Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Mycophenolate therapy in interstitial pneumonia with autoimmune features: a cohort study
Authors McCoy SS, Mukadam Z, Meyer KC ![]() , Kanne JP, Meyer CA
, Kanne JP, Meyer CA ![]() , Martin MD, Sampene E, Aesif SW, Rice LN, Bartels CM
, Martin MD, Sampene E, Aesif SW, Rice LN, Bartels CM
Received 5 May 2018
Accepted for publication 18 August 2018
Published 1 November 2018 Volume 2018:14 Pages 2171—2181
DOI https://doi.org/10.2147/TCRM.S173154
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Sara S McCoy,1 Zubin Mukadam,2 Keith C Meyer,2 Jeffrey P Kanne,3 Cristopher A Meyer,3 Maria D Martin,3 Emmanuel Sampene,4 Scott W Aesif,5 Laurie N Rice,6 Christie M Bartels1
1Division of Rheumatology, Department of Internal Medicine, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53705, USA; 2Division of Pulmonary and Critical Care, Department of Internal Medicine, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53705, USA; 3Department of Radiology, University of Wisconsin, Madison, WI 53792-3252, USA; 4Department of Biostatistics, School of Medicine and Public Health, University of Wisconsin, Madison, WI, USA; 5Department of Pathology, University of Wisconsin, Madison, WI 53792-3252, USA; 6Department of Pulmonology, SSM Health Dean Medical Group, Madison, WI 53715, USA
Objectives: International experts recently characterized interstitial pneumonia with autoimmune features (IPAF) as a provisional diagnosis for patients with interstitial lung disease who have characteristics of autoimmune disease but do not meet criteria for a specific autoimmune disease. We describe clinical characteristics of IPAF patients and examine responses to mycophenolate as a therapy for IPAF.
Methods: This retrospective cohort included adult patients meeting European Respiratory Society/American Thoracic Society classification criteria for IPAF. Sociodemographic, clinical, and pulmonary function test data were abstracted for patients with and without mycophenolate treatment and followed longitudinally from interstitial lung disease diagnosis for change in pulmonary function test results.
Results: We identified 52 patients who met criteria for IPAF. Of 52 IPAF patients, 24 did not receive mycophenolate and 28 did, with median time to mycophenolate treatment 22 months. Changes in FVC% and percentage predicted lung diffusion capacity for carbon monoxide (DLCO%) between the mycophenolate-treated and untreated groups were not significantly different (FVC% change P=0.08, DLCO% change P=0.17). However, there was a trend toward more rapid baseline decline of both FVC% and DLCO% in the mycophenolate-treated cohort before vs after mycophenolate therapy. The slope of both FVC% and DLCO% values improved after onset of mycophenolate exposure for the treated group, although this finding was not statistically significant.
Conclusion: Patients with IPAF might benefit from mycophenolate therapy. Larger prospective clinical trials are needed to evaluate the efficacy of mycophenolate for patients who meet criteria for IPAF.
Keywords: interstitial lung disease, autoimmune disease, connective tissue disease, mycophenolate
Introduction
Experts recently proposed the term “interstitial pneumonia with autoimmune features” (IPAF) as a research term for patients with an interstitial lung process consistent with idiopathic interstitial pneumonia (IP) combined with features of autoimmunity without meeting full diagnostic criteria for a specific connective tissue disease (CTD) diagnosis.1 Recently, the nomenclature and classification of criteria for IPAF were clarified by the European Respiratory Society/American Thoracic Society, allowing researchers to define and observe IPAF cohorts.1 A patient must meet criteria from two of the three prespecified domains to fulfill criteria for IPAF.1 These domains are clinical features of extrathoracic autoimmune disease, serologic evidence of autoimmune disease, and morphological criteria based on chest imaging, histopathology, or other multicompartment involvement.
Despite the clinical familiarity of concomitant interstitial lung disease (ILD) and nonspecific features of autoimmune disease, little information is available on treatment recommendations or clinical outcomes for patients with IPAF. In lung-dominant CTD, a disease similar but not identical to IPAF, mycophenolate, an immunosuppressive agent that impairs lymphocytogenesis, has shown therapeutic promise. In a retrospective cohort study of 19 patients with lung-dominant CTD, mycophenolate therapy was associated with trends toward improvement in pulmonary function test (PFT) values,2 but no comparison group was included. Other studies demonstrating benefits of mycophenolate have included a small number of patients with undifferentiated CTD and ILD within a broader pool of defined CTD-ILD patients.3 While mycophenolate has shown promise in treatment of various forms of CTD-ILD, little is known about the efficacy of mycophenolate as a therapy for IPAF.
We sought to identify and characterize a retrospective cohort of IPAF patients to examine the effectiveness of mycophenolate therapy. We present a cohort study of consecutive patients who fulfilled European Respiratory Society/American Thoracic Society classification criteria for IPAF.1 We examined longitudinal change in PFT and high-resolution computed tomography (HRCT) changes associated with mycophenolate therapy, hypothesizing that treatment with mycophenolate may attenuate lung function decline as reflected by PFT values and radiographic features on HRCT.
Methods
Inclusion/exclusion
This study was approved by the University of Wisconsin Health Sciences Institutional Review Board (IRB) with a waiver of individual informed consent for this minimal-risk retrospective study. Patient confidentiality was protected through approved IRB protocols. This was a retrospective-cohort study of adults ≥18 years old who met diagnostic criteria for IPAF.1 To create this academic system cohort, we utilized the electronic health record to identify patients who had both 1) positive antinuclear antibodies or any diagnosis of autoimmune disease (scleroderma, systemic lupus erythematosus, Sjögren’s syndrome/sicca, dermatomyositis, polymyositis, CTD, undifferentiated CTD) and 2) a diagnosis of ILD, nonspecific IP (NSIP), usual IP (UIP), lymphoid IP (LIP), diffuse alveolar damage, or organizing pneumonia (OP). Resultant records were reviewed and excluded if a patient met criteria for a definite autoimmune condition or had not seen both pulmonology and rheumatology departments through this system. Patients were reviewed to ensure they met the minimum of two of three required domains to satisfy the diagnosis of IPAF1 (Figure 1).
 | Figure 1 Exclusion and inclusion of patients who met criteria for IPAF diagnosis and saw both pulmonology and rheumatology departments within the University of Wisconsin health system. |
Medical records were extracted manually by MD reviewers (SM, ZM, and LR). Using a standardized tool for case review, we extracted all clinical and serologic data. Clinical experts performed HRCT and histopathological evaluations. Multicompartment involvement was defined as unexplained intrinsic airway disease (by PFT, HRCT, or histopathology), unexplained pulmonary vasculopathy (by presence of elevated mean pulmonary artery pressure ≥25 mmHg and pulmonary capillary-wedge pressure ≤15 mmHg), and unexplained pleural or pericardial effusion or thickening (diagnosed by HRCT or echocardiography).1
Outcomes/data collection
PFTs and 6-minute walk test (6MWT)
Our primary outcome of interest was a clinically significant change in PFTs. PFTs, including percentage predicted FVC and diffusion capacity of lungs for carbon monoxide (DLCO), were collected for all patients. PFT values averaged in 6-month time intervals were anchored on the patient’s date of first ILD diagnosis for longitudinal analysis of the entire cohort. Analysis of the treated group was anchored with baseline at therapy initiation. Distances for the 6MWT were extracted from each patient’s medical record to examine trends over time.
CT imaging
The first (baseline) and last HRCT were reviewed and scored independently by two of three blinded subspecialist thoracic radiologists (JPK, CAM, MDM) at five levels for total extent of disease, extent of reticulation, proportion of ground-glass opacity (GGO), and coarseness of reticulation as described by Goh et al.4 Discrepancies ≥20% for continuous scores or more than one grade for categorical scores were resolved independently by the third radiologist in blinded fashion. Morphologic patterns were determined independently by two radiologists (NSIP, OP, NSIP with OP overlap, LIP, other), with the third radiologist independently settling discrepancies. Our outcomes of interest included the global (total) extent of ILD, proportion of GGO, global extent of GGO and reticulation, and quantification of the coarseness of reticulation.
Pathology
All available pathological specimens were independently reviewed by a pathologist with specialty in ILD (SA). Specimens were categorized as NSIP, OP, NSIP with OP overlap, LIP, interstitial lymphoid aggregates with germinal centers, or diffuse lymphoplasmacytic infiltration (with or without lymphoid follicles).
Variable definitions
Our primary exposure of interest was mycophenolate. Mycophenolic acid use was considered equivalent to mycophenolate mofetil for the purposes of this study. Exposure start and end dates were recorded using electronic health-record prescription data. Additional clinical data extracted for analysis included age, sex, tobacco use, cardiovascular disease (defined as coronary artery disease, congestive heart failure, or cerebrovascular disease), gastroesophageal reflux (GER), malignancy, obstructive sleep apnea, pulmonary arterial hypertension, and pulmonary embolism. Clinical features abstracted included inflammatory arthritis, Raynaud’s phenomenon, digital fissures/ulcers, telangiectasias, digital edema, serositis, extensor surface rash, and immunosuppressive/immunomodulatory therapy (azathioprine, cyclophosphamide, leflunomide, methotrexate, and prednisone). Additional medications recorded include azithromycin, proton-pump inhibitors, and ranitidine. Laboratory data collection included blood counts, complement, creatinine, inflammatory markers, ANA, anti-Jo1, anticitrullinated cyclic protein antibodies, double-stranded DNA, MDA5, PM-Scl, rheumatoid factor (double or more the upper limit of normal), ribonucleoprotein antibodies, anti-Scl70, anti-Smith antibodies, anti-SSA/SSB antibodies, and other tRNA antibodies.
Statistical analysis
Differences in baseline characteristics by mycophenolate status were compared via χ2 or Fisher’s exact tests for categorical variables and Student’s t-test for continuous variables. We considered P<0.05 significant. Simple linear regression and t-tests of the calculated change variable were performed to evaluate differences in 6MWT and PFTs in patients within and between groups with and without mycophenolate exposure. Paired t-tests were performed to compare pre- and postmycophenolate slopes in the exposed groups. Patients were censored at time of death, lung transplant, or end of data availability (patient was lost to follow-up or last review date in January 2017). Kaplan–Meier survival analyses were performed to examine how mycophenolate exposure impacted survival, time to transplant, or time to PFT decline. Statistical analysis was performed using Stata version 14 (StataCorp LP, College Station, TX, USA) and GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Baseline characteristics
We identified 52 adult patients meeting criteria for IPAF, 33 (63%) female (aged 31–83 years). Of the 52 patients with IPAF, 24 (46%) were not treated with mycophenolate (unexposed) and 28 (54%) were treated with mycophenolate (exposed) at median 22 months from diagnosis (Table 1). The median duration of mycophenolate exposure was 41 months from ILD diagnosis (range 16 days to 93 months). The median dose of mycophenolate was 2,000 mg daily.
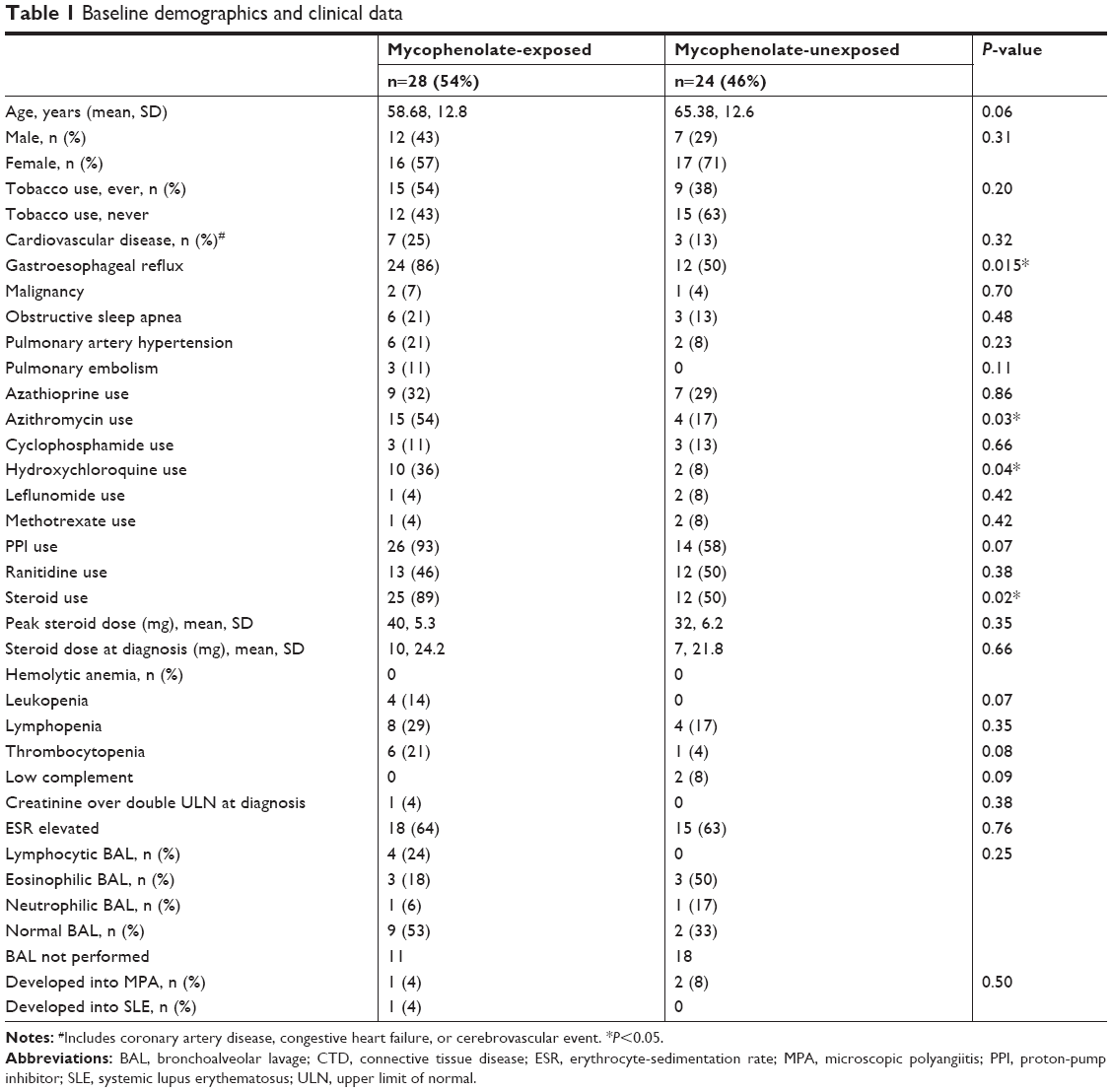 | Table 1 Baseline demographics and clinical data |
Clinical characteristics were generally similar between the mycophenolate-exposed and -unexposed groups, while some differences were noted. There were significantly higher numbers of patients with GER in the mycophenolate-exposed than the mycophenolate unexposed group. No other clinical characteristics, including cardiovascular disease, malignancy, obstructive sleep apnea, pulmonary arterial hypertension, and pulmonary embolism, were different between the two groups. The mycophenolate-exposed group was treated more frequently with azithromycin, hydroxychloroquine, and steroids. Laboratory features including hematologic abnormalities, complement, creatinine, and inflammatory markers were similar between the two groups. Bronchoalveolar lavage tended to be lymphocyte-predominant in the mycophenolate group, although this finding did not reach statistical significance. Four patients with IPAF went on to develop a confirmed autoimmune disease: microscopic polyangiitis (n=3) and SLE (n=1). IPAF criteria were compared between mycophenolate-exposed and unexposed groups within the three domains. The clinical domains were not significantly different between the mycophenolate-exposed and unexposed groups (Table 2). Within the serologic domain, autoantibodies were also similar between the mycophenolate-exposed and unexposed groups, with the exception of anti-RNP antibodies, which were more frequent in the mycophenolate-treated group (P=0.03). Multicompartment involvement did not vary between mycophenolate-exposed and unexposed groups. There was no difference in the manner by which IPAF patients achieved diagnostic criteria (Table 2). Within the mycophenolate-exposed group, five patients reported adverse effects that led to cessation or change of therapy: nausea (n=2), gastrointestinal upset (n=1), diarrhea (n=1), and myalgia (n=1).
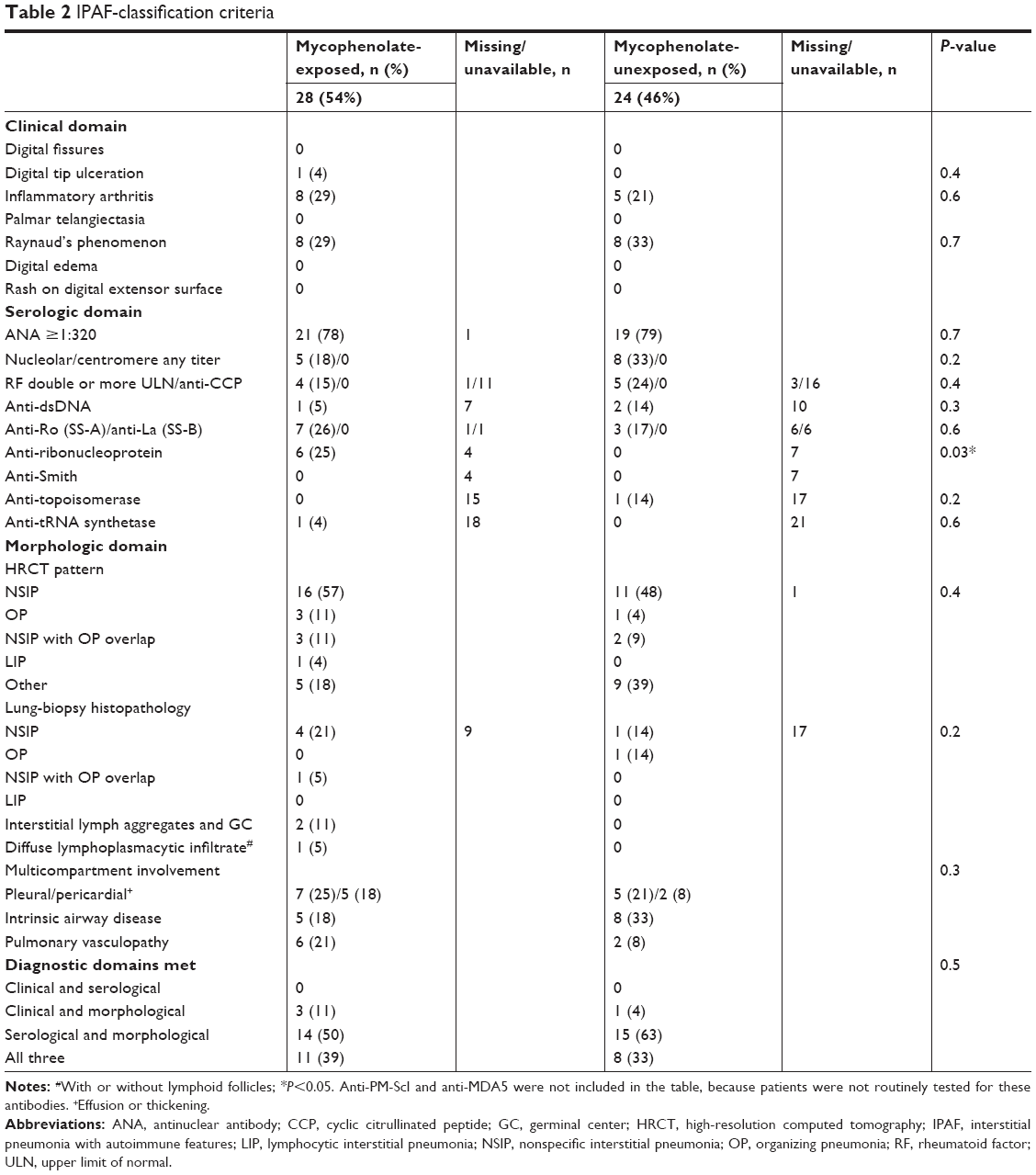 | Table 2 IPAF-classification criteria |
Baseline biopsy findings
Thirty patients had pathology analysis of lung tissue (of which 26 were available for review by our pathologist, and seven of these were explants or autopsy). Of the 26 biopsies, 19 from the mycophenolate-exposed group and seven from the unexposed group were available for review. Patterns of involvement were broad and in many cases mixed: NSIP (n=5), OP (n=1), NSIP with OP (n=1), LIP (n=0), interstitial lymphoid aggregates with germinal centers (n=2), UIP (n=1), UIP with atypical features (n=8), acute lung injury (n=4), interstitial lymphoplasmacytic inflammation (n=6), and normal (n=2). Interestingly, while many pathological specimens were best classified as of UIP pattern histologically, half (n=13) had atypical features, such as extensive peribronchiolar metaplasia. Among those who had GER, 12 had peribronchiolar metaplasia, whereas among those patients without GER, nine had peribronchiolar metaplasia (P=0.14).
PFT and 6MWT findings
Mycophenolate-treated compared to untreated
The mean average decline in FVC% and DLCO% between the mycophenolate-treated (n=28) and untreated (n=24) groups was not significantly different (FVC% change, P=0.08; DLCO% change, P=0.17; Table 3). However, there was a trend toward a more rapid decline in both FVC% and DLCO% in the mycophenolate-exposed group (Figure 2). The mycophenolate-unexposed group had relatively stable PFTs. Change in FVC% was compared between the group without GER and with GER. Change in FVC was stable in the no-GER group (FVC% change 0.06 in 24 months), whereas in the GER group the FVC% decreased (0.32 in 24 months, P=0.04).
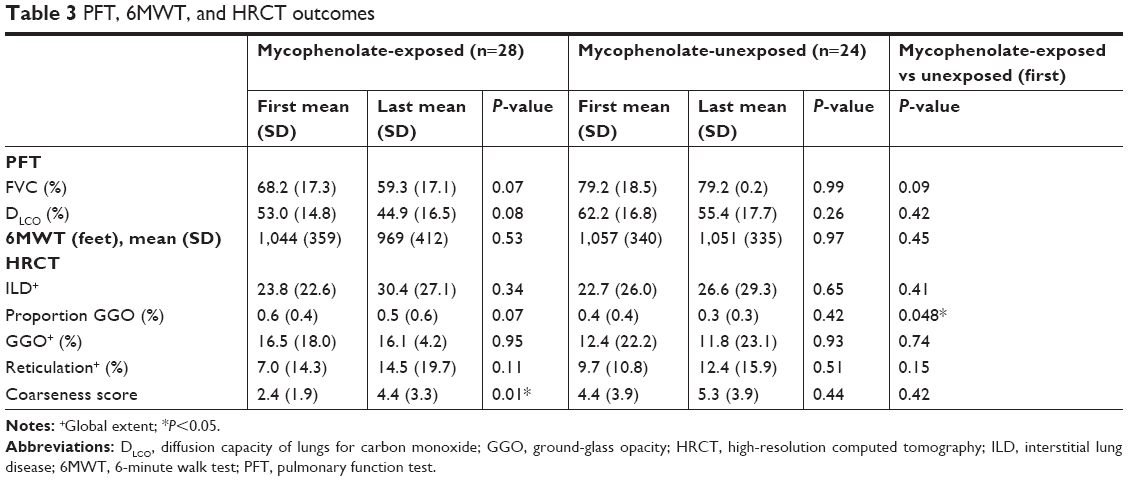 | Table 3 PFT, 6MWT, and HRCT outcomes |
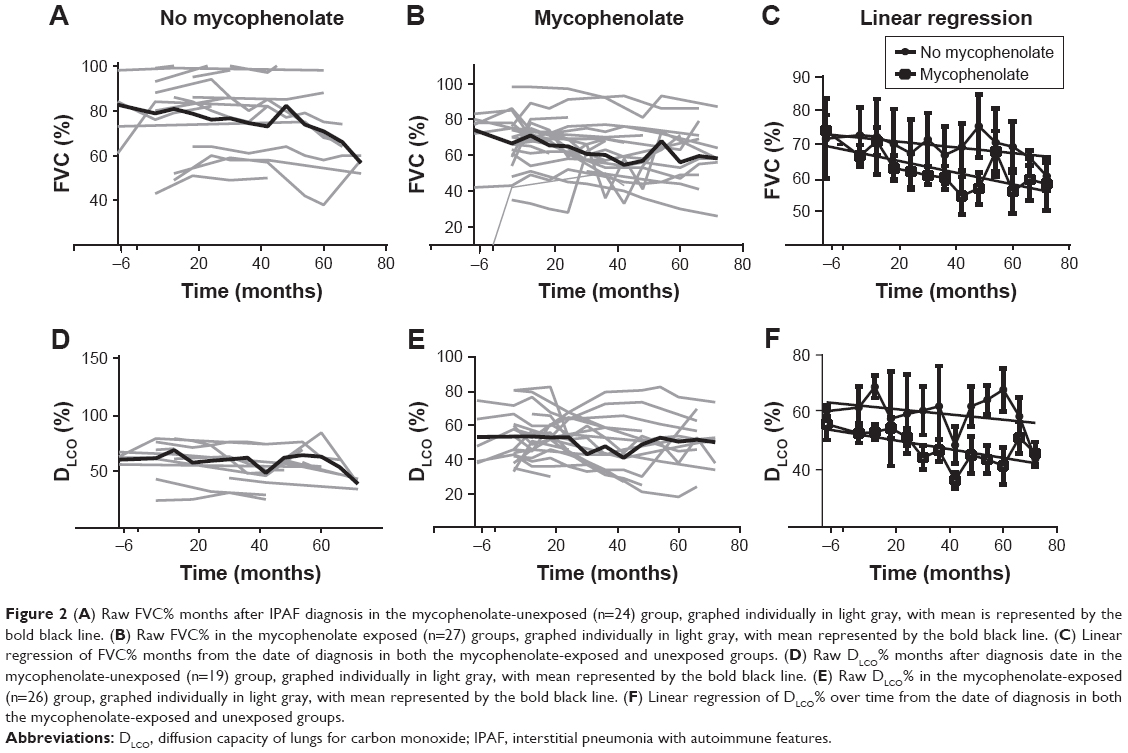 | Figure 2 (A) Raw FVC% months after IPAF diagnosis in the mycophenolate-unexposed (n=24) group, graphed individually in light gray, with mean is represented by the bold black line. (B) Raw FVC% in the mycophenolate exposed (n=27) groups, graphed individually in light gray, with mean represented by the bold black line. (C) Linear regression of FVC% months from the date of diagnosis in both the mycophenolate-exposed and unexposed groups. (D) Raw DLCO% months after diagnosis date in the mycophenolate-unexposed (n=19) group, graphed individually in light gray, with mean represented by the bold black line. (E) Raw DLCO% in the mycophenolate-exposed (n=26) group, graphed individually in light gray, with mean represented by the bold black line. (F) Linear regression of DLCO% over time from the date of diagnosis in both the mycophenolate-exposed and unexposed groups. |
Mycophenolate-treated group before compared to after initiation of treatment
Simple linear regression of DLCO% before and after mycophenolate therapy (n=23) showed improvement in DLCO% slope after mycophenolate therapy; however, this did not achieve statistical significance (P=0.55; Figure 3). Sensitivity analysis was performed with inclusion of only patients who had at least one DLCO% measurement before and after mycophenolate therapy (n=18), and the trend persisted (P=0.48). Paired t-tests in this group did not reach significance, but achieved P=0.06. Excluding patients who had received lung transplants from the analysis yielded a statistically significant improvement in slope of DLCO% before and after mycophenolate therapy (n=18, P=0.009). When transplant patients were included with last observation carried forward, the trend was also consistent, with the slope improving after mycophenolate therapy (P=0.75). Transplant compared to nontransplant mycophenolate-exposed patients had significantly lower baseline DLCO% (P=0.02).
 | Figure 3 (A) Raw FVC% in months before and after mycophenolate exposure among the mycophenolate-exposed. Data graphed individually (n=26) in light gray, and mean with SEM represented with the bold black line. (B) Linear regression of FVC% before and after mycophenolate exposure with SD. (C) DLCO% before and after mycophenolate exposure, graphed individually (n=26) in light gray and mean with SEM represented with the bold black line. (D) Linear regression of DLCO% before and after mycophenolate exposure with SD. |
Within the mycophenolate group, 26 of 28 patients had FVC% recorded around the time of mycophenolate initiation (Figure 3). The slope of mycophenolate-exposed patients improved after exposure, but this increase did not reach statistical significance (P=0.15). Sensitivity analysis was performed with inclusion of patients who had at least one time point before and after mycophenolate initiation (n=19), and the slope continued a trend toward improvement after mycophenolate exposure compared to before mycophenolate exposure (P=0.09). Paired t-tests in this group did not reach significance (P=0.97). If patients who went on to receive lung transplants were removed, analysis of FVC% demonstrated an improved slope after mycophenolate exposure, but this did not reach statistical significance (P=0.28). When transplant patients were included with last observation carried forward, the trend persisted with the slope improving after mycophenolate therapy (P=0.15). 6MWT did not change significantly over time nor vary significantly between mycophenolate groups.
Imaging outcomes
Baseline findings
One patient did not have HRCT available for radiology to review. Baseline HRCT patterns of ILD did not vary significantly between the mycophenolate-exposed and unexposed groups. However, mycophenolate-exposed groups more frequently had NSIP by HRCT (n=16, 57%; unexposed, n=11, 48%; P=0.6). HRCT scores in the mycophenolate-exposed group tended to have a higher global extent of GGO and lower extent of reticulation with less overall coarseness than the unexposed group, but trends did not reach statistical significance (Table 3). Baseline HRCT showed increased GGO in the mycophenolate-exposed than the unexposed group (P=0.048; Figure S1). In the mycophenolate-exposed group, six patients (21%) had hiatal hernia and four (14%) esophageal dilation. In the mycophenolate-unexposed group, seven patients (29%) had hiatal hernia and nine (38%) esophageal dilation. When transplant patients treated with mycophenolate were compared to nontransplant patients treated with mycophenolate, the first available HRCT scan had significantly more reticulation (P≤0.0001) and significantly less GGO (P=0.03).
Change in HRCT over time
When the last available HRCT scan was compared to the first available HRCT scan, coarseness increased significantly between first and last HRCT in the mycophenolate group (P=0.01). There was no significant difference in global ILD, GGO%, reticulation percentage, or overall proportion GGO between baseline and final HRCT in the mycophenolate-treated or untreated groups.
Other outcomes
Two deaths occurred in the mycophenolate-exposed group compared to five in the mycophenolate-unexposed group. Significantly more patients in the mycophenolate-exposed group (n=7) received a lung transplant than those in the mycophenolate-unexposed group (n=0; P=0.01). Kaplan–Meier curves based on time to decline (≥10 FVC% and ≥15% DLCO%), transplant, and death were not significantly different between the two groups. The log-rank test did not yield any statistically significant difference for this composite end point (P=0.41).
Discussion
In this study, we carefully identified a cohort of IPAF patients to evaluate the observed efficacy of mycophenolate therapy on PFTs, 6MWT, and HRCT. In comparison to other observational studies of IPAF, our overall demographic composition demonstrated similar though not identical frequency of clinical, laboratory, and morphologic characteristics.5–7
Although we did not find a difference in PFT change or mortality in mycophenolate-exposed compared to unexposed patients, mycophenolate-exposed patients showed a trend toward lower baseline PFT scores and greater decline in PFT scores that began preexposure. Patients who present with a more significant disease burden may be more likely to receive therapy with mycophenolate, especially if lung function appears to be declining progressively over time. It has been proposed that IPAF patients can be stratified into a lower-risk cohort with a favorable survival pattern, based on the presence of a clinical domain of IPAF diagnosis in combination with HRCT or histopathology to fulfill IPAF criteria.5 However, we found that mycophenolate-exposed and unexposed groups met the clinical and morphologic domains with similar frequency, making this a less likely cause of the difference in PFT trends between these two groups. Unexplained pulmonary vasculopathy, a proposed risk factor for mortality, was seen in six mycophenolate-exposed patients.8 Multicompartment involvement in IPAF has been proposed to be associated with increased mortality,5,8 but there was no difference in rates of multicompartment involvement between exposed and unexposed patients. Alternatively, mycophenolate exposure itself possibly led to decline in PFT scores. We do not favor this theory, because of the improvement seen after mycophenolate therapy within the mycophenolate-exposed group.
Interestingly, within the mycophenolate-exposed group, we were able to show that there was a trend toward improvement in PFT scores after initiation of mycophenolate. This difference reached statistical significance when those who required lung transplants were removed. These findings indicate that a subgroup of IPAF patients may benefit from mycophenolate therapy. HRCT in patients who required transplants demonstrated significantly more reticulation and less GGO than those who did not go on to require transplants. Patients who required transplants had significantly worse DLCO% on PFTs. Previous observational studies of IPAF have identified DLCO as a predictor of survival,5 which is consistent with previous studies of risk-factor identification in other diseases of pulmonary fibrosis.9,10 Histopathologically, IPAF patients frequently have coexistence of NSIP and UIP patterns;8 however, our findings may support patients with less reticulation and more GGO responding more robustly to mycophenolate therapy. Similar findings of GGO correlating positively with therapeutic response have been seen in systemic sclerosis-related ILD.11
We found that mycophenolate-exposed patients had increased GER frequency. This is potentially relevant, as our pathologist identified relatively frequent rates of peribronchiolar metaplasia. Peribronchiolar metaplasia has been reported in both aspiration and systemic sclerosis-related LD.12,13 It is also possible that aspiration may contribute to the pathogenesis of IPAF-related LD, supporting potential benefits of aggressive treatment of pathological GER in this population. Although we did not find any difference in GER frequency in patients with and without peribronchiolar metaplasia, there was a trend toward association with GER and peribronchiolar metaplasia. Similar to other studies that have demonstrated detrimental effects of GER on lung function,14,15 we found that GER was associated with greater reduction in FVC% over time, lending additional support to GER treatment.
Despite strengths including robust IPAF-inclusion definitions and data collection, this study has several limitations. First, because this was a retrospective-cohort study, we were limited to reporting associations, but unable to identify causal relationships. Second, EMR prescription does not ensure consistent exposure. Third, mycophenolate exposure might reflect indication bias, as patients were not randomized to receive mycophenolate therapy. We also considered that the trend toward improvement of PFTs after mycophenolate exposure could have reflected a survivor effect. However, when we performed sensitivity analyses with last value carried forward, this trend persisted. Nonetheless, despite several sensitivity analyses, multiple confounders undoubtedly exist in this observational retrospective-cohort study. Many patients were treated with other immunosuppressive therapies and varying doses of corticosteroids. However, the dose of corticosteroids at diagnosis and peak corticosteroid dose were similar between the two groups, so likely would not explain the described differences. Given the relatively low number of patients included in this cohort, we could not perform multivariate analysis. Moreover, as a tertiary referral center, our IPAF cohort may reflect a referral bias toward more severe disease.
Conclusion
Mycophenolate therapy might attenuate ILD progression in patients with IPAF, and findings suggest that it could be more beneficial in patients with more GGO and less reticulation on HRCT. Further study of mycophenolate in a prospective, randomized controlled trial for IPAF treatment is warranted to investigate further whether mycophenolate is a beneficial therapy for IPAF patients with clinically significant/progressive ILD.
Acknowledgments
This project described was supported by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. CMB receives unrelated peer-reviewed grant funding from Independent Grants for Learning and Change (Pfizer). The abstract of this paper was presented at the 2017 American College of Rheumatology Conference as a poster presentation/conference talk with interim findings. The abstract was published in the ACR’s Meeting Abstracts (ACRabstracts.org, abstract 1171).
Disclosure
The authors report no conflicts of interest in this work.
References
Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–987. | ||
Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest. 2010;138(2):251–256. | ||
Swigris JJ, Olson AL, Fischer A, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest. 2006;130(1):30–36. | ||
Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177(11):1248–1254. | ||
Oldham JM, Adegunsoye A, Valenzi E, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767–1775. | ||
Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: A single center experience. Respir Med. 2016;119:150–154. | ||
Ferri C, Manfredi A, Sebastiani M, et al. Interstitial pneumonia with autoimmune features and undifferentiated connective tissue disease: Our interdisciplinary rheumatology-pneumology experience, and review of the literature. Autoimmun Rev. 2016;15(1):61–70. | ||
Adegunsoye A, Oldham JM, Valenzi E, et al. Interstitial pneumonia with autoimmune features: value of histopathology. Arch Pathol Lab Med. 2017;141(7):960–969. | ||
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. | ||
Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–728. | ||
Yabuuchi H, Matsuo Y, Tsukamoto H, et al. Evaluation of the extent of ground-glass opacity on high-resolution CT in patients with interstitial pneumonia associated with systemic sclerosis: comparison between quantitative and qualitative analysis. Clin Radiol. 2014;69(7):758–764. | ||
Yousem SA, Faber C. Histopathology of aspiration pneumonia not associated with food or other particulate matter: a clinicopathologic study of 10 cases diagnosed on biopsy. Am J Surg Pathol. 2011;35(3):426–431. | ||
de Souza RB, Borges CT, Capelozzi VL, et al. Centrilobular fibrosis: an underrecognized pattern in systemic sclerosis. Respiration. 2009;77(4):389–397. | ||
Martinez CH, Okajima Y, Murray S, et al. Impact of self-reported gastroesophageal reflux disease in subjects from COPDGene cohort. Respir Res. 2014;15:62. | ||
Gavini S, Borges LF, Finn RT, et al. Lung disease severity in idiopathic pulmonary fibrosis is more strongly associated with impedance measures of bolus reflux than pH parameters of acid reflux alone. Neurogastroenterol Motil. 2017;29(5):e13001. |
Supplementary material
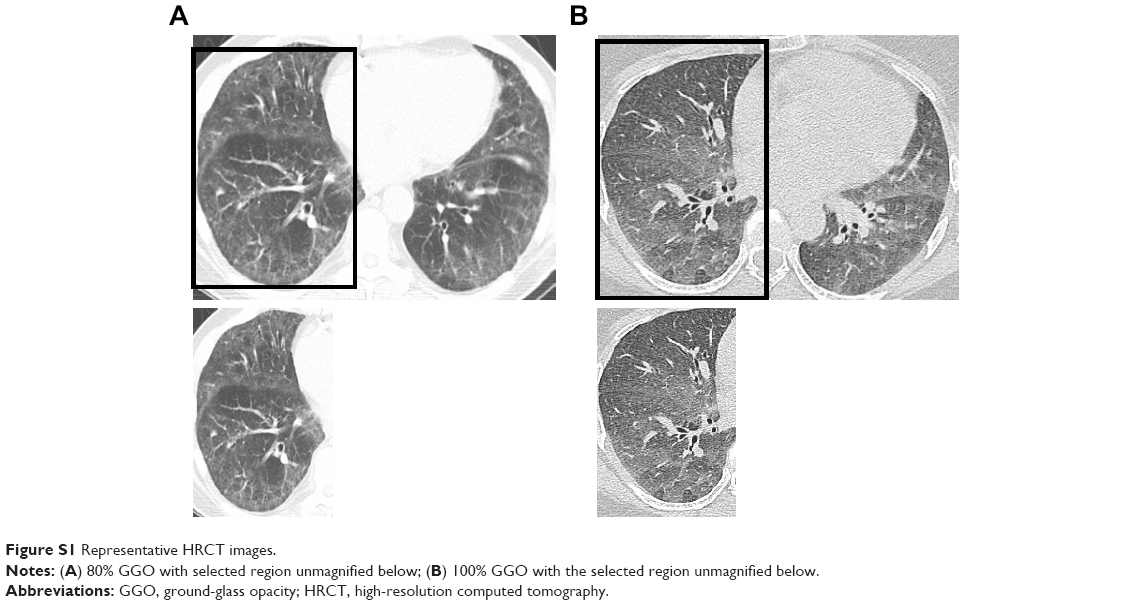 | Figure S1 Representative HRCT images. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.