Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanodrugs: pharmacokinetics and safety
Authors Onoue S, Yamada S, Chan H
Received 25 October 2013
Accepted for publication 17 December 2013
Published 20 February 2014 Volume 2014:9(1) Pages 1025—1037
DOI https://doi.org/10.2147/IJN.S38378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Satomi Onoue,1 Shizuo Yamada,1 Hak-Kim Chan2
1Department of Pharmacokinetics and Pharmacodynamics, School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka, Japan; 2Advanced Drug Delivery Group, Faculty of Pharmacy, The University of Sydney, Sydney, NSW, Australia
Abstract: To date, various nanodrug systems have been developed for different routes of administration, which include dendrimers, nanocrystals, emulsions, liposomes, solid lipid nanoparticles, micelles, and polymeric nanoparticles. Nanodrug systems have been employed to improve the efficacy, safety, physicochemical properties, and pharmacokinetic/pharmacodynamic profile of pharmaceutical substances. In particular, functionalized nanodrug systems can offer enhanced bioavailability of orally taken drugs, prolonged half-life of injected drugs (by reducing immunogenicity), and targeted delivery to specific tissues. Thus, nanodrug systems might lower the frequency of administration while providing maximized pharmacological effects and minimized systemic side effects, possibly leading to better therapeutic compliance and clinical outcomes. In spite of these attractive pharmacokinetic advantages, recent attention has been drawn to the toxic potential of nanodrugs since they often exhibit in vitro and in vivo cytotoxicity, oxidative stress, inflammation, and genotoxicity. A better understanding of the pharmacokinetic and safety characteristics of nanodrugs and the limitations of each delivery option is necessary for the further development of efficacious nanodrugs with high therapeutic potential and a wide safety margin. This review highlights the recent progress in nanodrug system development, with a focus on the pharmacokinetic advantages and safety challenges.
Keywords: nanoparticles, nanotechnology, nanotoxicity, solubilization, targeted delivery
Introduction
Recently, considerable attention has been directed toward nanoscience, and a number of efforts have been made for the development and commercial applications of new nanotechnology in both academic and industrial institutions.1–3 As defined by the Royal Society and Royal Academy of Engineering, “nanoscience” is the study of phenomena and manipulation of materials at atomic, molecular, and macromolecular scales, where the properties differ significantly from those at a larger scale; and “nanotechnologies” are the design, characterization, production, and application of structures, devices, and systems by controlling shape and size at the nanometer scale.4 The growing fields of nanoscience and nanotechnology have transformed many sectors of industry, with breakthrough applications in the areas of biotechnology, electronic, cosmetics, food sciences, and pharmaceutics. In particular, strategic application of nanotechnologies to pharmaceutical research and development has led to the successful development of nanodrugs, described as drug delivery systems developed to operate at the nanometer size range with novel engineered properties that provide medical benefits in the clinical treatment of several diseases.1
Research into the rational delivery and targeting of pharmaceutical, therapeutic, and diagnostic agents is at the forefront of projects in nanodrugs. The early efforts in nanodrugs were focused on improving the molecular properties of already available therapeutic and diagnostic agents, but more recently, nanotechnology proponents have attempted to apply new therapeutic and diagnostic modalities for improving the developability. The major targets in the development of nanodrugs are 1) specific drug targeting and delivery; 2) greater safety and biocompatibility; 3) faster development of new medicine with a wide safety margin; and 4) improved pharmacokinetic behavior.5 Theoretically, nanodrugs can easily pass through the fine capillary blood vessels and the lymphatic endothelium, and they might have longer circulation times in the blood and/or higher binding capability and accumulation at some target sites.6 In particular, nanotechnologies have been used to develop site-specific drug targeting, for the treatment of brain diseases.7 Nanodrugs might also produce less inflammatory and immune response in tissues compared with drugs of larger size.8 In spite of these attractive characteristics, nanodrugs sometimes induce oxidative stress, genetic damage, and the inhibition of cell division and cell death, depending on their physicochemical characteristics (such as particle surface, size, and chemical composition).9 Nanotoxicology is emerging as an important subdiscipline of nanoscience and nanotechnology because of the finding of increasing toxic effects of nanodrugs and nanomaterials on living organisms. However, the toxicology of nanoparticles is poorly understood as there are no sufficient methods to assess their safety. A better understanding of nanotoxicity and its mechanisms would be of great help to develop new nanodrugs with a wide safety margin. In this article, we review recent advances in nanodrug development and the pharmacokinetic/safety characteristics of nanodrugs.
General pharmacokinetic characteristics of nanodrugs
A number of efforts have been made in the development of nanodrugs over the past few decades, and the concept of nanodrugs has evolved considerably. There are various types of nanodrug systems, most designed with the drug encapsulated in a carrier (eg, dendrimers, liposomes, micelles, and polymeric nanoparticles). A nanodrug system can offer several pharmacokinetic advantages, such as specific drug delivery, high metabolic stability, high membrane permeability, improved bioavailability, and long duration of action (Table 1). Therefore, by altering the biopharmaceutic and pharmacokinetic properties of new drug candidates, nanodrug systems could be a promising approach to obtain the drug properties. The physicochemical properties of nanodrugs, such as size, surface charge, and hydrophobicity, affect their mucosal absorption characteristics, and smaller nanodrugs show higher transcellular uptake via follicle-associated epithelia than do larger ones.3,10–12 Nanoparticles can enter cells via endocytotic processes, including caveolar- and clathrin-mediated endocytosis, potocytosis, pinocytosis, and patocytosis.13 In contrast, larger particles can be quickly opsonized and removed from the bloodstream via the macrophages of the reticuloendothelial system (RES). In the formulation design of nanoparticles, it is necessary to minimize opsonization and prolong the circulation of nanoparticles in clinical use. This can be achieved by the surface coating of nanoparticles with hydrophilic polymers/surfactants, and/or the formulation of nanoparticles with biodegradable copolymers with hydrophilic segments, such as polyethylene glycol (PEG), polyethylene oxide, poloxamer, poloxamine, and polysorbate 80 (Tween 80).14 Nanodrugs with a positive surface charge can interact with the negative charges of mucin owing to abundant sulfate sialic acid and sugar moieties, resulting in enhanced transportation across mucus and increased internalization by epithelial cells.10 Strategic functionalization with some membrane permeation enhancers or ligands for receptors expressed on the cellular membrane may also promote the transcellular transport of entrapped drugs.11,12,15 In addition to transcellular transport, nanodrugs equipped with bioadhesive polymers or chelators could enhance the paracellular transport of entrapped drugs, via the regulation of tight junctions.16, 17 Surface modification of nanodrugs, with specific proteins, antibodies, and other biomolecules, can be used to design drugs that act selectively on particular tissues,18 and this approach has been employed to provide improved therapeutic potential and reduced side effects of some anticancer drugs. In general, drug delivery systems employing nanotechnologies are designed to be administered via injection, transdermally, or orally, although recent studies have demonstrated the promising outcomes with pulmonary administration of nanodrug systems.19,20 Inhaled particles undergo pulmonary clearance, such as mucociliary clearance and macrophage clearance, leading to a limited duration of action. However, nanoparticles have been praised for their advantageous drug delivery properties to the lung, including their avoidance of mucociliary and macrophage clearance and long residence times before degradation or translocation by epithelial cells takes place.21,22 Thus, the nanodrug approach should enhance the therapeutic potential of entrapped drugs and contribute to the acceleration of pharmaceutical development.
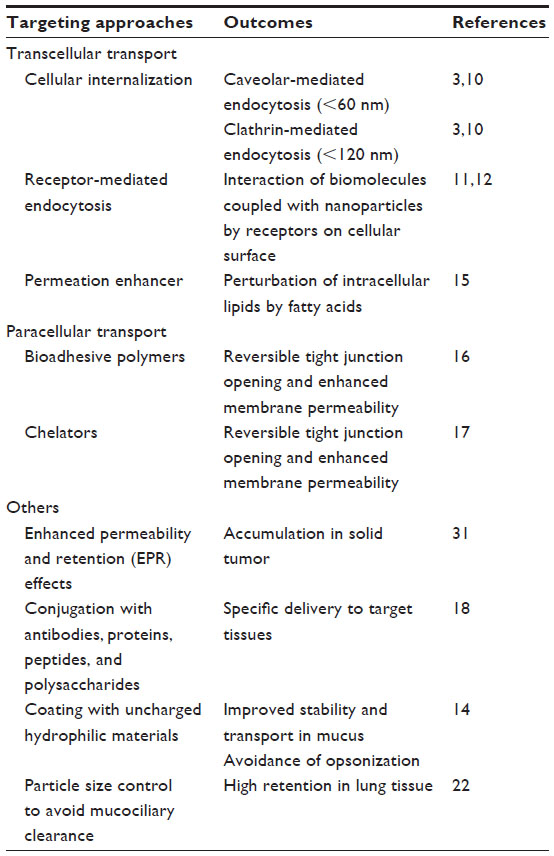 | Table 1 Targeted delivery of nanoparticles |
General safety characteristics of nanodrugs
Despite attractive functions and a bright future outlook for nanodrugs, there is increasing concern over their safety. Knowledge of the toxic effects of nanodrugs is limited but is rapidly growing. Nanoparticles are expected to be able to diminish the toxicity of chemotherapy drugs or other drugs with a narrow therapeutic index; however, a number of in vitro and in vivo studies have shown that some nanoparticles demonstrated toxicity in biological systems, causing cytotoxicity, an allergic response, or inflammation.23 Nanoparticles tend to produce reactive oxygen species (ROS) and free radicals, resulting in oxidative stress, inflammatory events, deoxyribonucleic acid (DNA) damage, multinucleus formation, and fibrosis.13 Another toxicity concern associated with nanodrugs is their accumulation within cells, particularly with continuous exposure or long-term use. The upper size limit for the toxicity of nanoparticles is not fully clarified; however, it has been thought to lie between 65 nm and 200 nm.24 Nanoparticle toxicity is extremely complex and multifactorial and depends on physicochemical properties, such as size, shape, and surface properties (charge, area, and reactivity). The size effect is likely to be more important for nanoparticle toxicity than the actual composition of the nanodrugs. The particle surface area can also be a better predictor of the toxic and pathological responses to nanoparticles than the particle mass dose. Although some nanodrugs were functionalized with cationic polymers, cationic formulations have been described to affect cell proliferation, differentiation, and proapoptotic genes, in human epithelial cells.13 The polycationic nature of these formulations sometimes induces both necrosis and apoptosis; therefore, in the design of drug carriers, issues of safety, toxicity, and availability have to be taken into account. The surface reactivity of nanoparticles can cause chemical damage to surrounding tissues. When inhaled, micron-sized particles tend to deposit in the central airways; however, inhaled nanoparticles can deposit in the lung periphery, causing much greater inflammation.25 In particular, needle-shaped carbon nanotubes, nanowires, and nanofibers might cause fibrotic lung disease and rare tumors, such as mesothelioma.26 The interaction of nanoparticles with skin also has received significant attention recently because of the increasing use of nanoparticles in stain-resistant clothing, cosmetics, and sunscreens. The dermal penetration of nanoparticles can be dependent on the physicochemical properties of the nanoparticles and skin condition. Limited in vivo studies have been conducted to address the issue of cutaneous nanotoxicity,25 and they demonstrated minimal irritancy potential and no evidence of irritation or allergic response for some nanoparticles. In contrast to these benign findings, many nanoparticles have been found to be cytotoxic and proinflammatory to dermal cell lines in vitro;23 therefore, additional in vivo studies of chronic dosing of nanodrugs would be needed for further clarification of nanotoxicity. Few studies have examined the systemic toxicity of nanoparticles, and most of these have been acute-toxicity studies investigating the 50% lethal dose (LD50) values of tested nanoparticles. Since the nanoparticles are commonly taken up by the RES, many of the target organs have been thought to be members of RES, such as the liver and spleen.27 However, nanoparticles used in biomedical applications are commonly coated with biocompatible materials to reduce opsonization and avoid the RES uptake, so the target organs for biomedical nanoparticles might be shifting away from the RES. The toxic potential of nanodrugs would be variable depending on administration routes, so an improved understanding of the risk factors related to nanodrugs and nanomaterials in the human body will aid the further development and exploitation of a variety of nanodrugs.
Dendrimers
Dendrimers, highly branched macromolecules with a specific size and shape, are a class of carriers of nanodrugs, in which the hydrophobic core and hydrophilic periphery exhibit micelle-like performance along with drug loading properties, in solution.28 Drug payloads can be either entrapped within the dendrimer scaffold via the generation of noncovalent complexes or linked to the dendrimer surface via covalent conjugation, so dendrimers can incorporate a lower amount of drugs than other carriers.29 Covalently constructed dendritic macromolecules have the advantage of more specific control over drug release and may be designed to limit drug release in the systemic circulation and to trigger release under tumor-specific conditions (Table 2).30 Dendritic polymers have recently been shown to improve the delivery of doxorubicin and other cytotoxic drugs to solid tumors and to reduce their accumulation in noncancerous tissues.31 Dendrimers have also been well proven as a tool in the solubilization of poorly soluble drugs, and poly(amidoamine) dendrimers and other polymeric dendrimers have been applied to flurbiprofen,32 methotrexate,33,34 and piroxicam35 for solubilization and targeted delivery. In particular, lactoferrin-conjugated dendritic nanocomposite exhibited an enhanced residence time in the systemic circulation and high lung delivery, possibly leading to reduced dosing frequency as well as nominal dose.18 Other target selectivity has also been demonstrated via the conjugation of targeting ligands, such as folate, arginylglycylaspartic acid (RGD) peptides, epidermal growth factor, vascular endothelial growth factor (VEGF), and monoclonal antibodies, to the dendrimer surface (Table 3).36 In spite of their attractive functions, most dendrimers demonstrate toxic and hemolytic activity because of their positively charged surface.37 However, anionic dendrimers and modified dendrimers with masking of the peripheral cationic group can exhibit diminished hemolytic activity. Thus, surface engineering of dendrimers should lead to improvement of their pharmacokinetic and safety properties, in the context of biomedical applications.
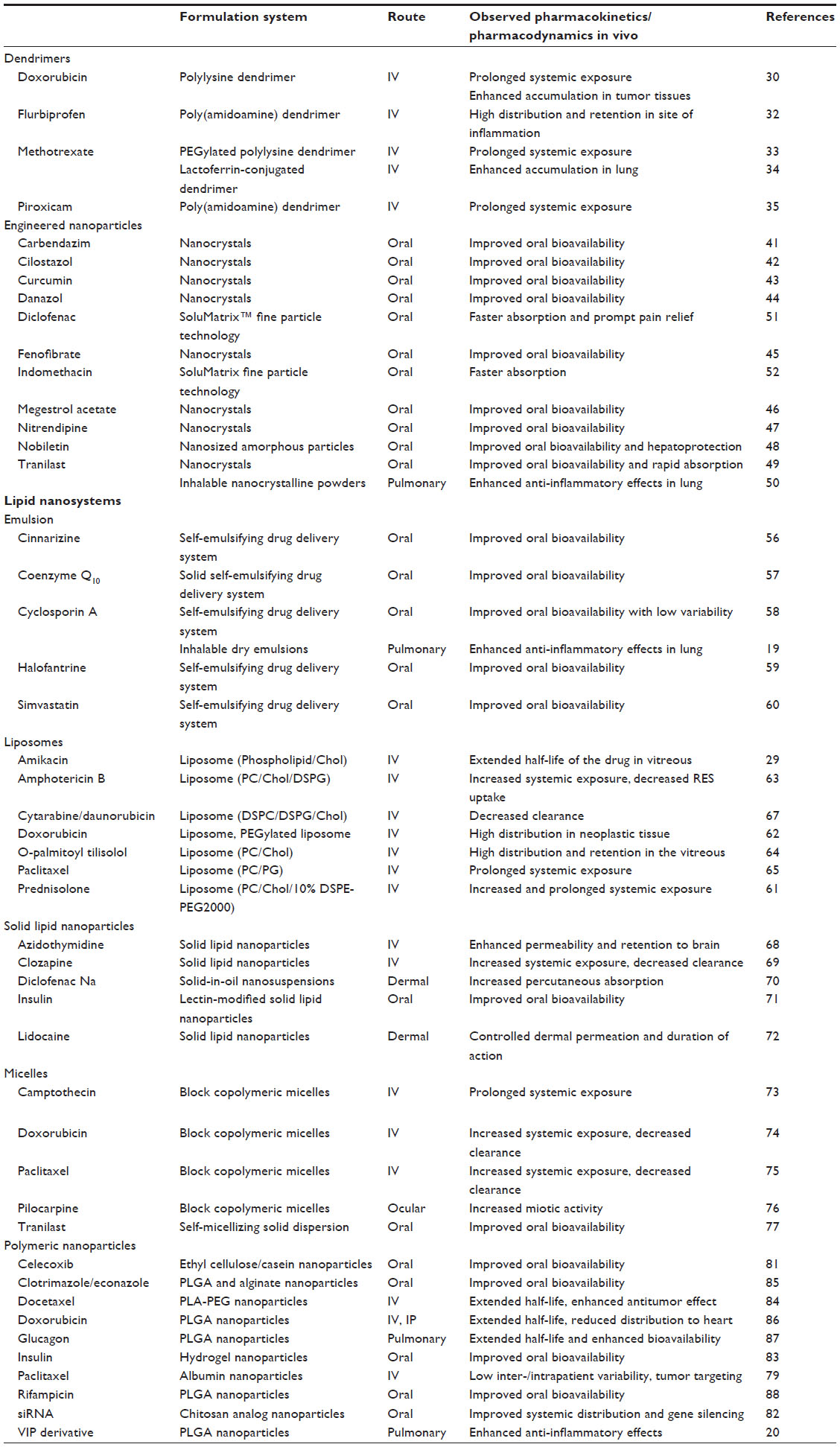 | Table 2 Nanodrugs and their biopharmaceutical characteristics |
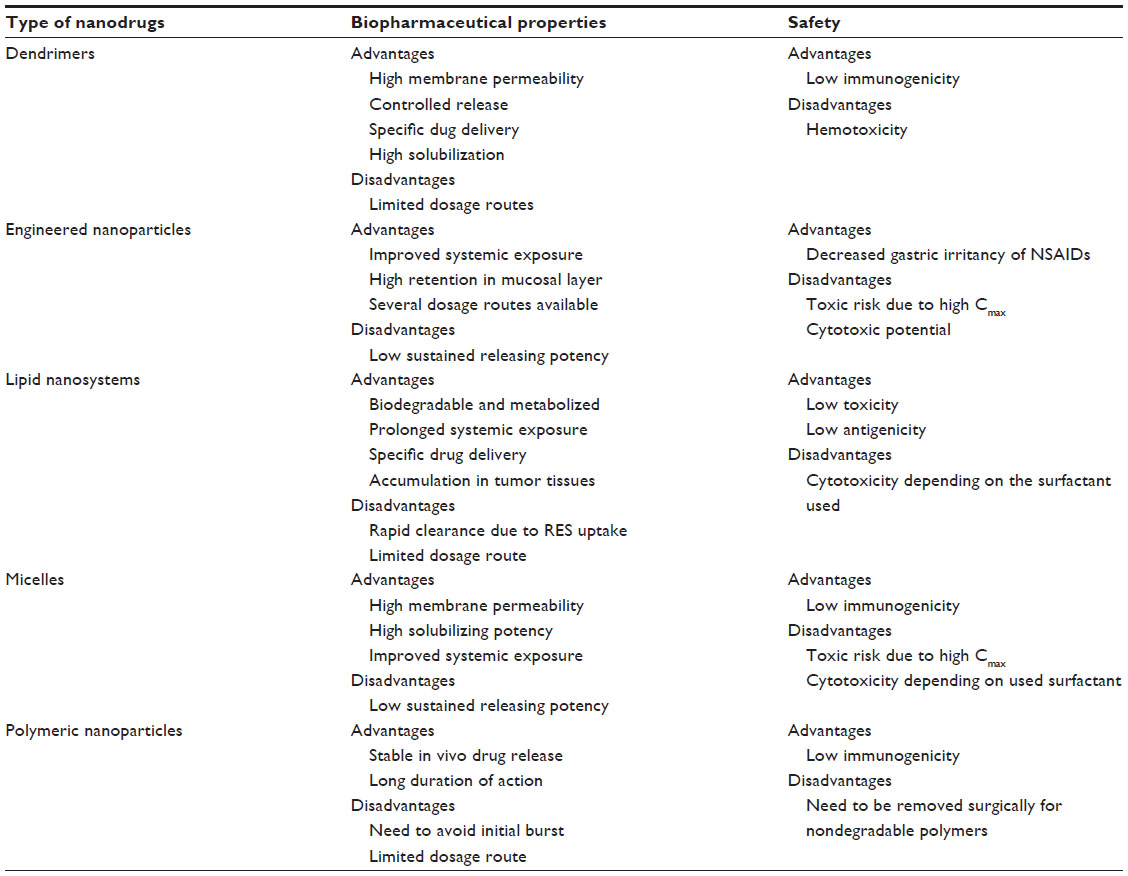 | Table 3 Biopharmaceutical and safety characteristics of nanodrugs |
Engineered nanoparticles (nanosized particles)
A particle size reduction approach is widely used to increase the dissolution rate, since the dissolution rate of a drug proportionally increases with increasing surface area of drug particles.38 As defined in the Prandtl boundary layer equation, the decrease of diffusion layer thickness brought by reducing particle size, particularly down to <5 μm, would result in accelerated dissolution. Based on this, drug nanocrystal technology has been the highlight in the pharmaceutical field, and the approaches developed to produce drug nanosuspensions mainly include the so-called “bottom-up” (controlled precipitation) and “top-down” types (wet-milling with beads, and high-pressure homogenization).39,40 In both bottom-up and top-down approaches, hydrophilic polymer and/or surfactant are typically used to stabilize a nanosuspension. The nanoparticles of drug are dispersed into inert carriers after a drying process, such as spray drying or lyophilization, and the resulting solidified nanocrystal formulations can be defined as nanocrystalline solid dispersions.
There have been numerous studies demonstrating the enhanced oral bioavailability and pharmacological effects of pharmaceuticals and neutraceuticals obtained via nanotechnologies (Table 2).41–52 In the nanosized formulation approach, maximum concentration (Cmax) and bioavailability were increased up to dozens of folds compared with conventional formulations with micrometer particle size. Interestingly, the neutral or acidic compounds, such as danazol,44 cilostazol,42 tranilast,49 and curcumin43 have shown better improvements in the pharmacokinetic parameters than have the basic compounds, via nanocrystal technologies. Recently, a lower-dose diclofenac submicron particle capsule was developed with the use of SoluMatrix™ technology (iCeutica, Philadelphia, PA, USA), and in 2013, the US Food and Drug Administration (FDA) approved this nanodrug for treatment of mild to moderate acute pain in adults.51 In the Phase I study, the oral nanoformulated diclofenac (35 mg) demonstrated faster absorption and similar Cmax compared with diclofenac at higher dose (50 mg), in healthy subjects, and it also provided effective analgesia in the Phase III clinical study, in patients with acute pain.53 The SoluMatrix™ technology was also applied to indomethacin, and a Phase I study demonstrated that the oral nanoformulated indomethacin exhibited a more rapid time to maximal concentration (Tmax) (1.1 hours) compared with indomethacin (2.0 hours), possibly leading to more rapid onset of action.52 The Cmax for nanoformulated indomethacin (40 mg) was found to be slightly higher compared with standard oral indomethacin (50 mg) in healthy subjects (3,115 ng/mL vs 2,759 ng/mL, respectively). Thus, nanoformulated systems can lower the dose of drugs, thus improving their safety and tolerability, while maintaining their effectiveness. In general, neutral and acidic drugs would be poorly soluble in gastric fluid, and the improved dissolution behavior of these chemicals under acidic conditions via nanotechnologies could lead to marked enhancement in their oral bioavailability. Nanoparticles generally exhibit mucoadhesion to biological mucosa, and the mucoadhesion effect also plays an important role in the enhancement of oral bioavailability. The transcellular transport of nanosized particles through the endothelial cells of the small intestine via endocytosis has also been believed to be one of the major mechanisms for the improved oral absorption, and the transcellular uptake could be influenced by several factors, such as particle size and surface charge (Table 1).16 The mechanisms for enhanced oral absorption can be mainly summarized as follows: 1) improved dissolution behavior; 2) bioadhesion to the intestinal wall; and 3) transcellular uptake. In addition to the enhanced dissolution and bioavailability, nanocrystals have provided further pharmaceutical benefits, including reproducibility of oral absorption, improved dose-bioavailability, proportionality, and increased patient compliance as a result of the reduction of the number of oral units to be taken.54 Theoretically, as well as for oral dosing, nanocrystal formulations can be employed for dermal, ocular, and pulmonary drug delivery. An inhalable nanocrystalline solid dispersion of tranilast was previously prepared by wet-milling technology and exhibited high inhalation performance and improved anti-inflammatory effects, in a rat model of airway inflammation, compared with crystalline tranilast with a larger particle size.50
Recently, there has been increasing concern about the potential nanotoxicity of nanosized particles, and the particles of major toxicological concern are those below 100 nm.54 Although larger nanometer particles (>200 nm) can only be internalized by macrophages, causing effects inside those cells, smaller nanoparticles (with a diameter of 150 nm or much smaller) can be internalized by any cell via pinocytosis. In this context, small nanoparticles can access any cell of the body, possibly resulting in a higher cytotoxic potential. Orally taken nanocrystals could cause pharmacokinetic transition, with higher Cmax and shorter Tmax, and the higher and more rapid systemic exposure of drugs might lead to some adverse effects. In contrast, it was shown that the gastric irritancy of oral nonsteroidal anti-inflammatory drugs (NSAIDs) could be decreased via nanocrystal technologies because the nanocrystals achieved distribution uniformity in the gastrointestinal fluid without high and prolonged local concentration.39
Lipid nanosystems
Lipid nanosystems, including emulsions, liposomes, and solid lipid nanoparticles, have been studied intensively to improve the biopharmaceutical properties and/or therapeutic index of drugs.1 With respect to the safety concerns over lipid nanosystems, lipid-based colloidal carriers are believed to be well tolerated in living systems since they are usually made of physiological compounds and, therefore, metabolism should decrease the risk of acute and chronic toxicity.55 Nevertheless, for the development of solid lipid nanoparticle emulsions with a wide safety margin, careful consideration should be taken of the potential toxicity of the emulsifiers.
Emulsions
In recent years, self-emulsifying drug delivery systems (SEDDSs) have been utilized to enhance the oral bioavailability of poorly water-soluble drugs, especially of highly lipophilic drugs.56–60 SEDDSs are isotropic mixtures of oil, surfactant, cosolvent, and solubilized drug, and the SEDDS approach requires fine dissolution and chemical stability of the drugs in the oil phase. These formulations can rapidly form oil in water (O/W) fine emulsions, when dispersed in the aqueous phase under mild agitation. SEDDSs are additionally classified as either self-microemulsification drug delivery systems (SMEDDSs) or self-nanoemulsification drug delivery systems (SNEDDSs), according to the size range of their oil droplets.38 SMEDDSs form microemulsions ranging in droplet size from 100 to 250 nm, and finer nanoemulsions, with a diameter of less than 100 nm, can be obtained using SNEDDSs. The SEDDS approach has been thought to be suitable for the Biopharmaceutics Classification System (BCS)38 class II drugs, the characteristics of which are low solubility and high permeability. Generally, the bioavailability of a BCS class II drug is rate-limited by its dissolution so that even a small increase in dissolution rate sometimes results in a large increase in bioavailability. Therefore, the rapid emulsification of these formulations in the gastrointestinal tract can provide both improved oral bioavailability and a reproducible plasma concentration profile (Table 2). The droplet size of the emulsion could influence the bioavailability of orally administered drugs. For instance, two SEDDSs formulations of cyclosporin A (Sandimmune® [Novartis Pharmaceuticals Corp, Basel, Switzerland], a coarse SMEDDS formulation, and Neoral® [Novartis], a fine SNEDDS formulation) are available on the market, and Neoral® is more rapidly and consistently absorbed than Sandimmune®, in humans. The strategic application of emulsion approaches to poorly soluble drugs could result in the rapid increase of systemic exposure, which might show unwanted side effects if the drugs have low therapeutic index (Table 3). To overcome this limitation, sustained-release SEDDS would be a viable dosage option to modulate high peak plasma concentrations of the administered drugs.16 In addition to the oral dosage form, an inhalable dry emulsion of cyclosporin A was proposed for the treatment of asthma, chronic obstructive pulmonary disease (COPD), and allograft rejection after pulmonary transplantation, and the insufflated dry emulsions showed higher potency than did cyclosporin A particles, in a rat model of acute airway inflammation.19
Liposomes
Liposomes, a type of microcapsule, enclose liquid compartments with a multilamellar structure consisting of lipid bilayers. A liposomal formulation can be prepared by the dehydration-rehydration method, the reverse-phase evaporation vesicle method, and by the proliposome method. Several pharmacokinetic challenges have been pointed out for conventional liposomes, including nonspecific uptake, within a few minutes to a few hours, by the RES; rapid clearance; and opsonization.1 These pharmacokinetic properties of liposomes depend on their physicochemical characteristics, such as size, surface charge, membrane lipid packing, steric stabilization, dose, and route of administration. A number of efforts have been made to overcome these drawbacks, and recent studies demonstrated that liposomes coated or grafted with hydrophilic polymers were efficacious for attenuating the opsonization of the liposomes and rapid clearance. Considerable attention has been drawn to PEG-modified liposomes since they have exhibited an increased systemic half-life for the encapsulated drug, based on significant reduction in nonspecific RES uptake.61 PEG-modified liposomes also have advantages in terms of passive targeting to tumors.3 Tumor vasculature is well characterized by a chaotic network of thin-walled, leaky vessels, so liposomes have the ability to cross into the interstitial spaces in viable tumor areas, with limited washout. This process is referred to as the enhanced permeability and retention (EPR) effect, and small PEG-modified liposomes with a diameter of 100–200 nm can permeate through the tumor vasculature, eventually leading to their accumulation in tumor tissue (Table 3). As a result of basic research in both academia and industry, liposomes have been widely used as pharmaceutical carriers in the past decade because of their attractive biopharmaceutical properties: 1) high encapsulation efficiency for both hydrophilic and hydrophobic therapeutic agents; 2) protection of encapsulated drugs from undesired effects of external conditions; 3) functionalization upon conjugation with specific ligands, for the targeting specific cells, tissues, and organs of interest; 4) prolonged systemic circulation with the use of inert and biocompatible polymers; and 5) controllable size and surface charge (Table 2).29,61–65 Currently, a number of liposomal formulations have obtained approval for the treatment of cancer, infections, and meningitis, including amphotericin B (Abelcet®; Sigma-Tau Pharmaceuticals, Inc., Gaithersburg, MD, USA) and doxorubicin (Doxil® [Janssen Pharmaceuticals, Inc., Titusville, NJ, USA] and Myocet® [Enzon Pharmaceuticals, Piscataway, NJ, USA]). Doxil® is the first FDA-approved nanodrug, pharmacokinetic characteristics of which are 1) prolonged drug circulation time and 2) avoidance of RES uptake, due to the use of PEGylated nanoliposomes.66 In humans, the area under the concentration-time curve (AUC) of plasma doxorubicin after the intravenous (IV) administration of Doxil® (50 mg/m2) is ~300-fold greater than that with free drug at same dose, and clearance and volume of distribution are drastically reduced, by at least 250- and 60-fold, respectively.66 CPX-351 (liposome-encapsulated cytarabine and daunorubicin) is currently under clinical development for the treatment of leukemia.67 Following IV administration of CPX-351 in patients with advanced leukemia, the clearance of cytarabine and daunorubicin was found to be less than 120 mL/h/m2 across all the dose levels (24–134 units/m2), which was markedly less than the clearance rates for unencapsulated daunorubicin (38,600 mL/h/m2) and cytarabine (134,000 mL/h/m2). The very low rate of clearance might be attributed to the apparent lack of a distribution phase for the encapsulated drugs. In addition to the therapeutic agents, liposomal delivery has been applied to imaging techniques, such as magnetic resonance imaging (MRI), positron or single-photon emission (computed) tomography (PET/SPECT), and fluorescence, at the forefront of medical diagnostics in preclinical and clinical settings, for the assessment of treatment efficacy.
Solid lipid nanoparticles
Solid lipid nanoparticles are described as colloidal nanoparticles of highly purified triglycerides, complex glyceride mixtures, monoglycerides, hard fats, or waxes stabilized by a surfactant and fabricated via a high-pressure homogenization and nanoemulsion technique. Solid lipid nanoparticles have recently emerged as an alternative to liposomal formulations, owing to various advantages: 1) improved physical stability; 2) modulated release of the loaded drugs; 3) relative low cost compared with phospholipids used for liposomes; and 4) easy scale-up and manufacturing (Table 3). In contrast, solid lipid nanoparticles have some disadvantages: 1) drug expulsion after recrystallization; 2) limited loading capacity, depending on the solubility of drugs in the oil phase; and 3) relatively high water content of the dispersions.16
A variety of pharmaceutical substances, such as small molecules, peptides, and proteins, can be applied to solid lipid nanoparticle systems with the aim of improving pharmacokinetic behavior, and many different routes are available for the administration of solid lipid nanoparticles, unlike for liposomes (Table 3).68–72 There are major challenges for the oral delivery of therapeutic peptides/proteins - to overcome the gastrointestinal barriers and protect the structure in the gastrointestinal tract; despite this, promising results in the oral delivery of insulin have been achieved with the use of the solid lipid nanoparticle approach.71 Solid lipid nanoparticles might thus be a promising approach for the formulation of other therapeutic peptides and proteins. As observed with the liposomal formulations, IV-administered solid lipid nanoparticles of drug also exhibited longer systemic circulation, due to decreased clearance69 and higher accumulation in the tissues,68 compared with that exhibited by the drug itself.
Micelles
Polymeric micelles are spherical nanostructures formed by supermolecular assembly of amphiphilic copolymers in aqueous environments, normally as a consequence of ion pair or hydrophobic interaction. Micellar nanoparticles have received considerable attention in contemporary drug delivery research since micellar formulations can achieve the protection of internal drugs from degradation, solubility enhancement, and target-specific delivery.1,73–77 Hydrophobic drugs tend to be entrapped in the semisolid core of micelles, and the core-shell structure can mimic the naturally occurring transport system. Therefore, micellar nanoparticles can improve the absorption and distribution of internal drugs and also, avoid opsonization and phagocytic clearance by RES uptake.78
Micellar nanoparticles can be a viable alternative to liposomal formulations, in terms of passive and active targeting of the disease site in the body. After the IV administration of micelles, their tissue distribution and clearance has tended to be highly altered compared with those of free drugs, possibly resulting in better clinical outcomes (Table 2).73–75 The micellar nanoparticles can also be applied to liquid eye drops, to attenuate the rapid elimination of the drugs from the precorneal area, offering longer duration of action.76 Recently, Onoue et al developed a self-micellizing solid dispersion of poorly water-soluble drugs, with the use of an amphiphilic block copolymer of 2-methacryloyloxyethyl phosphorylcholine (MPC) unit and a n-butyl methacrylate (BMA) unit ([poly(MPC-co-BMA)]).77 The new solid dispersion system, tranilast, showed significant improvement in dissolution behavior and rapid formation of micelles with a diameter of 100–150 nm, and there appeared to be accelerated absorption of tranilast, with ~50-fold enhancement of oral bioavailability in rats (Figure 1). Clinical development of several micellar nanoparticles is ongoing, with the aim of improving the pharmacokinetic behavior and reducing the potential side-effects of anticancer drugs.79 NK105, a polymeric micellar nanoparticle formulation of paclitaxel, is currently under clinical trial in the patients with gastric cancer and breast cancer.75 In a Phase I study, the AUC of NK105 at 150 mg/m2 (recommended Phase II dose) was ~15-fold larger than that of the conventional paclitaxel formulation at the dose of 210 mg/m2 (clinical dose for a 3-week regimen in Japanese patients). The volume of distribution and the clearance of NK105 were significantly lower than those of the conventional formulation, while the hematological and nonhematological toxicities of NK105 were mild and well manageable. Although the micellar nanoparticles have been thought to be a safe delivery system, there are some safety concerns, including possible side effects after rapid elevation of systemic exposure, and toxicity of the surfactant used. In particular, prior to clinical use, the safety of newly developed micellizing agents has to be checked carefully with respect to chronic dosing.
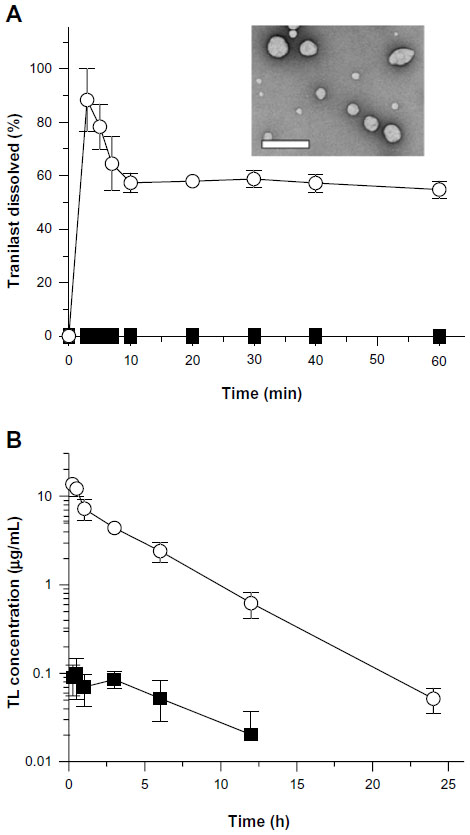 | Figure 1 Biopharmaceutical characteristics of self-micellizing solid dispersions. |
Polymeric nanoparticles
Polymeric nanoparticles can be defined as solid particles with a size in the range of 10–1,000 nm; they allow encapsulation of the drugs inside a polymeric matrix, protecting them from enzymatic and hydrolytic degradation.80 The polymeric nanoparticles can be prepared by several classical methods, including nanoprecipitation, emulsion-diffusion, double emulsification, emulsion-coacervation, and polymer-coating. Polymeric nanoparticles show some advantages with respect to other drug delivery systems for several types of pharmaceutical substance (small molecules, peptides, proteins and small interfering ribonucleic acid [siRNA]) (Table 2),20,81–84 which include 1) high stability during storage; 2) controlled release; 3) multiple available routes of administration; and 4) prolonged duration of action. Once the polymeric nanoparticles reach the target tissues, the drug may be released by desorption, diffusion through the polymer matrix or polymer wall, or nanoparticle erosion. To obviate the need for surgical retrieval of the exhausted depot, clearance of the dosage from the injection site requires the use of biodegradable polymers. Of the available biomaterials, poly(lactic-co-glycolic acid) (PLGA) is the most commonly used FDA-approved polymer for biodegradable and biocompatible controlled-release devices, with high versatility provided by the suitable selection of the polymer molecular weight, copolymerization, and functionalization.20,85–88 The number of commercial polymeric nanoparticles employing biodegradable carriers is growing and is expected to continue to do so, in line with the promise of further peptide-, protein-, and DNA/ribonucleic acid (RNA)-based drugs emerging from the biotechnology sector. A new polymeric nanoparticle of docetaxel, targeting the extracellular domain of prostate-specific membrane antigen, was developed for the treatment of patients with solid tumors.84 In the Phase I study, the plasma levels of docetaxel (30 mg/m2, IV) administered as polymeric nanoparticles of docetaxel were at least twofold higher than those of an equivalent dose of docetaxel solution, and the high plasma concentrations of docetaxel persisted for at least 48 hours. Albumin nanoparticle technology is particularly well adapted for applications with lipophilic drugs, and albumin-bound paclitaxel (Abraxane®; Celgene Corp, Summit, NJ, USA) was approved by the FDA for the treatment of metastatic breast cancer (2005) and non-small cell lung cancer (2012).79 Abraxane® is obtained by high-pressure homogenization of paclitaxel in the presence of serum human albumin, devoid of any solvent excipients. In humans, Abraxane® has exhibited good pharmacokinetics linearity over the various doses tested, up to 300 mg/m2, and inter-/intrapatient variability in the pharmacokinetic parameters was low. The volume of distribution for Abraxane® was found to be markedly higher than that of free paclitaxel solution, thus suggesting a greater extravascular distribution of Abraxane®. Hydrogel nanoparticles of insulin were also developed, comprising cross-linked materials with the ability to absorb a large amount of water without dissolving,83 and this new technology should allow the oral delivery of insulin and high clinical compliance. In general, polymeric nanoparticles exhibit low immunogenicity and low toxicity (Table 3). Polymeric nanoparticles are commonly coated with nonionic surfactants, and the presence of surfactants on the particle surface markedly reduces immunological interactions, such as opsonization, and also, the interactions between the surface chemical group of nanodrugs via van der Waals forces, hydrophobic interaction, or hydrogen bonding.89
Conclusion and future outlook
In pharmaceutical research and development, the important biopharmaceutical characteristics of drug candidates can be listed as 1) solubility; 2) membrane permeability; 3) metabolic stability; and 4) systemic pharmacokinetics and pharmacodynamics; these factors would have major impact on the drugability and developability of new pharmaceutical products. Nanodrug approaches might resolve the biopharmaceutical problems related to imprecise control of drug release, poor stability, limited pharmacokinetic behavior, and toxicity of the active ingredient. In recent years, as much as ~70% of new drug candidates have shown poor aqueous solubility, and ~40% of marketed drugs for oral use are identified to be practically insoluble in aqueous media (<100 μg/mL).38 Some nanodrug approaches have been found to be efficacious in improving the dissolution behavior of drugs with limited solubility (BCS class II/IV drugs), and current nanotechnologies and ongoing research should bring clinically useful nanodrug systems with improved pharmacokinetic profiles. Therefore, interest in nanodrugs has increased significantly in the last two decades, and several nanodrugs with reduced drug toxicity or enhanced drug efficacy have been successfully developed. In contrast, nanodrugs for targeted delivery are still under development. The suitable selection and further development of highly qualified targeting ligands, such as antibodies, peptides, or aptamers, might accelerate the development of the next generation of nanodrugs with high therapeutic potential. According to the report by Uchegbu and Siew,90 there were 6,242 entries on clinical trials for nanodrugs, and over half of these clinical trials originated from USA. The vast majority of clinical trials are studying cancer patients (~72%), and the other biomedical applications are in infectious diseases (~6%), imaging (~2%), and dental composites (~0.2%). Currently, the nanodrug systems are believed to be feasible and promising in cancer therapy since the nanodrug systems theoretically allow targeting of the particles to increase the concentration of the drug at the site of interest, while reducing the systemic side effects. However, in addition to cancer, there are many serious diseases (diabetes, COPD, dementia, etc) that need to be addressed, and these might eventually be treated effectively via nanotechnology, upon further maturation of the technology platform.
In addition to the nanoparticles presented in this review article, carbon nanotubes have recently emerged as a new option for possible use in methodologies for cancer treatment, bioengineering, and gene therapy. In spite of such attractive features, the toxicity of carbon nanotubes is a prime concern. A deeper understanding of the toxic mechanisms and related physicochemical properties is needed, and carbon nanotubes have to be further developed to optimize the drug payload and reduce their potential toxicity. Via such efforts in academia and the pharmaceutical industry, carbon nanotubes might be a viable and safe option as a nanodrug carrier in the future.
We might still be far from the ultimate goal of the nanodrug approach; however, further development and/or strategic use of suitable nanotechnologies should provide a bright future in the treatment of several diseases, upon successful improvement in the safety, efficacy, and the quality of drugs.
Disclosure
The authors report no conflicts of interest in this work.
References
Devalapally H, Chakilam A, Amiji MM. Role of nanotechnology in pharmaceutical product development. J Pharm Sci. 2007;96(10):2547–2565. | |
Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. Biocompatibility of engineered nanoparticles for drug delivery. J Control Release. 2013;166(2):182–194. | |
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615-627. | |
Yang W, Peters JI, Williams RO. Inhaled nanoparticles – a current review. Int J Pharm. 2008;356(1–2):239–247. | |
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | |
Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. | |
Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65-81. | |
Liu W, Yang XL, Ho WS. Preparation of uniform-sized multiple emulsions and micro/nano particulates for drug delivery by membrane emulsification. J Pharm Sci. 2011;100(1):75–93. | |
Nel A, Xia T, Mädler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311(5761):622–627. | |
Roger E, Lagarce F, Garcion E, Benoit JP. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine (Lond). 2010;5(2):287–306. | |
Jain A, Jain SK. In vitro and cell uptake studies for targeting of ligand anchored nanoparticles for colon tumors. Eur J Pharm Sci. 2008;35(5):404–416. | |
Francis MF, Cristea M, Winnik FM. Exploiting the vitamin B12 pathway to enhance oral drug delivery via polymeric micelles. Biomacromolecules. 2005;6(5):2462–2467. | |
Vega-Villa KR, Takemoto JK, Yáñez JA, Remsberg CM, Forrest ML, Davies NM. Clinical toxicities of nanocarrier systems. Adv Drug Deliv Rev. 2008;60(8):929-938. | |
des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. | |
Mori S, Matsuura A, Rama Prasad YV, Takada K. Studies on the intestinal absorption of low molecular weight heparin using saturated fatty acids and their derivatives as an absorption enhancer in rats. Biol Pharm Bull. 2004;27(3):418–421. | |
Shahbazi MA, Santos HA. Improving oral absorption via drug-loaded nanocarriers: absorption mechanisms, intestinal models and rational fabrication. Curr Drug Metab. 2013;14(1):28–56. | |
Chuang EY, Lin KJ, Su FY, et al. Calcium depletion-mediated protease inhibition and apical-junctional-complex disassembly via an EGTA-conjugated carrier for oral insulin delivery. J Control Release. 2013;169(3):296–305. | |
Kozlowska D, Foran P, MacMahon P, Shelly MJ, Eustace S, O’Kennedy R. Molecular and magnetic resonance imaging: The value of immunoliposomes. Adv Drug Deliv Rev. 2009;61(15):1402–1411. | |
Onoue S, Sato H, Ogawa K, et al. Inhalable dry-emulsion formulation of cyclosporine A with improved anti-inflammatory effects in experimental asthma/COPD-model rats. Eur J Pharm Biopharm. 2012;80(1):54–60. | |
Onoue S, Matsui T, Kuriyama K, et al. Inhalable sustained-release formulation of long-acting vasoactive intestinal peptide derivative alleviates acute airway inflammation. Peptides. 2012;35(2):182–189. | |
Mobley C, Hochhaus G. Methods used to assess pulmonary deposition and absorption of drugs. Drug Discov Today. 2001;6(7):367-375. | |
Bur M, Henning A, Hein S, Schneider M, Lehr CM. Inhalative nanomedicine – opportunities and challenges. Inhal Toxicol. 2009;21 Suppl 1:S137–S143. | |
Ai J, Biazar E, Jafarpour M, et al. Nanotoxicology and nanoparticle safety in biomedical designs. Int J Nanomedicine. 2011;6:1117–1127. | |
Drobne D. Nanotoxicology for safe and sustainable nanotechnology. Arh Hig Rada Toksikol. 2007;58(4):471–478. | |
Choi HS, Frangioni JV. Nanoparticles for biomedical imaging: fundamentals of clinical translation. Mol Imaging. 2010;9(6):291–310. | |
Wick P, Manser P, Limbach LK, et al. The degree and kind of agglomeration affect carbon nanotube cytotoxicity. Toxicol Lett. 2007;168(2):121–131. | |
Stern ST, McNeil SE. Nanotechnology safety concerns revisited. Toxicol Sci. 2008;101(1):4–21. | |
Liu M, Fréchet JMJ. Designing dendrimers for drug delivery. Pharm Sci Technolo Today. 1999;2(10):393-401. | |
Honda M, Asai T, Oku N, Araki Y, Tanaka M, Ebihara N. Liposomes and nanotechnology in drug development: focus on ocular targets. Int J Nanomedicine. 2013;8:495–503. | |
Kaminskas LM, McLeod VM, Kelly BD, et al. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine. 2012;8(1):103–111. | |
Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. | |
Asthana A, Chauhan AS, Diwan PV, Jain NK. Poly(amidoamine) (PAMAM) dendritic nanostructures for controlled site-specific delivery of acidic anti-inflammatory active ingredient. AAPS PharmSciTech. 2005;6(3):E536–E542. | |
Kaminskas LM, Kelly BD, McLeod VM, et al. Capping methotrexate α-carboxyl groups enhances systemic exposure and retains the cytotoxicity of drug conjugated PEGylated polylysine dendrimers. Mol Pharm. 2011;8(2):338–349. | |
Kurmi BD, Gajbhiye V, Kayat J, Jain NK. Lactoferrin-conjugated dendritic nanoconstructs for lung targeting of methotrexate. J Pharm Sci. 2011;100(6):2311–2320. | |
Prajapati RN, Tekade RK, Gupta U, Gajbhiye V, Jain NK. Dendimer-mediated solubilization, formulation development and in vitro-in vivo assessment of piroxicam. Mol Pharm. 2009;6(3):940–950. | |
Kaminskas LM, Porter CJ. Targeting the lymphatics using dendritic polymers (dendrimers). Adv Drug Deliv Rev. 2011;63(10–11):890–900. | |
Ziemba B, Matuszko G, Bryszewska M, Klajnert B. Influence of dendrimers on red blood cells. Cell Mol Biol Lett. 2012;17(1):21–35. | |
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | |
Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Drug nanocrystals: In vivo performances. J Control Release. 2012;160(3):418–430. | |
Chan HK, Kwok PC. Production methods for nanodrug particles using the bottom-up approach. Adv Drug Deliv Rev. 2011;63(6):406–416. | |
Jia L, Wong H, Wang Y, Garza M, Weitman SD. Carbendazim: disposition, cellular permeability, metabolite identification, and pharmacokinetic comparison with its nanoparticle. J Pharm Sci. 2003;92(1):161–172. | |
Jinno J, Kamada N, Miyake M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111(1–2):56–64. | |
Onoue S, Takahashi H, Kawabata Y, et al. Formulation design and photochemical studies on nanocrystal solid dispersion of curcumin with improved oral bioavailability. J Pharm Sci. 2010;99(4):1871–1881. | |
Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. | |
Hanafy A, Spahn-Langguth H, Vergnault G, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–426. | |
Sylvestre JP, Tang MC, Furtos A, Leclair G, Meunier M, Leroux JC. Nanonization of megestrol acetate by laser fragmentation in aqueous milieu. J Control Release. 2011;149(3):273–280. | |
Xia D, Cui F, Piao H, et al. Effect of crystal size on the in vitro dissolution and oral absorption of nitrendipine in rats. Pharm Res. 2010;27(9):1965–1976. | |
Onoue S, Nakamura T, Uchida A, et al. Physicochemical and biopharmaceutical characterization of amorphous solid dispersion of nobiletin, a citrus polymethoxylated flavone, with improved hepatoprotective effects. Eur J Pharm Sci. 2013;49(4):453–460. | |
Kawabata Y, Yamamoto K, Debari K, Onoue S, Yamada S. Novel crystalline solid dispersion of tranilast with high photostability and improved oral bioavailability. Eur J Pharm Sci. 2010;39(4):256–262. | |
Onoue S, Aoki Y, Kawabata Y, et al. Development of inhalable nanocrystalline solid dispersion of tranilast for airway inflammatory diseases. J Pharm Sci. 2011;100(2):622–633. | |
Manvelian G, Daniels S, Gibofsky A. The pharmacokinetic parameters of a single dose of a novel nano-formulated, lower-dose oral diclofenac. Postgrad Med. 2012;124(1):117–123. | |
Manvelian G, Daniels S, Altman R. A phase I study evaluating the pharmacokinetic profile of a novel, proprietary, nano-formulated, lower-dose oral indomethacin. Postgrad Med. 2012;124(4):197–205. | |
Gibofsky A, Silberstein S, Argoff C, Daniels S, Jensen S, Young CL. Lower-dose diclofenac submicron particle capsules provide early and sustained acute patient pain relief in a phase 3 study. Postgrad Med. 2013;125(5):130–138. | |
Shegokar R, Müller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399(1–2):129–139. | |
Martins S, Sarmento B, Ferreira DC, Souto EB. Lipid-based colloidal carriers for peptide and protein delivery – liposomes versus lipid nanoparticles. Int J Nanomedicine. 2007;2(4):595–607. | |
Larsen AT, Ohlsson AG, Polentarutti B, et al. Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur J Pharm Sci. 2013;48(1–2):339–350. | |
Onoue S, Uchida A, Kuriyama K, et al. Novel solid self-emulsifying drug delivery system of coenzyme Q10 with improved photochemical and pharmacokinetic behaviors. Eur J Pharm Sci. 2012;46(5):492–499. | |
Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–230. | |
Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160(1):25–32. | |
Thomas N, Holm R, Garmer M, Karlsson JJ, Müllertz A, Rades T. Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2013;15(1):219–227. | |
Teshima M, Fumoto S, Nishida K, et al. Prolonged blood concentration of prednisolone after intravenous injection of liposomal palmitoyl prednisolone. J Control Release. 2006;112(3):320–328. | |
Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761-769. | |
Tomii Y. Lipid formulation as a drug carrier for drug delivery. Curr Pharm Des. 2002;8(6):467-474. | |
Kawakami S, Yamamura K, Mukai T, et al. Sustained ocular delivery of tilisolol to rabbits after topical administration or intravitreal injection of lipophilic prodrug incorporated in liposomes. J Pharm Pharmacol. 2001;53(8):1157–1161. | |
Fetterly GJ, Straubinger RM. Pharmacokinetics of paclitaxel-containing liposomes in rats. AAPS PharmSci. 2003;5(4):E32. | |
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | |
Feldman EJ, Kolitz JE, Trang JM, et al. Pharmacokinetics of CPX-351; a nano-scale liposomal fixed molar ratio formulation of cytarabine: daunorubicin, in patients with advanced leukemia. Leuk Res. 2012;36(10):1283–1289. | |
Reddy LH, Sharma RK, Chuttani K, Mishra AK, Murthy RR. Etoposide-incorporated tripalmitin nanoparticles with different surface charge: formulation, characterization, radiolabeling, and biodistribution studies. AAPS J. 2004;6(3):e23. | |
Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release. 2005;107(2):215–228. | |
Piao H, Kamiya N, Hirata A, Fujii T, Goto M. A novel solid-in-oil nanosuspension for transdermal delivery of diclofenac sodium. Pharm Res. 2008;25(4):896–901. | |
Zhang N, Ping Q, Huang G, Xu W, Cheng Y, Han X. Lectin-modified solid lipid nanoparticles as carriers for oral administration of insulin. Int J Pharm. 2006;327(1–2):153–159. | |
Pathak P, Nagarsenker M. Formulation and evaluation of lidocaine lipid nanosystems for dermal delivery. AAPS PharmSciTech. 2009; 10(3):985–992. | |
Watanabe M, Kawano K, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Preparation of camptothecin-loaded polymeric micelles and evaluation of their incorporation and circulation stability. Int J Pharm. 2006;308(1–2):183–189. | |
Matsumura Y, Hamaguchi T, Ura T, et al. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br J Cancer. 2004;91(10):1775–1781. | |
Kato K, Chin K, Yoshikawa T, et al. Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer. Invest New Drugs. 2012;30(4):1621–1627. | |
Pepić I, Jalsenjak N, Jalsenjak I. Micellar solutions of triblock copolymer surfactants with pilocarpine. Int J Pharm. 2004;272(1–2):57–64. | |
Onoue S, Kojo Y, Suzuki H, et al. Development of novel solid dispersion of tranilast using amphiphilic block copolymer for improved oral bioavailability. Int J Pharm. 2013;452(1–2):220–226. | |
Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19(1):1–72. | |
Fanciullino R, Ciccolini J, Milano G. Challenges, expectations and limits for nanoparticles-based therapeutics in cancer: A focus on nano-albumin-bound drugs. Crit Rev Oncol Hematol. 2013;88(3):504–513. | |
Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1–2):113–142. | |
Morgen M, Bloom C, Beyerinck R, et al. Polymeric nanoparticles for increased oral bioavailability and rapid absorption using celecoxib as a model of a low-solubility, high-permeability drug. Pharm Res. 2012;29(2):427–440. | |
Zhang J, He C, Tang C, Yin C. Ternary polymeric nanoparticles for oral siRNA delivery. Pharm Res. 2013;30(5):1228–1239. | |
Chaturvedi K, Ganguly K, Nadagouda MN, Aminabhavi TM. Polymeric hydrogels for oral insulin delivery. J Control Release. 2013;165(2):129–138. | |
Hrkach J, Von Hoff D, Mukkaram Ali M, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. | |
Pandey R, Ahmad Z, Sharma S, Khuller GK. Nano-encapsulation of azole antifungals: potential applications to improve oral drug delivery. Int J Pharm. 2005;301(1–2):268–276. | |
Reddy LH, Murthy RS. Pharmacokinetics and biodistribution studies of Doxorubicin loaded poly(butyl cyanoacrylate) nanoparticles synthesized by two different techniques. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2004;148(2):161–166. | |
Onoue S, Kuriyama K, Uchida A, Mizumoto T, Yamada S. Inhalable sustained-release formulation of glucagon: in vitro amyloidogenic and inhalation properties, and in vivo absorption and bioactivity. Pharm Res. 2011;28(5):1157–1166. | |
Sharma A, Sharma S, Khuller GK. Lectin-functionalized poly (lactide-co-glycolide) nanoparticles as oral/aerosolized antitubercular drug carriers for treatment of tuberculosis. J Antimicrob Chemother. 2004;54(4):761-766. | |
Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020-1037. | |
Uchegbu IF, Siew A. Nanomedicines and nanodiagnostics come of age. J Pharm Sci. 2013;102(2):305–310. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.