Back to Journals » Infection and Drug Resistance » Volume 11
Mutations in the gyrA, parC, and mexR genes provide functional insights into the fluoroquinolone-resistant Pseudomonas aeruginosa isolated in Vietnam
Authors Nguyen KV, Nguyen TV, Nguyen HTT, Le DV ![]()
Received 28 July 2017
Accepted for publication 22 November 2017
Published 28 February 2018 Volume 2018:11 Pages 275—282
DOI https://doi.org/10.2147/IDR.S147581
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
Kinh Van Nguyen,1,2,* Trung Vu Nguyen,1,3,* Hang Thi Thuy Nguyen,4 Duyet Van Le1
1Clinical Laboratories, National Hospital for Tropical Diseases, 2Infectious Department, Hanoi Medical University, 3Microbiology Department, Hanoi Medical University, 4Department of Microbiology, National Geriatric Hospital, Hanoi, Vietnam
*These authors contributed equally to this work
Introduction: Pseudomonas aeruginosa has many mechanisms of resistance to fluoroquinolones. The main mechanism is to change the effect of two enzymes that open the DNA helix – the enzyme DNA gyrase (gyrA) and the topoisomerase IV (parC). In addition, mutations that render the MexAB-oprM pump (mexR) dysfunctional, leading to its overexpression, also enhance resistance to fluoroquinolones. In this study, we aim to detect point mutations of gyrA, parC, and mexR genes that are predicted to be associated with fluoroquinolone resistance in 141 fluoroquinolone-resistant clinical isolates of P. aeruginosa isolated in Vietnam during 2013–2016.
Methods: We tested minimum inhibitory concentrations (MICs) of fluoroquinolone antibiotics in 141 clinical isolates of P. aeruginosa using the VITEK 2 Compact System, followed by PCR assay, to detect and clone the fluoroquinolone resistance-determining region (FRDR) of gyrA, parC, and mexR. Point mutations were analyzed through Sanger sequencing, and the correlation between genetic mutations and phenotypic resistance of 141 clinical isolates was undertaken.
Results: Fluoroquinolone-resistant substitution mutations such as Ile for Thr83 and Met for Thr133 in gyrA, Leu for Ser87 in parC, and Val for Glu126 in the repressor of mexR were mainly detected. Comparative analytical data indicated that amino acid alterations within the gyrA and parC genes are highly associated with resistance to ciprofloxacin (CIP) and levofloxacin (LEV) in the isolates, whereas alterations in the efflux regulatory mexR gene are not highly consistent with resistance in these isolates. Moreover, fluoroquinolone-resistant clinical isolates of P. aeruginosa were mainly isolated from pus and sputum specimens.
Conclusion: In clinical isolates of P. aeruginosa, a high correlation was observed between MICs of CIP and LEV and alterations in gyrA and parC genes. However, mutations occurring in mexR did not highly correlate with the antibiotic resistance of the bacterium.
Keywords: fluoroquinolone-resistant Pseudomonas aeruginosa, fluoroquinolone resistance-determining region, FRDR, mutation, gyrA, parC, mexR
Introduction
Pseudomonas aeruginosa is a gram-negative bacterium and a clinically significant pathogen that causes opportunistic infections – particularly nosocomial infections such as pneumonia, wound infections, sepsis, and gastrointestinal infections.1,2 The bacterium is highly resilient in the external environment, causing major infections and severe clinical symptoms due to virulence factors involved in the infective process. Carbapenems, aminoglycosides, and fluoroquinolones are the three classes of important antibiotics that are used as potent agents for the treatment of Pseudomonas infections.3–5 However, excessive use of broad-spectrum antibiotics has resulted in the emergence of highly resistant strains of P. aeruginosa. Data from previous studies indicate that multidrug-resistant P. aeruginosa strains can tolerate carbapenems, aminoglycosides, and fluoroquinolone drugs. As a result, the potency of these antibiotics against serious infections of P. aeruginosa gets diminished.4,6–9
The fluoroquinolone drugs – ciprofloxacin (CIP) and levofloxacin (LEV) – have several targets such as DNA gyrase (type II topoisomerase), topoisomerase IV, and the efflux pump regulatory protein encoded by mexR.5,10–12 Fluoroquinolone resistance is presented mainly by mutations in the fluoroquinolone resistance-determining region (FRDR) of gyrA, which codes for DNA gyrase subunits, and parC, which codes for topoisomerase IV subunits. Consequently, mutations within the topoisomerase II (gyrA), topoisomerase IV (parC), and the efflux regulatory (mexR) genes are the main mechanisms of fluoroquinolone resistance in gram-negative bacteria.13–15 Amino acid alterations found in gyrA, parC, and mexR are associated with high-level fluoroquinolone resistance in gram-negative bacteria, including P. aeruginosa.5,11,15–17
Several studies on fluoroquinolone resistance have revealed that the prevalence of mutations in gyrA, parC, and mexR for P. aeruginosa is significantly localized to countries such as South Korea, Taiwan, People’s Republic of China, and Japan.5,11,16,17 However, data on fluoroquinolone-resistant clinical isolates of P. aeruginosa in Vietnam are not well established. In this study, mutations of gyrA, parC, and mexR in fluoroquinolone-resistant clinical isolates of P. aeruginosa from Vietnam are investigated, and the correlation between fluoroquinolone minimum inhibitory concentrations (MICs) and point mutations in gyrA, parC, and mexR are compared.
Materials and methods
Clinical isolates of P. aeruginosa
We collected 141 clinical isolates of P. aeruginosa from 13 national hospitals located throughout Vietnam during 2013–2016. Of these, 21 isolates were susceptible to both CIP and LEV (MIC of CIP≤1 µg/mL, MIC of LEV≤2 µg/mL), 10 isolates had intermediate resistance to both CIP and LEV (MIC of CIP=2 µg/mL, MIC of LEV=4 µg/mL), and 110 isolates were highly resistant to both CIP and LEV (MIC of CIP≥4 µg/mL, MIC of LEV≥8 µg/mL). To test the antibiotic susceptibility of all clinical isolates of P. aeruginosa, MICs of fluoroquinolone antibiotics were determined at the Bacterial Laboratory (National Hospital for Tropical Diseases, Vietnam) using the VITEK 2 Compact System (BioMerieux, Marcy I’Etoile, France).
DNA extraction and PCR amplification of partial sequences of gyrA, parC, and mexR
Qiagen kits (QIAamp DNA Mini Kit, Qiagen Sciences, Germantown, MD, USA) were used for extraction of total DNA from P. aeruginosa isolates in accordance with the manufacturer’s protocol. Briefly, cell pellets were resuspended in 200 µL of the given lysis buffer (Buffer AL [>8 mM EDTA and >0.5% SDS]) containing 20 µL proteinase K, and incubated at 56°C for 30 minutes. Thereafter, 4 µL RNase was added and the sample was treated in accordance with the manufacturer’s protocol (for microfuge-scale preparations).
The PCR amplification of gyrA, parC, and mexR of P. aeruginosa was done in a 50-μL reaction volume. The PCR mixture was prepared in a 0.2 mL thin-walled tube, containing 25 ng template DNA (total DNA isolated from P. aeruginosa isolates), 30 pmol primers, and 0.2 mM dNTP mix (containing equimolar quantities of dATP, dCTP, dGTP, and dTTP). The primers gyr-A1 (5′–GTGTGCTTTATGCCATGAG–3′) and gyr-A2 (5′–GGTTTCCTTTTCCAGGTC–3′) were used to amplify 287 bp of the FRDR of the gyrA gene. The primers par-C1 (5′–CATCGTCTACGCCATGAG–3′) and par-C2 (5′–AGCAGCACCTCGGAATAG–3′) were used to amplify 267 bp of the FRDR of the parC gene. The primers mex-R1 (5′–CTGGATCAACCACATTTACA–3′) and mex-R2 (5′–CTTCGAAAAGAATGTTCTTAAA–3′) were used to amplify 503 bp of the FRDR of the mexR gene.10 The final volume was made up with 10X Pyrococcus furiosus (Pfu) buffer and distilled water. In addition, 1 μL Pfu DNA polymerase was added to the reaction mixture after denaturation at 100°C for 2 minutes. The PCR was carried out in a Genius PCR Thermal Cycler (Techne) with a 105°C heated lid. The amplification parameters included 35 cycles of initial heat activation at 95°C for 45 seconds, annealing at 51°C for 30 seconds, elongation at 72°C for 30 seconds, and a final 72°C elongation step for 10 minutes.
Sanger sequencing and partial sequence analysis of gyrA, parC, and mexR
Dideoxy sequencing of PCR products was undertaken using BigDye™ Terminator Chemistry v. 3.1 (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s instructions. As described previously, the forward and reverse primers for gyrA, parC, and mexR were used as sequencing primers using the ABI 3130 Bio-analyzer (Applied Biosystems).
Original nucleotide sequences of gyrA, parC, and mexR were obtained from the GenBank nucleotide sequence database with accession numbers L29147, AB003428, and U23763, respectively. Target regions of these three genes were analyzed for all 141 clinical isolates of P. aeruginosa, using ATGC 7.2 (supported by the Japanese ACC program). The DNA sequences were then compared with original nucleotide sequences of gyrA (accession number L29147), parC (accession number AB003428), and mexR (accession numberU23763) genes in the GenBank data for wild-type P. aeruginosa.
Results
Antibiotic resistance of P. aeruginosa to CIP and LEV
In total, 141 clinical isolates of P. aeruginosa were collected from different specimens and all of these isolates were then tested for antimicrobial susceptibility to both CIP and LEV. Considering the susceptibility breakpoints suggested by the Clinical and Laboratory Standard Institute in 2015 (CLSI 2015), the majority of isolates (134/141 strains) were mainly collected from pus, sputum, urine, and blood – of which 21 strains were found to be susceptible, 10 strains were intermediate, and 102 strains were highly resistant to both CIP and LEV. In addition, the minority of clinical isolates (7/141 strains) were collected from peritoneal fluid, bronchial lavage, and cerebrospinal fluid, with no strain found to be susceptible to CIP and LEV – all of these isolates were revealed to be highly resistant to both CIP and LEV (Table 1).
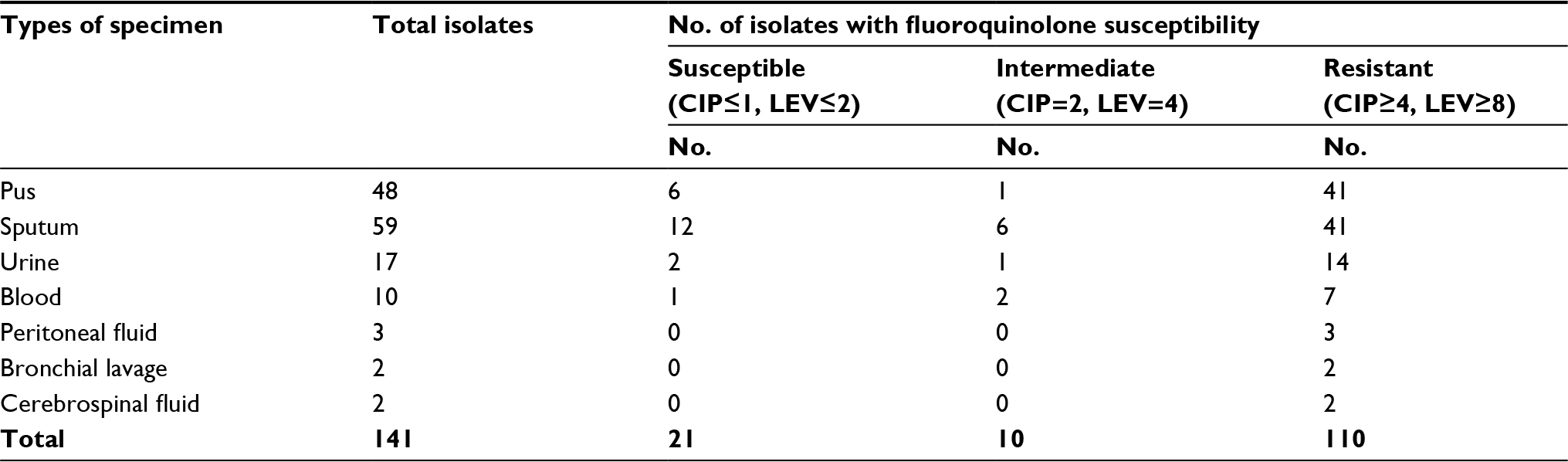 | Table 1 Results of a screen for fluoroquinolone susceptibility from 141 clinical isolates of P. aeruginosa Abbreviations: CIP, ciprofloxacin; LEV, levofloxacin. |
Amino acid alterations in gyrA, parC, and mexR
Partial DNA fragments of gyrA, parC, and mexR were amplified using PCR, and nucleotide sequences of all the PCR-amplified fragments were determined using Sanger sequencing. Alterations found in 141 clinical isolates of P. aeruginosa were classified into eight groups according to the pattern of amino acid changes detected in gyrA, parC, and mexR as follows (Table 2): Group I had no mutations in all three genes; Group II mutations only occurred in gyrA; Group III mutations only occurred in parC; Group IV mutations only occurred in mexR; Group V mutations occurred in both gyrA and parC; Group VI mutations appeared in all three genes; Group VII mutations occurred in gyrA and mexR; and, finally, Group VIII mutations appear in both parC and mexR.
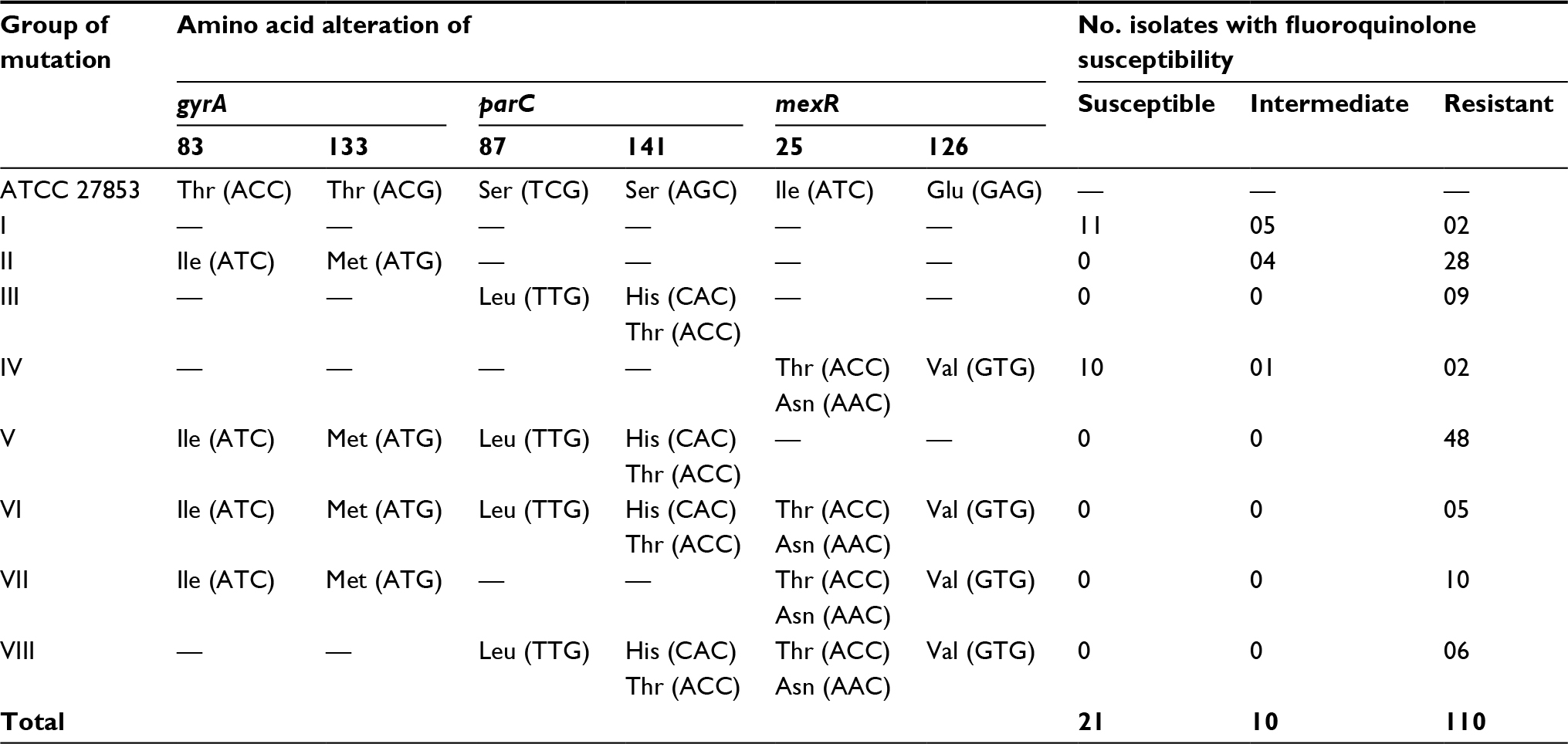 | Table 2 Comparison of amino acid alterations in gyrA, parC, and mexR with fluoroquinolone susceptibility of P. aeruginosa isolates |
Mutations found in the topoisomerase II, topoisomerase IV, and the efflux regulatory mexR genes of P. aeruginosa isolates were then compared with the corresponding nucleotide sequences of the gyrA, parC, and mexR genes of P. aeruginosa ATCC 27896. Amino acid replacements found in gyrA, parC, and mexR are listed in Table 2. Of these eight mutation groups, the majority of the P. aeruginosa clinical isolates, resistant to CIP and LEV, were found in groups V (48 isolates), II (28 isolates), VII (10 isolates), and III (09 isolates). The results showed that some of the P. aeruginosa clinical isolates possessing a single amino acid substitution were either susceptible or intermediately resistant to CIP and LEV. Susceptible P. aeruginosa isolates were found only in groups I (11 isolates) and IV (10 isolates), whereas intermediately resistant isolates were found in groups I (five isolates), II (four isolates), and IV (one isolate). In addition, the results showed that mutations – either only in gyrA or in both gyrA and parC – were most frequently found in the P. aeruginosa isolates resistant to CIP and LEV. In contrast, mutations found in mexR were consistent with only a small proportion of the P. aeruginosa isolates resistant to CIP and LEV.
Clinical isolates of P. aeruginosa, sorted by amino acid alterations found in the target genes (gyrA, parC, and mexR), and the fluoroquinolone susceptibility pattern of clinical isolates of P. aeruginosa are shown in Tables 2 and 3. The comparison of the DNA sequences of P. aeruginosa ATCC 27853 with the gyrA DNA sequences in 141 P. aeruginosa isolates showed that an alteration in the gyrA gene at codon 83 (Thr83→Ile) was found in 76 clinical isolates of P. aeruginosa, harboring gyrA mutations (groups II, V, VI, and VII), as shown in several studies. However, a novel single-nucleotide mutation was also found in gyrA at codon 133 (Thr133→Met) in a total of 51 clinical isolates of P. aeruginosa. Further analysis of the data revealed that a total of 93 fluoroquinolone-resistant isolates had mutations in gyrA – of which, 47 isolates possessed only a single-nucleotide mutation at codon 83 (Thr83→Ile), 19 isolates were found to have a novel single-nucleotide alteration at codon 133 (Thr133→Met), and the remaining isolates possessed double mutations in gyrA (Thr83→Ile and Thr133→ Met).
The main amino acid alterations found in parC mainly occurred at codon 87 (Ser87→Leu), as reported in previous studies. Moreover, other minor novel mutations were determined in parC at codon 141 (Ser141→His) in one isolate and (Ser141→Thr) in another isolate (Table 2). Furthermore, results revealed that the single amino acid alteration in parC at codon 87 (Ser87→Leu) was observed in 66 fluoroquinolone-resistant clinical isolates, and a single-nucleotide mutation was found at codon 141 (Ser141→Thr), whereas double mutations at codons 87 and 141 (Ser87→Leu and Ser141→His) were detected in another clinical isolate of P. aeruginosa (Table 3).
 | Table 3 Source of isolation and amino acid mutations of gyrA, parC, and mexR in correlation with fluoroquinolone susceptibility of clinical isolates of P. aeruginosa |
Common mutations in the efflux regulatory mexR gene were mainly detected at codon 126 (Glu126→Val). Other alterations in mexR for fluoroquinolone resistance were observed only in the fluoroquinolone-resistant isolates at codon 25 (Ile25→Thr and Ile25→Asn); moreover, these mutations were mentioned in previous studies. None of the mutations found in these clinical isolates correlated with low and intermediate MIC values of the fluoroquinolones tested. However, the single amino acid substitution occurring at codon 126 (Glu126→Val) of mexR was found in 16 fluoroquinolone-resistant isolates, 10 susceptible isolates, and only one intermediately resistant isolate of P. aeruginosa (Tables 2 and 3).
Drug resistance of P. aeruginosa clinical isolates to CIP and LEV in relation to gyrA, parC, and mexR mutations and distribution of specimen types
Results obtained from mutation data of gyrA, parC, and mexR were compared with the antibiotic susceptibility data of P. aeruginosa clinical isolates and distribution of specimen types among these isolates in order to elucidate the correlation between amino acid alterations and phenotypic resistance to the fluoroquinolone drugs (CIP and LEV), and original specimens of isolations are shown in Table 3. In the 141 P. aeruginosa isolates, no amino acid alteration in gyrA was identified in susceptible isolates. Mutations of gyrA were found only in the resistant isolates. Moreover, the comparison indicated that mutations occurring in gyrA were consistent with the phenotypic resistance of most P. aeruginosa isolates to fluoroquinolone drugs. In addition, mutations occurring in parC were reported to correlate well with fluoroquinolone-resistant clinical isolates. However, none of the mutations in parC was detected in susceptible isolates of P. aeruginosa. In contrast, amino acid alterations of mexR were found in both susceptible and resistant isolates. Furthermore, the results indicated that differences in the relative resistance of P. aeruginosa clinical isolates to fluoroquinolone drugs were indistinguishable.
Of the 141 clinical isolates of P. aeruginosa collected during 2013–2016 from 13 national hospitals across nine cities in Vietnam, most of the P. aeruginosa strains were isolated from different types of specimen (Table 3), and the highest numbers of isolates (59 and 48) were obtained from sputum and pus, respectively, whereas only 2–17 isolates were collected from cerebrospinal fluid, bronchial lavage, peritoneal fluid, urine, and blood. Data showed that amino acid substitutions at codons 83 and 133 of gyrA and at codons 87 and 141 of parC were most frequently identified in all types of specimen – excepting mutations in parC of the resistant clinical isolates collected from cerebrospinal fluid and bronchial lavage. In addition, amino acid substitutions at codon 25 of mexR were mainly detected in highly resistant isolates of P. aeruginosa, whereas the mutation at codon 126 of mexR was detected in both susceptible and resistant isolates, and it was not detected in all specimen types. The correlation between mutations occurring in mexR and the antibiotic resistance of clinical isolates remained inconsistent.
Discussion
Single amino acid alteration in target genes is one of the common pathways for antibiotic resistance in bacteria.18 Previous studies have indicated that molecular resistance mechanisms of P. aeruginosa to fluoroquinolone drugs are mainly due to mutations in the FRDR of gyrA, parC, and mexR.5,10–12 Several known mutations in the FRDR of these genes have been described in previous studies such as a single-nucleotide mutation at codon 83 (Thr83→Ile) of gyrA, codon 87 (Ser87→Leu) of parC, and codon 126 (Glu126→Val) of mexR, in which most studies demonstrated that clinical isolates of P. aeruginosa comprising mutations at codon 83 (gyrA) and 87 (parC) are highly associated with fluoroquinolone resistance.5,11,12 In contrast, other studies that reported mutations occur at codon 126 of mexR did not show correlation with the resistance to fluoroquinolone drugs.10
In this study, the FRDR of these genes were amplified by PCR, and the full nucleotide sequences of these PCR-amplified fragments demonstrated that the nucleotide sequences are intact. Our results showed several point mutations in all three genes and that most of these mutations were found in fluoroquinolone-resistant clinical isolates of P. aeruginosa. In contrast, fewer mutations were detected in the susceptible clinical isolates, including a mutation at codon 126 of mexR. These results indicated that amino acid alterations within the DNA gyrase (gyrA) and topoisomerase IV (parC) genes are highly associated with resistance to CIP and LEV in the isolates, whereas alterations in the efflux regulatory mexR gene are not consistent with resistance to CIP and LEV in these isolates. In addition, this study found that mutations occurring in gyrA are consistent with highly fluoroquinolone-resistant P. aeruginosa isolates. Moreover, amino acid alterations, frequently found in parC, show a high correlation with fluoroquinolone resistance in most of the resistant P. aeruginosa isolates. These findings indicate that DNA gyrase and type II topoisomerase enzymes may be primary physiological targets of fluoroquinolone drugs, thereby establishing alterations in gyrA, parC, and other target enzymes. Although mutations are observed in gyrA, parC, and mexR, the accumulation of mutations in the mexR gene does not correlate with the resistance phenotype of P. aeruginosa isolates. In addition, our data revealed that the accumulation of mutations in the gyrA gene accompanied by the simultaneous presence of mutations in the parC gene correlate with the development of high-level fluoroquinolone resistance in clinical isolates of P. aeruginosa (Table 2). Moreover, P. aeruginosa possesses a low-permeability outer membrane, leading to very slow antibiotic diffusion.19 Therefore, active efflux systems play an important role in decreasing intracellular concentrations of antimicrobial agents.20,21 Previous studies indicated there are several repressors involved in controlling expression of mexAP-oprM operon in P. aeruginosa, including mexR, nalD, nalC, and cpxR.22 Mutations in these genes code for repressor proteins in order to generate defective form, typically leading to enhanced multidrug resistance in P. aeruginosa due to overexpression of the mexAB-oprM operon. In this study, mutations in the mexR gene are not only found in resistant isolates but also appear in susceptible strains, indicating that single or double alterations occurring in the mexR gene may not change the antibiotic susceptibility of P. aeruginosa clinical isolates; moreover, this suggests that these mutations may not alter the mexR binding site in the promoter region but can introduce resistance when co-occurring with mutations in other promoters such as nalD, nalC, or cpxR.
Previous studies have reported that major amino acid alterations in gyrA frequently occur at codon 83 (Thr83→Ile).5,10,11,23 However, novel mutations in gyrA at codon 133 (Thr133→Met) were first reported in this study. In addition, minor mutations are found in gyrA at codon 87 (Asp87→Asn, Asp87→Gly, Asp87→His, and Asp87→Tyr) and at codon 106 (Glu106→Leu) as described in a previous study.11 Our data indicate that amino acid substitutions are frequently detected in gyrA at codons 83 and 133 (Thr83→Ile and Thr133→Met), and most of the P. aeruginosa clinical isolates possessing these mutations are consistent with fluoroquinolone resistance. Furthermore, the results showed that mutations in gyrA occurring either at codon 87 or 133 were both associated with highly resistant isolates. The finding of a novel mutation at codon 133 in gyrA suggests that genetic modification of antibiotic resistant gene of clinical strains of P. aeruginosa isolated in Vietnam was considerably different from clinical isolates in other countries. Similar amino acid substitutions frequently observed in parC at codon 87 (Ser87→Leu) are always present only in the resistant isolates, as reported in several studies,5,11 and other minor novel mutations detected at codon 141 (Ser141→His and Ser141→Thr). Although mutations at codon 141 in parC were rare (presented in a few clinical isolates), the present data show that P. aeruginosa clinical isolates possessing these mutations are consistent with fluoroquinolone resistance.
Interestingly, results of this study showed that the number of clinical isolates of P. aeruginosa with amino acid alterations in both gyrA and parC was higher than that of strains carrying single-nucleotide mutations in gyrA, as well as that amino acid alterations in parC account for a small proportion of clinical isolates. This finding indicates that gyrA is the primary target for fluoroquinolones, and that point mutations occurring in parC have been shown to enhance the level of drug resistance of the isolates.
Although the number of clinical isolates carrying mutations in gyrA is higher than that in parC, the relevant correlation between amino acid mutations in the gyrA and the parC genes and fluoroquinolone resistance in the P. aeruginosa isolates cannot be demonstrated. Major amino acid alterations in the mexR gene are mainly identified at codon 126 (Glu126→Val), and other minor mutations are detected at codon 25 (Ile25→Thr and Ile25→Asn); these mutations were reported in previous investigations. Earlier studies have shown that mutations in mexR do not show any drug resistance. Thus, it maybe speculated that mutations at codons 25 and 126 in mexR alone may not change the antibiotic susceptibility of fluoroquinolone-resistant clinical isolates of P. aeruginosa, but may promote the levels of resistance if accompanied by mutations that occur in other repressors such as nalA, nalD, or CpxR. Moreover, a combination of alterations in mexR and gyrA or in mexR and parC does not improve fluoroquinolone resistance, suggesting that mutations co-occurring in mexR and gyrA or in mexR and parC do not always indicate resistance to fluoroquinolones. The study, while trying to identify novel mutations consistent with fluoroquinolone resistance, recognizes that the alteration frequently found at codon 133 of gyrA (Thr133→Met) is highly correlated with fluoroquinolone resistance. Further, minor novel alterations were found in parC and mexR, but they do not support any evidence consistent with fluoroquinolone resistance.
A total of 141 clinical isolates of P. aeruginosa were collected from 13 independent hospitals across nine cities in Vietnam; most of these strains were mainly isolated from pus and sputum specimens, but only a few isolates were collected from cerebrospinal fluid, bronchial lavage, peritoneal fluid, urine, and blood, with mutations found most frequently at codons 83 and 133 in gyrA, at codon 87 in parC, and at codons 25 and 126 in mexR. It is now known that P. aeruginosa is an opportunistic bacterium and is found everywhere in the soil, water, and the air or on the human body, especially in the hospital environment (nosocomial infection). Combined with favorable conditions such as wound, incision, or penetration into the respiratory system in patients, they then have a favorable environment to develop and cause disease. In addition, P. aeruginosa strains were mainly isolated from patients in the Emergency Department and General Surgery Department. The reason for this is that most patients in the departments normally have very serious medical conditions, high mortality risk, and have to face treatment interventions such as surgery, mechanical ventilation, endotracheal intubation, surgical drainage, and nasogastric intubation – all with a very high risk of infection. Therefore, the rate of resistance clinical isolates of P. aeruginosa collected from pus and sputum is highest in clinical specimens.
In conclusion, most of the clinical isolates of P. aeruginosa collected in Vietnam were mainly isolated from pus and sputum specimens, with amino acid alterations frequently occurring at codon 83 (Thr83→Ile and Thr133→Met) in gyrA and at codon 87 (Ser87→Leu) in parC consistent with high-level resistance to CIP and LEV in clinical isolates of P. aeruginosa isolated from Vietnam, whereas the mutation detected at codon 126 (Glu126→Val) in mexR does not correlate with the antibiotic resistance of the bacterium. Substitution mutations co-occurring in gyrA and parC show higher-level fluoroquinolone resistance compared with single-nucleotide mutations occurring in gyrA.
Acknowledgment
This research was supported in part by a grant from the Ministry of Science and Technology of Vietnam (grant no. KC.10.18/11–15).
Disclosure
The authors report no conflicts of interest in this work.
References
Bălăşoiu M, Bălăşoiu AT, Mănescu R, Avramescu C, Ionete O. Pseudomonas aeruginosa resistance phenotypes and phenotypic highlighting methods. Curr Health Sci J. 2014;40(2):85–92. | ||
Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171(11):1209–1223. | ||
Mesaros N, Nordmann P, Plésiat P, et al. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect. 2007;13(6):560–578. | ||
El Solh AA, Alhajhusain A. Update on the treatment of Pseudomonas aeruginosa pneumonia. J Antimicrob Chemother. 2009;64(2):229–238. | ||
Wang YT, Lee MF, Peng CF. Mutations in the quinolone resistance-determining regions associated with ciprofloxacin resistance in Pseudomonas aeruginosa isolates from Southern Taiwan. Biomarkers Genomic Med. 2014;6(2):79–83. | ||
Rizek C, Fu L, Dos Santos LC, et al. Characterization of carbapenem-resistant Pseudomonas aeruginosa clinical isolates, carrying multiple genes coding for this antibiotic resistance. Ann Clin Microbiol Antimicrob. 2014;13:43. | ||
Morita Y, Tomida J, Kawamura Y. Responses of Pseudomonas aeruginosa to antimicrobials. Front Microbiol. 2014;4:422. | ||
Lee YJ, Liu HY, Lin YC, Sun KL, Chun CL, Hsueh PR. Fluoroquinolone resistance of Pseudomonas aeruginosa isolates causing nosocomial infection is correlated with levofloxacin but not ciprofloxacin use. Int J Antimicrob Agents. 2010;35(3):261–264. | ||
Pakyz AL, Lee JA, Ababneh MA, Harpe SE, Oinonen MJ, Polk RE. Fluoroquinolone use and fluoroquinolone-resistant Pseudomonas aeruginosa is declining in US academic medical centre hospitals. J Antimicrob Chemother. 2012;67(6):1562–1564. | ||
Gorgani N, Ahlbrand S, Patterson A, Pourmand N. Detection of point mutations associated with antibiotic resistance in Pseudomonas aeruginosa. Int J Antimicrob Agents. 2009;34(5):414–418. | ||
Lee JK, Lee YS, Park YK, Kim BS. Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int J Antimicrob Agents. 2005;25(4):290–295. | ||
Jalal S, Ciofu O, Hoiby N, Gotoh N, Wretlind B. Molecular mechanisms of fluoroquinolone resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob Agents Chemother. 2000;44(3):710–712. | ||
Redgrave LS, Sutton SB, Webber MA, Piddock LJ. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22(8):438–445. | ||
Fàbrega A, Madurga S, Giralt E, Vila J. Mechanism of action of and resistance to quinolones. Microb Biotechnol. 2009;2(1):40–61. | ||
Srikumar R, Paul CJ, Poole K. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-oprM multidrug efflux system of Pseudomonas aeruginosa. J Bacteriol. 2000;182(5):1410–1414. | ||
Yang X, Xing B, Liang C, Ye Z, Zhang Y. Prevalence and fluoroquinolone resistance of Pseudomonas aeruginosa in a hospital of South China. Int J Clin Exp Med. 2015;8(1):1386–1390. eCollection 2015. | ||
Nakano M, Deguchi T, Kawamura T, et al. Mutations in the gyrA and parC genes in fluoroquinolone-resistant clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1997;41(10):2289–2291. | ||
Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13(1):42–51. | ||
Hancock RE. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin Infect Dis. 1998;27 (Suppl 1):S93–S99. | ||
Ruiz J. Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J Antimicrob Chemother. 2003;51(5):1109–1117. | ||
Dreier J, Ruggerone P. Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front Microbiol. 2015;6:660. | ||
Tian ZX, Yi XX, Cho A, O’Gara F, Wang YP. CpxR activates MexAB-OprM efflux pump expression and enhances antibiotic resistance in both laboratory and clinical nalB-type isolates of Pseudomonas aeruginosa. PLoS Pathog. 2016;12(10):e1005932. | ||
Mouneimné H, Robert J, Jarlier V, Cambau E. Type II topoisomerase mutations in ciprofloxacin-resistant strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1999;43(1):62–66. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.