 34 nm in water and about 320 nm in biological media, with a mostly neutral zeta potential within the ±20 mV range, leading to outstanding efficiency in siRNA delivery and targeted gene downregulation, while exhibiting minimal toxicity.
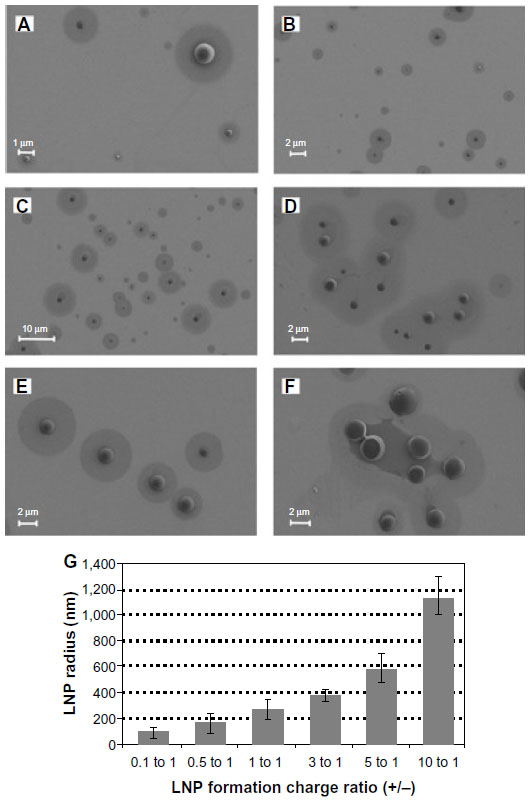 | Figure 6 Lipitoid-siRNA complexes at varying charge ratios. |
Cellular uptake mechanisms
Inhibiting the internalization pathway of the lipitoid-siRNA complexes
Rational design of effective siRNA carrier systems can be possible only through knowledge of their cellular entry routes. The cellular uptake of delivery vehicles is infrequently studied, and it is often assumed that siRNA vehicles will be endocytosed and transported to the lysosome, following the “proton-sponge hypothesis”.8,36,37 Recent data have revealed that this is not always the case and that some very widely studied transfection agents follow an uptake mechanism which bypasses endosomes and the lysosome,38–40 differing from the assumed internalization route. To reveal whether the cellular uptake mechanism of the lipitoid-siRNA complexes depends on lysosomal encapsulation and escape, we probed the internalization pathway of the best performing complexes in the presence of a set of chosen inhibitors acting at certain stages of various possible cell internalization pathways. GFP-expressing U2OS cell lines were targeted with GFP siRNA, and the inhibitors were used as described in the literature (see Materials and methods). The outcome of all inhibition tests was measured by quantification of the expressed GFP level by flow cytometry (Figure 7).
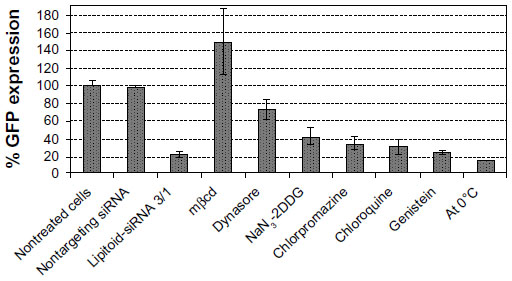 | Figure 7 Inhibitors of internalization and their effects on lipitoid-siRNA complex uptake and subsequent silencing of GFP expression. |
The most potent inhibitor was mbcd, which is known to deplete cholesterol in lipid rafts.41 mbcd inhibition resulted in complete elimination of GFP silencing, suggesting a lipid raft-mediated uptake. The increase in GFP expression was dramatic and higher than the expected maximum level of 100% upon inhibiting the transfection with mbcd, which is a commonly observed trend with mbcd blocking.16 To confirm that internalization is lipid raft mediated, we utilized dynasore, a reversible, noncompetitive inhibitor of dynamin1 and dynamin2 GTPase. The use of dynasore also resulted in dramatic inhibition of the LNP transfection activity as evident from the mere 25% silencing of GFP expression (Figure 7). Considering that GTPase activity of dynamin is essential for the budding of the caveolae from the plasma membrane, this result suggests that lipitoid-siRNA complexes are internalized through a dynamin- and lipid raft/caveolae-dependent pathway. NaN3 and 2DDG were used in tandem to block ATP synthesis in the cell to test for the involvement of energy-dependent endocytosis. Uptake inhibition in the presence of 0.1% NaN3 and 50 mM 2DDG had little effect on the internalization of the complexes, suggesting that internalization is not energy-dependent. Chlorpromazine, a cationic amphiphatic drug, was used to block clathrin-mediated endocytosis. Chlorpromazine, which incorporates into the lipid layer of the plasma membrane and increases lipid fluidity, hampering formation of invaginations,33 is a cationic amphiphatic drug typically used to block clathrin-mediated endocytosis. Clathrin-mediated endocytosis was ruled out as the primary route of entry by the inefficiency of chlorpromazine as an inhibitor. Chloroquine is a widely used lysosomal inhibitor that acts by inhibiting the acidification of organelles. The use of chloroquine did not have a dramatic effect on LNP internalization, suggesting that neither lysosomal inhibition nor endosome escape plays a significant role on the internalization efficiency of lipitoid-siRNA LNPs. This result is in agreement with previous studies conducted with lipitoid-DNA uptake studies.32 The use of genistein, a nonspecific tyrosine kinase inhibitor, which mainly inhibits clathrin-mediated endocytosis, further confirms a nonclathrin-mediated cellular uptake of lipitoid-siRNA nanoplexes. The widely assumed “proton-sponge” hypothesis for the internalization of cationic siRNA delivery lipids is thus unlikely to be a primary mechanism for the internalization of lipitoid-siRNA complexes. The proton-sponge hypothesis is further ruled out by inhibition experiments performed by incubating the cells at 0°C. The low temperature did not significantly block the internalization of these complexes, revealing that even when cellular processes are generally slowed and endocytosis is significantly reduced, the lipitoid-siRNA complex internalization is not halted.
In combination, these inhibition results suggest that there may be more than one possible route of internalization of these complexes. The major route may be a nonclathrin, nonendocytosis, lipid raft/caveolae-dependent, and dynamin-mediated process. Alternatively, the lipitoid-siRNA based LNPs may also utilize an uptake route that bypasses both the lysosome and the endosome, which would consequently not be dependent on endosomal escape mechanisms.
The results of this study reveal that lipitoid-siRNA nanospheres do not follow the classical endosome internalization pathway, which requires a lysosomal escape route. Instead, siRNA-loaded lipitoid-based LNPs are internalized through a clathrin-independent, dynamin and lipid raft-dependent pathway of internalization, which bypasses the lysosome. This route of internalization has recently been proposed as a distinct pathway of entry into the cell and requires the involvement of caveolae, dynamin, and actin.42 In agreement with our results, a recent finding with latex beads reveals that as the particle size increases from 50 nm to around 500 nm, the route of internalization shifts from clathrin-mediated to caveolae-mediated uptake, bypassing the lysosome.43
Caveolae are invaginations on the plasma membranes of the cells, enriched in cholesterol and sphingolipids, and are not typically involved in classical endocytosis.44,45 Since they are involved in cell signaling pathways, however, caveolae are also rich in some cell-signaling receptors such as the folate receptor.45 Although caveolae are present in most normal tissues, they are overexpressed in some solid tumors.46 Since the discovery of tissue-specific, overexpressed caveolar molecules, it has been proposed that caveolae may be employed as a useful site for targeted gene delivery in vivo.46 The lipitoid-siRNA nanospheres may ultimately be suitable candidates for targeting caveolae-rich cancer cells, owing to their efficiency and route of cellular uptake.
Conclusion
The rational design of siRNA delivery systems can only be possible if the factors that influence the efficiency of these systems are properly described. In order to gain more knowledge about the structural characteristics of an efficient siRNA delivery system, we investigated the cellular uptake and gene silencing activity of lipitoid DMPE-(NaeNmpeNmpe)3 in complex with siRNA oligos.
Here we show that the lipitoid forms spherical complexes with siRNA, termed LNPs, in which the complex size strongly depends on the ionic strength of the medium. This parameter affects not only the size but also the morphology of the complexes, thereby leading to alterations in transfection efficiency. When LNPs formed in sterile distilled water are transferred into the biological growth medium OptiMEM, the spheres become enlarged by an order of magnitude, resulting in low transfection efficiency. When the lipitoid-siRNA LNPs are formed directly in the biological medium OptiMEM, the spheres are still an order of magnitude larger than the LNPs formed in distilled water; however, they result in higher silencing efficiency and virtually no cytotoxicity.
Further investigation of the lipitoid-siRNA LNP system in OptiMEM revealed that the stoichiometry of lipitoid to RNA (expressed as positive to negative charge ratio) was another critical parameter, affecting not only the size and morphology of the formed complexes, but also their transfection efficiency and cytotoxicity. Lipitoid-siRNA based LNPs formed at the charge ratio 3/1 in OptiMEM were determined to be the best performing complexes for this system. These LNPs, with average radii of 320±11 nm in OptiMEM, were not the smallest sized LNPs that could be obtained through the self-assembly of lipitoid and siRNA, but they resulted in up to >90% silencing efficiency, with virtually no cytotoxicity. As a result, the lipitoid-siRNA LNPs were shown to be a defined system that could be fine-tuned and designed for optimized bioactivity. In vivo administration will require additional optimization, including a balance of attaining particle sizes that are sufficiently large to permit effective silencing benefiting from an increased blood circulation time, but sufficiently small to avert systemic difficulties such as diminished transport through the leaky tumor vasculature.47
The route of internalization of these LNPs into the cell was shown to bypass the lysosome, possibly through a lipid raft/caveolae-mediated and dynamin-dependent route, which does not utilize clathrin or endocytosis. This result suggests a possible explanation for the high efficiency of the LNPs, and indicates that an endosomal escape mechanism may not be manifest for every system. In fact, uptake mechanisms may be distinct for different systems, and the route of internalization should be probed instead of assumed. The observed route of internalization for the lipitoid-siRNA LNPs has recently been proposed as a distinct pathway of entry into the cell and requires the participation of caveolae, dynamin, and actin.42 This result may be beneficial in designing strategies that are based on targeting the caveolae, which are known to be overexpressed in certain tumorigenic tissues.
Currently, investigations of carrier systems are frequently performed in water; however, our study reveals that both the physical and the biological behavior of the siRNA cargo complexes are significantly different in biological medium in comparison to water. It is evident from our results that selecting for the smallest cargo vehicles may be counterproductive, as efficiency of transfection may increase with increasing LNP size. The route of cellular internalization may be different for each system, as shown for the LNPs presented here, and it should not be assumed that every siRNA delivery vehicle will be endocytosed and then transferred to the lysosome, from which the siRNA will escape. The results presented in this study will facilitate the rational design of siRNA delivery systems, exhibiting further enhancements in efficiency and cell-type specificity.
Acknowledgments
Portions of this work were performed as a User Project at the Molecular Foundry, Lawrence Berkeley National Laboratory, which is supported by the Office of Science, Office of Basic Energy Sciences of the Department of Energy under Contract No DE-AC02-05CH11231. The support of the National Science Foundation (NSF), through award CHE-#1152317 to KK, is gratefully acknowledged. This work would not be possible without the Horizon Travel Award for Research, the Margaret and Herman Sokol Travel/Research Award, and MacCracken Full Fellowship awarded by New York University to YUK.
Disclosure
The authors report no conflicts of interest in this work.
References
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. | |
Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15(2):188–200. | |
Haussecker D. The business of RNAi therapeutics. Hum Gene Ther. 2008;19(5):451–462. | |
Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60–71. | |
Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6(9):1130–1146. | |
Resnier P, Montier T, Mathieu V, Benoit JP, Passirani C. A review of the current status of siRNA nanomedicines in the treatment of cancer. Biomaterials. 2013;34(27):6429–6443. | |
Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10(7):521–535. | |
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. | |
Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10(9):3223–3230. | |
Akinc A, Goldberg M, Qin J, et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol Ther. 2009;17(5):872–879. | |
Patil ML, Zhang M, Taratula O, Garbuzenko OB, He H, Minko T. Internally cationic polyamidoamine PAMAM-OH dendrimers for siRNA delivery: effect of the degree of quaternization and cancer targeting. Biomacromolecules. 2009;10(2):258–266. | |
Aigner A. Cellular delivery in vivo of siRNA-based therapeutics. Curr Pharm Des. 2008;14(34):3603–3619. | |
Andersen MØ, Howard KA, Paludan SR, Besenbacher F, Kjems J. Delivery of siRNA from lyophilized polymeric surfaces. Biomaterials. 2008;29(4):506–512. | |
Beh CW, Seow WY, Wang Y, et al. Efficient delivery of Bcl-2-targeted siRNA using cationic polymer nanoparticles: downregulating mRNA expression level and sensitizing cancer cells to anticancer drug. Biomacromolecules. 2009;10(1):41–48. | |
Breunig M, Hozsa C, Lungwitz U, et al. Mechanistic investigation of poly(ethylene imine)-based siRNA delivery: disulfide bonds boost intracellular release of the cargo. J Control Release. 2008;130(1):57–63. | |
Crombez L, Aldrian-Herrada G, Konate K, et al. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol Ther. 2009;17(1):95–103. | |
Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc Chem Res. 2008;41(6):749–759. | |
Semple SC, Akinc A, Chen J, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28(2):172–176. | |
Huang CY, Uno T, Murphy JE, et al. Lipitoids – novel cationic lipids for cellular delivery of plasmid DNA in vitro. Chem Biol. 1998;5(6):345–354. | |
Lobo BA, Vetro JA, Suich DM, Zuckermann RN, Middaugh CR. Structure/function analysis of peptoid/lipitoid:DNA complexes. J Pharm Sci. 2003;92(9):1905–1918. | |
Utku Y, Dehan E, Ouerfelli O, et al. A peptidomimetic siRNA transfection reagent for highly effective gene silencing. Mol Biosyst. 2006;2(6–7):312–317. | |
Akinc A, Zumbuehl A, Goldberg M, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26(5):561–569. | |
Love KT, Mahon KP, Levins CG, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107(5):1864–1869. | |
Chen D, Love KT, Chen Y, et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J Am Chem Soc. 2012;134(16):6948–6951. | |
Sahay G, Querbes W, Alabi C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653–658. | |
Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J Am Chem Soc. 1992;114(26):10646–10647. | |
Sun J, Zuckermann RN. Peptoid Polymers: A Higher Designable Bioinspired material. ACS Nano. 2013:7;4715–4732. | |
Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev Res. 1995;35(1):20–32. | |
Schiffmacher AT, Keefer CL. Optimization of a lipitoid-based plasmid DNA transfection protocol for bovine trophectoderm CT-1 cells. In Vitro Cell Dev Biol Anim. 2012;48(7):403–406. | |
Zamai L, Canonico B, Luchetti F, et al. Supravital exposure to propidium iodide identifies apoptosis on adherent cells. Cytometry. 2001;44(1):57–64. | |
Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105(33):11613–11618. | |
Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN. A combinatorial approach to the discovery of efficient cationic peptoid reagents for gene delivery. Proc Natl Acad Sci U S A. 1998;95(4):1517–1522. | |
Masotti A, Mossa G, Cametti C, et al. Comparison of different commercially available cationic liposome-DNA lipoplexes: Parameters influencing toxicity and transfection efficiency. Colloids Surf B Biointerfaces. 2009;68(2):136–144. | |
Schneider G, Decher G. Functional core/shell nanoparticles via layer-by-layer assembly. investigation of the experimental parameters for controlling particle aggregation and for enhancing dispersion stability. Langmuir. 2008;24(5):1778–1789. | |
van Asbeck AH, Beyerle A, McNeill H, et al. Molecular parameters of siRNA–cell penetrating peptide nanocomplexes for efficient cellular delivery. ACS Nano. 2013;7(5):3797–3807. | |
Behr JP. The proton sponge: a trick to enter cells the viruses did not exploit. Chimia Int J Chem. 1997;51:34–36. | |
Boussif O, Lezoualc’h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92(16):7297–7301. | |
Gabrielson NP, Pack DW. Efficient polyethylenimine-mediated gene delivery proceeds via a caveolar pathway in HeLa cells. J Control Release. 2009;136(1):54–61. | |
Chung YC, Cheng TY, Young TH. The role of adenosine receptor and caveolae-mediated endocytosis in oligonucleotide-mediated gene transfer. Biomaterials. 2011 Jul;32(19):4471–4480. | |
Perez AP, Cosaka ML, Romero EL, Morilla MJ. Uptake and intracellular traffic of siRNA dendriplexes in glioblastoma cells and macrophages. Int J Nanomedicine. 2011;6:2715–2728. | |
Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161(4):673–677. | |
Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005;6(2):112–126. | |
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. | |
Thomsen P, Roepstorff K, Stahlhut M, van Deurs B. Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol Biol Cell. 2002;13(1):238–250. | |
Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. | |
Carver LA, Schnitzer JE. Caveolae: mining little caves for new cancer targets. Nat Rev Cancer. 2003;3(8):571–581. | |
Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4(1):81–89. |
Supplementary materials
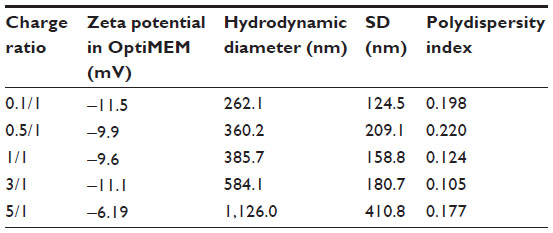 | Table S1 Zeta potentials and hydrodynamic diameters of LNPs formed at varying charge ratios |
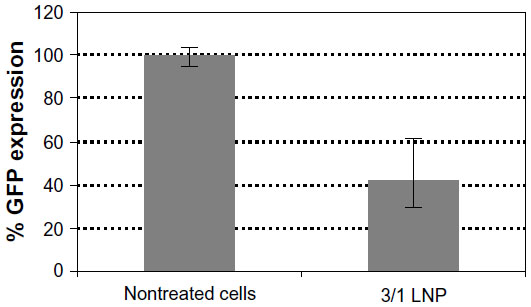 | Figure S1 Transfection of NM2C5 cells with siRNA-lipitoid LNPs. |
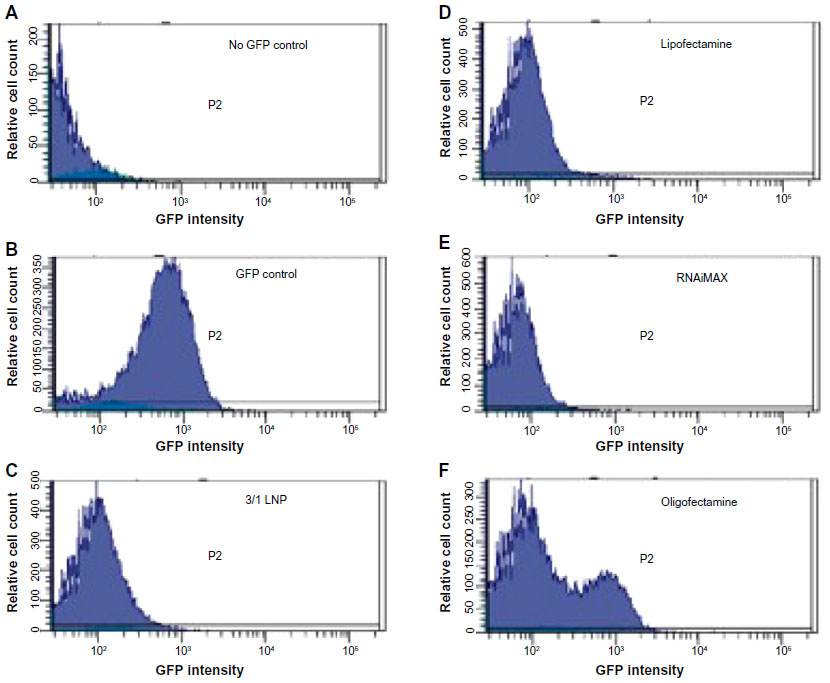 | Figure S2 Silencing activity of LNPs is comparable to commercial transfection reagents. |
 | Figure S3 The effects on siRNA-lipitoid complexes 5 minutes after transferred into OptiMEM (A), 10 minutes after transferred into OptiMEM (B), and 60 minutes after transferred into OptiMEM (C). |
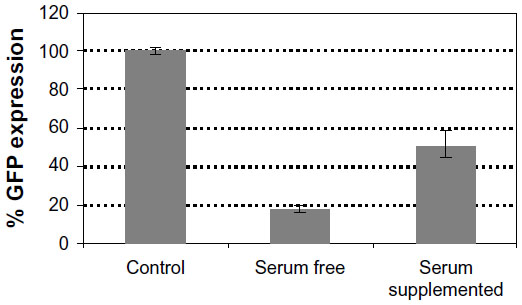 | Figure S4 The effect of the presence of serum at the time of transfection on the silencing efficiency of LNPs. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.