Back to Journals » International Journal of Nanomedicine » Volume 13
Nanodelivery systems and stabilized solid-drug nanoparticles for orally administered medicine: current landscape
Authors Kermanizadeh A, Powell LG, Stone V ![]() , Møller P
, Møller P
Received 16 June 2018
Accepted for publication 9 August 2018
Published 16 November 2018 Volume 2018:13 Pages 7575—7605
DOI https://doi.org/10.2147/IJN.S177418
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ali Kermanizadeh,1,2 Leagh G Powell,1 Vicki Stone,1 Peter Møller2
1NanoSafety Research Group, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, UK; 2Section of Environmental Health, Department of Public Health, University of Copenhagen, Copenhagen, Denmark
Abstract: The use of nanoparticles as a means of targeted delivery of therapeutics and imaging agents could greatly enhance the transport of biologically active contents to specific target tissues, while avoiding or reducing potentially undesired side effects. Generally speaking, the oral route of administration is associated with good patient compliance, as it is convenient, economical, noninvasive, and does not require special training. Here, we review the progress of the utilization of nanodelivery-system carriers or stabilized solid-drug nanoparticles following oral administration, with particular attention on toxicological data. Mechanisms of cytotoxicity are discussed and the problem of extrapolating knowledge to human scenarios highlighted. Additionally, issues associated with administration of drugs via the oral route are underlined, while strategies utilized to overcome these are highlighted. This review aims to offer a balanced overview of strategies currently being used in the application of nanosize constructs for oral medical applications.
Keywords: nanomedicine, oral route of delivery, nanomaterials, nano-stabilized solid drugs
Introduction
Nanotechnology is based on the understanding and exploitation of matter at the nanoscale (1–100 nm)1 in the integration of the nanostructures into larger systems for real-world applications. Nanoparticles (NPs) are approximately 1,000 times smaller than the average human cell. It is this small size (larger surface area) and other unique physiochemical properties that make them extremely desirable for medical applications.1–4 Over the last two decades, nanotechnology has gained prominence in modern biology, with the field of nanomedicine becoming one of the most prominent and vital.4 Nanomedicine allows for drug particles or delivery devices to be manipulated at the nanoscale for improved delivery to different parts of the body, while at the same time retaining the valuable pharmacological properties of the drug.5 However, efficient drug delivery and the excellent release potential of these delivery systems may be hindered by possible untoward side effects. The unique properties that make NPs so desirable might also contribute to their potential toxicity.6 Therefore, safety evaluation of NPs with intended medical applications is of utmost importance.
In recent years, vast progress has been made in the synthesis of a variety of materials that can be used as nanovehicles (carbons, synthetic polymers, micelles, liposomes/vesicles, and drug–polymer polysaccharide conjugates, to name a few).7–10 Currently, commercial nanomedicines are predominantly generated using two main strategies: a nanocarrier for drug molecules and stabilized solid-drug NPs (Figure 1). These strategies have the potential to target a single cell type, and represent revolutionary tools for effective medicine delivery. For example, NP surfaces can be chemically designed to influence site-specific drug delivery, as well as the kinetics of drug release.9,12 In addition to drug delivery, the discovery of quantum effects (size-dependent properties) that result in materials with specific emission, absorption, or light-scattering spectra has expanded potential applications in imaging and diagnosis.11 Further advantages of the use of NPs can include improved drug stability and longer shelf-life9 compared with traditional formulations. Each of these properties allows researchers to design and use NPs in medical applications that can enhance on conventional molecular approaches.13
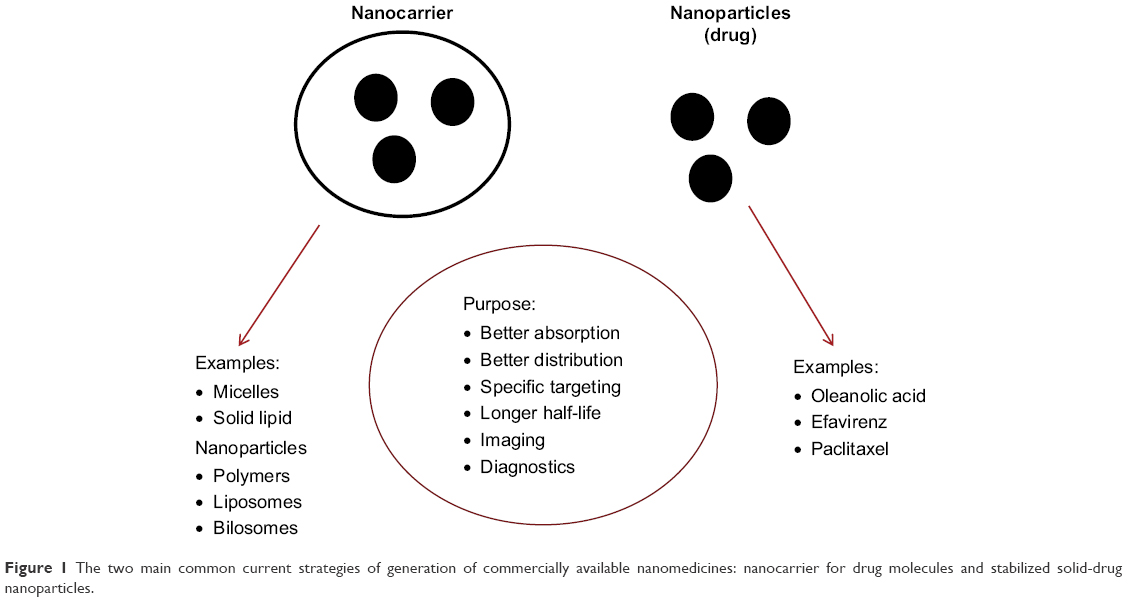 | Figure 1 The two main common current strategies of generation of commercially available nanomedicines: nanocarrier for drug molecules and stabilized solid-drug nanoparticles. |
Since drug delivery via the oral route is the focus of this article, it is important to consider potential local effects in the gastrointestinal tract (GIT). The GIT mucosa forms a dynamic physical and enzymatic barrier between the body and the luminal environment, which not only contains nutrients but also is laden with potentially hostile microorganisms and toxins. Therefore, the GIT mucosa has evolved to allow efficient transport of nutrients across the epithelium while rigorously excluding passage of harmful molecules and organisms. The exclusionary properties of the GIT mucosa present a complex set of problems for oral drug delivery.14 Another significant hurdle for oral delivery of many drugs is their inherently poor stability in the gastric environment.4,15,16 At the site of therapeutic absorption, NPs can be heavily affected in transport through the digestive tract, due to varying pH, transit time, pressure and bacterial content. The distinct features of each part of the digestive tract can introduce challenges to the application of therapeutics to the gut. Nonetheless, oral delivery is still the most common method for drug administration, due to its simplicity, noninvasive nature, and good patient compliance. Following ingestion, translocation of particles into and across the GIT mucosa can occur via four mechanisms: 1) endocytosis, through enterocytes, 2) the M-cell-rich layer of Peyer’s patches (small-intestine lymphoid aggregates), 3) persorption, where particles can translocate through a “hole” left in the epithelium when enterocytes shed from the villous tip, and 4) the paracellular route, where NPs pass through weakened tight junctions of the epithelial cell layer.17–20 Moreover, NP translocation and interactions with tissues in the GIT can be influenced by size, morphology, hydrophilic–hydrophobic balance, and surface functionalization of the particles in question. For example, it has been suggested that negatively charged materials exhibit poor bioavailability, due to electrostatic repulsion and mucus entrapment.21 Once in the submucosal tissue, NPs can enter the lymphatics and the blood capillaries.12
The remainder of the manuscript examines the progress made in utilization of nanoscale carriers or stabilized solid-drug NPs in medical applications following oral route of administration. The search criteria included a combination of the following terms: “nanomedicine”, “nanoparticles”, “nanomaterials”, “in vivo”, “liposomes”, “micelles”, “solid drug nanoparticles”, “oral route”, “oral exposure”, “oral administration”, “gastrointestinal tract”, “nanodelivery” “nanocarrier”, “nanovehicles”, “nanoconstructs”, “nanoemulsions”, “adverse effects”, “toxicity”, “cytotoxicity”, “drug delivery”, “therapeutics”, “imaging”, “systemic”, and “Caco-2 cells”. The last literature search was conducted on August 24, 2017. Due to the inevitable limitations and exclusionary nature of any literature search, studies that did not include the search terms in the title or did not provide adequate information in the abstract might have been unintentionally omitted. However, all attempts have been made to construct a comprehensive review and offer a balanced overview of the success of the current strategies for the use of nanosize constructs for oral medical applications. Due to the complexity of the topic being reviewed and discussed, we have intentionally aimed to make each study section comprehensive. Nevertheless, this does not include the methodology utilized for the manufacture of the constructs or loading and encapsulation efficacy of the active pharmaceutical ingredients. The review focuses on in vivo studies (predominantly in rodents), rather than in vitro experimentation, in order to capture the products that are potentially closer to market (assuming that initial screening and developmental work is conducted in vitro). As such, the reader could presuppose that the discussion focuses on “relatively safe” constructs. However, on occasion, this was not the case, ie, constructs loaded with chemicals designed to induce tumor-cell death. The review does not incorporate general NP oral toxicology/biokinetics, but instead focuses on particles with intended medical applications. The main body of the review has been structured and divided based on World Health Organization ICD11 classification of disease22 and the intended therapeutic use of the nanomedicine.
Diseases of the digestive system
Helicobacter pylori infects more than half the world’s population and is responsible for most cases of inflammatory gastritis, peptic ulcers, and gastric cancer.23 Currently, the standard treatment for H. pylori infection involves two antibiotics (clarithromycin combined with amoxicillin or metronidazole) and a proton-pump inhibitor, termed “triple therapy”.23 Unfortunately, bacteria-eradication rates with triple therapy have decreased significantly over the last 20 years as a consequence of increased bacterial resistance to these antibiotics. Intriguingly, various free fatty acids, including linolenic acid (LLA) have been shown to contain antibacterial activity against a broad range of bacteria, including H. pylori. However, the in vivo inhibition of H. pylori by the free fatty acids is problematic, fundamentally due to their poor solubility and the tendency for them to be oxidized and esterified in the GIT. To address these concerns, in a recent study LLA was packed inside a liposome nanocarrier composed of L-α-phosphatidylcholine and cholesterol (LipoLLA; ~100 nm). First, the H. pylori bactericidal activity of LipoLLA was evaluated in vitro. The authors established the bactericidal concentration as 65 and 80 μg/mL (LipoLLA and LLA, respectively). Interestingly, despite the strong bactericidal activity, the LipoLLA showed negligible toxicity to human gastric carcinoma cells following 24-hour exposure up to concentrations of 900 μg/mL. H. pylori mainly resides within the adherent mucus layer, close to the epithelial surface of the stomach. Therefore, for effective antibacterial treatment, permeation of the drug across the mucus layer and its retention on the stomach wall are critical.23 To assess this, male C57BL/6 mice (~30 g) were exposed to 1.2 mg RhB-labeled LipoLLA for 4 or 24 hours via oral gavage. Quantification of gastric retention of the liposome construct was noted as 69 μg at 4 hours posttreatment, decreasing to 34 μg at 24 hours. Next, in vivo therapeutic efficacy of LipoLLA was investigated in an H. pylori-infected C57BL/6 mouse model. Therapeutic efficacy was evaluated by enumeration and comparison of H. pylori in the stomachs of animals in different treatment groups and controls. The data demonstrated that in the mice treated with LipoLLA, the bacterial burden was 5.5×104 CFU/g in comparison to 108 in the control infected mice. Overall, the LipoLLA treatment reduced H. pylori burden ~2.5-fold compared to the control infected animals, whereas triple therapy reduced bacterial burden only ~1.5-fold. Additionally, mRNA expression of IL1β, IL6, and TNFα was significantly reduced in infected mice treated with LipoLLA (no acute inflammatory response to LLA liposome). Finally, the in vivo toxicity of the drug formulation was assessed in uninfected mice exposed to 2.4 mg of the materials per day over a 5-day period. Gastric tissue treated with LipoLLA maintained an undisturbed structure with a clear layer of epithelial cells. Furthermore, similar to the negative control, there was no apparent increase in gastric epithelial apoptosis in LipoLLA-treated mice (TUNEL staining). The absence of any detectable gastric histopathology or toxicity within a 5-day treatment period suggested that orally administered LipoLLA is relatively safe at the specified dose. Overall, the data denote that the nanoformulation discussed in this study could hold potential for effective and safe treatment of H. pylori infections.23
In a 2015 study, the aim was to develop nanoconstructs for better oral delivery of the acid-labile and poorly water-soluble drug lansoprazole (Lpz), for gastric ulcer therapy.24 Acid-related disorders, such as peptic ulcer and gastroesophageal reflux disease, occur frequently in elderly individuals. Proton-pump inhibitors (eg, Lpz) are among the most effective drugs for the treatment of ulcer-related disorders. The two constructs were composed of positively charged Eudragit RS100 (~205 nm) or negatively charged poly(lactic-co-glycolic acid) (PLGA; ~220 nm). Male Wistar rats (300 g) were exposed to the formulations (5 mg Lpz/kg) via the oral route 1 hour after administration of ethanol was used to induce gastric ulceration. The animals were killed 4 hours after NP administration. First, the data showed that the NPs were localized in both ulcerated and nonulcerated regions after oral administration. Moreover, the results indicated that the negatively charged particles adhered more readily to the ulcerated region, whereas the positively charged NPs predominantly localized in the nonulcerated region of the stomach. It was also demonstrated that all the drugs were eliminated within 6 hours after oral administration of Lpz solution (unformulated control), while the NP formulations showed sustained and prolonged Lpz concentrations up to 24 hours. The elimination half-life and mean residence time for the Eudragit RS100 (4.6 and 8.2 hours, respectively) and PLGA (4.7 and 9 hours) were significantly longer than those for Lpz solution (1.4 and 1.9 hours). Finally, the ulcer-healing efficacy of Lpz NPs was evaluated at 7 days postexposure in ulcer-induced Wistar rats. The results showed that the gastric ulceration gradually healed within 1 week after oral administration of Lpz solution and Lpz NPs. The oral administration of Lpz solution healed 72% of gastric ulcers, while oral administration of the positively and negatively charged NPs healed 96% and 93% of gastric ulcers, respectively.24 These results clearly demonstrate improved targeting and efficacy of Lpz when administered in an NP form compared to free drug.
Inflammatory bowel disease (IBD) is an umbrella of diseases of which ulcerative colitis and Crohn’s disease are the most prevalent. If overt inflammation is not adequately suppressed, chronic relapsing disorders of the GIT can develop into colorectal cancer.25 The current anti-inflammatory strategy consists of systematic administration of therapeutics at high doses, often associated with significant adverse side effects. CD98 is highly upregulated and expressed in colonic tissue with active colitis and Crohn’s disease.25 Additionally, the glycoprotein has been shown to be highly expressed on intestinal macrophages and plays an important role in macrophage activation. In recent years, RNA interference using siRNA has become a powerful tool for the silencing of disease-related genes. However, exploration of the therapeutic potential of siRNA has been extremely slow, due to the absence of carriers for targeted in vivo delivery.25 If not adequately controlled, the utilization of siRNA can be dangerous, as it can easily cross tissue and cell barriers to enter the cytoplasm and cause the degradation of mRNA.25 In a novel study, a targeted CD98 siRNA (siCD98)-conjugated maleimide, urocanic acid, and polyethylene glycol (PEG) NP-delivery system was utilized for specific targeting of macrophages in the inflamed colon after oral administration.25 The NPs were noted to be 150–250 nm in size. First, the cytotoxicity of the constructs was investigated in Colon-26 cells and murine macrophages following a 6-hour (important to note that this is a relatively short exposure) in vitro treatment (100 nmol/L siRNA). The authors did not observe any alteration in cell viability in response to the NPs. The tissue biodistribution of siCD98-functionalized NPs was investigated using ex vivo tissue sections. The data demonstrated significant quantities of NPs accumulated in colonic epithelial cells after 6 hours of incubation. A T-lymphocyte-transfer mouse model of chronic colitis is considered the prototypical and best-characterized model of chronic colitis.25 The anti-inflammatory effect of functionalized NPs was further investigated in this in vivo model. RAG−/− mice (animals that do not have mature lymphocyte populations, ~30 g) were injected with a flow-cytometry-purified CD4+CD45RBhigh T-cell population. This resulted in the characteristic tissue loss associated with IBD. The mice were exposed via gavage twice per week for 5 consecutive weeks, with or without the encapsulated siCD98-functionalized NPs (1 mg/kg siCD98 or scrambled siRNA on each occasion). Oral administration of siCD98-loaded NPs significantly reduced weight loss at week 9 compared with untreated mice or mice treated with scrambled siRNA-loaded NPs. Furthermore, the data demonstrated that myeloperoxidase (MPO) activity in the colon was increased in mice that had received a T-cell transfer. MPO levels were reduced by up to 65% in the siCD98-loaded NP-treated group compared with untreated animals (reduced inflammatory response). mRNA expression of TNFα, IL6, and IL12 was also decreased in the treated mice (60%, 80%, and 32% respectively), suggesting reduced inflammation. Finally, the study investigated whether orally delivered functionalized siCD98-loaded NPs could mitigate the clinical manifestation of dextran sodium sulfate (DSS)-induced acute colitis in the mouse model. After 6 days, mice in the DSS control group had a body-weight loss of 18%, which was roughly twice that of mice in the treatment groups. Moreover, histopathological examination of colon tissue from the DSS control group exhibited clear signs of inflammation, including epithelial disruption, goblet-cell depletion, and significant infiltration of inflammatory cells into the mucosa. In contrast, inflammation in tissue from siCD98-loaded NP-treated mice was markedly lower. Overall, the study showed promising evidence of successful use of a siCD98-conjugated maleimide, urocanic acid, and PEG NP-delivery system as a means of effective IBD therapy.25
Another popular strategy in the treatment of IBD is the specific targeting of immune cells, which are highly abundant in inflamed regions of the GIT. This strategy is gathering popularity due to better understanding of the kinetics of increased adhesion of small-particulate drug carriers to inflamed tissue. This strategy was explored using surface-modified amorphous silica NPs. The NPs were covalently bound to 5-amino salicylic acid and characterized as ~150 nm in diameter. In these trials, male BALB/c mice (~25 g) were exposed to 2,4,6-trinitrobenzenesulfonic acid to induce colitis. During the treatment period, animals were orally exposed to doses up to 100 mg/kg body weight of salicylic acid in various formulations once daily for 6 consecutive days. Animals were killed 24 hours after the last drug/particle administration. Histological examination of mice exposed to fluorescently labeled NPs showed selective accumulation in the inflamed tissue (quantitative analysis exhibited sixfold-higher adhesion of NPs to diseased tissue in comparison to healthy intestine). After induction of experimental colitis, clinical activity indices increased dramatically within 24 hours, indicating intestinal inflammation. The decrease in clinical activity became visible on day 6 for both the NP- and free salicylic acid-treated mice. Similar to observations made regarding the clinical activity index, colon weight:length ratios after drug treatment were lower in NP-treated animals comparison to the colitis control. For both formulations, MPO activity was also significantly reduced, with higher therapeutic effects for NPs at 25 or 50 mg/kg than free drug, even at 100 mg/kg. The selectivity of the NP formulation was further supported by the observation that locally applied modified NPs showed significant and therapeutic efficiency.26
As indicated, such IBDs as ulcerative colitis and Crohn’s disease are both characterized by an overexpression of inflammatory cytokines and mediators, namely TNFα, IL6, and NFκB.27 In a recent study, raloxifene (potent suppressive of the transcription of NFκB) was incorporated into nanomicelles (~70 nm) composed of poly(styrene-co-maleic acid).28 These micelles were used as a delivery platform to improve raloxifene’s water solubility, protecting it from first-pass metabolism and efflux mechanisms. In this study, female BALB/c mice (~20 g) were administered with 3% DSS in drinking water for 14 days to induce colitis. At day 7, the mice were orally treated, either by free raloxifene or the micelle formulation, for 3 days with a dose equivalent to 5 mg/kg. The authors showed that compared to the controls, weight gain was clearly visible after 14 days in the raloxifene-treated animals. Additionally, the average colon length of DSS-treated mice was 7.4 cm compared with 12.3 and 11.5 cm in free raloxifene- and nanomicelle-treated mice respectively. Histological examination of the DSS-treated mice indicated highly inflamed colon tissue with extensive mononuclear cell infiltration. Colon tissue obtained from both free raloxifene- and micelle-treated groups clearly demonstrated tissue recovery. Finally, analysis of plasma collected from the DSS-treated mice showed increased IL6 levels of 194% compared to unexposed mice. This was reduced to 88% and 53% following administration of the free-drug suspension and nanoformulation compared to the DSS-treated animals, suggesting that the micellar delivery system was more efficient in the reduction of inflammatory cytokines.28
Diseases of the liver
Oleanolic acid is a naturally derived triterpene used clinically in the treatment of hepatitis (inflammatory disease of liver, autoimmune or secondarily induced by drugs, toxins, or alcohol).29 However, like many other hydrophobic drugs, it has very poor bioavailability. In a 2005 trial, oleanolic acid nanosuspensions (~285 nm spheres) were prepared and utilized in a male Kunming mouse model (~20 g). First, carbon tetrachloride (CCl4) was utilized to induce hepatocellular damage in the exposed mice, evidenced by increased serum ALT and hepatic malondialdehyde content. Oral pretreatment with 100 mg/kg oleanolic acid before CCl4 administration led to a 61% and 43% reduction in ALT rise in serum and liver malondialdehyde content, respectively. After pretreatment with oleanolic acid NPs, ALT values were 80% and 66%, respectively. The higher absorption of nanosize oleanolic acid may account for the enhanced hepatoprotective properties of oleanolic acid NPs.29
Norcantharidin is a demethylated analog of cantharidin that is very effective in hepatic carcinoma chemotherapy. However, its clinical applications are limited, due to some serious side effects, which include nephrotoxicity and inflammation.30 In a recent study, male and female Sprague Dawley rats and CD1 mice were orally exposed to 5 mg/kg of a formulation containing polyvinylpyrrolidone-coated norcantharidin chitosan (CS) NPs (~140 nm). The data showed that oral administration of the NPs had greater bioavailability of around 173% in comparison to the traditional drug formulation. After oral dosing, total residue concentrations in the tissues were highest at 1–3 hours and mostly distributed to the liver and the kidneys. The quantity of the parent drug eliminated in the feces of the nanoformulation was 32 times greater than that of the traditional drug (reduced toxicity in hepatic cells due to decreased norcantharidin buildup).30
An active ingredient of Rabdosia rubescens, oridonin has been used for the treatment of hepatic carcinomas.31 However, poor aqueous solubility, low oral bioavailability, and rapid clearance limit clinical application of the compound. The principle that wheat-germ agglutinin-modified lipid–polymer hybrid NPs (WGA-LPNs) can promote cellular uptake after oral delivery via receptor-mediated endocytosis was used to develop a delivery system for improved oral delivery of the prodrug. The liposomes were prepared in an aqueous phase containing 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, PEG2,000, and phosphatidylcholine (Lipoid S100) (~120 nm). For pharmacokinetic analysis, male Sprague Dawley rats (~220 g) were administered via oral gavage different formulations at a dose of 10 mg/kg for up to 24 hours. In order to assess the efficacy of the delivery system, a tumor-xenograft model was established by inoculation of HepG2 cells subcutaneously into the right-armpit region of male BALB/c nude mice (~20 g). Once tumor volume reached approximately 200 mm3, the animals were administered formulations every 2 days via oral gavage for 24 days 5-fluorouracil (5FU) solution (22.5 mg/kg), oridonin suspension (7.5 mg/kg), LPNs, or WGA-LPNs (7.5 mg/kg). Tumor volume and body weight were recorded every 2 days during treatment. The data showed that the relative serum bioavailability of WGA-LPNs increased ninefold compared with free suspension. Next, efficacy evaluation showed the modified liposome-delivery system to be highly effective against the tumor. The therapeutic properties (a reduction in tumor volume and reduced body-weight loss) of the liposomes were significantly higher than both the free oridonin formulation and 5FU. Overall, the data demonstrated that the liposome formulation had great potential as an oral delivery vehicle for oridonin, due to its ability to permeate the intestinal mucus layer, allowing for significantly improved pharmacokinetics, and effective antitumor activity.32
Diseases of the circulatory system
Nisoldipine is a calcium-channel blocker used to treat hypertension.33 However, the oral bioavailability of the drug is only about 5% of the overall administered dose, due to very low aqueous solubility and high presystemic metabolism.34 In an attempt to improve the solubility and oral bioavailability of the compound, 100 nanosize compositions with varying concentrations of different oils (25%–70% w:w), surfactants (30%–75% w:w), and cosurfactants (0–25% w:w) were prepared. From the initial panel, two nanocarrier formulations were selected as potential candidates for effective drug delivery based on solubility, stability over time, droplet size (one of the most important characteristics for stability evaluation and in vivo absorption), particle-size distribution, and negative ζ-potential. The two nanocarriers were termed ACP19 (Acrysol EL135 60%, PEG 30%, and Capmul MCM 10%; ~100 nm) and PCT08 (peppermint oil 60%, Capmul MCM 35%, and Transcutol 5%; ~700 nm). The bioavailability of nisoldipine was investigated in male Wistar rats (~250 g). The two formulations and free drug suspension were administered to the animals at a dose of 5 mg/kg by oral gavage. The data demonstrated that the nisoldipine plasma concentrations in Wistar rats treated with self-emulsified formulations were significantly higher than those treated with the drug in suspension. The bioavailability of ACP19 and PCT08 was 2.5- and 2.2-fold greater than the drug in suspension, respectively. Moreover, the maximum nisoldipine concentration in plasma of ACP19 and PCT08 was 76 and 69 ng/mL respectively, compared to the drug in suspension (23 ng/mL). The study concluded that the optimized ACP19 formulation enhances the solubility and bioavailability of nisoldipine.34
Olmesartan medoxomil is a potent angiotensin II receptor antagonist clinically used for the treatment and management of hypertension.35 However, the drug exhibits low and inconsistent oral bioavailability in humans, primarily due to its high lipophilicity. In a recent study, a cationic self-nanoemulsifying oily formulation composed of oleic acid (32 mg/mL), Tween 40 (55 mg/mL), and Transcutol HP (44 mg/mL; ~50–100 nm) was manufactured to enhance the oral bioavailability of the cargo. In these experiments, unisex Wistar rats (~300 g) were orally exposed to the formulation at doses of 20 mg/kg for up to 72 hours. Pharmacokinetic data exhibited significantly superior oral bioavailability, both in terms of extent and rate of absorption by the nanoformulation compared to the free drug suspension (4.9-fold improvement in plasma concentrations and 0.96-fold reduction in time to maximum concentration).35
Simvastatin (Smv) is an inhibitor of 3-hydroxy-3-methyl coenzyme A reductase, which can lower plasma cholesterol concentrations.36 The compound is currently used clinically to treat hyperlipidemia and various cardiovascular diseases.36 However, Smv has extremely poor water solubility, resulting in oral bioavailability of <5%. In a 2015 study, two nanosize formulations were manufactured to encapsulate Smv to enhance its oral bioavailability. These included a liposome composed of soybean lecithin and cholesterol (~110 nm) and an Smv-polyamidoamine dendrimer (~10 nm). Male Sprague Dawley rats with a body weight of 150 g were orally administered Smv 140 mg/kg body weight for 10 hours. The data showed that the maximum plasma concentrations of Smv in the dendrimer-complex- and the liposome-treated groups were 2.2 and 3.8 times higher, respectively, compared to the free Smv-exposed animals. Furthermore, both the dendrimer and liposome formulations dramatically enhanced the oral bioavailability of Smv (2.5- and 3.7-fold increase, respectively). The study provided evidence of an effective and complementary strategy for the improved oral administration of Smv.36
Hydroxysafflor yellow A (HSYA) is the active component of Carthamus tinctorius, a plant routinely utilized in Chinese medicine for treatment of various cerebrovascular and cardiovascular diseases.37 HSYA has also been demonstrated to have a substantial antagonistic effect on the platelet-activating factor receptor, as well as promising neuroprotective action. However, the compound is extremely hydrophobic with low oral bioavailability. In order to increase oral absorption, the drug was encapsulated in a caprylic/capric triglyceride bean-phospholipid NP construct (~50 nm). Male Sprague Dawley rats (~200 g) were orally exposed to 25 mg/kg of the NPs over a period of 24 hours. The data demonstrated that drug plasma concentrations were enhanced 23-fold following oral administration of HYSA NPs compared to traditional HSYA solution.37
Curcumin is known to contain beneficial properties, which include but are not limited to anti-inflammatory, anticoagulation, antithrombus, antioxidant, and antiangiogenic attributes.38 However, curcumin has extremely poor oral bioavailability, which has hampered its utilization in more advanced clinical formulations. In a 2012 investigation, a polybutylcyanoacrylate-NP system was introduced to improve its oral bioavailability. The nanostructures were found to be in the range of 40–400 nm with a mean size of 100 nm. The pharmacokinetics of NPs were addressed in a male Wistar rat model (~200 g) following oral administration of 50 mg/kg of the formulation. The data demonstrated that the polybutylcyanoacrylate NPs enhanced the relative bioavailability of curcumin more than eightfold compared to the traditional suspension, with ileum and colon being the main absorption sites in the GIT.39
In another study, an NP construct composed of curcumin and negative polyanion tripolyphosphate (~110 nm) was developed to increase the solubility, permeability, dissolution, and bioavailability of the polyphenol. The pharmacokinetic properties of the NPs were compared with a curcumin suspension (dissolved in 0.5% aqueous solution of sodium carboxymethylcellulose) in Sprague Dawley rats (~300 g) after an oral dose of 100 mg/kg. The authors showed the relative bioavailability of curcumin NPs was 450% of the free chemical in suspension. Interestingly, there was no significant difference in plasma availability between the two formulations, with total clearance at around 12 hours postexposure. The significantly augmented absorption was explained by increased affinity between the NPs and the epithelial cell membrane in the GIT, due to better dispersibility of curcumin in the NP formulation.40
In a similar study, PLGA and polyvinyl alcohol (PVA)-based NPs (~190 nm) were utilized to improve the bioavailability of curcumin. Here, male Sprague Dawley rats (~150 g) were exposed to 100 mg/kg of the NPs by oral gavage. Pharmacokinetic data demonstrated that NP encapsulation increased the intestinal absorption of the polyphenol and was predominantly available as metabolite curcumin glucuronide in plasma. Interestingly, the analysis of the curcuminoids in the liver and kidneys 1.5 hours postexposure did not show any increased accumulation in these organs for the NP formulations.41
Drug adsorption into a nanostructured carrier with a large surface area might be a useful approach for improving the dissolution rate and absorption of poorly water-soluble drugs. In addition, the use of hydrophilic or pH-sensitive polymers might further enhance the biological efficacy of poorly water-soluble drugs. In a 2013 study, an amphiphilic polymeric solubilizer (Soluplus) was applied to a colloidal silica nanomatrix (termed SCCSN) for enhanced solubility and oral absorption of three water-insoluble drugs (atorvastatin calcium [an HMG-CoA reductase inhibitor], dutasteride [a 5α-reductase inhibitor], and sorafenib tosylate [an inhibitor of several tyrosine protein kinases]). The matrix was characterized using scanning electron microscopy and showed a primary particle size of about 10 nm. Male Sprague Dawley rats (~200 g) were exposed via the oral route to 2 mg/kg of the complex. All SCCSNs had rapid dissolution rates, with all three drugs reaching maximum solubility within 30 minutes postexposure. For atorvastatin calcium, dutasteride, and sorafenib tosylate, the maximum saturated concentration of SCCSN was significantly higher than that of the unloaded version of each drug, with approximately 2.8-, 326- and 46-fold increases in solubility, respectively. The data suggested that the SCCSN system could be a promising approach to improve oral absorption of poorly water-soluble drugs. It is important to state that no toxicity data were provided in the study.42
Diabetes mellitus
Oral delivery of insulin is still a challenge, due to rapid enzymatic degradation in the stomach, inactivation and digestion by enzymes in the intestinal lumen, and poor permeability across the intestinal epithelium because of its high molecular weight and lack of lipophilicity.43 The oral delivery of insulin can prove highly advantageous, as it transports the drug directly to the liver, its primary site of action via portal circulation. In an interesting study, insulin solid-lipid NPs (SLNs) modified with stearic acid–octaarginine (~160 nm) were prepared to improve the stability and bioavailability of the hormone. SLNs have a large specific surface area of physiologically compatible lipid matrix and can protect against the harsh GIT environment, principally by encapsulation of proteins within the nanostructures. Furthermore, the carrier itself can be taken up by epithelial cells or the lymphoid tissues in Peyer’s patches. In this experiment, NPs with and without stearic acid–octaarginine were administered to the duodena of fasted diabetic Sprague Dawley rats (~200 g) at an insulin dose of 25 IU/kg. The standard insulin solution as a control showed no hypoglycemic effect. The unmodified SLNs had maximum blood glucose lowering of 73% at 2 hours, while the stearic acid–octaarginine NPs showed significant hypoglycemic effect: maximum blood glucose lowered to 30% at 1.5 hours.43
Due to its important role in glucose metabolism, GLP1 and its derivatives have been recognized as drug candidates for the treatment of type 2 diabetes.44 However, GLP1 is rather poor in crossing the intestinal epithelium and extremely susceptible to enzymatic degradation. In a 2012 study, a silica-based (silica particles) and Eudragit (polymer to enable delivery) pH-sensitive nanomatrix (~300 nm) was prepared in an attempt to provide a strategy for oral delivery of the peptide prodrug. In these experiments, male Sprague Dawley rats (~200 g) were orally administered with the constructs at a dose of 1 mg/kg. In a subset of the trials, the hypoglycemic efficacy of the formulation was investigated in a group of animals receiving 2 g/kg glucose intraperitoneally after oral exposure to the NPs. The data showed that oral administration of the NPs had a significant hypoglycemic effect that was comparable to the same dose administered via the intraperitoneal route (77% in comparison with the intraperitoneal GLP1 group). Next, it was demonstrated that the bioavailability of the oral nanomatrix in comparison to the intraperitoneally injected GLP1 was 36%. Finally, toxicological assessment of oral administration of NPs revealed no histological abnormalities in the stomach, duodenum, jejunum, ileum, cecum, or colon. This seems to suggest no associated acute GIT injuries or morphology changes following NP exposure at the administered dose. Overall, the study provided evidence of the manufacture of a novel pH-sensitive (important for drug release) GLP1 nanomatrix with high hypoglycemic efficacy and very little toxicity, which could prove beneficial as an oral antidiabetic agent for the treatment of type 2 diabetes mellitus.45
Exendin 4 is an incretin (hormone that stimulates insulin production) analog found in lizard saliva and belongs to a new class of antidiabetic drugs. Exenatide is a synthetic version of exendin 4 that mimics the glycoregulatory activity of the mammalian hormone GLP1.46 To date, the only route of administration is subcutaneous injection. Oral delivery of exenatide would clearly be beneficial to diabetic patients, as it would improve patient compliance and alleviate pain and discomfort from repeated dosing. In a recent study, goblet-cell-targeting sodium triphosphate and CS NPs (~150 nm) were manufactured to encapsulate exenatide. In these experiments, two animal models were utilized. First, a male Sprague Dawley rat model (~250 g, 50 μg/kg) was used to analyze the pharmacokinetic profile of oral exposure to the drug. Additionally, a diabetic male mouse model (30 μg/kg) was utilized to assess the pharmacological effects of the exenatide-loaded NPs. The data demonstrated that the oral administration of modified CS NPs resulted in maximum plasma levels of the drug – 2,238±156 pg·h/mL with relative bioavailability of 6.5% – 1.7-fold higher than unmodified CS NPs. Moreover, in vivo fluorescence imaging of fluorescein isothiocyanate-labeled exenatide NPs at 30 minutes and 1, 2, or 4 hours showed significant accumulation of the particles in the stomach, small intestine, and liver. Finally, the pharmacological effects of the drug-loaded NPs displayed comparatively high hypoglycemic effects at 30 minutes and 1 hour postadministration. The data suggested that the CS NPs could prove to be highly beneficial in type 2 diabetes patients.47
In a recent study, polylactic acid (PLA)-b-Pluronic (P85)-b-PLA vesicles (~180 nm) were developed as a novel carrier for oral insulin delivery. In these experiments, male Kunming mice (~25 g) were fasted for 15 hours before administration of three intraperitoneal injections of alloxan to induce diabetes. The diabetic mice were exposed to insulin vesicles at doses of 50, 100, and 200 IU/kg. For comparison, free insulin (5 IU/kg) was injected subcutaneously into the diabetic mice. Oral administration of insulin-loaded NPs resulted in minimum blood glucose concentration after 2.5 hours, which was 15% of the initial glucose level. Furthermore, the blood glucose concentration increased slowly to 32% of initial blood concentrations after 10.5 hours and was maintained for an additional 14 hours (32.5% of initial blood glucose concentration at 24.5 hours). In contrast, the minimum glucose concentration (11% of initial glucose level) was observed 2.5 hours after subcutaneous injection of free insulin. This indicated that the hypoglycemic effect for the oral administration of vesicles was maintained over a longer time than for the standard subcutaneous injection of free insulin.48
Neoplasms
Oral chemotherapy is an attractive proposal for many reasons, one of the most important being the avoidance of injections and improved patient compliance and quality of life. In addition, it allows for prolonged exposure of anticancer drugs to cancerous cells by maintaining effective plasma drug concentration, thus increasing drug efficacy and reducing adverse effects, as well as facilitating chronic treatment regimens. However, oral delivery of anticancer drugs is extremely challenging, due to GIT physiological barriers: presystemic metabolism, low aqueous solubility, and poor intestinal membrane permeability.
Docetaxel (Dtx) is a potent anticancer drug utilized in the treatment of breast cancer, non-small-cell lung cancer, prostate cancer, and gastric adenocarcinomas.49 Currently, an intravenous formulation of Dtx is commercially available. However, it contains high concentrations of Tween 80, which has been associated with severe hypersensitivity reactions.50 Oral chemotherapy would have some apparent advantages, including offering a noninvasive route of administration and being considerably more cost-effective. In addition, oral administration of anticancer drugs might provide a prolonged systemic exposure profile with smaller fluctuations in drug concentrations, which may lead to lower toxicity and improved efficacy. In a recent study, Tween 80 or D-α-tocopheryl PEG1,000 succinate (TPGS 1000) surface-modified tristearin liposomes (~200 nm) were prepared and evaluated for their pertinence as oral delivery systems for Dtx in male Sprague Dawley rats. The animals were exposed to the liposomes via oral gavage (20 mg/kg). The data demonstrated the intestinal absorption and relative oral bioavailability of Dtx was significantly improved in TPGS-emulsified liposomes, in all probability due to better inhibition of drug efflux. Next, the toxic potential (pathological alterations) of Dtx-loaded liposome exposure was evaluated in the intestinal mucosa of the exposed animals. The investigators did not observe any evidence of tissue damage in the sections of the jejunum examined.51
As touched upon, Dtx is considered one of the most important antitumor drugs in clinical use. However, similar to numerous other drugs discussed in this review, it has very poor solubility in water. To address this and other related issues associated with the use of the commercially available formulation (currently administered via the intravenous route) in a 2012 study, Dtx-loaded lecithin NPs were manufactured (~360 nm). In this set of trials, male Sprague Dawley rats (~250 g) were orally exposed to 20 mg/kg of the drug-loaded NP formulation for up to 12 hours. The maximum plasma concentration of drug from the NPs was fourfold higher than that of the Dtx solution. Additionally, maximum plasma concentration was reached at later time points for the NP formulation compared to the free drug, suggesting a sustained-release profile. The authors postulated that the increased oral bioavailability of Dtx in the NPs might have been attributable to several factors. First, the uptake of NPs may have been enhanced by the relatively small particles, particularly in gut-associated lymphoid tissue, whereby they could potentially have bypassed the liver first-pass metabolism. Second, the sustained-release property of the NPs increased the circulation time of the drug, which prolonged Dtx residence in systemic circulation. Third, encapsulation of Dtx into the NPs might have protected the drug from degradation. Finally, the lecithin lipids might have augmented permeation of the prodrug through the intestinal membrane.52
To improve the oral bioavailability of doxorubicin (Dox; a class of chemotherapy drug [anthracyclines] that functions by slowing or stopping growth of cancer cells by blocking type 2 topoisomerase 2), polymeric nanomicelles composed of hyaluronic acid-linked β-cyclodextrin were developed in a recent study. The micelles were then coated with CS to increase the biocompatibility and biodegradability of the construct. In most physiological fluids, CS is positively charged, which can elicit adhesion to GIT mucus. Particle size was measured by transmission electron microscopy (TEM) and dynamic light scattering and was around 250 nm, with particles stable up to 30 days. Pharmacokinetic investigation of the micelles in a male Wistar rat model (~250 g) following oral administration of the composites containing 40 mg/kg Dox demonstrated that CS-coated nanomicelles increased maximum plasma concentration up to fourfold. Furthermore, CS-coated micelles were retained longer in the GIT (~3 hours in comparison to ~2 hour for Dox solution). This study clearly demonstrated the benefits of the use of the nanomicelles in terms of biodegradability, biocompatibility, and bioadhesion as a means of improving the oral bioavailability of anticancer medication.53
Paclitaxel (Ptx) is highly efficacious drug in the treatment of non-small-cell lung cancer, breast cancer, ovarian carcinoma, and head and neck cancers. Despite its efficacy, the traditional methodologies for drug delivery are associated with severe side effects, such as hypersensitivity, nephrotoxicity, and neurotoxicity.54 In a recent investigation, Ptx-stabilized Pluronic F127 nanocrystals (~150 nm) were used in an attempt to reduce the side effects associated with traditional drug administration. In these studies, 4T1 cells (murine breast cancer) were subcutaneously injected into the stomachs of BALB/c mice (~20 g), with tumor volumes of the xenograft mice being monitored every other day. In the oral administration studies, the nanocrystals were administered on a daily gavage regime. First, the toxicity of the formulations was assessed in intravenously injected animals. The administration of Ptx in Cremophor (commercially available formulation) resulted in six of six mice dying following exposure to 30 mg/kg. However, no mortality was observed up to doses of 60 mg/kg for the Ptx nanocrystals. Following the oral administration of the drug, no antitumor activity was observed in mice treated with the commercially available Ptx suspension. Nevertheless, oral administration of the nanocrystals significantly inhibited tumor growth. Unsurprisingly, oral administration of the drug resulted in less efficacy compared to the intravenous route. Overall, the data suggest that the nanocrystals achieved higher efficiency of drug loading, with few side/toxic effects in comparison to the currently commercially available Cremophor-loaded formulations following oral exposure.55
Unfortunately, most anticancer drugs have very poor oral bioavailability, as is the case for Ptx. In a 2010 study, vitamin E TPGS 1000-emulsified PLGA NPs were manufactured for oral delivery of Ptx. The materials characterized were spherical and around 300 nm in diameter. First, in a series of in vitro cytotoxicity experiments, MCF7 human breast cancer cells were exposed to the Ptx-loaded NP suspension or a commercially available formulation at drug concentrations of 0.025–50 mg/mL for 24–72 hours. The data demonstrated that compared with the commercially available formulation of the same dose, Ptx in NPs was more effective for antiproliferative activity on MCF7 cells (though these differences were relatively small). Next, the in vivo pharmacokinetics of Ptx in either its current clinical formulation or in NPs were investigated in male Sprague Dawley rats (~250 g) at a dose of 10 mg/kg of the prodrug. The authors showed that the drug concentration in plasma for the commercially available formulation was rather low, with an oral bioavailability of only 2.4%. In contrast, for the drug in the TPGS 1000-emulsified NPs, blood concentration was significantly increased to 8,510 ng·h/mL, which was around tenfold higher (24%). Finally, the sustainable therapeutic time for the NPs was dramatically increased to 88 hours, around 13 times that of the commercially available formulation. Overall, the data demonstrated the formulation of emulsified PLGA NPs was a promising approach for oral Ptx administration. However, it should be stated that intravenous injections are the primary administration route for the commercially available formulation in current anticancer strategies, and hence direct comparison between the two delivery formulations is not necessarily appropriate.56
In a 2012 study, a soy phosphatidylcholine–glycerol dioleate-based liquid crystalline NP system (~85 nm) was developed as an oral delivery platform for Ptx. In vivo pharmacokinetics of the drug formulations were investigated in Sprague Dawley rats after oral administration of a commercially available drug and loaded NPs at a Ptx dose of 5 mg/kg for up to 24 hours. The data showed that plasma concentrations of the drug were 2.1-fold higher for the NPs compared to the commercially available Ptx. Furthermore, absolute bioavailability of Ptx delivered by the NP carrier and the traditional formulation was calculated as 13% and 6.5%, respectively. The findings of this study suggested that the NP-delivery system may be an auspicious candidate for improving the oral bioavailability of the prodrug.57
N3-O-toluyl-fluorouracil (TFU), a prodrug composition of 5FU, has demonstrated promising in vitro and in vivo anticancer efficacy.58 However, the drug is extremely hydrophobic and has poor bioavailability after oral administration. To address these issues in a recent study, TFU-loaded cationic liposomes were utilized to enhance the oral bioavailability of the drug. The liposomes were manufactured from soy lecithin and shown to be around 180 nm. In these experiments, the biodistribution of the formulations was investigated in a Kunming mouse model (~28 g). Animals were exposed to 50 mg/kg TFU liposomes via the oral route. The authors showed that the GIT bioavailability of cationic TFU liposomes was doubled compared to the drug in suspension. The enhanced monodispersion of the liposomes in the GIT was suggested as the primary contributing factor to the increased levels of drugs in the circulation.59
A series of self-assembled nanostructures with prodrug conjugates based on commercially available capecitabine (N-pentyloxycarbonyl-50-deoxy-5-fluorocytidine) were developed by substituting the pentyl group at the N4 position with three different hydrophobic alkyl chains: palmityl (5FCPal; 700 nm), oleyl (5FCOle; 255 nm), and phytanyl (5FCPhy; 164 nm). These substitutions were intended to enhance the efficacy of the commercially available formulation. The efficacy of the new formulations was evaluated in female BALB/c mice (~20 g) injected with 4T1 cells into the third mammary fat pad. Daily drug treatment was commenced at 3 days postinoculation. Animals were exposed to 1 mmol of the nanoconstructs or the commercially available drug for 6, 10, 13, or 17 days via oral gavage and the size of developed tumors measured. The data demonstrated that after 17 days, the 5FCOle-treatment group possessed significantly smaller tumors than all other treatment groups (around a fifth the size of the tumors in both the 5FCPhy- and capecitabine-treatment groups and a tenth the size of tumors in the untreated control group). Interestingly, spleens in the 5FCOle-treatment group were normal in size, while spleens from the other treatment groups were significantly enlarged. This is indicative of a strong inflammatory response induced by the tumor. The data from this study provided convincing evidence for the significant potential of 5FCOle-modified NPs delivered orally in the treatment of progressive breast cancer.60
The Notch signaling pathway is crucially important in the regulation of the development and tissue homeostasis of stem cells and known to be deregulated in inflammatory intestinal disease and colon cancer. The majority of clinical studies aiming to inhibit Notch have principally been focused on the use of γ-secretase inhibitors, which impede the proteolytic processing and activation of the Notch receptor.61 In a recent study, surface-modified silica NPs were manufactured and utilized for the specific delivery of N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT; γ-secretase inhibitor) to overcome challenges in oral administration caused by drug degradation at low pH and reduced permeability through the mucus layers. In the study, silica NPs were surface modified with polyethylenimine (PEI), PEG, and folic acid (FA) (~400 nm) with the aim of better facilitating binding to negatively charged cells and reducing uptake by the reticuloendothelial system. First, in vitro cytocompatibility of all produced NPs was assessed in colon cancer Caco2 cells for 48 hours and demonstrated particles to be atoxic up to 50 μg/mL. Oral exposure to the NPs on 3 consecutive days (100 mg/kg) in male FVB/N mice (~25 g) was used to investigate uptake in the small intestine and colon. The data showed that numbers of goblet cells in the colons of mice given DAPT/FA-PEG-PEI mesoporous silica NPs were significantly higher than DAPT alone or the empty NPs. Modified NPs were also able to increase the expression of MUC2 (gene encoding a member of the mucin protein family) in the colon. Interestingly, a similar trend of improvement and goblet-cell hyperplasia with PEG-PEI-coated NPs was seen in the small intestine. These data were further supported by an increased level of stool hydration in mice fed DAPT alone or DAPT/FA-PEG-PEI mesoporous silica NPs (indicative of Notch-inhibition-induced cell switch from water-absorbing colonocytes to mucus-producing goblet cells). The authors concluded that the modified NPs were efficient carriers for drug delivery and targeted Notch inhibition following oral administration (particularly in the colon).62
Probucol is a routinely utilized drug with antioxidant and anti-inflammatory properties. It has also been reported that the drug can reduce the activity of MMP9 and decrease the expression of MCP1.63 These inflammatory factors are essential in certain features of cancer metastasis. In a recent study, a probucol nanoassembly was manufactured to increase the bioavailability of the drug (probucol is highly hydrophobic). The NP assembly (termed DNP) comprised probucol and Triton X-100 and was prepared via the self-assembly technique (~30 nm). The efficacy of the DNP formulation following oral administration was assessed in male Sprague Dawley rats (~200 g). The animals were administered 200 mg/kg probucol in the DNP formulation or the free drug. Additionally, the distribution of fluorescently labeled NPs was quantified in the intestinal tract using an ex vivo imaging system. After oral administration, fluorescence intensity was highest in the duodenum, jejunum, and ileum. At each investigated time point, the probucol concentration in different intestinal segments was significantly higher for the DNP than the free prodrug suspension (4 hours post-DNP exposure, drug concentrations in the duodenum, jejunum, and ileum had increased by factors of 220, 195, and 233 respectively). Furthermore, TEM analysis showed DNP in endosomes or lysosomes of the enterocytes lining the intestinal villi. Next, oral pharmacokinetic data showed that the plasma concentration of probucol from DNP was much higher (~13-fold) than the free probucol suspension. In a subsequent experiment, the biodistribution of DNP in major organs was determined in female BALB/c mice (~20 g). DNP or the probucol suspension was administered by oral gavage (200 mg/kg) and mice autopsied 4 hours postadministration. Ex vivo imaging of the major organs showed that DNP was distributed mainly in the liver and lungs, with concentrations much higher compared to the free probucol suspension. A series of in vitro experiments demonstrated that the invasion activity of 4T1 cells was inhibited by probucol and DNP, with inhibitory rates of 59% and 32%, respectively. Additionally, no cytotoxicity was detected in 4T1 cells following 24-hour treatment of DNP at concentrations of 0.004–4 μg/mL. Subsequently, the focus was placed on the establishment of the effectiveness of DNP in inhibition of lung metastasis in a rodent orthotopic breast cancer model. DNP treatment significantly improved the survival rate of mice relative to free probucol administration. Moreover, considerably more metastatic nodules were noted in the lungs of animals in the free probucol and control treatment groups, but rarely observed in the DNP-treated mice. The average number of visually detected metastatic nodules per lung was 12 in the control and free probucol groups, yet this was reduced to one in the DNP-treated animals. Finally, the in vivo toxic potential of the nanoassemblies was investigated in BALB/c mice administered orally (200 mg/kg per day) with the different formulations daily for 20 days. At this juncture, all major organs were removed for histological evaluation. The authors did not observe any significant histopathological alterations in DNP-treated animals compared to the controls. Therefore, it was concluded that the DNP presented good biocompatibility at the doses and time points investigated. Overall, the study provided compelling evidence for the efficacy of the DNP formulation as a probucol-delivery platform and could be extremely valuable as a therapeutic strategy.64
DIM-D (1,1-bis[3′-indolyl]–1-[p-chlorophenyl] methane) is a novel semisynthetic derivative of diindolylmethane, a potent anticancer molecule that induces apoptosis, triggers cell-cycle arrest, and inhibits cell hyperproliferation in skin, bladder, and breast cancer cell lines. Notably, DIM-D obstructs ultraviolet B-induced generation of ROS. Further, the drug protects against oxidative damage evident by DNA damage, lipid peroxidation, and protein carbonylation, which usually precedes mutagenesis and photocarcinogenesis within ultraviolet B-exposed skin cells. However, DIM derivatives are principally impermeable to biological membranes. In a recent study, the oral route of delivery was explored as a potential avenue for skin deposition of DIM-D following exposure of three lipid-based delivery systems. These consisted of: formulation 1, DIM-D (10%), Captex oil (40%), Capmul MCM (20%), and Kolliphor HS15 (40%), ~40 nm; formulation 2, DIM-D (10%) diacylglycerol (Enova) oil (40%), Cremophor RH40 (25%), Labrasol (15%), and vitamin E TPGS 1000 (10%), ~84 nm; and formulation 3, DIM-D (0.1%), Compritol (3.75%), Miglyol (1.25%), Tween 80 (3.5%), and Pluronic F68 (1.5%), ~115 nm. In these experiments, male Sprague Dawley rats (~300 g) were administered doses of 60 mg/kg orally of different formulations. Exposure to all formulations resulted in maximum blood concentrations of DIM-D at 2 hours postadministration, with levels highest for formulation 2 (6.2 μg/mL), which was significantly higher than formulation 1 (0.6 μg/mL) and formulation 3 (0.3 μg/mL). Formulations 2 and 1 were 51.9- and 4.6-fold higher than the free drug solution, respectively. Next, the data demonstrated that formulation 1 resulted in the most significant deposition of DIM-D into the skin; however, all three formulations enhanced the amount of DIM-D deposited in the skin. Finally, in vivo toxicity assessment of the different DIM-D formulations intended for prolonged usage was undertaken. In these experiments, SKH1 hairless mice were orally exposed at a dose of 100 mg/kg for 1 month, with hearts, lungs, kidneys, and livers harvested and subjected to histological analysis. Examination of each organ showed no histological changes between DIM-D-treated tissue and the negative control. In summary, the data from the study demonstrated effective enhanced oral bioavailability and skin deposition of DIM-D in the nanoformulations compared to the free drug solution.65
Oxaliplatin (Oxa) is a third-generation platinum antitumor compound, which exerts its effects by interfering with DNA replication and transcription machinery through nuclear DNA-adduct formation. Clinically, Oxa is often utilized as a first-line chemotherapy strategy in combination with 5FU for treatment of advanced colorectal cancer.66 Presently, both drugs are administered by the intravenous route and exhibit poor oral bioavailability. However, a recent study aimed to develop an oral delivery system for Oxa and 5FU. This was achieved by constructing an ion-pairing complex of Oxa with a deoxycholic acid derivative (Nα-deoxycholyl-L-lysyl-methylester [DCK], a permeation enhancer) before preparation of a multiple water-in-oil-in-water nanoemulsion incorporating Oxa-DCK and 5FU. The construct was characterized and shown to be spherical and <50 nm in size. In the first experiment, male Sprague Dawley rats (~200 g) were orally exposed to 10 mg/kg of the nanoemulsion or appropriate controls. The authors showed that intestinal absorption of Oxa improved significantly after being incorporated in the nanoemulsion, resulting in a 1.66-fold increase in maximum plasma concentrations. Furthermore, oral absorption of Oxa was significantly enhanced by complex formation with DCK, and its maximum plasma concentrations were increased by 306% compared to the free drug. Next, CT26 cells (mouse colon fibroblasts) were subcutaneously grafted into the flanks of male BALB/c mice (~20 g). On day 14, when tumors became evident, the mice were orally exposed to the nanoemulsion formulation daily (10 mg/kg per animal) for 18 days. After the treatment, tumor mass in the nanoemulsion-treated group was reduced by 43% compared to the control animals. Finally, to determine the effect of oral combination treatment using the nanoemulsion (Oxa-DCK and 5FU)-delivery system on tumor-growth inhibition, proliferating-cell density and apoptosis in tissues were investigated. It was demonstrated that the nanoemulsion treatment resulted in a profound decrease in proliferating-cell density, as well as increased tumor apoptosis. The authors concluded that the oral administration of the nanoemulsion comprising Oxa-DCK and 5FU produced a synergistic effect on tumor-growth inhibition due to improved oral bioavailability of Oxa and 5FU.66
In a recent study, a nanomicelle (corn oil, Labrasol, and Transcutol-P) drug-delivery carrier of tamoxifen and naringenin (flavonoid with anticancer and anti-inflammatory properties) was developed. Loaded micelles were characterized and shown to be around 70 nm. Female Wistar rats (~250 g) were orally administered with the nanocarriers containing equivalent doses of tamoxifen at 10 mg/kg and naringenin 20 mg/kg body weight for up to 72 hours. In a concurrent lymphatic uptake study, the exposed animals were anesthetized before cannulation of the mesenteric lymph duct. Additionally, the antitumor potential of the formulation was assessed in a 7,12-dimethylbenz[a]-anthracene-induced female rat breast cancer model at a dose of 45 mg/kg for 3 consecutive weeks at weekly intervals. The drug regimen was started after 10 weeks of 7,12-dimethylbenz[a]-anthracene exposure, followed by repeated oral dosing (once every 3 days) of the nanoemulsion. Furthermore, levels of ALT and AST in plasma, as well as hepatic glutathione and thiobarbituric acid-reactive substance alterations, were measured as a sign of drug-induced hepatotoxicity. The data showed that exposure to the micelles resulted in significant augmentation in drug maximum plasma concentrations after a single oral dose (11.5-fold increase). Additionally, the time that the drug was present at maximum concentration in serum was reduced by 0.5-fold. Next, the data demonstrated the maximum lymphatic uptake for the NPs was 121.5 μg/mL and occurred in the first 3 hours, which was 3.8-fold higher compared to the free drug suspension. Examination of liver damage revealed the loaded nanoemulsion to be atoxic at the doses administered. Finally, the in vivo anticancer activity of the nanoemulsion showed a reduction in tumor size of 85% compared to the free tamoxifen–naringenin suspension, which reduced tumor size by only 30%.67
20(S)-protopanaxadiol (PPD) is an aglycone metabolite that has exhibited powerful pleiotropic anticancer effects. The drug is currently undergoing phase I clinical trials in China.68 However, similar to several other anticancer agents, the compound has low oral absorption and is metabolized extensively in the liver. In a recent study, PPD was encapsulated in cubic NPs (~150 nm) prepared by fragmentation of glyceryl monooleate and poloxamer 407. The NPs were then linked to piperine (alkaloid with the potential to affect the bioavailability of other compounds) to generate a novel drug-delivery system. In these experiments, male Sprague Dawley rats (~200 g) were administered with oral doses equivalent to 2 mg/kg of PPD for up to 12 hours. The data demonstrated that the maximum concentration of free PPD after oral administration was 73.45 ng/ml and the time to maximum concentration was about 85 minutes. However, the maximum plasma concentration after oral administration of PPD cubosomes loaded with piperine was 142 ng/mL and the time to reach the maximum concentration was 110 minutes (oral bioavailability of 248% compared to the free drug). The data suggested that the increased bioavailability of PPD-cubosomes loaded with piperine was due to an increase in absorption and inhibition of PPD-NP metabolism, rather than improved release of PPD. Cubic NPs may prove to be a promising oral carrier for anticancer drugs with poor oral absorption and means of reducing hepatic metabolism.68
The MDM2 oncogene plays a critical role in the development and progression of cancer through p53-dependent and p53-independent pathways.69 Ginseng is a natural product with a long history of chemopreventive use. The anticancer properties of ginseng have largely been attributed to the presence of saponins, which are termed “ginsenosides”. However, similar to other natural compounds, the therapeutic applications of these compounds are extremely limited, due to low aqueous solubility and instability under harsh GIT conditions, extensive metabolism, and rapid elimination. In a recent study, a novel ginsenoside, 25-OCH3-PPD (GS25; functions by inhibition of MDM2) isolated from Panax notoginseng was successfully encapsulated into PEG-PLGA NPs (~45 nm). The in vivo pharmacokinetics of GS25 and GS25 NPs were first assessed in male CD1 mice (~20 g). Additionally, PC3 human prostate cancer cells were subcutaneously injected into the left inguinal area of male CD1 mice. All animals were monitored for activity, physical condition, body weight, and tumor growth for 3 weeks. NPs were administered by oral gavage at a dose of 20 or 100 mg/kg/day 5 days a week for 4 weeks, while GS25 in PEG/ethanol/saline was administered at a similar dose for the same period. The data showed that compared to intravenous injection of 20 mg/kg GS25, oral administration of GS25 (100 mg/kg) caused a very low peak value of drug concentration (0.9 μg/mL), due to low bioavailability (~15%). The NP encapsulation of GS25 resulted in dramatically altered plasma concentration times (20 and 100 mg/kg oral doses of GS25 NPs increased to 21.4 and 93.1 h·μg/mL). Compared to oral administration of GS25 at a dose of 100 mg/kg, drug concentrations of 20 and 100 mg/kg GS25 NPs were three- and ninefold higher, respectively. On a similar theme, compared to free GS25, GS25 NP half-life was extended to >7 hours. Furthermore, tissue biokinetic data showed that drug accumulation was increased by the nanodelivery system in the liver, lungs, kidneys, spleen, heart, brain, and pancreas. To investigate the effects of NP encapsulation on tumor uptake further, pharmacokinetic studies of GS25 and NP formulation were performed in nude mice bearing PC3 xenograft tumors. Consistently with the pharmacokinetic profiles in CD1 mice, the encapsulation of GS25 increased plasma drug concentrations. More importantly, there was a substantial increase in the tumor uptake of the NPs compared to GS25 alone. To determine the optimal therapeutic doses of GS25 and GS25 NPs, initial maximum-tolerated-dose studies were performed in CD1 mice. The data showed no toxicity to the mice at a dose of up to 400 mg/kg. Further, histological examination of various tissue samples from treated animals showed no pathological alterations. Next, the anticancer efficacy of GS25 and GS25 NPs was evaluated using the PC3 xenograft model of human prostate cancer. The authors demonstrated that 100 mg/kg GS25 had moderate effects on tumor growth (4-week oral treatment led to approximately 41% inhibition of PC3 tumor growth). However, treatment for the same period using 20 and 100 mg/kg GS25 NPs inhibited the growth of PC3 xenograft tumors by approximately 75% and 87% respectively. Finally, expression levels of MDM2 and other apoptosis-related proteins were examined in vivo. It was noted that NP encapsulation enhanced the effects of GS25 on the expression of MDM2, poly-ADP-ribose polymerase, and Bax. In summary, the study provided compelling evidence for an efficient oral nanodelivery system as an MDM2 inhibitor with potential practical applications in cancer therapy.70
Harmine is a β-carboline compound that has been attributed with antitumor properties. However, the compound has very poor oral bioavailability. In a strategy to improve the suitability of the compound in medical applications, the drug was encapsulated in N-trimethyl CS-coated liposomes (soybean phosphatidylcholine–cholesterol 20:5 w:w; ~170 nm). In this study, Sprague Dawley rats (~250 g) were exposed to the liposomes at doses of 60 mg/kg. The data showed that the elimination rate of free suspension of the compound was faster than that of liposome formulation (by up to 4 hours). In addition, the bioavailability of harmine through oral administration was enhanced by 60% in the nanoformulation compared to the free drug. One possible explanation for enhancement of the bioavailability of encapsulated drugs is the opening of tight junctions in intestinal epithelial cells caused by the N-trimethyl CS coating.71
Infectious and parasitic diseases
Luteolin is believed to have antimicrobial, anti-inflammatory and antimutagenic properties. However, despite its beneficial biological activities, the flavone has low oral bioavailability due to very poor water solubility. In a recent study, glycerol monostearate and Tween 80 luteolin-loaded NPs (~50–120 nm) were manufactured to attempt to overcome this issue. Male Sprague Dawley rats were orally exposed to the NPs at a dose of 20 mg/kg. The data demonstrated that the drug encapsulated in the NP formulation had significantly higher solubility and bioavailability in plasma compared to the native drug suspensions. Moreover, the time taken for clearance of drug incorporated within the NPs was significantly decreased.72
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most commonly prescribed globally for their antipyretic, analgesic, and anti-inflammatory effects. However, it has been reported that the use of NSAIDs may cause severe adverse effects, including the development of ulcers, bleeding, and perforation in the GIT. Based on the principle that antioxidant compounds and free-radical scavengers heal NSAID-induced inflammation, in a recent study a redox NP (RNPO) formulation with the ROS-scavenging potential of nitroxide radicals was developed. The RNPO was composed of a core micelle (~40 nm), prepared by the self-assembly of methoxy-PEG-poly[4-(2,2,6,6-tetramethylpiperidine-1-oxyl)oxymethylstyrene] and an amphiphilic block polymer possessing nitroxide radicals as side chains of the hydrophobic segment. The antioxidant potential of the NPs was assessed in a male ICR (~30 g) mice model in which the animals were pretreated (oral gavage) with 13.34 mg/mL of NPs 1 hour before induction of small-intestine inflammation following the administration of indomethacin (Ind). The data demonstrated significant histological damage in both the jejunum and ileum of mice treated with Ind, manifested predominantly as focal and upper villous necrosis. However, histology of small intestines from RNPO-treated mice showed almost no damage and appeared similar to the control mice. Unsurprisingly MPO activity was significantly increased in the small intestines of Ind-treated mice, suggesting increased neutrophil invasion in the ileum region. MPO levels decreased significantly in the jejuna and the ilea of mice pretreated with RNPO. Overall, the authors demonstrated that RNPO pretreatment efficiently scavenged superoxide anions in an Ind-induced small-intestine-inflammation model. As such, it was suggested that oral administration of RNPO prior to oral exposure to NSAIDs might be beneficial in the treatment of small-intestine injury in patients on regular NSAID medication.73
AIDS is traditionally treated with nucleoside reverse-transcriptase inhibitors (NRTIs); however, this antiretroviral therapy has limited bioavailability and biodistribution (inherent hydrophilic nature, hence their effectiveness in accessing virus sanctuaries is limited).74 To overcome these limitations, in a 2013 study NRTIs were loaded into liposome nanocarriers to modulate drug biodistribution. The authors investigated the antiviral efficacy of the nucleoside analogs of dideoxycytidine and didanosine in nanoassemblies (1-hydroxybenzotriazole hydrate, O-(7-azabenzotriazol-1-yl)-N,N,N,N′-tetramethyluronium hexafluoroborate, dideoxycytidine, and diisopropylethylamine; 100–300 nm) on HIV. These nanoassemblies were further modulated with PEG coupled to either cholesterol or squalene. Wistar rats (~200 g) were treated with the different formulations at a dose of 45 mg/kg via oral gavage. The nanoformulation showed greater absorption and higher diffusion into the liver, spleen, bone marrow, thymus, and brain (all tissue associated with HIV infection). Furthermore, nanoformulation treatment of infected blood mononuclear cells in vitro resulted in enhanced antiviral efficacy compared to the parent NRTIs, with a two- to threefold decrease in 50% effective dose and a near doubling in drug-selectivity-index score.74
Combination pharmacotherapy is commonly administered for treatment of AIDS. However, it is often difficult simultaneously to deliver all active components of the drugs to targeted cells and maintain an effective concentration using current delivery methods.75 Nanocarriers could potentially entrap different types of drugs and deliver the pharmaceutics to desired destinations. In a recent study, this strategy was utilized for highly active antiretroviral therapy via the oral route. A bolaamphiphilic prodrug termed ZPDD was prepared via the combination of azidothymidine (Azt) and didanosine (Ddi). The nanoscale self-assemblies (~174 nm) were then used to target macrophages, both in vitro and in vivo. Male Kunming mice (~25 g) were exposed to ZPDD self-assemblies at 100 mg/kg per animal. Furthermore, the anti-HIV action of ZPDD self-assemblies was explored in HIV1-infected MT4 (human T-cell leukemia-derived lymphocyte) cells. It was noted that ZPDD had high antiviral activity with EC50 of 5 nM, equal to the EC50 of Azt. Oral administration of ZPDD self-assemblies resulted in significant dose (up to 20 nmol/g) accumulation in the spleen, lymph nodes, thymus, liver, and lungs, with high macrophage accumulation. Interestingly, the molar concentration of Azt was much higher than that of ZPDD in the targeted tissue. The authors attributed these findings to the rapid degradation of the ZPDD self-assemblies. Finally, the study demonstrated that the bioavailability of ZPDD was 31% in comparison to the same dose administered via the intravenous route. The relatively low oral bioavailability may be related to the unstable properties of the assemblies in the stomach, enzyme damage, or insufficient absorption into the bloodstream. Overall, the data demonstrated that the self-assembly could deliver two types of drugs simultaneously to targeted tissue at a relatively high dose. However, it had poor bioavailability following oral exposure.75
Efavirenz (Efv) is considered first-line therapy for new cases of HIV. However, the drug is expensive, and this is a genuine issue in developing countries. In a very interesting recent study, emulsion-templated freeze-drying was utilized for the manufacture of solid Efv NPs with the aim of the development of a cheaper, more effective oral anti-HIV formulation. After an initial screening of 49 different stabilizers, one formulation was identified as the most effective in terms of low cytotoxicity (epithelial colorectal cells) and greatest cellular permeability (70:20:10 weight ratio of Efv, PVA, and α-tocopherol–PEG succinate). Particles were characterized extensively, and confirmed to be around 300 nm in size. In these experiments, male Wistar rats were exposed to 10 mg/kg of the traditional or nanoformulated Efv, with blood samples collected at 15, 30, 45, 60, 120, 180, 240, and 300 minutes postexposure. The data demonstrated that at an equivalent dose, significantly higher plasma maximum concentration was achieved for the nanoformulation compared to the traditional medicine: maximum concentration (478 vs 126 ng/L) and minimum concentration (379 vs 80 ng/L). In summary, the data showed that the identified nanoformulation might prove to be a viable option for the scalable manufacture of cheaper Efv.76
One of the main impediments to the effective treatment of HIV is the numerous viral sanctuaries, ie, lymph nodes or the intestine, which are often inaccessible to traditional anti-HIV compounds (ie, the prodrug Ddi). In an attempt to address this issue in a 2011 study, a multilamellar dipalmitoylphosphatidylcholine vesicles were utilized to encapsulate Ddi (~100 nm). For long-term storage and subsequent experiments, the vesicles were freeze-dried. First, phytohemagglutinin P-activated peripheral blood mononuclear cells were treated with increasing concentrations of loaded vesicles. Following a 1-hour incubation, cells were infected with an HIV1 LAI strain. Viral replication was measured by quantifying reverse-transcriptase activity. In parallel, the cytotoxicity of the compounds was evaluated in uninfected phytohemagglutinin P-activated peripheral blood mononuclear cells. The data demonstrated that the vesicles were effective against the HIV1 LAI with an ED50 of 9.9 μM and had relatively low cytotoxicity (LD50 >100 μM) compared to the unencapsulated drug with ED50 of 1.2 μM and LD50 of 40 μM. Next, Wistar rats (~200 g) were exposed to 45 mg Ddi/kg via the oral route. Pharmacokinetic and biodistribution data showed that at 4 and 8 hours postadministration, drug plasma concentrations were six- and fivefold higher, respectively, for the nanoformulation compared to the free molecule. The drug-loaded liposomes presented a higher terminal elimination half-life (2.37 hours) than the free drug (0.76 hours). Next, evaluation of drug biodistribution at 24 hours postexposure with radiolabeled loaded liposomes revealed a higher distribution of the liposomal formulation (most visible in the intestine, spleen, and testicles). Moreover, radioactivity recovered in plasma was fourfold higher after liposomal administration than the free drug. Finally, urinary excretion of the liposome drug-delivery system was increased compared to that of free drug whereas the feces excretion was very similar for both treatments. In conclusion, the data showed preservation of Ddi encapsulated in the liposomes with enhanced antiviral activity, blood half-life, and intestinal accumulation.77
As mentioned previously, the therapeutic efficacy of a drug administered by the oral route strongly depends on the administered dose, GIT absorption, and its bioavailability. To improve the oral bioavailability of Efv in another recent study, the drug was encapsulated within poly-ε-caprolactone PVA NPs (~250 nm). Oral pharmacokinetics of Efv were assessed in male Wistar rats (~300 g). The animals were administered the drug at equivalent doses of 20 mg Efv/kg via oral gavage. The authors observed that the encapsulation of Efv in the manufactured NP construct significantly increased the oral bioavailability of the drug (4.8-fold) when compared with pure Efv NPs (prepared by employing the same methodology without the addition of the poly-ε-caprolactone).78
Primaquine (Pq) is a widely used antimalarial drug. To date, it is the only available drug for combating the relapsing form of malaria. However, the drug has serious side effects, including nausea, vomiting, methemoglobinemia, leukopenia, leukocytosis, and abdominal cramps.79 In a recent set of trials, Pq-loaded positively charged PVA SLNs (Pq-SLNs; 236 nm) were utilized to enhance drug efficacy and half-life with the aim of reducing toxicity and better patient compliance. The ζ-potential of the SLNs changed from −6.54 mV to 23 mV by binding positively charged CS as a surface modifier. To investigate the efficacy of the drug formulations, Swiss albino mice (~25 g) were infected with blood containing parasitized Plasmodium berghei ANKA. Following 2 hours’ incubation, the test animals were orally exposed with two doses of either Pq-SLNs or free Pq (2 or 5 mg/kg/day). Drug administration was repeated at 24, 48, and 72 hours postinfection. The authors noted that treatment with Pq-SLNs at a dose of 2 mg/kg/day resulted in chemosuppression of 94% in comparison to 72% in mice treated with free Pq at a similar dose. Additionally, it was observed that formulation of Pq in the nanoemulsion had effective antimalarial activity against P. berghei at a 25% lower dose compared with the conventional oral formulation. In summary, the study demonstrated that the SLNs were small enough to allow direct absorption into the GIT. This, coupled with a positive surface charge, might allow for high hepatic absorption. The drug is highly effective against hypnozoites (liver stage of all malaria species), which would explain why the formulation was highly efficacious, even at a reduced dosage.79
Amphotericin B (AmB) is one of the most effective antifungal drugs available for the treatment of systemic fungal infections, such as candidiasis or aspergillosis. Furthermore, it is prescribed for the treatment of visceral leishmaniasis. The traditional intravenous administration of AmB in the current marketed formulations results in high drug levels in target organs, but is also associated with significant accumulation in the kidneys, which is highly problematic, as the drug is a known nephrotoxin. In a recent study, the effects of administration of AmB in an N-palmitoyl-N-monomethyl-N,N-dimethyl-N,N,N-trimethyl-6-O-glycol CS (GCPq) self-assembling NP system via the oral route (as a means of protecting the drug from gastric degradation and selective transportation to target organs, hence diminishing kidney exposure) was investigated. The AmB-GCPq resulted in the solubilization of AmB crystals and the formation of highly stable NPs (35–200 nm). First, the biokinetics of the drug were investigated in a male CD1 mouse model (~30 g). The authors showed that for all formulations (traditional and GCPq), AmB plasma concentrations increased slowly and were sustained for 8 hours postadministration. The drug was mainly tissue-bound and accumulated in the liver, spleen, and lungs. However, AmB-GCPq administration resulted in higher plasma levels when compared to AmB in dextrose (oral bioavailability of AmB-GCPq NPs was 25%). Furthermore, significantly higher levels of AmB were found in the livers of AmB-GCPq-exposed animals. Next, the efficacy of oral administration of the GCPq NPs was investigated in three separate disease states (visceral leishmaniasis, candidiasis, and aspergillosis) using standard models of the disease in different murine models. For all experiments, animals were dosed with 1 or 5 mg/kg of different drug formulations. It was noted that the AmB-GCPq NPs targeted organs of pathology, resulting in effective treatment in preclinical disease models (particularly evident for aspergillosis and systemic candidiasis). The study provided preliminary evidence of the effectiveness of the use of particular NPs for specific targeting of drugs to key organs via the oral route.80
Triptolide (Tp) is a compound extract derived from traditional Chinese medicine. Tp has been documented to possess a wide array of unique bioactive properties, ie, anti-inflammatory, anticystogenesis, and anticancer activities.81 However, the clinical use of Tp has been heavily hampered, due to its high toxicity to the hepatic, renal, digestive, and reproductive systems. The unique pharmacokinetics of Tp signify that it gains fast access to blood circulation and is rapidly metabolized with extensive tissue distribution, which all contribute to its toxic nature. In a 2014 study, two nanoscale delivery systems were utilized to extend the delivery time for the chemical and decrease the fluctuations of Tp in plasma with the aim of potentially reducing the adverse side effects associated with traditional oral delivery formats. The authors used Cremophor RH40, Lipoid E80, and Transcutol HP to formulate a nanostructured lipid carrier composed of solid and liquid lipids termed NLC (~230 nm) and an entire SLN (~180 nm). Firstly, the pharmacokinetics of the loaded carriers were examined in male Sprague Dawley rats (~300 g). The rodents were orally administered the Tp-NLCs, Tp-SLNs, or free Tp at a dose of 544 μg per animal. Time to maximum concentration and half-life of free Tp in plasma were calculated as 12 and 42 minutes, respectively. However, maximum concentration of Tp was delayed to 43 minutes and 27 minutes for Tp-NLCs and Tp-SLNs, respectively. The half-life was very similar for the two nanoformulations, calculated at around 90 minutes. In subsequent subacute-toxicity studies, rats were exposed to free Tp and the two carrier systems at two doses (500 and 650 μg/kg). The drugs were administered daily by oral route for 28 days. Firstly, statistically significant decreases in weight gains were noted with the high doses of free Tp and Tp-SLN in the second week. This was appraised as GIT toxicity of Tp. After the last measurement, the decrease in weight gains in the Tp-NLC group was significant compared to the free Tp group and was considerably smaller than that of the Tp-SLN group at the high dose utilized. Next, levels of ALT and AST in plasma were measured as a sign of liver damage and found to be significantly elevated following exposure to Tp. Exposure to Tp-NLC and Tp-SLN showed no marked elevation at 500 μg/kg. However, AST and ALT levels were both increased in animals exposed to 650 μg/kg Tp-SLNs and were comparable to the Tp controls. In contrast, oral exposure to 650 μg/kg Tp-NLC was not associated with liver damage measured by plasma AST and ALT alterations. Increased serum creatinine and blood urea nitrogen concentrations in the blood can be indicative of renal dysfunction. At the high dose of 650 μg/kg, both Tp- and Tp-SLN-exposed animals had higher creatinine and blood urea nitrogen than the control group, while Tp-NLC exposure showed no significant renal toxicity. Malondialdehyde as a by-product of lipid peroxidation was demonstrated to be elevated following exposure to the free Tp and the Tp-NLC formulation at the higher dose utilized in these experiments (although the reliability of the assay is questionable). Finally, histopathological changes in the liver, spleen, and kidneys were observed in animals exposed to high doses of Tp and Tp-SLNs. This pathology was manifested as fatty degeneration in hepatocytes, dead cells phagocytosed in the spleen, macrophages in the white pulp, and clear proximal tubular dilations in the kidneys. However, in Tp-NLC-exposed rats, no apparent changes were found. It is important to state that the histopathology was not quantified in this study and the observations only descriptive in nature.81
Andrographolide (Adg) is the main active ingredient of the herbaceous plant Andrographis paniculata, commonly used in Chinese medicine and with acknowledged antibacterial and anti-inflammatory properties. Unfortunately, the clinical use of Adg is limited, due to its extremely poor solubility following oral administration. In an attempt to increase the bioavailability of the drug in a recent set of trials, an Adg nanosuspension (Adg-NS) was prepared via wet-media milling and freeze-drying D-α-tocopheryl PEG1,000 succinate and sodium lauryl sulfate. The prepared NPs were shown to be around 215 nm in size. Pharmacokinetics analysis of the NS in male Sprague Dawley rats (~250 g) following oral exposure of 40 mg/kg Adg resulted in significantly higher mean plasma concentrations compared to commercially available dripping pills. Next, a carrageenan-induced paw-edema model was utilized in a male Kunming mouse model (~25 g) to investigate the anti-inflammatory potential of the NS. The animals were treated once daily by oral gavage with aspirin (150 mg/kg), coarse Adg powder, Adg dripping pills, or the freeze-dried Adg-NS (all at 40 mg/kg) for 4 days. On the fourth day, 30 minutes after the last administration, 1% carrageenan was injected into the right hind paws of the mice and paw volume measured over time. The authors noted that the NPs were extremely effective in reducing paw swelling (swelling reduced from 30 minutes postexposure, 52.5% decreased swelling after 1 hour) and comparable to aspirin. Additionally, a significant reduction in plasma levels of nitric oxide, IL1, and TNFα levels and an increase in superoxide dismutase activity were observed in the sera of mice treated with the Adg-NS. Overall, the data from the study seemed to indicate that Adg incorporated in the nanoformulation has enhanced oral bioavailability and biological efficacy.82
Baicalein (Bcl) is a flavonoid sourced from Scutellaria baicalensis. The metabolite has been reported to have anticancer, antioxidant, antiviral, and anti-inflammatory properties. However, poor intestinal solubility and oral bioavailability have dramatically limited its clinical efficacy. Therefore, nanoemulsions composed of hemp oil, PEG monooleate, and sodium oleate (~90 nm) were developed to improve the intestinal stability and permeability of Bcl. In these experiments, Sprague Dawley rats (~250 g) were administered the nanoemulsion at a dose of 25 mg/kg via oral gavage for up to 12 hours. The authors demonstrated that the Bcl NPs yielded higher blood Bcl concentrations at all time points. The maximum plasma concentration was 42 μg/mL, with oral bioavailability of nanoemulsions calculated to be increased by 523% and 242% relative to the free suspensions and conventional emulsions, respectively.83
Miltefosine has shown potential to be effective against certain developmental phases of Schistosoma mansoni. In a recent study, the effectiveness of miltefosine as a potential antischistosomal drug was investigated in a selection of lipid nanocapsules composed of Solutol and Labrafac. This was followed by coating of the liposomes with either dicetyl phosphate (anionic), hexadecyltrimethylammonium bromide (cationic), oleic acid, or CS (~50 nm). In these experiments, S. mansoni-infected male Swiss albino mice were exposed to a single 20 mg/kg oral dose of miltefosine, either as an aqueous solution or the same concentration encapsulated in the lipid nanocapsules, with the animals killed 7 days posttreatment. Administration of the miltefosine-encapsulated formulations reduced the mean worm burden to different extents, with the hexadecyltrimethylammonium bromide (42%) and the oleic acid (88%) being the most effective. Additionally, lipid nanocapsule treatment was associated with ameliorated liver pathology and extensive damage to the worm tegument (outer body of the parasite). The data seem to suggest that the miltefosine nanocapsules can enhance therapy of S. mansoni, even after a single oral dose.84
In a 2013 study, vaccination with encapsulated outer-membrane vesicles (OMVs) (naturally released from the cell) of Shigella flexneri was utilized as a protection strategy to protect mice against an experimental infection with shigellosis. In these experiments, BALB/c mice (~20 g) were immunized with either free OMVs or OMVs encapsulated in polyanhydride NPs (~150 nm) via the oral route at doses of 50 or 100 μg OMVs. On week 8, the animals were infected intranasally with a lethal dose of the bacteria. The authors showed that all immunized animals remained in good health compared to controls (exhibiting no respiratory difficulties or changes in body temperature). Furthermore, a clear adjuvant effect of the encapsulation of OMVs into NPs was detected (IgG1 levels). Interestingly and importantly, following challenge with the lethal dose of S. flexneri, oral immunization with free OMVs or NPs provided high levels of protection (80% and 100%).85
Diseases of the nervous system
Levodopa (L-dopa) has been the standard drug for treatment of Parkinson’s disease. It is a precursor to the neurotransmitters dopamine, norepinephrine, and epinepherine. L-dopa can cross the blood–brain barrier (BBB), whereas dopamine is unable to do so.86 Therefore, L-dopa can be used to increase effective dopamine concentrations in the treatment of Parkinson’s disease. The utilization of nanocarrier systems would allow for delivery of drugs across the intact BBB for this purpose. In a recent study, the toxicity assessment of a layered double hydroxide (LDH) L-dopa-delivery system with controllable ion-exchange capacity and pH-dependent solubility was undertaken. The delivery system was developed with 16% loading of L-dopa into a zinc–aluminum layered hydroxide nanocomposite. The synthesized LDH construct demonstrated pH-dependent sustained release with improved thermal stability. In these trials, Sprague Dawley rats (~250 g) were exposed to the nanocomposites once daily for 28 days at doses of 5 or 500 mg/kg body weight via the oral route. First, animals treated with zinc–aluminum LDH intercalated with L-dopa over 28 days showed no signs of discomfort or mortality. Food and water intake was unaffected during the study period. Additionally, no signs of vomiting, diarrhea, paralysis, convulsion, irritation, bleeding, or breathing difficulties were observed. However, analysis of the biochemical parameters showed elevated levels of AST in nanocomposite-treated animals in comparison to the controls, indicative of hepatotoxicity following exposure to the zinc–aluminum LDH intercalated with L-dopa at the higher dose. Next, urea, electrolyte, and creatinine levels were analyzed after the 28-day oral dosing of the rats. The data demonstrated that all electrolyte and urea levels were within the same range as the control groups. Finally, histological examination of the liver showed well-preserved hepatocyte morphology: a well-maintained lobular array around the central vein, radiating sinusoids, and normal portal triads. It is very possible that mild liver injury manifested as elevated ALT levels occurred without any clinical symptoms. However, small inflammatory changes were noted in kidneys of treated animals, notably as leukocyte infiltrations in the tissue. Overall, the data suggest that oral administration of the LDH construct at doses up to 500 mg/kg resulted in only mild clinical toxicity. The data clearly signified the necessity for additional and more detailed examination of the acute hepatic and renal adverse effects after oral dosing with the nanocomposite.87
It is understood that excessively generated reactive oxygen species are associated with age-related neurodegenerative disease. In a recent study, the scavenging potential of reactive oxygen species in the brain was investigated following oral administration of RNPO (40 nm), prepared by self-assembly of redox polymers possessing antioxidant nitroxide radicals in a male senescence-accelerated-prone mouse model. In these experiments, NPs were delivered via the oral route (300 mg/kg) for 30 days. The data demonstrated that levels of oxidative stress in the brain of exposed mice were remarkably reduced, which in turn resulted in the amelioration of cognitive impairment with increased numbers of surviving neurons. In order to assess the safety of the redox polymer nanotherapeutics, body weight, vital organ weight, blood pressure, and histological features of vital organs were measured. The authors observed no significant differences in body weight or histopathological alterations in major organs (liver, spleen, kidney, lung, testicles, or heart) of NP-exposed animals.88
There is a degree of uncertainty in regard to the effectiveness of the colloidal systems designed for central nervous system targeting (ability to bypass the BBB) following oral exposure. To address this issue, two murine models (male albino Wistar Hannover rats [~200 g] and C57BL/6 mice [~25 g]) were exposed to specifically manufactured fluorescently labeled-PLGA NPs (termed g7-NPs). The NPs were characterized and demonstrated to be 140–200 nm in size. Following an acclimatization period, the animals were orally exposed (rats, 10 mg/animal; mice, 3 mg/animal) to the NPs for 180 minutes. The authors reported a clear and preferential localization of NPs around the neuronal nuclei, which showed the ability of g7-NPs to cross the BBB. It is important to note that the principal body of data generated in this study was qualitative in nature with no physiologically relevant large doses. Therefore, further work is required to corroborate the findings within this investigation.89
Diseases of musculoskeletal and connective tissue
Arthritis is a chronic inflammatory disease involving irreversible damage to the joints of the body. Currently, a broad variety of therapeutics are available for the treatment of arthritis, including the use of analgesics, glucocorticoid, and NSAIDs. Unfortunately, it is challenging to create highly efficient oral delivery systems with site-specific therapeutic release capacity. To address some of these challenges, male Sprague Dawley rats (~250 g) were orally administered (2.5, 5, and 10 mg/kg) Ind-containing nanocarriers (assembly of β-cyclodextrin-conjugated PEI; 205 nm). The NPs exhibited a spherical shape and were positively charged. The data demonstrated that the Ind plasma concentration in the nanomedicine group was significantly higher than that of the traditional medicine-administered group. Tissue-distribution studies showed high levels of Ind in the heart, spleen, kidney, and lungs. Importantly, treatment with Ind NPs at 5 and 10 mg/kg almost completely inhibited carrageenan-induced acute inflammation and complete Freund’s adjuvant-induced arthritis (enhanced therapeutic activity of the NP, even at a low dosage). The authors showed the bioavailability of Ind can be dramatically enhanced by utilization of the nanoformulation in comparison to commercially available Ind tablets. It was postulated that the formulation might serve as a highly efficient and effective delivery platform for the oral delivery of water-insoluble therapeutics in clinical settings.90
Bisphosphonates that inhibit osteoclast resorption are used widely in the treatment and prevention of osteoporosis. In general, bisphosphonates are eliminated by urinary excretion and have very low oral bioavailability.91 Oral administration of this class of drugs is associated with a range of undesirable side effects, including diarrhea, nausea, vomiting, esophageal inflammation, and the development of ulcers. One such drug, risedronate, is poorly absorbed and associated with a range of toxic effects when taken orally. It is understood that oral risedronate can form an insoluble complex with calcium and magnesium in food. Therefore, there is a strong need to develop more efficient drug-delivery systems for risedronate with less propensity to cause esophageal irritation and better bioavailability. In a 2014 study, a CS-coated liposome was developed to improve the intestinal drug absorption of the drug. The liposomes were prepared via the freeze-drying method using the combination of the lipids 1,2-distearoryl-sn-glycero-3-phosphocholine and distearoryl-sn-glycero-3-(phospho-rac-[1-glycerol]), with subsequent coating of the liposomes with CS (~950 nm). In these experiments, male Sprague Dawley rats weighing about 250 g were orally exposed to the liposomes at a dose equivalent to 5 mg/kg risedronate. The authors noted that the CS-coated liposome formulation increased the amount of drug excreted in the urine threefold in rats compared with the free drug formulation. The use of CS-coated liposomes may prevent or considerably reduce the interaction with food, thus reducing the esophageal irritation associated with the traditional formulations of the drug. The findings from this study suggested that CS-coated liposomes could improve the oral bioavailability of risedronate.93
Parathyroid hormone (PTH) is one of the major hormones regulating calcium-ion concentrations in the blood.93 Anabolic functions are stimulated by intermittent exposure to PTH, resulting in differentiation and proliferation or reduced apoptosis of osteoblasts.94 Furthermore, catabolic functions are stimulated by continuous PTH exposure and associated with increased receptor activator of NFκB-ligand expression and inhibition of osteoprotegerin expression.94 Teriparatide, a recombinant human PTH (1–34), is currently used clinically to treat osteoporosis in postmenopausal women and hypogonadal osteoporosis in men. However, the drug is currently administered once daily via subcutaneous injection and is often associated with poor patient compliance. In an attempt to improve patient acquiescence, an enteric microcapsule containing an ionic nanocomplex of PTH (1–34) and lysine-linked deoxycholic acid of the drug was manufactured (~35 nm). In these experiments, bilateral ovariectomy was performed in female Sprague Dawley rats (~200 g) to induce osteoporosis. The antiosteoporotic effects of the orally administered nanocomplex were evaluated in these animals following exposure of the carriers at doses of 50 μg/kg. Twelve weeks after the treatments, the left and right tibiae were removed. The data showed that the rats treated with oral PTH–lysine-linked deoxycholic acid-loaded enteric microcapsules had increased bone-mineral density (159%), bone-volume fraction (175%), and trabecular numbers (174%) compared with the controls. It appears that the novel nanocomplex oral delivery system described could be a promising treatment modality for osteoporosis due to enhanced osteogenesis and trabecular connectivity.94
Other or unspecified diseases
In order to overcome the challenge of drug insolubility, C57/BL6 mice were orally exposed to solidified (lyophilized) monomethoxy-PEG-PLA micelles encapsulating the hydrophobic molecule 6-coumarin (~20 nm). The micelles were administered at 30 mg/mL for 2 hours. TEM analysis demonstrated that labeled micelles were present in significant numbers in the small intestines of exposed animals. Additionally, in a concurrent in vitro experiment, endocytosis of the micelles by Caco2 cells was demonstrated to be clathrin-mediated and both energy- and temperature-dependent, which is indicative of specific receptor binding. The authors summarized that the particular micelles used in this study could be beneficial in oral delivery of poorly soluble small molecules.95
For any oral medicine to be effective, it must negotiate and bypass multiple physiological barriers, including the avoidance of enzymatic degradation in the harsh gastric environment or overcoming the generally poor permeability across the intestinal epithelium. Additionally, the tendency for immunotolerance being favored in the gut means that effective oral immunization in general is very challenging. To address these limitations in a topical study, tetanus toxoid encapsulated glucomannan-O-carboxymethyl–distearyl phosphatidylethanolamine conjugate bilosomes (a colloidal delivery system incorporating bile salts into the vesicular lipid bilayer membrane; ~150 nm) were manufactured. The bilosomes were prepared by a combination of sodium deoxycholate (bile acid), Span 80 (surfactant), and cholesterol (0.1:2:1 ratio). Male BALB/c mice were immunized with five flocculation units of tetanus toxoid via oral gavage. The animals further received a second immunization on day 22. At the end of day 35, the glucomannan bilosomes elicited a significantly higher systemic immunoresponse (measured by serum IgG levels) compared to all controls. Additionally, the bilosomes were capable of inducing a strong mucosal immunoresponse (soluble IgA titer in salivary and intestinal secretions) as well as a cell-mediated immunoresponse (IL2 and IFNγ measured in the spleen).96
PLA-b-Pluronic-b-PLA NPs are highly biodegradable and biocompatible and could represent an interesting potential drug-delivery option. In a recent study, these carriers were synthesized from commercially available Pluronic P85 and biodegradable PLA. The NPs were fully characterized (mean diameter of about 170 nm) before being analyzed for toxicity in a Kunming mouse model (~30 g). The animals were exposed to 8, 40, or 200 μg/animal of empty nanocarriers for a period of 28 days via oral gavage. The authors did not observe any behavioral, clinical, biochemical, or histological toxicity in the heart, liver, or kidneys at any of the doses investigated. However, it should be stated that the doses utilized in the study were rather low for oral exposure; therefore, a note of caution is required. This is of even more importance considering the study lacked biokinetic data.97
In a 2010 study, the toxicity and biodistribution of PLGA NPs with potential medical applications were assessed following oral delivery. Characterization data showed the NPs to be very polydispersed in solution with a size range of 300 nm to 1 μm. To investigate biodistribution and NP-induced histopathology, female BALB/c mice (~20 g) were exposed to 4 mg of empty carriers via oral gavage on a single (24 hours) or repeated regime (every day for 5 days). The animals were killed on day 1, 5, and 7 posttreatment. The brain, heart, kidneys, liver, lung, spleen, intestines, pancreas, stomach, and thymus were collected from each animal. From the data, it was evident the particles accumulated predominantly in the liver at around 40% of the administered dose, followed by the kidneys (~30%), heart (~10%), and brain (~10%) throughout the exposure period. Finally, no significant histopathological changes were noted for the NPs at the doses and time points investigated.98
The absorption and distribution of four rhodamine B isothiocyanate-incorporated silica oxide NPs with potential for medical applications was undertaken. The NPs were either 20 or 100 nm with a positive or negative charge. In these experiments, female athymic nude mice (~20 g) were orally exposed to NPs at a dose of 2 mg per animal. Whole-body optical fluorescence imaging of mice demonstrated fluorescence signals mainly in the GIT. Additionally, ex vivo optical images of major organs from exposed mice showed distribution of the NPs to the lungs, liver, pancreas, and kidneys. NPs with a diameter of 20 nm showed higher uptake in the lungs compared to the larger particles. However, following oral administration of the silica NPs, no surface-charge-associated differences were observed in terms of translocation and distribution. Interestingly, at 10 hours postadministration, the majority of 20 nm NPs were cleared from the organs and detectable only in the kidneys. On the other hand, the 100 nm NPs were visualized mainly in the liver. Finally, the dissociation of rhodamine B isothiocyanate from the NPs was investigated in vitro in highly acidic conditions (mimicking the low pH in the stomach [1.2]). It was noted that there was no release from the larger 100 nm NPs, whereas minimal dissociation of rhodamine B isothiocyanate was observed from the 20 nm NPs. This finding is best explained by the larger exposed surface area of the smaller NPs in the highly acidic surroundings.99
Currently, the only available management in food allergy is strict avoidance of the culprit food. However, in recent years, allergen immunotherapy has been investigated to protect food-allergic individuals from severe reactions.100 The strategy is based on continuous low-dose administration of the allergen to provide protection against the IgE-governed adverse responses to the allergen. In a recent study, polyanhydride NPs (150 nm) loaded with a peanut-protein extract (PE) were evaluated for oral immunotherapy potential in peanut-sensitized mice (CD1, ~20 g). The animals were sensitized by oral administration of a mixture of peanut butter and 5 μg cholera toxin on days 1, 7, 15, and 21. On days 25, 28, and 35 the animals received a single oral dose of 1 mg PE, either suspended in purified water or incorporated in the NPs. Finally, on days 44 and 45, animals were challenged by an injection of 2 mg PE intraperitoneally, in order to incite an anaphylactic shock in the sensitized animals. Anaphylaxis was evaluated by means of symptom scores and mast-cell protease 1 levels. The data demonstrated that 20 minutes after challenge, piloerection and cyanosis were similar for animals treated with either PE or PE-NPs. Moreover, animals from the unsensitized group were found completely static with a high difficulty for simple movement, whereas treated animals were able to move or react against pain or pressure. Moreover, PE-NP treatment was associated with significantly lower levels of mast-cell protease 1 and a significantly higher survival rate after challenge.100 Table 1 summarizes all the studies highlighted in this review, their intended medical application, and therapeutic activity.
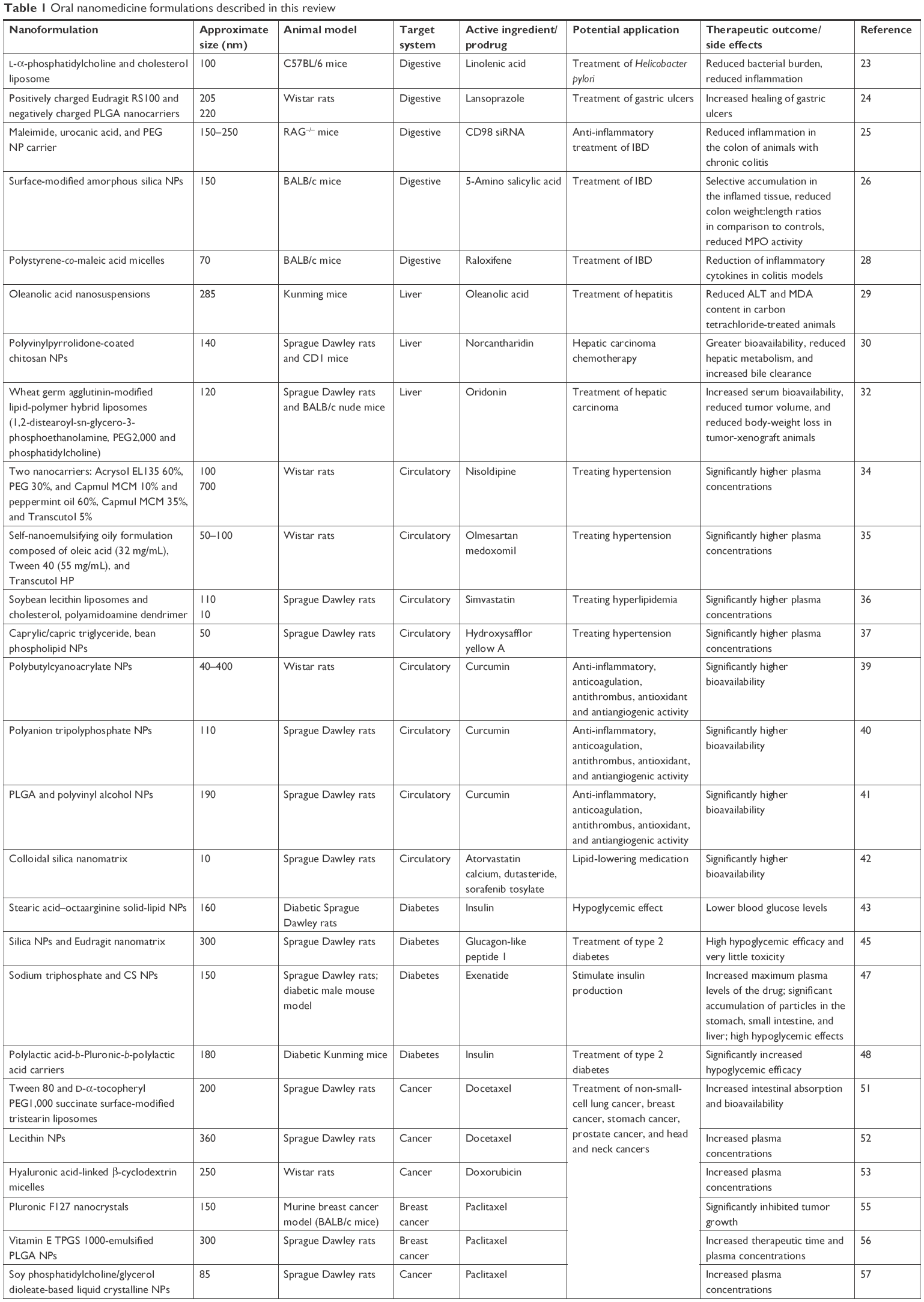 | Table 1 Oral nanomedicine formulations described in this review |
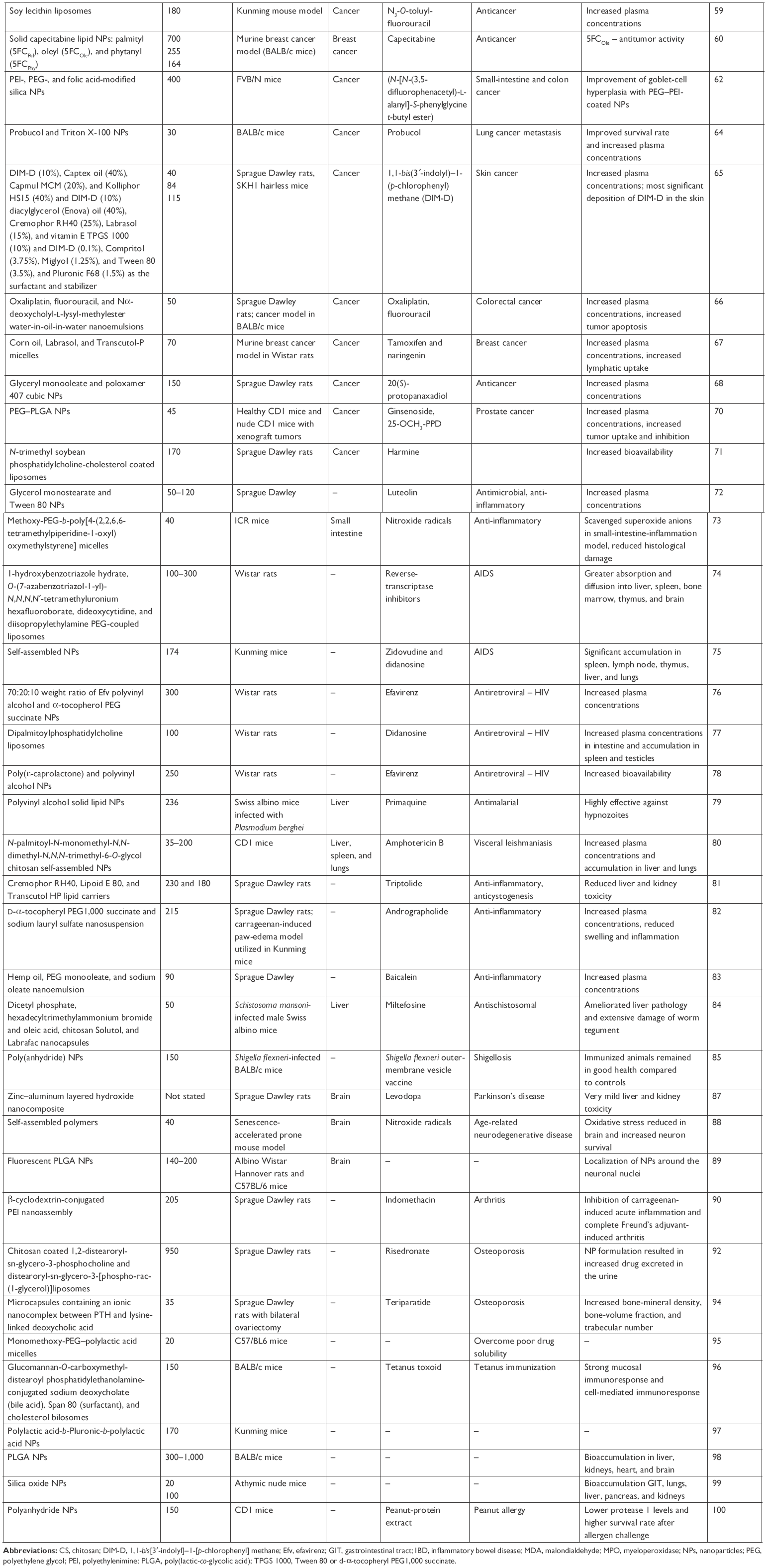 |
Summarizing remarks
For a successful oral nanodelivery platform, the system must allow for efficient transport of the NP–drug complex or the drug alone following release from the NP to the desired destination, with intestinal epithelial cells being extremely important in the efficacy of transport of orally administered drugs into systemic circulation. In general, hydrophobic high-molecular-weight drugs are relatively poor at penetration of GIT physical barriers, leading to lower systemic bioavailability.101 Moreover, microfibrils between two epithelial cells provide a tight junction, also acting as a physical barrier for orally administered formulations.102 Furthermore, the presence of a multitude of metabolic enzymes (eg, proteases/peptidases, trypsin, chymotrypsin, elastase, carboxypeptidases) and the acidic environment of the stomach can affect the drug and/or NPs. Another principal problem for oral nanomedicines is the phenomenon of first-pass metabolism in the liver, which causes the therapeutic to be metabolized and changed extensively before reaching systemic circulation.103 As highlighted, the GIT is an extremely complex system, with numerous barriers to a drug/dosage system, thereby affecting drug and NP stability, uptake, and thus bioavailability. From the literature, it is clear that emptying of gastric contents is an additional important factor in drug circulation and accounts for much of the variation in absorption. The ability to control the release of the prodrug would be highly advantageous to negate erratic drug absorption. It is important to note that due to the fast rate of gastric emptying, it is often difficult for the drug to achieve maximum absorption through the oral route.103 Factors that influence gastric emptying include but are not limited to when the last meal was consumed, the age and sex of the individual, and the nature of the specific meals consumed around the time that the medicine is administered.104 It seems logical to consider the uptake mechanisms of a given formulation. For example, if the construct cargo is released in close proximity to the enterocytes, its uptake will involve the usual dietary absorption mechanisms and the drug will be transported directly to the liver via the portal vein. An alternative physiological route of transport could be the gut’s lymphatic vessels. These vessels link to the mesenteric lymphatic network, and any material in the lymphatic systemic will ultimately end up in the systemic blood circulation. This direct-uptake route for nanoconstructs and their transport to gut-associated lymphatic tissue could potentially be advantageous for oral drug-delivery approaches.
In addition to the physiologically harsh environment and the formidable physical barriers of the GIT, there are a number of important physicochemical/pharmaceutical factors attributed to the drug itself that further influence the permeability and bioavailability of a nanoformulation. The first of these limiting factors is the solubility of the formulation at a physiologically relevant pH.105 Unsurprisingly, highly soluble drugs are not associated with bioavailability issues. However, low/poor bioavailability is common with orally administered forms of hydrophobic and slowly absorbed drugs. It has been reported that up to 50% of all new chemical constituents developed in drug discovery programs have poor aqueous solubility and are thus considered unsuitable for clinical testing.106 If the rate of dissolution of the drug is significantly slower than the rate of absorption, then dissolution of the drug is considered the rate-limiting step and the formulation might not necessarily be a viable candidate as an effective oral medicine. Furthermore, it has been suggested that drug dissolution in intestinal fluid is required to allow partitioning of drug molecules across the epithelial cell membrane through passive diffusion.107 As touched upon and highlighted in numerous studies, the molecular size of drugs significantly influences their permeation ability through biological membranes. To overcome (at least partially) the various limitations discussed herein, nanoformulations are being developed to enable the stimuli-responsive release of drugs (eg, pH, temperature, light) by combining advancements in materials chemistry. Due to all the aforementioned complications, to date only a handful of oral nanoformulations are commercially available. These include Emend (antiemetic, US Food and Drug Administration [FDA] approval 2003), Rapamune (organ rejection, FDA approval 2002), Tricor (lipid regulation, FDA approval 2004), Megace ES (anorexia and cachexia, FDA approval 2005), and Renagel (chronic kidney disease, FDA approval 2000).108
Toxicological considerations
In a physiological environment, NPs acquire a new identity, which might alter their size, surface charge, and aggregation, along with a constantly changing and devolving deposited protein corona. It is noteworthy that a particle’s acquired biological identity can be dramatically different from the material designed in the laboratory. The biological attributes afforded by the corona can affect physiological and toxicological responses to nanoformulations in vivo. Both the required medical and toxic effects of nanosize delivery systems are fundamentally governed by their physicochemical characteristics, including their bioavailable surface area, specific surface properties (eg, chemistry and charge), solubility, and shape.109 These factors influence their ability to enter and deposit in tissue that might be inaccessible to larger counterparts.110 Furthermore, nanocarriers can alter the physicochemical properties of the cargo drugs, peptides, and genes, as well as altering the solubility and pharmacokinetic disposition upon delivery compared to unmodified drugs.24,34,48,55,66 Therefore, orally administered nanomedicines can be potentially hazardous to human health (the need to address the balance between improved delivery/efficacy and potential nanotoxicity). These adverse effects are principally caused by the nanoparticulate delivery directly interacting with biological surroundings or as a result of unwanted bioaccumulation in the case of insoluble carriers. Interestingly and importantly, from a toxicological perspective, in the majority of studies that were scrutinized in this review, safety evaluation was either entirely lacking for the manufactured formulation or toxicity testing was restricted to simple in vitro models or acute in vivo studies. This might be one of the reasons that despite considerable therapeutic promise demonstrated by many of the manufactured nanoconstructs in experimental settings, the use and translation of nanomedicines from laboratory to clinical settings have been extremely slow. It is imperative that appropriate and comprehensive risk-reduction strategies be devised and incorporated for all materials proposed for medical applications. To address this issue, the development of risk-assessment frameworks that address the toxicological potential of nanomedicines is of crucial importance (ie, H2020 project BIORIMA). Any preclinical toxicological assessment should include in vitro and in vivo long-term physiologically relevant exposure of the delivery formats, although this would have to be tailored and designed to take into account the intended therapeutic application of the medicine. Currently, the majority of toxicological strategies are influenced by route of delivery rather than mode of action.
It is also important to acknowledge that it is extremely difficult to fully understand how a given material/formulation behaves in the gut or evaluate its potential toxicity in the GIT in vivo. Nevertheless, bioaccumulation/biokinetics and toxicity assessment of the vehicle/prodrug in target organs is very possible. As touched upon, it is worth reiterating that is very possible that hazardous nanomedicines may have been effectively screened out in research than preceded the current preclinical investigations in animal models discussed in this review.
Other considerations
It is important to recognize that rodents have a different metabolism than humans and might exhibit a different metabolic rate compared with humans for certain drugs (eg, rats have a higher metabolic clearance of Efv compared to humans). These interspecies differences must be considered in the evaluation of the pharmacokinetic benefits and toxic potential of given formulations.111 As an additional complication, nanocarriers can alter the physicochemical properties of a variety of drugs, peptides, genes, and pharmaceutical changes in stability, solubility, and pharmacokinetic disposition can occur upon delivery, which need to be considered.
Future perspectives
The utilization of nanomedicines in conjunction with biological systems – namely solid drug NPs, drugs, proteins, and DNA – has the potential to have an outstanding impact in treating various diseases, as well as various applications in tissue regeneration, biological sensors, and drug delivery. Additionally, combinations of imaging and therapeutic agents incorporated into the same nanoformulation will undoubtedly be beneficial in the future. Considerable efforts have been to ameliorate the bioavailability-related issues of oral drug delivery and increasing therapeutic efficacy, which will hopefully in time result in improved end-user compliance. The literature clearly demonstrates the potential of nanosize carrier systems over traditional approaches (at the molecular level) for oral delivery. Among the most successful of such approaches are the development of nanocrystals, nanoemulsions, and polymeric NPs. In addition, interest in self-emulsifying drug-delivery systems, dendrimers, and micelles is increasing.
Overcoming the physical barriers in the GIT is a significant hindrance to the successful design of nanostructures for the oral route of administration. To this end, modification of particle-size distribution and surface area is a critical parameter in terms of the capacity of NPs to interact with the intestinal mucosa. It is crucial that nanoformulations are stable in biological fluids (particularly in the acidic microenvironment of the stomach) to avoid the premature release of cargo and unwanted toxic effects. The surface chemical composition will undoubtedly influence the stability of the nanocarrier, as well its interaction and transport across the epithelial barrier. Finally, the specific targeting/affinity of the nanomedicine for membrane receptors may be beneficial (although principally difficult) in overcoming the biological barriers.
Despite the plethora of advantages that nanomedicine offers, systems still have limitations, one of which is potential to affect human health adversely, which needs to be thoroughly addressed and is still regarded as an afterthought in the majority of studies scrutinized in this review. A wide array of complex issues involving physiological, physicochemical, and molecular processes need to be considered for a better understanding of potential clinical toxicities of nanomedical formulations. It might sound obvious, but nanomedical preparations with great potential in animal models require further assessment in human clinical testing, which is associated with its own set of challenges. In addition, clinical translational studies, including pharmacokinetic profiles and long-term toxicity, should be performed to address fundamental questions in regard to the clinical feasibility of oral nanomedicine.
Acknowledgment
This work has been partially supported by H2020 funded project BIORIMA (Grant code - 760928).
Disclosure
The authors report no conflicts of interest in this work.
References
da Silva AL, Cruz FF, Rocco PRM, Morales MM. New perspectives in nanotherapeutics for chronic respiratory diseases. Biophys Rev. 2017;9(5):793–803. | ||
Wen R, Banik B, Pathak RK, Kumar A, Kolishetti N, Dhar S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv Drug Deliv Rev. 2016;99(Pt A):52–69. | ||
Pathak K, Raghuvanshi S. Oral bioavailability: issues and solutions via nanoformulations. Clin Pharmacokinet. 2015;54(4):325–357. | ||
Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today. 2014;9(2):223–243. | ||
Jm Y, Yj L, Qiu LY, Jin Y. Polymeric nanoparticles of cholesterol-modified glycol chitosan for doxorubicin delivery: preparation and in-vitro and in-vivo characterization. J Pharm Pharmacol. 2009;61:713–719. | ||
Johnston H, Brown D, Kermanizadeh A, Gubbins E, Stone V. Investigating the relationship between nanomaterial hazard and physicochemical properties: Informing the exploitation of nanomaterials within therapeutic and diagnostic applications. J Control Release. 2012;164(3):307–313. | ||
Ding C, Li Z. A review of drug release mechanisms from nanocarrier systems. Mater Sci Eng C Mater Biol Appl. 2017;76:1440–1453. | ||
Kermanizadeh A, Jantzen K, Brown DM, Møller P, Loft S. A flow cytometry-based method for the screening of nanomaterial-induced reactive oxygen species production in leukocytes subpopulations in whole blood. Basic Clin Pharmacol Toxicol. 2018;122(1):149–156. | ||
Lanone S, Andujar P, Kermanizadeh A, Boczkowski J. Determinants of carbon nanotube toxicity. Adv Drug Deliv Rev. 2013;65(15):2063–2069. | ||
Vega-Vila KR, Takemoto JK, Yanez JA, Remsberg CM, Forrest ML, Davies NM. Clinical toxicities of nanocarrier systems. Adv Drug Deliv Rev. 2008;60:929–938. | ||
Shen X, Li T, Chen Z, et al. Luminescent/magnetic PLGA-based hybrid nanocomposites: a smart nanocarrier system for targeted codelivery and dual-modality imaging in cancer theranostics. Int J Nanomedicine. 2017;12:4299–4322. | ||
Kermanizadeh A, Villadsen K, Østrem RG, Jensen KJ, Møller P, Loft S. Integrin targeting and toxicological assessment of peptide-conjugated liposome delivery systems to activated endothelial cells. Basic Clin Pharmacol Toxicol. 2017;120(4):380–389. | ||
Grimaldi AM, Incoronato M, Salvatore M, Soricelli A. Nanoparticle-based strategies for cancer immunotherapy and immunodiagnostics. Nanomedicine. 2017;12(19):2349–2365. | ||
Silva AC, Santos D, Ferreira D, Lopes CM. Lipid-based nanocarriers as an alternative for oral delivery of poorly water-soluble drugs: peroral and mucosal routes. Curr Med Chem. 2012;19(26):4495–4510. | ||
de Sio L, Caracciolo G, Placido T, et al. Applications of nanomaterials in modern medicine. Rendiconti Lincei. 2015;26(S2):231–237. | ||
Tatham LM, Rannard SP, Owen A. Nanoformulation strategies for the enhanced oral bioavailability of antiretroviral therapeutics. Ther Deliv. 2015;6(4):469–490. | ||
Braakhuis HM, Kloet SK, Kezic S, et al. Progress and future of in vitro models to study translocation of nanoparticles. Arch Toxicol. 2015;89(9):1469–1495. | ||
Richard J. Challenges in oral peptide delivery: lessons learnt from the clinic and future prospects. Ther Deliv. 2017;8(8):663–684. | ||
Powell JJ, Faria N, Thomas-Mckay E, Pele LC. Origin and fate of dietary nanoparticles and microparticles in the gastrointestinal tract. J Autoimmun. 2010;34(3):J226–J233. | ||
Panyala A, Chinde S, Kumari SI, Grover P. Assessment of genotoxicity and biodistribution of nano- and micron-sized yttrium oxide in rats after acute oral treatment. J Appl Toxicol. 2017;37(12):1379–1395. | ||
Fröhlich E, Roblegg E. Oral uptake of nanoparticles: human relevance and the role of in vitro systems. Arch Toxicol. 2016;90(10):2297–2314. | ||
World Health Organisation International classification of disease, 11th version -. Available from: http://www.who.int/classifications/icd/en/. Accessed July 18, 2018. | ||
Thamphiwatana S, Gao W, Obonyo M, Zhang L. In vivo treatment of Helicobacter pylori infection with liposomal linolenic acid reduces colonization and ameliorates inflammation. Proc Natl Acad Sci U S A. 2014;111(49):17600–17605. | ||
Alai M, Lin WJ. Application of nanoparticles for oral delivery of acid-labile lansoprazole in the treatment of gastric ulcer: in vitro and in vivo evaluations. Int J Nanomedicine. 2015;10:4029–4041. | ||
Xiao B, Laroui H, Viennois E, et al. Nanoparticles with surface antibody against CD98 and carrying CD98 small interfering RNA reduce colitis in mice. Gastroenterology. 2014;146(5):1289–1300. | ||
Moulari B, Pertuit D, Pellequer Y, Lamprecht A. The targeting of surface modified silica nanoparticles to inflamed tissue in experimental colitis. Biomaterials. 2008;29(34):4554–4560. | ||
Bevivino G, Monteleone G. Advances in understanding the role of cytokines in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2018;12(9):907–915. | ||
Greish K, Taha S, Jasim A, et al. Styrene maleic acid encapsulated raloxifene micelles for management of inflammatory bowel disease. Clin Transl Med. 2017;6(1):28. | ||
Chen Y, Liu J, Yang X, Zhao X, Xu H. Oleanolic acid nanosuspensions: preparation, in-vitro characterization and enhanced hepatoprotective effect. J Pharm Pharmacol. 2005;57(2):259–264. | ||
Ding XY, Hong CJ, Liu Y, et al. Tissue distribution, and metabolites of a polyvinylpyrrolidone coated norcantharidin chitosan nanoparticle formulation in rats and mice, using LC-MS/MS. Int J Nanomedicine. 2012;7:1723–1735. | ||
Zhang HP, Li GQ, Guo WZ, et al. Oridonin synergistically enhances JQ1-triggered apoptosis in hepatocellular cancer cells through mitochondrial pathway. Oncotarget. 2017;8(63):106833–106843. | ||
Liu Y, Liu J, Liang J, et al. Mucosal transfer of wheat germ agglutinin modified lipid-polymer hybrid nanoparticles for oral delivery of oridonin. Nanomedicine. 2017;13(7):2219–2229. | ||
Nekkanti V, Rueda J, Wang Z, Betageri GV. Comparative evaluation of proliposomes and self micro-emulsifying drug delivery system for improved oral bioavailability of nisoldipine. Int J Pharm. 2016;505(1–2):79–88. | ||
Krishnamoorthy B, Habibur Rahman SM, Tamil Selvan N, et al. Design, formulation, in vitro, in vivo, and pharmacokinetic evaluation of nisoldipine-loaded self-nanoemulsifying drug delivery system. J Nanoparticle Res. 2015;17(1):34. | ||
Beg S, Sharma G, Thanki K, Jain S, Katare OP, Singh B. Positively charged self-nanoemulsifying oily formulations of olmesartan medoxomil: Systematic development, in vitro, ex vivo and in vivo evaluation. Int J Pharm. 2015;493(1–2):466–482. | ||
Qi R, Zhang H, Xu L, et al. G5 PAMAM dendrimer versus liposome: A comparison study on the in vitro transepithelial transport and in vivo oral absorption of simvastatin. Nanomedicine. 2015;11(5):1141–1151. | ||
Lz L, Tong CQ, Yu J, Han M, Gao JQ. Mechanism of enhanced oral absorption of hydrophilic drug incorporated in hydrophobic nanoparticles. Int J Nanomedicine. 2013;8:2709–2717. | ||
Perrone D, Ardito F, Giannatempo G, et al. Biological and therapeutic activities, and anticancer properties of curcumin. Exp Ther Med. 2015;10(5):1615–1623. | ||
Sun M, Zhao L, Guo C, et al. Evaluation of an oral carrier system in rats: bioavailability and gastrointestinal absorption properties of curcumin encapsulated PBCA nanoparticles. J Nanoparticle Res. 2012;14(2):705. | ||
Sun W, Zou Y, Guo Y, et al. Construction and characterization of curcumin nanoparticles system. J Nanoparticle Res. 2014;16(3):2317. | ||
Harigae T, Nakagawa K, Miyazawa T, et al. Metabolic fate of poly-(lactic-co-glycolic acid)-based curcumin nanoparticles following oral administration. Int J Nanomedicine. 2016;11:3009–3022. | ||
Kim MS. Soluplus-coated colloidal silica nanomatrix system for enhanced supersaturation and oral absorption of poorly water-soluble drugs. Artif Cells Nanomed Biotechnol. 2013;41(6):363–367. | ||
Zhang ZH, Zhang YL, Zhou JP, Hx L. Solid lipid nanoparticles modified with stearic acid-octaarginine for oral administration of insulin. Int J Nanomedicine. 2012;7:3333–3339. | ||
Liu J, Yang K, Xiao W, et al. GLP-1 receptor agonists stimulate ANGPTL8 production through the PI3K/Akt pathway in a GLP-1 receptor-dependent manner. Peptides. 2018;106:83–90. | ||
Qu W, Li Y, Hovgaard L, et al. A silica-based pH-sensitive nanomatrix system improves the oral absorption and efficacy of incretin hormone glucagon-like peptide-1. Int J Nanomedicine. 2012;7:4983–4994. | ||
Tran KL, Park YI, Pandya S, et al. Overview of glucagon-like peptide-1 receptor agonists for the treatment of patients with type 2 diabetes. Am Health Drug Benefits. 2017;10:178–188. | ||
Li X, Wang C, Liang R, et al. The glucose-lowering potential of exenatide delivered orally via goblet cell-targeting nanoparticles. Pharm Res. 2015;32:1017–1027. | ||
Xiong XY, Li QH, Li YP, Guo L, Li ZL, Gong YC. Pluronic P85/poly(lactic acid) vesicles as novel carrier for oral insulin delivery. Colloids Surf B Biointerfaces. 2013;111:282–288. | ||
Rafiei P, Haddadi A. Pharmacokinetic consequences of PLGA nanoparticles in Docetaxel drug delivery. Pharm Nanotechnol. 2017;5(1):3–23. | ||
Picard M. Management of Hypersensitivity Reactions to Taxanes. Immunol Allergy Clin North Am. 2017;37(4):679–693. | ||
Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495–504. | ||
Hu K, Cao S, Hu F, Feng J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine. 2012;7:3537–3545. | ||
Liu Y, Zhai Y, Han X, et al. Bioadhesive chitosan-coated cyclodextrin-based superamolecular nanomicelles to enhance the oral bioavailability of doxorubicin. J Nanoparticle Res. 2014;16(10):2587. | ||
Eloy JO, Petrilli R, Chesca DL, Saggioro FP, Lee RJ, Marchetti JM. Anti-HER2 immunoliposomes for co-delivery of paclitaxel and rapamycin for breast cancer therapy. Eur J Pharm Biopharm. 2017;115:159–167. | ||
Liu F, Park JY, Zhang Y, et al. Targeted cancer therapy with novel high drug-loading nanocrystals. J Pharm Sci. 2010;99(8):3542–3551. | ||
Zhao L, Feng SS. Enhanced oral bioavailability of paclitaxel formulated in vitamin E-TPGS emulsified nanoparticles of biodegradable polymers: in vitro and in vivo studies. J Pharm Sci. 2010;99(8):3552–3560. | ||
Zeng N, Gao X, Hu Q, et al. Lipid-based liquid crystalline nanoparticles as oral drug delivery vehicles for poorly water-soluble drugs: cellular interaction and in vivo absorption. Int J Nanomedicine. 2012;7:3703–3718. | ||
Liu C, Liu D, Bai F, Zhang J, Zhang N. In vitro and in vivo studies of lipid-based nanocarriers for oral N3-o-toluyl-fluorouracil delivery. Drug Deliv. 2010;17(5):352–363. | ||
Liu D, Liu C, Zou W, Zhang N. Enhanced gastrointestinal absorption of N3-O-toluyl-fluorouracil by cationic solid lipid nanoparticles. J Nanoparticle Res. 2010;12(3):975–984. | ||
Sagnella SM, Gong X, Moghaddam MJ, et al. Nanostructured nanoparticles of self-assembled lipid pro-drugs as a route to improved chemotherapeutic agents. Nanoscale. 2011;3(3):919–924. | ||
Nasrin A, Hassan M, Ye P. Inhibition of Notch signaling pathway using γ-secretase inhibitor delivered by a low dose of Triton-X100 in cultured oral cancer cells. Biochem Biophys Res Commun. 2018;495(3):2118–2124. | ||
Desai D, Prabhakar N, Mamaeva V, et al. Targeted modulation of cell differentiation in distinct regions of the gastrointestinal tract via oral administration of differently PEG-PEI functionalized mesoporous silica nanoparticles. Int J Nanomedicine. 2016;11:299–313. | ||
Kawamoto R, Yamashita A, Nishihira K, et al. Different inflammatory response and oxidative stress in neointimal hyperplasia after balloon angioplasty and stent implantation in cholesterol-fed rabbits. Pathol Res Pract. 2006;202(6):447–456. | ||
Zhang Z, Cao H, Jiang S, et al. Nanoassembly of probucol enables novel therapeutic efficacy in the suppression of lung metastasis of breast cancer. Small. 2014;10(22):4735–4745. | ||
Boakye CH, Patel K, Patel AR, et al. Lipid-based oral delivery systems for skin deposition of a potential chemopreventive DIM derivative: characterization and evaluation. Drug Deliv Transl Res. 2016;6(5):526–539. | ||
Pangeni R, Choi SW, Jeon OC, Byun Y, Park JW. Multiple nanoemulsion system for an oral combinational delivery of oxaliplatin and 5-fluorouracil: preparation and in vivo evaluation. Int J Nanomedicine. 2016;11:6379–6399. | ||
Sandhu PS, Kumar R, Beg S, et al. Natural lipids enriched self-nano-emulsifying systems for effective co-delivery of tamoxifen and naringenin: Systematic approach for improved breast cancer therapeutics. Nanomedicine. 2017;13(5):1703–1713. | ||
Jin X, Zhang Z, Sun E, et al. Enhanced oral absorption of 20(S)-protopanaxadiol by self-assembled liquid crystalline nanoparticles containing piperine: in vitro and in vivo studies. Int J Nanomedicine. 2013;8:641–652. | ||
Zhang Z, Li M, Wang H, Agrawal S, Zhang R. Antisense therapy targeting MDM2 oncogene in prostate cancer: effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. PNAS. 2003;100:11636–11641. | ||
Voruganti S, Qin JJ, Sarkar S, et al. Oral nanoparticle preparation, characterization, in vitro and in vivo anti-prostate cancer activity, and mechanisms of action. Oncotarget. 2015;6:21379–21394. | ||
Chen WL, Yuan ZQ, Liu Y, et al. Liposomes coated with N-trimethyl chitosan to improve the absorption of harmine in vivo and in vitro. Int J Nanomedicine. 2016;11:325–336. | ||
Dang H, Meng MHW, Zhao H, et al. Luteolin-loaded solid lipid nanoparticles synthesis, characterization, & improvement of bioavailability, pharmacokinetics in vitro and vivo studies. J Nanoparticle Res. 2014;16(4):2347. | ||
Sha S, Vong LB, Chonpathompikunlert P, Yoshitomi T, Matsui H, Nagasaki Y. Suppression of NSAID-induced small intestinal inflammation by orally administered redox nanoparticles. Biomaterials. 2013;34(33):8393–8400. | ||
Hillaireau H, Dereuddre-Bosquet N, Skanji R, et al. Anti-HIV efficacy and biodistribution of nucleoside reverse transcriptase inhibitors delivered as squalenoylated prodrug nanoassemblies. Biomaterials. 2013;34(20):4831–4838. | ||
Jin Y, Xin R, Tong L, du L, Li M. Combination anti-HIV therapy with the self-assemblies of an asymmetric bolaamphiphilic zidovudine/didanosine prodrug. Mol Pharm. 2011;8(3):867–876. | ||
Mcdonald TO, Giardiello M, Martin P, et al. Antiretroviral solid drug nanoparticles with enhanced oral bioavailability: production, characterization, and in vitro-in vivo correlation. Adv Healthc Mater. 2014;3(3):400–411. | ||
Skanji R, Andrieux K, Lalanne M, et al. A new nanomedicine based on didanosine glycerolipidic prodrug enhances the long term accumulation of drug in a HIV sanctuary. Int J Pharm. 2011;414(1–2):285–297. | ||
Tshweu L, Katata L, Kalombo L, et al. Enhanced oral bioavailability of the antiretroviral efavirenz encapsulated in poly(epsilon-caprolactone) nanoparticles by a spray-drying method. Nanomedicine. 2014;9(12):1821–1833. | ||
Serrano DR, Lalatsa A, Dea-Ayuela MA, et al. Oral particle uptake and organ targeting drives the activity of amphotericin B nanoparticles. Mol Pharm. 2015;12(2):420–431. | ||
Omwoyo WN, Ogutu B, Oloo F, et al. Preparation, characterization, and optimization of primaquine-loaded solid lipid nanoparticles. Int J Nanomedicine. 2014;9:3865–3874. | ||
Zhang C, Peng F, Liu W, et al. Nanostructured lipid carriers as a novel oral delivery system for triptolide: induced changes in pharmacokinetics profile associated with reduced toxicity in male rats. Int J Nanomedicine. 2014;9:1049–1063. | ||
Qiao H, Chen L, Rui T, et al. Fabrication and in vitro/in vivo evaluation of amorphous andrographolide nanosuspensions stabilized by d-α-tocopheryl polyethylene glycol 1000 succinate/sodium lauryl sulfate. Int J Nanomedicine. 2017;12:1033–1046. | ||
Yin J, Xiang C, Wang P, Yin Y, Hou Y. Biocompatible nanoemulsions based on hemp oil and less surfactants for oral delivery of baicalein with enhanced bioavailability. Int J Nanomedicine. 2017;12:2923–2931. | ||
Eissa MM, El-Moslemany RM, Ramadan AA, Amer EI, El-Azzouni MZ, El-Khordagui LK. Miltefosine lipid nanocapsules for single dose oral treatment of Schistosomiasis Mansoni: a preclinical study. PLoS One. 2015;10(11):e0141788. | ||
Camacho AI, Irache JM, de Souza J, Sánchez-Gómez S, Gamazo C. Nanoparticle-based vaccine for mucosal protection against Shigella flexneri in mice. Vaccine. 2013;31(32):3288–3294. | ||
Lewitt PA. Levodopa therapy for Parkinson’s disease: Pharmacokinetics and pharmacodynamics. Mov Disord. 2015;30(1):64–72. | ||
Kura AU, Cheah PS, Hussein MZ, et al. Toxicity evaluation of zinc aluminium levodopa nanocomposite via oral route in repeated dose study. Nanoscale Res Lett. 2014;9(1):261. | ||
Chonpathompikunlert P, Yoshitomi T, Vong LB, Imaizumi N, Ozaki Y, Nagasaki Y. Recovery of cognitive dysfunction via orally administered redox-polymer nanotherapeutics in SAMP8 mice. PLoS One. 2015;10(5):e0126013. | ||
Tosi G, Ruozi B, Belletti D, et al. Brain-targeted polymeric nanoparticles: in vivo evidence of different routes of administration in rodents. Nanomedicine. 2013;8(9):1373–1383. | ||
Che L, Zhou J, Li S, et al. Assembled nanomedicines as efficient and safe therapeutics for articular inflammation. Int J Pharm. 2012;439(1–2):307–316. | ||
Kimura T. A new family of nonsiliceous porous hybrids from bisphosphonates. J Nanosci Nanotechnol. 2013;13(4):2461–2470. | ||
Jung IW, Han HK. Effective mucoadhesive liposomal delivery system for risedronate: preparation and in vitro/in vivo characterization. Int J Nanomedicine. 2014;9:2299–2306. | ||
Skrajnowska D, Bobrowska-Korczak B, Tokarz A. Disorders of mechanisms of calcium metabolism control as potential risk factors of prostate cancer. Curr Med Chem. 2017;24(38):4229–4244. | ||
Hwang SR, Seo DH, Byun Y, Park JW. The anti-osteoporotic effects of the orally administered nano-complex were evaluated these animals following oral exposure at doses of 50 μg/kg. Int J Nanomedicine. 2016;11:4231–4246. | ||
Abramov E, Cassiola F, Schwob O, et al. Cellular mechanism of oral absorption of solidified polymer micelles. Nanomedicine. 2015;11:1993–2002. | ||
Jain S, Harde H, Indulkar A, Agrawal AK. Improved stability and immunological potential of tetanus toxoid containing surface engineered bilosomes following oral administration. Nanomedicine. 2014;10:431–440. | ||
Yp L, Sun LZ, Yao LH, Xiong XY, Zl L. Acute and subacute toxicity studies of Pluronic P85/poly (lactic acid) nanoparticles in mice. Micro Nano Lett. 2013;8:796–800. | ||
Semete B, Booysen L, Lemmer Y, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine. 2010;6:662–671. | ||
Lee CM, Lee TK, Kim DI, et al. Optical imaging of absorption and distribution of RITC-SiO2 nanoparticles after oral administration. Int J Nanomedicine. 2014;9:243–250. | ||
Brotons-Canto A, Gamazo C, Martín-Arbella N, et al. Evaluation of nanoparticles as oral vehicles for immunotherapy against experimental peanut allergy. Int J Biol Macromol. 2018;110:328–335. | ||
Heish HL, Lee CH, Lin KC. Development of Yam Dioscorin-loaded nanoparticles for paracellular transport across human intestinal Caco-2 cell monolayers. J Agric Food Chem. 2018;7:1175–1183. | ||
Lechuga S, Ivanov AI. Disruption of the epithelial barrier during intestinal inflammation: quest for new molecules and mechanisms. Biochim Biophys Acta Mol Cell Res. 1864;2017:1183–1194. | ||
Cheng Y, El-Kattan A, Zhang Y, Ray AS, Lai Y. Involvement of drug transporters in organ toxicity: the fundamental basis of drug discovery and development. Chem Res Toxicol. 2016;29(4):545–563. | ||
Talattof A, Price JC, Amidon GL. Gastrointestinal motility variation and implications for plasma level variation: oral drug products. Mol Pharm. 2016;13(2):557–567. | ||
Lopes CM, Bettencourt C, Rossi A, Buttini F, Barata P. Overview on gastroretentive drug delivery systems for improving drug bioavailability. Int J Pharm. 2016;510(1):144–158. | ||
Mcdermott J, Scholes P. Formulation design space: a proven approach to maximize flexibility and outcomes within early clinical development. Ther Deliv. 2015;6(11):1268–1278. | ||
Shekhawat PB, Pokharkar VB. Understanding peroral absorption: regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm Sin B. 2017;7(3):260–280. | ||
Weissig V, Pettinger TK, Murdock N. Nanopharmaceuticals (part 1): products on the market. Int J Nanomedicine. 2014;9:4357–4373. | ||
Nguyen VH, Lee BJ. Protein corona: a new approach for nanomedicine design. Int J Nanomedicine. 2017;12:3137–3151. | ||
Yang L, Kuang H, Zhang W, Aguilar ZP, Wei H, Xu H. Comparisons of the biodistribution and toxicological examinations after repeated intravenous administration of silver and gold nanoparticles in mice. Sci Rep. 2017;7(1):3303. | ||
Huh Y, Smith DE, Feng MR. Interspecies scaling and prediction of human clearance: comparison of small- and macro-molecule drugs. Xenobiotica. 2011;41(11):972–987. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.