Back to Journals » Clinical Interventions in Aging » Volume 11
Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries
Authors Bagyinszky E, Youn YC ![]() , An SSA
, An SSA ![]() , Kim S
, Kim S ![]()
Received 1 July 2016
Accepted for publication 4 September 2016
Published 17 October 2016 Volume 2016:11 Pages 1467—1488
DOI https://doi.org/10.2147/CIA.S116218
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Eva Bagyinszky,1,* Young Chul Youn,2 Seong Soo A An,1 SangYun Kim3,*
1Department of BioNano Technology, Gachon University, Gyeonggi-do, 2Department of Neurology, College of Medicine, Chung-Ang University, Seoul, 3Department of Neurology, Seoul National University Budang Hospital, Gyeonggi-do, South Korea
*These authors contributed equally to this work
Abstract: Alzheimer’s disease (AD), the most common form of senile dementia, is a genetically complex disorder. In most Asian countries, the population and the number of AD patients are growing rapidly, and the genetics of AD has been extensively studied, except in Japan. However, recent studies have been started to investigate the genes and mutations associated with AD in Korea, the People’s Republic of China, and Malaysia. This review describes all of the known mutations in three early-onset AD (EOAD) causative genes (APP, PSEN1, and PSEN2) that were discovered in Asian countries. Most of the EOAD-associated mutations have been detected in PSEN1, and several novel PSEN1 mutations were recently identified in patients from various parts of the world, including Asia. Until 2014, no PSEN2 mutations were found in Asian patients; however, emerging studies from Korea and the People’s Republic of China discovered probably pathogenic PSEN2 mutations. Since several novel mutations were discovered in these three genes, we also discuss the predictions on their pathogenic nature. This review briefly summarizes genome-wide association studies of late-onset AD and the genes that might be associated with AD in Asian countries. Standard sequencing is a widely used method, but it has limitations in terms of time, cost, and efficacy. Next-generation sequencing strategies could facilitate genetic analysis and association studies. Genetic testing is important for the accurate diagnosis and for understanding disease-associated pathways and might also improve disease therapy and prevention.
Keywords: mutation, Asia, presenilin, amyloid precursor protein, genetics
Introduction
Alzheimer’s disease (AD), the most common form of senile dementia, is a major health problem.1 AD is a complex disease, and several genetic and/or environmental factors can contribute to disease progression. The main hallmarks of AD are the amyloid beta (Abeta or Aβ) peptides and the neurofibrillary tangles. The number of AD patients is rising rapidly as the lifetime expectancy increases. In Asia, the population is growing rapidly, especially in India and the People’s Republic of China. Aging of population is also an important current issue. According to 2010 statistics, the elderly constituted 11% of the total population. This percentage is expected to reach 16.5% and 22% by 2030 and 2050, respectively. The fastest increase in the number of elderly individuals has been observed in East Asian countries. More than half (60%) of all patients diagnosed with dementia live in Asian countries. The People’s Republic of China, Japan, and India are among the top seven countries with the highest number of dementia patients.2
Genetics of AD
The two main types of AD can be classified as early-onset AD (EOAD) and late-onset AD (LOAD). EOAD or autosomal dominantly inherited AD occurs before the age of 60–65 years. Three main genes are involved in EOAD, APP, PSEN1, and PSEN2, encoding amyloid precursor protein, presenilin-1, and presenilin-2, respectively. EOAD is relatively rare, with 5%–10% of all AD cases occurring before the age of 65 years. Mutations in all of these three genes could result in enhanced Abeta production and deposition.3 APP is a transmembrane (TM) protein, which can play a role in synaptic plasticity. Three enzymes, the alpha-, beta-, and gamma-secretases, have cleavage sites in APP protein. Abnormal cleavage of APP by gamma- and beta-secretases could result in impairment in the Abeta production. It was suggested that Abeta peptide may be important in synaptic vesicle regulation,4 but the oligomer form of peptide can be involved in neurotoxicity. Abeta42 and Abeta40 peptides and Abeta42/40 ratio have been established as the most important biomarkers for AD. However, even the small alterations in the Abeta42/40 levels could result in pathogenic phenotypes, by enhancing their oligomerization ability and stabilizing their oligomer forms, resulting in toxicity for synapses and nerve cells.5
Presenilins (PSEN1 and 2) are the components of gamma-secretase complex. They can be involved in memory, survival of neurons, and synaptic mechanisms. Involvement of PSENs in AD may be heterogeneous. Gamma-secretase dysfunctions have been established as the most common mechanisms in PSEN1–2 mutants, resulting in the impairment of APP cleavage and neuronal toxicity.1,3 Elevated Abeta42 could be associated with disease progression but not in all AD cases.6 Some mutations were revealed as “loss-of-function” variants, since they destroy the gamma-secretase activity. These mutations were associated with increased Abeta42/40 levels and impairment in synaptic plasticity.7 Ben-Gedalya et al8 revealed that some PSEN1 (especially proline exchange with other amino acids) variants could prevent the interaction of PSEN with the chaperone, called cyclophilin B, resulting in PSEN degradation or abnormal PSEN aggregation in endoplasmic reticulum. LOAD usually occurs later in life, after 65 years, and its prevalence increases with age. The genetic background of LOAD remains unclear because no exact causative genes have been reported yet. However, genome-wide association studies (GWAS) have identified several genes that might increase the risk of AD onset. The apolipoprotein E (APOE) E4 allele appears to be the main risk factor for LOAD, but it does not define all AD cases.1,3,9
This review focuses on EOAD and summarizes all mutations found in the three genes associated with EOAD in Asian families and patients. It also briefly summarizes the Asian GWAS, performed on the putative risk factor genes. In addition, the review describes methods that could be useful in genetic testing from the genotyping to next-generation sequencing (NGS).
APP, PSEN1, and PSEN2 mutations in Asian countries that might contribute to EOAD
Several mutations were discovered in APP, PSEN1, and PSEN2 that could contribute to disease progression. Most of these mutations are associated with familial EOAD, in which the inheritance pattern is autosomal dominant and follows Mendelian rules. However, several de novo cases of AD were reported in patients without any family history of dementia. The majority of pathogenic mutations were found in PSEN1 gene. Several PSEN1 mutations could be associated with early-onset AD, which occurs at the age <40 years, and with rapid and aggressive dementia progression. Mutations in APP and PSEN2 are quite rare but are possible causative factors for EOAD. Pathogenic mutations could result in disease onset at the age of 40–65 years.3 In the most Asian countries, as in Europe, genetics of EOAD is not well characterized, and only a few reports are available regarding mutations in EOAD causative genes. However, emerging research projects in Japan, Korea, and the People’s Republic of China are assessing the genetic background of patients with early-onset dementia. Table 1 lists all EOAD-associated mutations discovered in Asian countries. The information in this table comes from the two main databases that summarize the mutations found in EOAD-associated genes. The AD and frontotemporal dementia (FTD) mutation database10 was edited by Cruts et al and summarized all known mutations associated with the following two main types of dementia: AD and FTD. The Alzheimer’s Research Forum (http://www.alzgene.org/) is an up-to-date database that summarizes all findings and articles related to AD, including genetic mutations.10 In APP, 35 variants were discovered in the exon 16 and 17, and of them, 9 variants were (also) reported in Asia (Figure 1). Due to the Alzheimer’s Research Forum database, 227 variants were discovered in PSEN1 all around the world. Of them, 51 variants were (also) discovered in Japan, Korea, the People’s Republic of China, or Malaysia (Figure 2). In PSEN2, 38 missense and frameshift mutations were reported, and until 2014, no pathogenic mutation was found in PSEN2 in any Asian countries. However, recent studies revealed novel and known variants in PSEN2 gene in Korean and Chinese patients (Figure 3).11–13
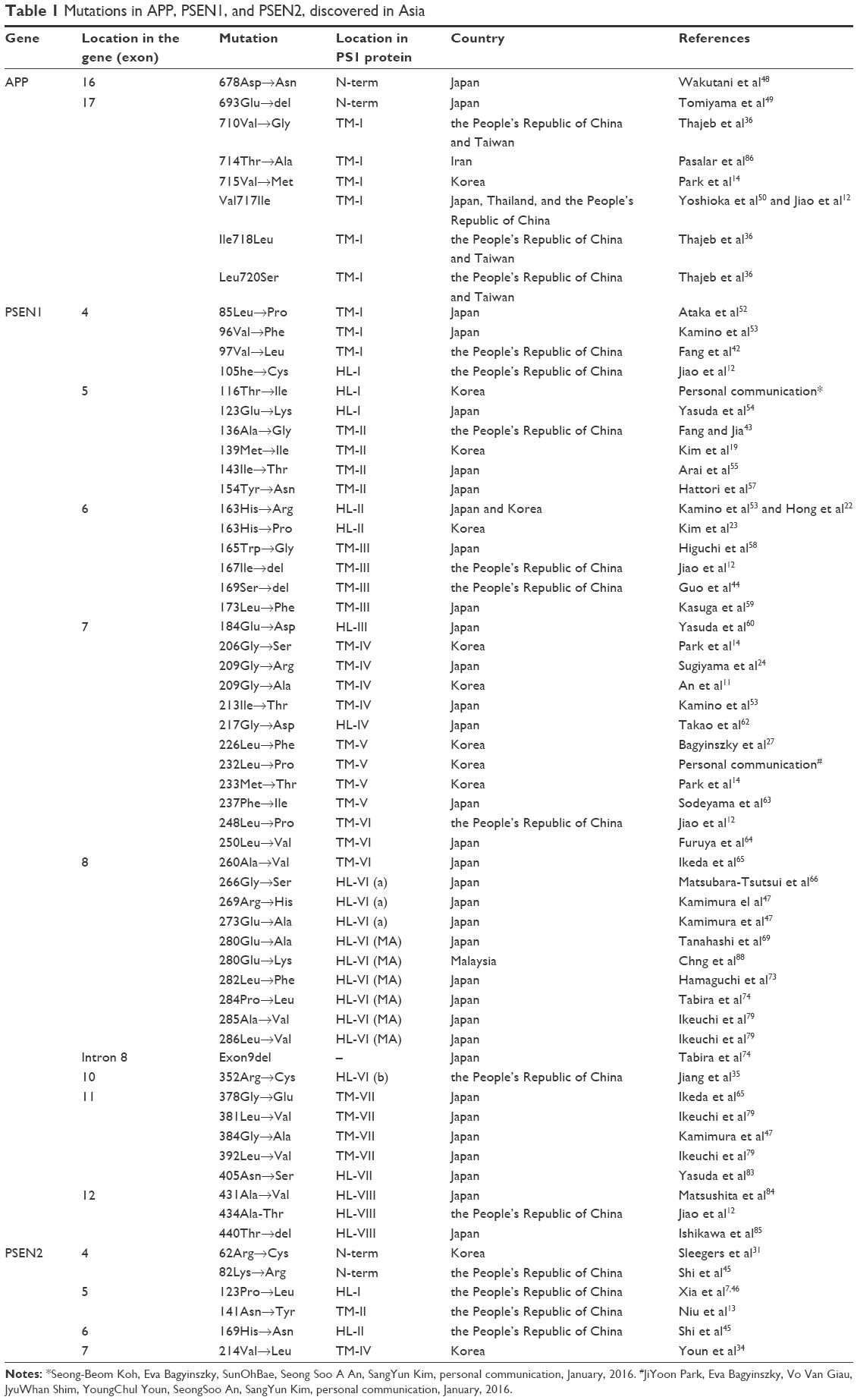 | Table 1 Mutations in APP, PSEN1, and PSEN2, discovered in Asia |
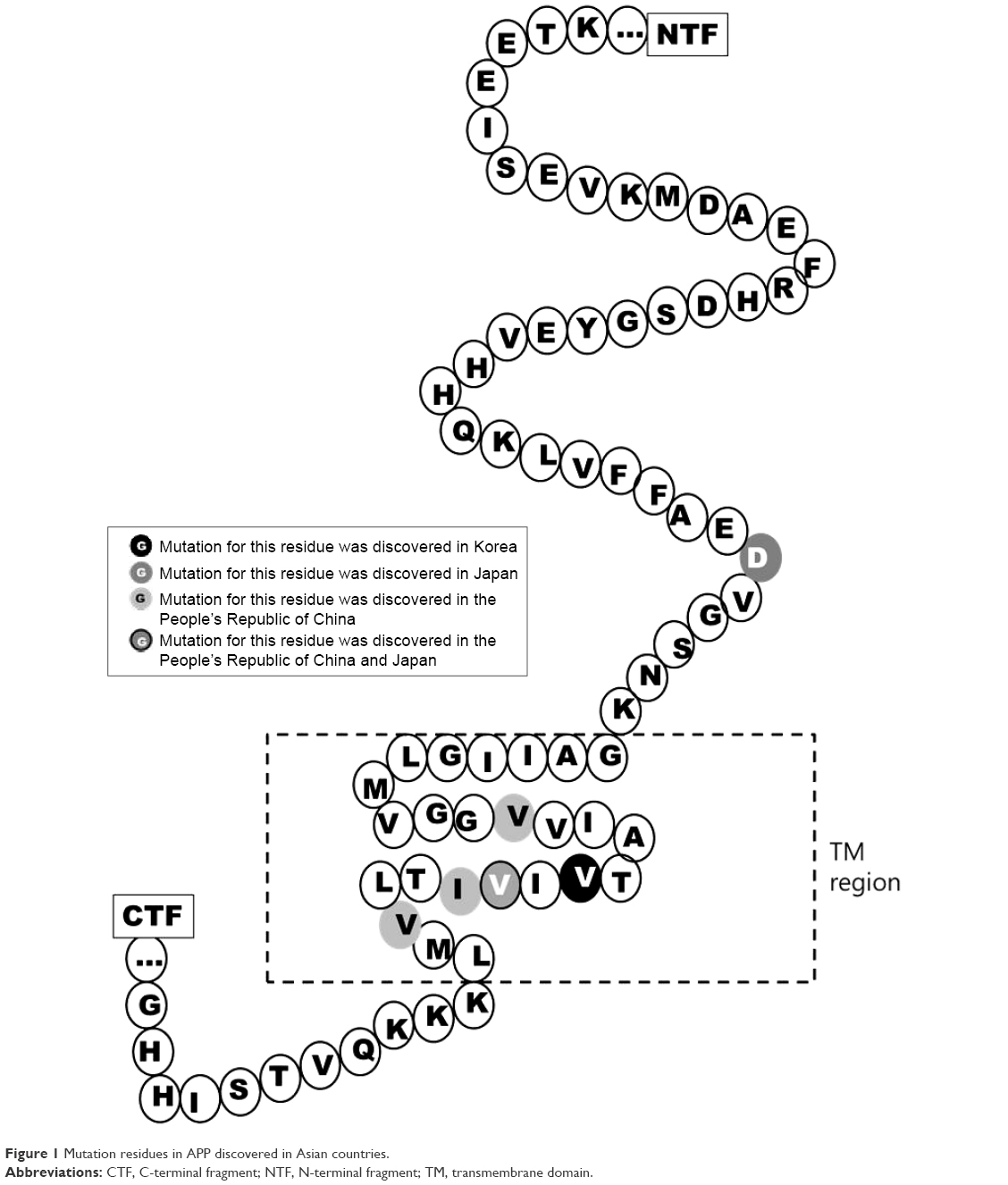 | Figure 1 Mutation residues in APP discovered in Asian countries. |
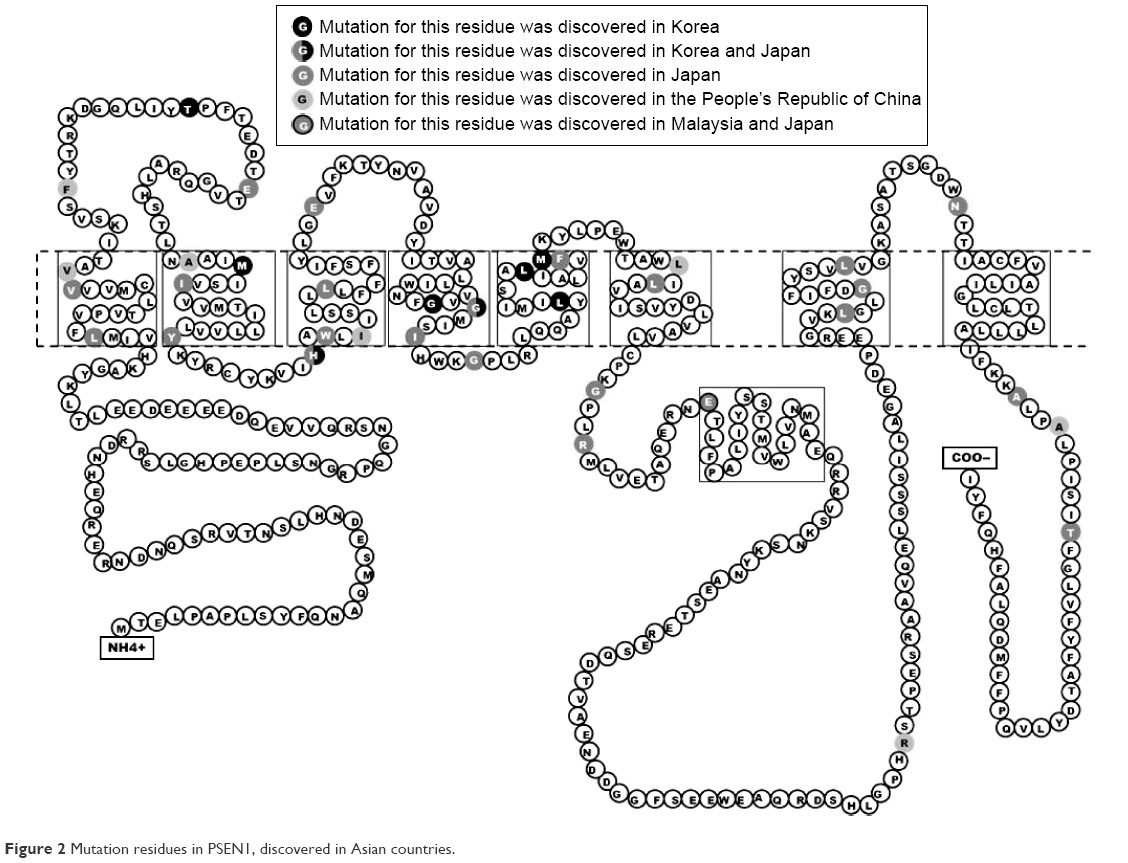 | Figure 2 Mutation residues in PSEN1, discovered in Asian countries. |
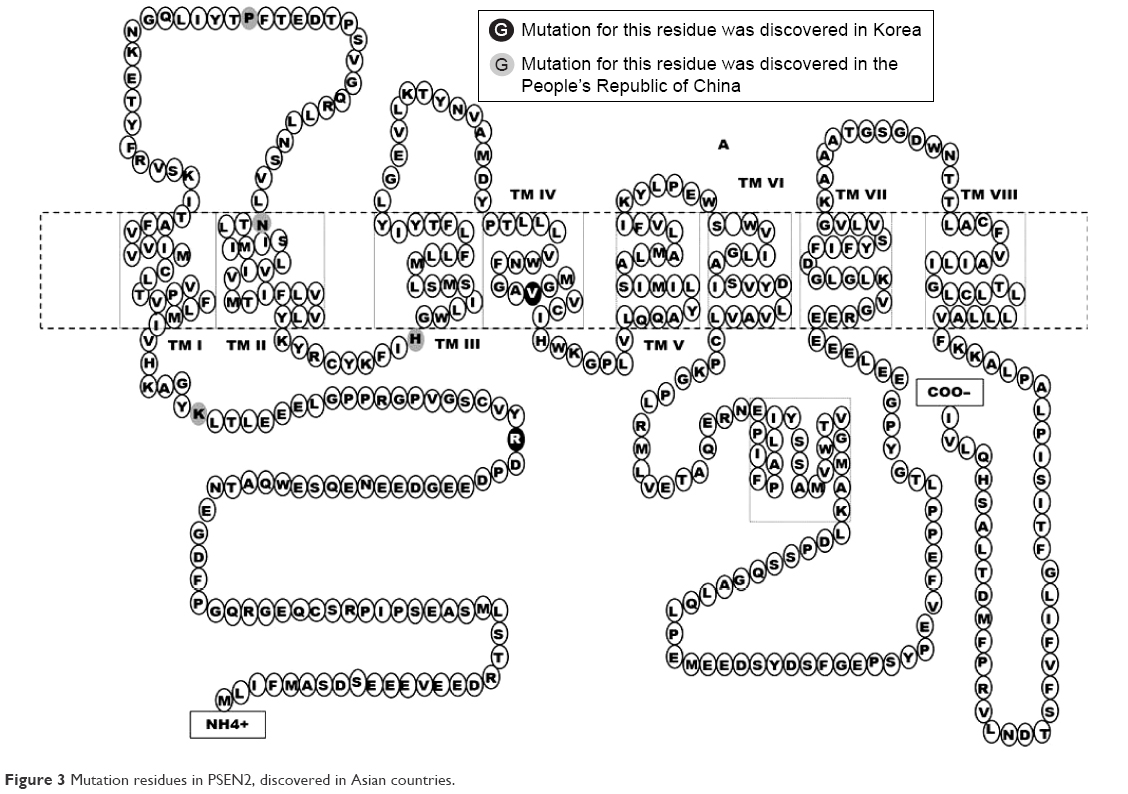 | Figure 3 Mutation residues in PSEN2, discovered in Asian countries. |
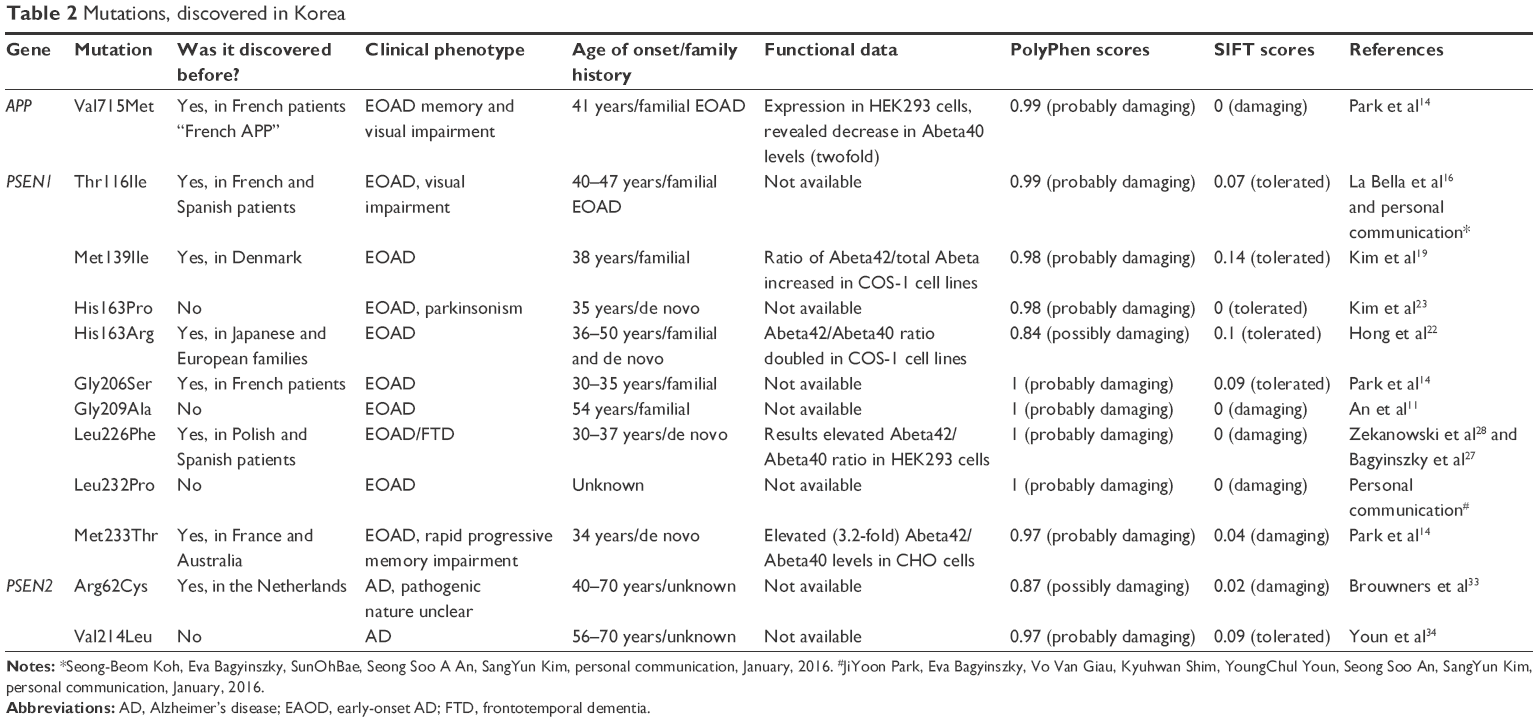
Mutations discovered in Korea
The genetic background of EOAD in Korea is not well characterized (Table 2). Until 2010, only five mutations were discovered: Val715Met in APP,6 Met139Ile, His163Arg, Gly206Ser, and Met233Thr in PSEN1, and no mutations in PSEN2 have been reported yet. Our research group found four additional novel or known PSEN1 mutations in Korean AD patients, including Thr116Ile (known), His163Pro (novel), Leu226Phe (known), and Leu232Pro (novel). We also discovered the following two PSEN2 mutations in Korea for the first time: Val214Leu (novel) and Arg62Cys (known). Since Korea is one of the fastest “aging countries” in the world, the number of AD, including EOAD, patients will rise fast. In the future, it might be possible to find additional novel/known variants in the EOAD causative genes.
 | Table 2 Mutations, discovered in Korea |
Mutations in APP
Val715Met (GTG→ATG) was discovered in a 41-year-old male patient in Korea who had a positive family history of dementia.14 The symptoms were memory and visual impairment, bradykinesia, and epilepsy. This is the first and currently the only pathogenic mutation in APP, which was found in Korea. Because the mutation was first discovered in a French family (with Italian descendants) with progressive memory decline, the mutation is also called “French APP”. The age of onset was the same in French and Korean patients.15 The Korean patient might be associated with familial EOAD, because other family members (uncles from the father’s side) also developed dementia. APP Val715Met was expressed in human embryonic kidney 293 (HEK293) cell lines, and a significant decrease in Abeta40 levels (twofold) was observed. Abeta42 levels did not change, but the ratio of Abeta42/Abeta40 increased (1.8-fold). These findings suggested that this mutation might destroy the Abeta40 cleavage site of gamma-secretase.15
Mutations in PSEN1
Thr116Ile (ACC→ATC) was identified in a Korean patient (age of onset, 42 years) for the first time in Asia. This patient showed a strong positive family history of disease; her mother was diagnosed with dementia. The patient’s three sisters agreed to genetic testing, and they have been screened for Thr116Ile. One of the sisters (37 years of age at the time of analysis) carried the mutation; she had not been clinically diagnosed with AD yet, but symptoms of memory impairment (forgetfulness) have already appeared in her early 30s (Seong-Beom Koh, Eva Bagyinszky, SunOhBae, Seong Soo A An, SangYun Kim, personal communication, January, 2016). Thr116Ile has been described in French and Italian patients before with familial or de novo cases of EOAD, with similar age of onset.16,17 For Thr116, an additional pathogenic mutation, Thr116Asn, was discovered, which was also associated with EOAD.18 Because Thr116 is located in the conservative HL-I region, a Thr→Ile exchange might disturb significantly the PSEN1 protein structure.
Met139Ile (ATG→ATC) was found in a female patient. The mutation has been described before but with a different codon change (ATG→ATA) in a European patient. Codon 139 might be an important residue in PSEN1 in the TM-II region of PSEN1, given that the following three additional mutations have been described: Met139Lys, Met139Thr, and Met139Val.19,20 In the Korean patient, memory problems started at the age of 38 years. She had a positive family history of dementia; her mother (deceased at the age of 45 years) and her elder sister had a similar type of memory decline. When PSEN1 Met139Ile (ATG→ATA) was expressed in COS-1 cell lines, the Abeta42/total Abeta ratio increased.21 His163Arg (CAT→CGT) is a quite frequently occurring AD-associated mutation, since it has been reported in 15 familial or sporadic EOAD cases in several parts of the world. In Korea, Hong et al described the mutation in a female patient who was hospitalized because of memory disturbances and personality changes at the age of 36 years.22 Symptoms started in her late 20s. Strong family history of disease was observed in her family; of the 22 family members tested, nine members were affected. The mutation was found in several Japanese familial and sporadic AD cases (five families), with the age of onset ranging from 47 to 50 years. When PSEN1 His163Arg was expressed in COS-1 cell lines, twofold increase was observed in Abeta42/Abeta40 ratio.21 The histidine at 163th position in PSEN1 might be an important residue in the protein. An additional variant, His163Tyr, was discovered, and in 2012, a novel mutation, His163Pro (CAT→CCT), was reported in a Korean female patient with clinically diagnosed EOAD and parkinsonism. She also experienced seizures, myoclonus, and cerebellar ataxia. This might be a de novo case of AD, since no affected family members were found. Even though the mutation was found in only one patient, evidence for its pathogenic nature is strong; it has not been found in healthy controls. The two additional AD causative mutations of His163 could also enhance the evidence on the pathogenic nature of His163Pro. In addition, the mutation is located in the border of TM and loop region, where the rigid proline exchange might disturb protein structure and function.23 Gly206Ser (GGT→AGT) was reported in Korean and European families, with the age of onset ranging from 30 to 35 years. Patients showed progressive episodic memory and visuospatial impairment, ataxia, acalculia, and aphasia. Gly206 is an important residue in PSEN1 protein, and the following three additional pathogenic mutations have been reported: Gly206Asp, Gly206Ala, and Gly206Va.l14,17 Gly209Ala (GGA→GCA) was recently discovered in a Korean EOAD patient, with the age of onset at 54 years. This might be a de novo case of EOAD, because no additional affected family members were found. The extra –CH3 group in alanine could result in extra stress inside the PSEN1 helix.11 Further studies about this mutation are needed, because it is located in the TM region, where additional Gly→Ala exchanges were described (the nearest of them is the Gly206Ala), which was also validated as pathogenic mutation. Similarly to G206A, Gly209 might also be an important residue in PSEN1, since additional mutations, such as Gly209Arg, Gly209Glu, and Gly209Val, have been described.24–26 Leu226Phe (CTC→TTC) is a known mutation, discovered in Korea for the first time by our research group.27 The mutation was detected in an AD patient, with the age of onset at 37 years, and appeared to be de novo case of AD. The age of death was 44 years.28 This variant has been reported in AD patients in Poland with the age of onset ranging from 30 to 44 years. The Polish proband patient was initially diagnosed with FTD, but postmortem studies revealed AD-associated neuropathology. In the EOAD Korean patient, additional symptoms, such as nonfluent aphasia and parkinsonism, were detected. An additional mutation (Leu226Arg) has been described for codon 226. In silico modeling was performed with PSEN1 Leu226Phe. The findings revealed that the interaction between the benzene rings of Tyr225 and Phe226 could disturb the TM-V region of PSEN1. In addition, the mutation might enhance hydrophobic interactions between PSEN1 protein and its putative binding partners.28–30 Met233Thr (ATG→ACG) was described in six families from France, Australia, and Korea. Mutation appeared in both familial and de novo cases of young onset AD that occurred in the 30s or early 40s. The Korean patient did not have a family history of dementia, with the age of disease onset at 34 years. The patient showed rapid progressive memory impairment; the score of Mini–Mental State Examination dropped drastically 2 years after the first symptoms appeared. Two other mutations, Met233Leu and Met233Val, have been described for codon 233.14 When the Met233Thr was expressed in Chinese hamster ovary (CHO) cells, the Abeta40/total Abeta ratio decreased sharply (2.2-fold), and the Abeta42/total Abeta ratio increased (3.2-fold).30 Leu232Pro (CTC→CCC) is a novel mutation recently discovered by our research group. Further investigations, clinical studies, and genetic testing of family members are needed to determine whether the mutation could be involved in AD progression. However, several pathogenic Leu→Pro exchanges have been reported in PSEN1, suggesting that this mutation may be involved in EOAD progression, and the nearest is the Leu235Pro. Since the mutation is located in the TM region, the rigid proline might result in significant disturbances. Proline, which was established as a helix breaking molecule, could result in serious torsion in the TM region (JiYoon Park, Eva Bagyinszky, Vo Van Giau, Kyuhwan Shim, YoungChul Youn, Seong Soo An, SangYun Kim, personal communication, January, 2016).
Mutations in PSEN2
Arg62Cys (CGC→TGC) was discovered in Asia for the first time by our research group. The mutation was identified in a dementia patient. Memory impairment, personality changes, and disorientation appeared at the age of 49 years. He had no family history of dementia (Kyung Won Park, Eva Bagyinszky, SeunOh Bae, Seong Soo An, SangYun Kim, personal communication, January, 2016). Arg62Cys is a known mutation in PSEN2, categorized as a “pathogenic nature unclear” variant in the two main AD mutation databases. The mutation was identified in LOAD patients and in healthy controls but with a higher frequency in AD patients. The mutation might be a risk factor for AD. Genetic–environmental interactions might also affect the onset of disease.31–33 Val214Leu (GTG→TTG) was also discovered by our research group. Val214Leu was one of the first PSEN2 mutations identified in Asia. In addition, it is the first mutation identified in the TM-IV region of PSEN2. We discovered the mutation in the following two unrelated patients: a 70-year-old patient with AD-type dementia and a 56-year-old patient with memory impairment. The exact family history is unknown in both cases. The absence of Val214Leu in normal controls and the results of an in silico analysis suggested that this mutation could result in abnormalities in the PS2 structure. Because leucine is more hydrophobic than valine, the mutation might result in extra stress inside the TM domain or between the TM and surrounding proteins and could be a possibly pathogenic variant of PSEN2.34
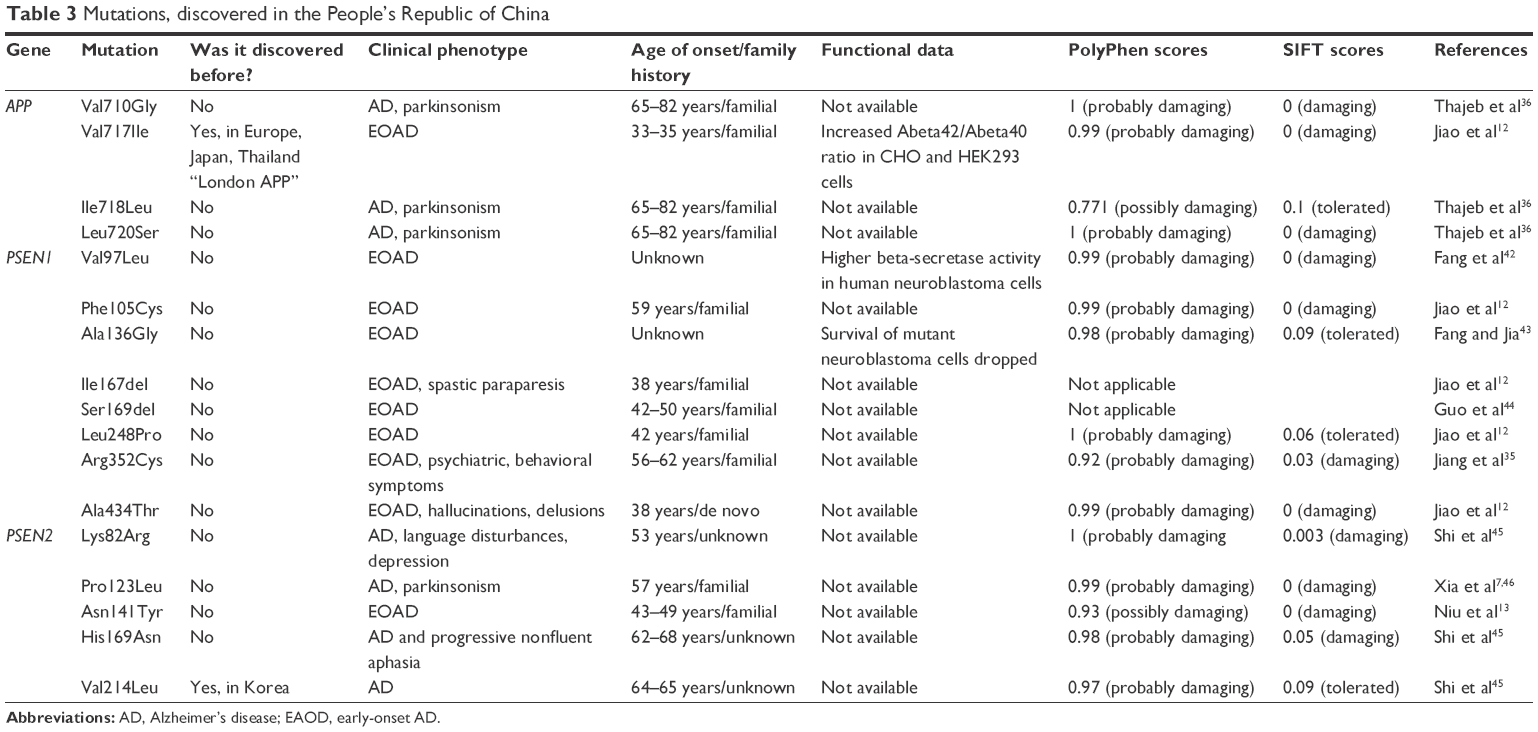
Mutations discovered in the People’s Republic of China and Taiwan
The genetic background of Chinese (and Taiwanese) AD patients was not well characterized. Initially, three mutations were discovered in APP gene and three mutations were discovered in PSEN1 gene. However, several recent studies reported additional novel mutations.12,13,35 Most of the variants, discovered in Chinese families and patients, are novel mutations, which have not been described in any other population (Table 3).
 | Table 3 Mutations, discovered in the People’s Republic of China |
Mutations in APP
The following four APP mutations have been found in 10 Chinese/Taiwanese patients: Val710Gly (one patient), Ile718Leu (eight patients), and Leu720Ser (three patients, of whom, the mutation occurred with Ile718Leu in two patients). The symptoms of disease started with progressive dementia in all patients, which appeared 2–9 years before the clinical diagnosis of AD. Additional symptoms, such as oral tendency and parkinsonism, were present in some patients. Family history of dementia was negative in all tested patients, with the age of onset ranging from 65 to 82 years. These mutations are located in the TM region of APP; they might be critical for APP processing and might increase or decrease Abeta42 and Abeta40 levels in the brain.36
Val717Ile (GTC→ATC, “London APP”) was reported in several EOAD cases. In Asia, it was identified in five Japanese families and one Thai family.37 This mutation was recently discovered in Chinese families, with the age of onset between 33 and 45 years, but clinical phenotypes of mutations were different in these families. In the first family, symptoms started with memory impairment and personality changes, followed by progressive memory loss and psychiatric symptoms. In the second family, disease progression was slower. In the proband patient, memory impairment started at the age of 45 years. Cognitive impairment was observed at the age of 50 years.32 This mutation was analyzed in several cell lines. CHO and HEK293 experiments suggested that Val717Ile increases the Abeta42/Abeta40 ratio.12,38 Decreased Abeta40 levels were reported after analyzing this mutation in HEK293, COS cell.39,40
Mutations in PSEN1
Val97Leu (GTG→TTG) was described for the first time in a Chinese family with EOAD. Because the mutation is not present in normal controls, it might be a novel mutation involved in familial EOAD.41 To validate the pathogenic nature of this mutation, PSEN1 Val97Leu was expressed in human neuroblastoma (SH-SY5Y) cells, and the Abeta concentration was monitored with ELISA and radioimmunity methods. Beta-secretase activity was also monitored. The data showed that intracellular and extracellular Abeta production was higher in the mutant cells, suggesting that the mutation is pathogenic.42 Phe105Cys (TTT→TGT) was discovered in a Chinese patient, who showed memory impairment at the age of 59 years. Family history was positive, since her sibling carried the same mutation, and the dementia was fully developed in her father (died at the age of 60 years). Patients showed typical amnestic symptoms.12 Ala136Gly (GCT→GGT) was discovered in a Chinese family. No data are available on the age of onset or on the clinical phenotypes of disease. Experiments were performed to establish the pathogenic nature of the mutation. Ala136Gly was expressed in human neuroblastoma cells. The survival of the mutant cells decreased significantly, suggesting that this mutation could have deleterious effects.43 Ile167del (delTTA) was initially diagnosed in a Chinese female patient, for whom the memory loss started at the age of 38 years. Later, she developed spastic paraparesis, personality changes, and disorientation. Family history was positive, since her mother died with AD. Her two siblings were also positive for this mutation, and they showed similar symptoms.12 Ser169del (delTCA.TCT) was described in a Chinese family with EOAD. Symptoms were early memory impairment (42–50 years), followed by cognitive dysfunction, memory decline, apraxia, and disorientation. Two additional mutations, Ser169Pro and Ser169Leu, which are involved in EOAD with rapid disease progression, were described for codon 169. Ser169del might be associated with disturbances in posttranslational modifications, in the protein structure or in the interactions with other proteins because of the missing –OH group.40 Leu248Pro (CTC→CCC) was found in a female patient, where the memory impairment started at the age of 42 years. Family history was positive, since her deceased father was diagnosed with AD and her younger brother was also affected by this mutation and showed similar clinical symptoms.44 Arg352Cys (CGC→TGC) was reported in three members of a Han family. This mutation might not segregate with the disease onset, since this mutation was also found in an unaffected individual. However, the unaffected family member might be presymptomatic and could present dementia-associated phenotypes in the future, with the age of disease onset at 56–62 years, and clinical phenotypes of disease were memory loss and psychiatric- and behavioral symptoms.35 Ala434Thr (GCT→ACT) was discovered in a 38-year-old patient, who was initially misdiagnosed with schizophrenia, because she showed hallucinations and delusions, followed by memory problems and cognitive disturbances. Family history was positive, because her mother was also diagnosed with AD, but the memory impairment was the main symptom in her.12
Mutation in PSEN2
In 2014, a novel PSEN2 mutation, Asn141Tyr (AAC→TAC), was discovered in a Han Chinese family. The mutation was identified in two affected family members, who were clinically diagnosed with EOAD. The proband was a female patient who developed memory impairment at the age of 43 years. Symptoms started with the inability to handle financial matters and find personal items. Two years later, the memory impairment became worse, and she was unable to perform her job. Several of her family members (grandfather, mother, and two aunts) died with dementia. Her sister developed memory decline at the age of 49 years and later developed speech difficulties and disorientation. In the later disease stages, she developed paranoia, visual hallucinations, and agitation. The location of mutation is similar to the Volga-German mutation (Asn141Ile), and this asparagine to tyrosine change might affect the Abeta42/Abeta40 ratio.13 Later, additional mutations have been described in Chinese patients. K82R (AAA→AGA) was found in a female patient at the age of 50 years. She developed AD with depression and language impairment. Atrophy was observed in the posterior region and hippocampus, and amyloid deposition appeared in different brain areas, such as frontal and parietal lobes and striatum.45 Pro123Leu (CAA→CTA) was found in a Chinese family, where the pedigree revealed several affected family members over four generations. At the proband patient, disease started at the age of 57 years, with personality changes and memory problems. Cognitive impairment with parkinsonism and myoclonic jerks were quite common phenotypes among the patients. However, mutation did not segregate with the disease, since several asymptomatic relatives also carried the mutation.46 His169Asn (CAT-AAT) was identified in two Chinese individuals, who were unrelated. One of the patients had familial form of AD in her late 60s, when she started with progressive memory loss. Her brother was also diagnosed with AD. The other patient was diagnosed with sporadic FTD at the age of 62 years. His symptoms were initially language impairment and personality changes, later followed by cognitive decline.45 Val214Leu (GTG→TTG) was also found in two unrelated AD patients, who developed disease phenotype in their 60s. One of the patients had familial form of AD, but segregation could not be confirmed. The second patient developed sporadic early-onset AD.45
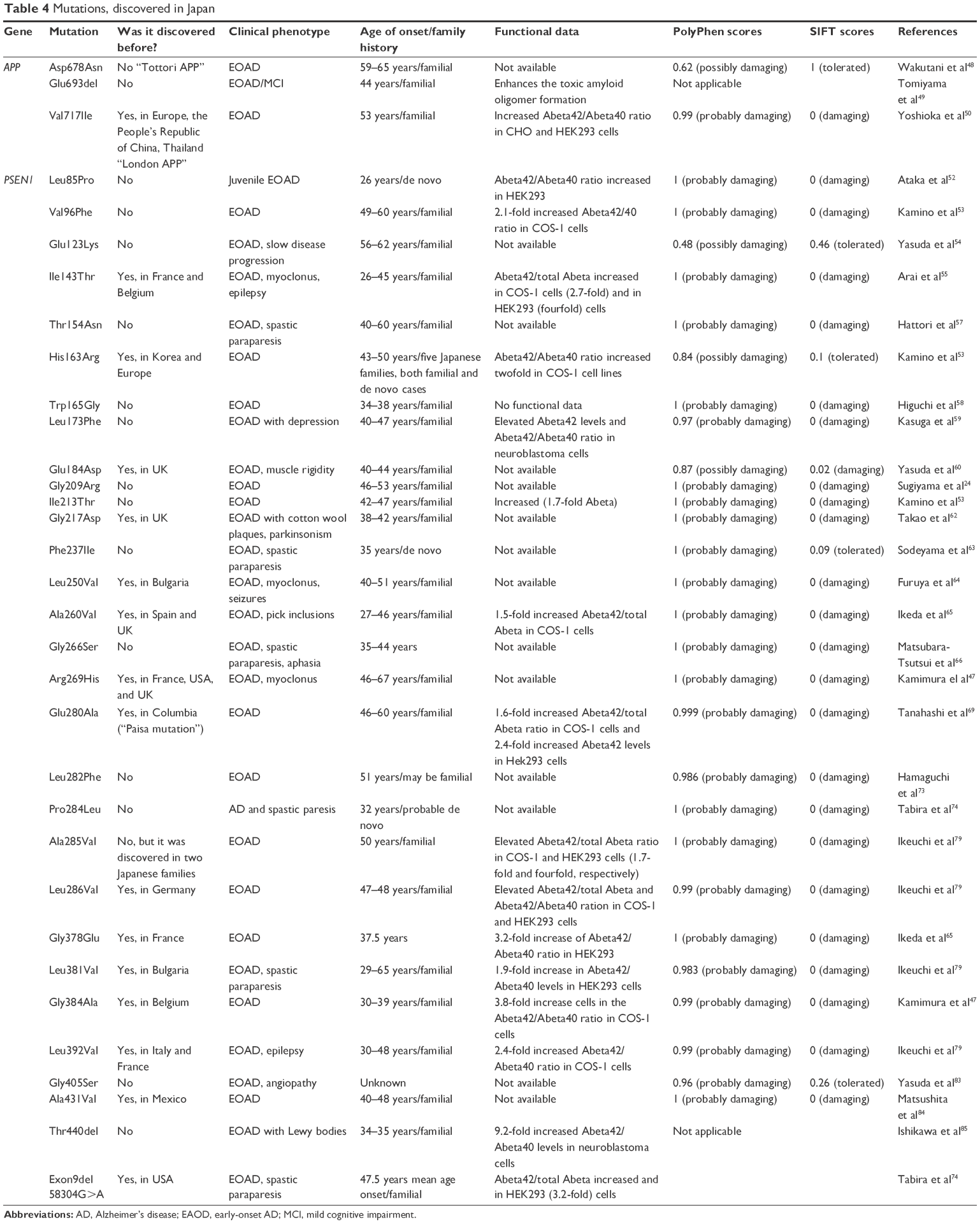
Mutations discovered in Japan
The genetics of EOAD in Japan has been relatively well studied, and several mutations have been identified in APP and PSEN1 (Table 4). To date, no PSEN2 mutations have been found in Japanese patients. However, silent mutations were reported.47
 | Table 4 Mutations, discovered in Japan |
Mutations in APP
Asp678Asn (GAC→AAC) or “Tottori APP” was described in a Japanese family, with the age of onset ranging from 59 to 65 years. The proband patient was hospitalized at the age of 65 years; she had amnesia, disorientation, and personality changes (loss of interest in activities and aggression). The symptoms started 6 years before the diagnosis. Seven years after the diagnosis, cortical hippocampal atrophy appeared, and the dementia worsened; the patient lost her ability to perform daily activities and needed assistance. The patient’s sister had dementia, which started with amnestic symptoms (inability to recognize her family). Over the next 15 years, she experienced severe cognitive decline (she needed personal care) with oral dyskinesia. The patient’s brother developed dementia symptoms when he was in his late 50s. The mother had no signs of dementia (died at 85 years). The father died in an accident at the age of 64 years, and it was unknown if he had any signs of dementia.48 Glu693del (delAGA) was found in two Japanese families. The mutation might be involved in AD and mild cognitive impairment in homo- and heterozygous stages, respectively. The mean age of onset of AD with this mutation is 44 years. The findings suggest that the mutation makes Abeta more resistant to proteolytic degradation. In addition, Abeta in mutant rats showed a unique aggregation pattern, forming oligomers instead of fibrils. It was suggested that soluble Abeta oligomers might be more toxic than the fibrillar Abeta aggregates.49 Val717Ile (GTC→ATC) was also found in a Japanese family.50 The mean age of onset of AD with this mutation is 53 years. Symptoms start with personality changes and disturbances in memory, emotions, or judgment, but dementia is predominant.51
Mutations in PSEN1
Leu85Pro (CTC→CCC) was discovered in a Japanese patient with de novo emergence of juvenile EOAD with spastic paraparesis. The age of onset was 26 years, but the patient had become withdrawn and unmotivated at an earlier age. PSEN1 Leu85Pro was expressed in HEK293 cells, and Abeta40 and Abeta42 concentrations were measured with ELISA. Western blotting was performed to assess changes in APP expression, but no difference was found between the expression of wild-type PSEN1 and that of mutant PSEN1. The secretion of Abeta42 increased, and the ratio of Abeta42/40 was higher in cells expressing mutant PSEN1 than in controls.52 Val96Phe (GTC→TTC) has been described in a Japanese family, with the age of onset ranging from 49 to 60 years. When PSEN1 Val96Phe was expressed in COS-1 cell lines, the Abeta42/total Abeta ratio increased (1.6-fold) in the mutant cells.21,53 Glu123Lys (GAG→AAG) was detected in a Japanese family, with the age of onset ranging from 56 to 62 years. Patients showed cognitive decline and memory deficits.54 Ile143Thr (ATT→ACT) was described for the first time in Belgian familial EOAD patients with cognitive decline, myoclonus, and epilepsy. The mutation was associated with an increased ratio of Abeta42/total Abeta. Abeta accumulation was also high, which suggests that the mutation correlates with PSEN1 dysfunction, leading to disease progression.55 The clinical phenotype of Japanese patients with the Ile143Thr mutation was similar to that of the Belgian patients. Codon 143 might be an important locus in PSEN1, given that the following additional mutations have been described: Ile143Met, Ile143Phe, Ile143Asn, and Ile143Val, and all of them have been confirmed as pathogenic mutations.56 Thr154Asn (TAT→AAT) has been described in a Japanese patient diagnosed with EOAD with spastic paraparesis. The first symptoms appeared in her 30s, and dementia developed when she was in her 40s. Her mother had a similar disease progression, but onset occurred later: spastic paraparesis and dementia appeared in her 40s and 60s, respectively. An additional pathogenic mutation at codon 154, Thr154Cys, has been identified.57 Trp165Gly (TGG→GGG) was discovered in a Japanese family with EOAD, with the age of onset ranging from 34 to 38 years. No detailed information is available on the patient’s clinical symptoms.58
Leu173Phe (TTG→TTC) has been described in a Japanese family. The patients exhibited depression and psychiatric symptoms before dementia progression. When Leu173Phe was expressed in neuroblastoma cells, the level of Abeta42 and the Abeta42/Abeta40 ratio increased.59 Glu184Asp (GAA→GAC) was found in a Japanese family with EOAD (age of onset at 40–44 years). The family members showed typical dementia symptoms, such as memory impairment, muscle rigidity, and disorientation. Disease duration was ~8.7±1.5 years. Three of the four patients with the mutation died at the age of 51 years. Postmortem examinations revealed gliosis, a high degree of neurodegeneration, senile plaques, and neurofibrillary tangles. The degree of amyloid angiopathy in the brain parenchyma and the leptomeninges regions was higher than in a typical AD patient.60 The mutation has also been described in a Caucasian family (in the UK) with EOAD and a similar age of onset.61 Gly209Arg (GGA→AGA) has been reported in a Japanese family, with the age of onset at 45–53 years. The symptoms were memory impairment, amnestic aphasia, disorientation, and personality change, without parietal focal symptoms, such as apraxia and agnosia. The mutation results in the exchange of small, hydrophobic glycine with large, polar arginine. The different phenotypes might be the result of different pathomechanisms, other genetic factors, or genetic–environmental interactions.24 Ile213Thr (ATT→ACT) was described in a Japanese family with EOAD, with the age of onset ranging from 42 to 48 years. When PSEN1 Ile213Thr was expressed in COS-1 cells, the Abeta42/total Abeta ratio increased (1.7-fold), but no detailed information was available on its pathogenic nature.21,53 Gly217Asp (GGT→GAT) was described in a Japanese family with EOAD and parkinsonism. Neuropathological changes, such as amyloid plaques, “cotton wool plaques”, amyloid angiopathy, tangles, and gliosis, were also detected.62 Phe237Ile (TTT→ATT) has been described in a Japanese patient with EOAD and spastic paraparesis without any family history. The first symptoms appeared when he was 31 years. Memory impairment and cognitive dysfunctions developed rapidly. Spastic paraparesis appeared when he was 35 years.63 Leu250Val (TTG→GTG) was found in a Japanese family and in a Caucasian (Bulgarian) family. In the patients, EOAD appeared with myoclonus (with tonic–clonic seizures), with the age of onset ranging from 40 to 51 years. Additional PSEN1 mutations, such as Leu153Val, Leu250Ser, and Glu280Ala, were associated with myoclonus, but tonic–clonic seizures appear only in patients with Leu250Val.64 Ala260Val (GCT→CCT) has been described in European and Japanese EOAD patients, with the age of onset ranging from 27 to 46 years. In the first Japanese pedigree (three patients), neuropathological studies detected senile plaques, tangles, neuronal loss, and Pick-like inclusions. In the second pedigree, the disease onset occurred later (mean, 51 years), but disease progression was faster.65 When PSEN1 Ala260Gly was expressed in COS-1 cells, the Abeta42/total Abeta ratio increased (1.5-fold).21 Gly266Ser (GGT→AGT) was reported in six patients in two generations of a Japanese family. Patients were diagnosed with AD, spastic paraparesis, and aphasia. The affected family members showed rapid disease progression after the age of 50 years. Diagnosis was complicated because the patients did not show typical AD symptoms, since tauopathy and prion disease-like symptoms were present. The patients were negative for mutations in the prion (PRNP) and Tau (MAPT) genes, suggesting that they had an AD variant.66 Arg269His (CGT→CAT) has been associated with EOAD and LOAD in American, European, and Japanese families. The mean age of disease onset is 50.5 years. The mutation was described by Gómez-Tortosa et al67 in an early dementia patient, with amyloid deposition particularly strong in the temporal cortex. Larner et al reported Arg269His in a patient with a positive family with LOAD. The examined patient exhibited the first symptoms of memory disturbances when he was 66 years of age.68 Her sister was also diagnosed with AD. Their grandmother, mother, and several aunts (5/6) were also diagnosed with AD or dementia. A few additional mutations were found in patients with LOAD. The study suggested that patients with LOAD should be tested for the main EOAD risk factor genes.68 Glu280Ala (GAA→GCA) or “Paisa mutation” has been detected in Japanese, European, and Columbian patients,69 with the age of onset ranging from 30 to 45 years. In Japan, Glu280Ala has been detected in familial AD cases.69 In Columbian families, it segregated with the disease. Glu280Ala might be independent from APOE alleles.70 PSEN1 Glu280Ala was expressed in different cell lines, including COS-1,21 N2a,71 and HEK293.72 The experiments established the pathogenic nature of the mutation. In COS-1 cells, the ratio of Abeta42/total Abeta (1.6-fold) increased. In HEK293 cells, the level of Abeta42 (2.4-fold) and the ratio of Abeta42/Abeta40 (2.1-fold) increased; no change was detected in the Abeta40 levels. In N2a cells, the amount of Abeta42 (2.7-fold) and the ratio of Abeta42/Abeta40 (2.4-fold) increased significantly, and a small increase in the level of Abeta40 (1.3-fold) was detected.21,71,72 Leu282Phe (CTT→TTT) was identified in a Japanese family, with the main age of onset at 51 years. Patients were diagnosed with progressive memory impairment. Given the family history, this might be a case of familial AD. The following two other mutations have been found at codon 282: Leu282Arg and Leu282Val. Both are associated with EOAD.73
Pro284Leu (CCA→CTA) was identified in a Japanese patient with AD and spastic paresis. The first movement problems started at the age of 32 years and became more serious. She died when she was 54 years. Postmortem studies found several cotton wool plaques and neurofibrillary tangles or amyloid angiopathy in her brain. Kuru-like plaques also appeared, but the prion-staining was negative.74 Ala285Val (GCT→GTT) has been described in two Japanese families, with the mean age of onset at 50.5 years. Patients did not show any sign of myoclonus, seizures, or paratonia. Increased Tau levels were found in the cerebrospinal fluid (CSF) of patients.75 Ikeda et al have also reported this mutation.65 The age of onset was similar, but disease progression was rapid. When PSEN1 Ala285Val was expressed in COS-1 cells, the Abeta42/total Abeta ratio increased (1.7-fold).21 When expressed in HEK293 cells, the Abeta42/total Abeta ratio increased (fourfold), and the Abeta40/total Abeta and Abeta38/total Abeta ratios decreased (1.3-fold and twofold, respectively).76 Leu286Val (CTC→GTC) has been described in a Japanese patient and in a Caucasian (German) family with EOAD, with the mean age of onset at 47 and 48 years, respectively.77,78 When PSEN1 Leu286Val was expressed in COS-1 and HEK293 cell lines, increases in the Abeta42/total Abeta ratio (1.5-fold) and Abeta42/Abeta40 ratio (2.1-fold) were detected, respectively.21,77 Gly378Glu (GGA→GAA) was initially described in France78 and in Japan,79 with the mean age of onset at 37.5 years, and all patients were diagnosed with AD. Cerebral amyloid angiopathy also appeared in some patients. When PSEN1 Gly378Glu was expressed in HEK293 cells, the Abeta42/Abeta40 ratio increased (3.2-fold).74 Leu381Val (CTT→GTT) was first described in a Bulgarian family. The patients were diagnosed with AD and spastic paraparesis, with the mean age of onset at 30.5 years. Abeta-positive cotton wool plaques appeared in the brain.76 In Japan, the mutation was found by Ikeuchi et al.79 When PSEN1 Leu381Val was expressed in HEK293 cells, the Abeta42/Abeta40 ratio increased (1.9-fold).79 Gly384Ala (GGA→GCA) was initially discovered in a Caucasian family, with the age of onset ranging from 30 to 39 years.56 In Japan, the mutation has been described in a family, with the age of onset ranging from 31 to 37 years. Autopsy confirmation suggested that the patients had AD: they had senile plaques and tangles inside their brain.69 The mutation has been analyzed in several studies. When PSEN1 Gly384Ala was expressed in COS-1 cells, significant increases (3.8-fold) in the Abeta42/Abeta40 ratio were detected.21 When expressed in CHO cells, the Abeta42/total Abeta ratio increased (3.8-fold),30 while in HEK293 cells, Abeta40 and the Abeta42/Abeta40 ratio decreased and increased significantly, respectively.76,80,81
Leu392Val (CTG→GTG) has been reported in several cases, with the mean age of onset at 42.5 years.17,79,82 Transfection into COS-1 cells increased the Abeta42/Abeta40 ratio (2.4-fold).16 An increase in the Abeta42/Abeta40 ratio (2.9-fold) was also detected after transfection into HEK293 cells.79 Asn405Ser (AAC→AGC) was found in one patient whose family history was unknown. The patient showed rapid and aggressive AD progression. The mutation was absent in healthy controls. The patient has several senile plaques and tangles in the brain (temporal cortex), but cerebral amyloid angiopathy also appeared in several areas. The mutation caused disturbances in the motor neuronal systems, leading to spastic paraparesis.83 Ala431Val (GCA→GTA) was found in a Japanese patient who was first diagnosed with mild cognitive impairment, followed by AD onset in the next 16 months. Tau and phospho-Tau levels increased in the CSF, and metabolic deficits were detected in several parts of the brain, including the posterior and medial temporal regions.84 T440del (delACC) was found in a 52-year-old male patient with early-onset dementia and parkinsonism. The family history was positive: the father and grandfather also suffered from a similar type of dementia. A serious degree of neuronal loss was detected in the brain; cotton wool plaques, cerebral amyloid angiopathy, and Lewy bodies were also present.85
Deletion of exon 9 is associated with a point mutation, 58304G→A, located in the splice acceptor consensus site in intron 8. This variant can affect the PSEN1 transcript splicing, by in-frame skipping of exon 9 and the junction of exon 8 and 9. This mutation was discovered in Japanese and Caucasian patients, resulting in EOAD with spastic paraparesis, with the mean age of onset at 47.5 years. Cotton wool plaques were also detected in the brain of patients. Mutation was expressed in HEK293 cell lines, where elevated Abeta42 levels and Abeta42/40 ratio were detected.74
Mutations discovered in other Asian countries
With the exception of Korea, the People’s Republic of China, Taiwan, and Japan, only a few reports are available on EOAD-associated mutations in Asian countries. Two APP mutations were discovered in Thailand and Iran, and recently, a novel PSEN1 mutation was reported in a Malaysian family (Table 5).
 | Table 5 Mutations, discovered in other Asian countries |
In Iran, an APP mutation, Thr714Ala (ACA→GCA or “Iranian APP”), has been reported in an autosomal dominant case of AD, with the mean age of onset at 55 years, but no detailed information was available regarding the clinical phenotypes.83 This mutation has also been reported in two other families. One of the patients, from Denmark, had atypical signs of AD; epilepsy also appeared. His father was a suspected AD patient, but the diagnosis was unclear. The age of onset ranged from 47 to 55 years. It has also been detected in a Polish (Caucasian) family, with the age of onset at 44–45 years. The symptoms were memory impairment, disorientation, changes in behavior, and aphasia.86,87 Val717Ile (GTC→ATC; London APP) has been identified in one Thai family, but no detailed information on the patients was available.37
No additional reports are available regarding EOAD-associated mutations in other East Asian countries. Studies of Malaysian patients have started recently. PSEN1 Glu280Lys (GAA→AAA) is a novel mutation, recently discovered by our research group in three siblings from Malaysia who were diagnosed with early-onset dementia. Strong evidence indicates that this mutation is involved in AD or dementia; it was found in three patients who likely had a positive family history of dementia. The existence of two additional pathogenic mutations at codon 280 (Glu280Gly and Glu280Ala) indicates that Glu280 is an important residue in PSEN1.88
Asian GWAS of LOAD
As mentioned before, the genetic background of LOAD is not well defined. Several genes have been proposed as risk factors for LOAD. In addition, nongenetic factors (environmental and lifestyle) could also play a role in AD that develops after the age of 65 years.89 Several meta-analyses and GWAS have been performed to identify genes and sequence variants that increase the risk for LOAD. These studies tested several mutations and polymorphisms by using high-throughput assays.90
APOE has three main alleles, with variations at codons 112 and 158. In the normal allele, called the E3 allele, codon 112 encodes cysteine, and codon 158 encodes arginine. The other two APOE alleles, E2 and E4, contain cysteine and arginine, respectively, at both sites. Several studies have confirmed the strong association between the E4 allele and AD.91 ApoE protein is the main cholesterol transporter in the brain; by binding to the Abeta peptide, it might contribute to AD progression. ApoE might inhibit Abeta clearance and induce neuroinflammation. The APOE E4 allele might be involved in LOAD, but it might not define all LOAD cases.91
Association studies were performed to find additional risk factor genes for AD. In Korea, Chung et al have performed GWAS; they found an association between AD and variants in PICALM, CR1, and BIN1.92,93 In the People’s Republic of China, several reports have described the relationship between different genes and LOAD. Jin et al have studied the CR1 gene and found an association between a polymorphism (rs6656401) and AD in the Han Chinese population.94 Liu et al have analyzed variants of several genes in Han Chinese subjects.95 Associations were found between LOAD and variants of three genes, MOBP (rs1768208), EIF2AK3 (rs7571971), and MAPT, in the presence of the APOE E4 allele.94 Tan et al have studied the MS4A and CD33 loci in the northern Han population and suggested that both loci might be involved in LOAD.96 Liu et al have performed GWAS on rs744373 in BIN1 and rs3851179 in PICALM in East Asians,97,98 and both genes showed a significant association with AD. Shang et al found an association between a variant (rs597668) of EXOC3L2 and AD.99 Several additional studies have reported associations between AD and variants of different genes, such as PLD3,100 SORCS1,101 NLRP3,102 and ARRB2.103 A few genetic studies have also been published in Japan on GWAS and different genes that might be involved in LOAD. PICALM and SORL1104,105 might contribute to LOAD. However, other Japanese studies have failed to find an association between AD and other genes, such as TREM2,106 CLU,107 GAB2,108 and PGBD1.109
Methods for genetic testing: from PCR-based methods to NGS
Genetics plays an important role in AD onset; several genes were suggested to be involved in disease onset. To identify the genes and mutations involved in AD progression, we should know the direct sequence of the causative and risk factor genes for the disease. Direct sequencing should be performed for the mutations in the following three main causative genes: APP, PSEN1, and PSEN2. Genomic DNA can be extracted from blood, tissues, and bone marrow, and specific DNA isolation kits have been developed for it. To identify the variants in them, PCR and standard sequencing methods can be performed.3 However, the mutations in these genes are relatively rare, and there may be similarities between the phenotypes of different neurodegenerative disorders. For the different types of early-onset dementia (such as frontotemporal dementia, prion diseases, and Parkinson’s disease), the phenotypes may be similar especially in the earlier stages. Pathological overlap may be possible, which makes the disease diagnosis more difficult.110
AD is one of the most complex disorders, in which several genes were identified to be involved, especially in the late-onset disease form. Even APOE is the main susceptibility factor for LOAD, but it does not define all cases of AD, which occur after the age of 65 years. As mentioned earlier, the genetic background of LOAD is currently unclear. Standard sequencing is still a widely used technology, but several problems are available in this approach, such as the cost, the error rates are relatively high, and it is time-consuming. Several genes and polymorphisms have been identified as risk factors for AD by GWAS.3 Recently, NGS approaches have been developed, which could provide a more accurate, fast, and cost-effective process to discover additional AD causative or risk factor genes. Several types of technologies are available, such as sequencing by synthesis (SBS), SBS by novel nucleotide analogs, homopolymer sequencing, double-ended sequencing, polony sequencing, single-nucleotide sequencing, and sequencing by hybridization or ligation.111 These approaches are useful in the searching for genes with Mendelian inheritance pattern, and they are also able to identify the common variants, which might be associated with the sporadic forms of AD.112 NGS can also be useful to analyze the transcriptomic and epigenetic changes, associated with the disease, and they might be helpful to understand the disease-associated pathways.113
Estimation of role of novel mutation
To verify the pathogenic nature of mutations in the EOAD causative genes, Guerreiro et al designed an algorithm. Mutations can be categorized as definitely pathogenic, probably pathogenic, possibly pathogenic, or nonpathogenic (putative risk factor) variants. In this study, segregation analysis was performed, and the affected family members should also be tested for the mutation. They also examined the residue, whether any additional mutation was found for it. Association studies are needed, since it was also important to define the pathogenic nature. Mutations should be screened in large number of healthy individuals. If the mutation appears in them, it might refute its pathogenic nature; however, it could be possible that these individuals could develop disease phenotype in the future.114 In Korea, the Korea Centers for Disease Control and Prevention (KCDC; http://www.cdc.go.kr) performed whole genome sequencing in 622 healthy individuals and screened them for pathogenic mutations. The Exome Aggregation Consortium database (http://exac.broadinstitute.org/) was designed for the same reason; they performed exome sequencing on >60,000 healthy individuals. This database is an easy-to-use database, which could be used for clinical and population genetic studies.
Guerreiro et al115 also suggested checking whether the mutations could affect the Abeta levels. Several mutations in PSEN1 and PSEN2 were tested in cell models, such as COS-1, N2A, and HEK293 cells, and coexpressed with APP to determine whether this mutation could result in increased Abeta42, decreased Abeta40 levels, or increased Abeta 42/Abeta40 ratio.21,53 These cell models could accurately determine the role of mutation in the disease progression. However, these cell model studies are expensive and time-consuming and were not performed for all discovered mutations (Tables 2–5).
In silico modeling is important, especially at the novel mutations, since it would be helpful to get hypothetical insight on the role of abnormal protein in disease progression. Recently, online prediction programs, such as PolyPhen2 and SIFT, were developed to estimate the damaging nature of mutations, by comparing several structure and function-based properties of proteins.116–118 In addition, three-dimensional modeling could also be helpful to estimate the role of mutations in disease progression. However, depending on the in silico predictions is not sufficient for full clinical prognosis, but these predictions could enhance the estimation of the pathogenic nature of mutations.119
Conclusion
Several neurodegenerative disorders can be distinguished, such as AD, FTD, amyotrophic lateral sclerosis, Parkinson’s disease, dementia with Lewy bodies, Huntington’s disease, and prion diseases. Currently, there is no effective treatment for neurodegenerative dementia, but potential therapeutic approaches might be successful in early disease stages. The main problem is that disease diagnosis, especially differential diagnosis, before the appearance of clinical symptoms is complicated. There is no unique marker for each disorder. However, combining proteomic and genetic markers might improve disease diagnosis. Determining the genetic background of dementia would also improve the accuracy of disease diagnosis. Several genes have been described as causative or risk factor genes for dementia.3,115
Several genetic studies on EOAD patients were performed in Japanese, Korean, and Chinese populations. Similar studies have not been investigated yet in South Asian countries (Thailand and Malaysia). Hence, our investigators may start multinational efforts for screening AD-related mutations in collaborations with them. Comparing the EOAD-associated mutations between Caucasian and Asian patients, >30 novel Asian mutations were found in APP, PSEN1, and PSEN2 (http://www.alzforum.org/mutations). In APP gene, novel mutations were discovered in Japanese or Chinese/Taiwanese patients, such as Asp678Asn (Tottori APP), Ile718Leu, and Leu720Ser. Val715Met (French APP) and the Val717Ile (London APP), which were initially discovered in British or French Caucasians, were also detected in Korean and Japanese EOAD patients, respectively.12,14,36,48 In PSEN1, several novel mutations have been discovered in Korean, Malaysian, and Chinese EOAD patients, such as His163Pro, Leu232Pro, Leu248Pro, and Glu280Lys.12,23,41,88 PSEN1 H163R, the most frequently described AD causative mutation among French, German, and Turkish Caucasians, has appeared in Korea and Japan.22,47,53,69 Additional mutations, such as Leu226Phe, Thr116Ile, Met233Thr (Korea), Ile143Thr, and Arg269His (Japan), were also reported in Korea and Japan.16,17,27,28,55,66–68 In the People’s Republic of China, most of the PSEN1 variants, such as Ile167del, Ser169del, and Leu248Pro, were newly discovered.12,43,44 PSEN2 mutation was rare in Asian countries; however, novel Val214Leu, His169Asn, and Asn141Tyr mutations were found. PSEN2 Arg62Cys mutation, which was initially discovered in Belgian Caucasians, was found among Korean.31–34,46
Because the overall population and aging population in most Asian countries are increasing, genetic testing of patients with AD and other types of dementia is important in the diagnosis of dementia. Several genes could be directly or indirectly involved in the onset of AD. These genes should be tested, and other candidates should be identified in order to obtain a clearer understanding of the cascade associated with disease progression. It was suggested that neurodegenerative disorders arise from a common molecular pathway. Clinical diversity might be the result of different genetic mutations that directly or indirectly affect disease progression or protect against the disease. However, determining the initial disease-associated process is also a key issue for diagnosis. Screening to identify potential candidate genes associated with the disease is important.112,120 NGS technologies can facilitate the discovery of novel disease-associated genes and identify candidate genes involved in the onset of EOAD or LOAD. Simultaneous analysis of different genes may improve differential diagnosis, which is essential in the development of preventative and therapeutic strategies. Genetic profiling could also help the disease risk prediction, since it might allow the start of prevention strategies in the presymptomatic stages.110
Acknowledgments
This work was supported by grants from the KCDC, Republic of Korea (2015-0169), and the Korea Health Technology R&D Project (HI14C3331) through the Korea Health Industry Development Institute, Korea Ministry of Health & Welfare.
Disclosure
The authors report no conflicts of interest in this work.
References
Ridge PG, Ebbert MT, Kauwe JS. Genetics of Alzheimer’s disease. Biomed Res Int. 2013;2013:25. | ||
Catindig JA, Venketasubramanian N, Ikram MK, Chen C. Epidemiology of dementia in Asia: insights on prevalence, trends and novel risk factors. J Neurol Sci. 2012;321(1–2):11–16. | ||
Bagyinszky E, Youn YC, An SS, Kim SY. The genetics of Alzheimer’s disease. Clin Interv Aging. 2014;9:535–551. | ||
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12(12):1567–1576. | ||
Kuperstein I, Broersen K, Benilova I, et al. Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. | ||
Szaruga M, Veugelen S, Benurwar M, et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J Exp Med. 2015;212(12):2003–2013. | ||
Xia D, Watanabe H, Wu B, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85:967–981. | ||
Ben-Gedalya T, Moll L, Bejerano-Sagie M, et al. Alzheimer’s disease-causing proline substitutions lead to presenilin 1 aggregation and malfunction. EMBO J. 2015;34:2820–2839. | ||
Alonso Vilatela ME, López-López M, Yescas-Gómez P. Genetics of Alzheimer’s disease. Arch Med Res. 2012;43:622–631. | ||
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–1344. | ||
An SS, Bagyinszky E, Kim HR, et al. Novel PSEN1 G209A mutation in early-onset Alzheimer dementia supported by structural prediction. BMC Neurol. 2016;16:71. | ||
Jiao B, Tang B, Liu X, et al. Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol Aging. 2014;35:.e1–.e6. | ||
Niu F, Yu S, Zhang Z, et al. A novel mutation in the PSEN2 gene (N141Y) associated with early-onset autosomal dominant Alzheimer’s disease in a Chinese Han family. Neurobiol Aging. 2014;35(10):.e1–.e5. | ||
Park HK, Na DL, Lee JH, Kim JW, Ki CS. Identification of PSEN1 and APP gene mutations in Korean patients with early-onset Alzheimer’s disease. J Korean Med Sci. 2008;23:213–217. | ||
Ancolio K, Dumanchin C, Barelli H, et al. Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val-715 ◊ Met betaAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96:4119–4124. | ||
La Bella V, Liguori M, Cittadella R, et al. A novel mutation (Thr116Ile) in the presenilin 1 gene in a patient with early-onset Alzheimer’s disease. Eur J Neurol. 2004;11(8):521–524. | ||
Raux G, Guyant-Marechal L, Martin C, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J Med Genet. 2005;42(10):793–795. | ||
Romero I, Jorgensen P, Bolwig G, et al. A presenilin-1 Thr116Asn substitution in a family with early-onset Alzheimer’s disease. Neuroreport. 1999;10(11):2255–2260. | ||
Kim HJ, Kim HY, Ki CS, Kim SH. Presenilin 1 gene mutation (M139I) in a patient with an early-onset Alzheimer’s disease: clinical characteristics and genetic identification. Neurol Sci. 2010;31(6):781–783. | ||
Boteva K, Vitek M, Mitsuda H, et al. Mutation analysis of presenillin 1 gene in Alzheimer’s disease. Lancet. 1996;347:130–131. | ||
Murayama O, Tomita T, Nihonmatsu N, et al. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci Lett. 1991;265(1):61–63. | ||
Hong KS, Kim SP, Na DL, et al. Clinical and genetic analysis of a pedigree of a thirty-six-year-old familial Alzheimer’s disease patient. Biol Psychiatry. 1997;42:1172–1176. | ||
Kim J, Bagyinszky E, Chang YH, et al. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: clinical, neuroimaging, and neuropathological findings. Neurosci Lett. 2012;530(2):109–114. | ||
Sugiyama N, Suzuki K, Matsumura T, et al. A novel missense mutation (G209R) in exon 8 of the presenilin 1 gene in a Japanese family with presenile familial Alzheimer’s disease. Hum Mutat. 1999;14:90. | ||
Rogaeva EA, Fafel KC, Song YQ, et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology. 2001;57(4):621–625. | ||
Poorkaj P, Sharma V, Anderson L, et al. Missense mutations in the chromosome 14 familial Alzheimer’s disease presenilin 1 gene. Hum Mutat. 1998;11:216–221. | ||
Bagyinszky E, Park SA, Kim HJ, Choi SH, An SS, Kim SY. PSEN1 L226F mutation in a patient with early-onset Alzheimer’s disease in Korea. Clin Interv Aging. In press 2016. | ||
Zekanowski C, Golan MP, Krzyśko KA, et al. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: genetic and bioinformaticstic assessment. Exp Neurol. 2006;200:82–88. | ||
Zekanowski C, Styczyńska M, Pepłońska B, et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol. 2003;184(2):991–996. | ||
Sato T, Dohmae N, Qi Y, et al. Potential link between amyloid beta-protein 42 and C-terminal fragment gamma 49-99 of beta-amyloid precursor protein. J Biol Chem. 2003;278(27):24294–24301. | ||
Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127(pt 4):1641–1649. | ||
Ertekin-Taner N, Younkin LH, Yager DM, et al. Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology. 2008;70:596–606. | ||
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40(8):562–583. | ||
Youn YC, Bagyinszky E, Kim H, Choi BO, An SS, Kim S. Probable novel PSEN2 Val214Leu mutation in Alzheimer’s disease supported by structural prediction. BMC Neurol. 2014;14:105. | ||
Jiang HY, Li GD, Dai SX, et al. Identification of PSEN1 mutations p.M233L and p.R352C in Han Chinese families with early-onset familial Alzheimer’s disease. Neurobiol Aging. 2015;36(3):.e3–.e6. | ||
Thajeb P, Wang P, Chien CL, Harrigan R. Novel polymorphisms of the amyloid precursor protein (APP) gene in Chinese/Taiwanese patients with Alzheimer’s disease. J Clin Neurosci. 2009;16:259–263. | ||
Finckh U, Kuschel C, Anagnostouli M, et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6(2):85–89. | ||
Herl L, Thomas AV, Lill CM, et al. Mutations in amyloid precursor protein affect its interactions with presenilin/gamma-secretase. Mol Cell Neurosci. 2009;41(2):166–174. | ||
De Jonghe C, Zehr C, Yager D, et al. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998;5(4):281–286. | ||
Tamaoka A, Odaka A, Ishibashi Y, et al. APP717 missense mutation affects the ratio of amyloid beta protein species (A beta 1-42/43 and a beta 1-40) in familial Alzheimer’s disease brain. J Biol Chem. 1994;269(52):32721–32724. | ||
Jia J, Xu E, Shao Y, Jia J, Sun Y, Li D. One novel presenilin-1 gene mutation in a Chinese pedigree of familial Alzheimer’s disease. J Alzheimers Dis. 2005;7(2):119–124. | ||
Fang B, Jia L, Jia J. Chinese Presenilin-1 V97L mutation enhanced Abeta42 levels in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2006;406(1–2):33–37. | ||
Fang BY, Jia JP. The effect of two newly Chinese presenilin-1 mutations on the sensitivity to trophic factor withdrawal in human neuroblastoma cells. Zhonghua Yi Xue Za Zhi. 2007;87:336–340. | ||
Guo J, Wei J, Liao S, Wang L, Jiang H, Tang B. A novel presenilin 1 mutation (Ser169del) in a Chinese family with early-onset Alzheimer’s disease. Neurosci Lett. 2010;468(1):34–37. | ||
Shi Z, Wang Y, Liu S, et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement Geriatr Cogn Disord. 2015;39(1–2):32–40. | ||
Xia M, Chen S, Shi Y, et al. Probable novel PSEN2 Pro123Leu mutation in a Chinese Han family of Alzheimer’s disease. Neurobiol Aging. 2015;36(12):.e13–.e18. | ||
Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160:76–81. | ||
Wakutani Y, Watanabe K, Adachi Y, et al. Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(7):1039–1042. | ||
Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. J Neurol Neurosurg Psychiatry. 2008;63(3):377–387. | ||
Yoshioka K, Miki T, Katsuya T, Ogihara T, Sakaki Y. The 717Val->Ile substitution in amyloid precursor protein is associated with familial Alzheimer’s disease regardless of ethnic groups. Biochem Biophys Res Commun. 1991;178:1141–1146. | ||
Lindquist SG, Nielsen JE, Stokholm J, et al. Atypical early-onset Alzheimer’s disease caused by the Iranian APP mutation. J Neurol Sci. 2008;268(1–2):124–130. | ||
Ataka S, Tomiyama T, Takuma H, et al. A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch Neurol. 2004;61(11):1773–1776. | ||
Kamino K, Sato S, Sakaki Y, et al. Three different mutations of presenilin 1 gene in early-onset Alzheimer’s disease families. Neurosci Lett. 1996;208:195–198. | ||
Yasuda M, Maeda K, Hashimoto M, et al. A pedigree with a novel presenilin 1 mutation at a residue that is not conserved in presenilin 2. Arch Neurol. 1999;56(1):65–69. | ||
Arai N, Kishino A, Takahashi Y, et al. Familial cases presenting very early onset autosomal dominant Alzheimer’s disease with I143T in presenilin-1 gene: implication for genotype-phenotype correlation. Neurogenetics. 2008;9:65–67. | ||
Cruts M, Backhovens H, Wang SY, et al. Molecular genetic analysis of familial early-onset Alzheimer’s disease linked to chromosome 14q24.3. Hum Mol Genet. 1995;4(12):2363–2372. | ||
Hattori S, Sakuma K, Wakutani Y, et al. A novel presenilin 1 mutation (Y154N) in a patient with early onset Alzheimer’s disease with spastic paraparesis. Neurosci Lett. 2004;368(3):319–322. | ||
Higuchi S, Yoshino A, Matsui T, et al. A novel PS1 Mutation (W165G) in a Japanese family with early-onset Alzheimer’s disease. Alzheimers Rep. 2000;3:227–231. | ||
Kasuga K, Ohno T, Ishihara T, et al. Depression and psychiatric symptoms preceding onset of dementia in a family with early-onset Alzheimer disease with a novel PSEN1 mutation. J Neurol. 2009;256:1351–1353. | ||
Yasuda M, Maeda K, Ikejiri Y, Kawamata T, Kuroda S, Tanaka C. A novel missense mutation in the presenilin-1 gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci Lett. 1997;232(1):29–32. | ||
Janssen JC, Beck JA, Campbell TA, et al. Early onset familial Alzheimer’s disease: mutation frequency in 31 families. Neurology. 2003;60(2):235–239. | ||
Takao M, Ghetti B, Hayakawa I, et al. A novel mutation (G217D) in the Presenilin 1 gene (PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol. 2002;104(2):155–170. | ||
Sodeyama N, Iwata T, Ishikawa K, et al. Very early onset Alzheimer’s disease with spastic paraparesis associated with a novel presenilin 1 mutation (Phe237Ile). J Neurol Neurosurg Psychiatry. 2001;71:556–557. | ||
Furuya H, Yasuda M, Terasawa K, et al. A novel mutation (L250V) in the presenilin 1 gene in a Japanese familial Alzheimer’s disease with myoclonus and generalized convulsion. J Neurol Sci. 2003;209:75–77. | ||
Ikeda M, Sharma V, Sumi SM, et al. The clinical phenotype of two missense mutations in the presenilin I gene in Japanese patients. Ann Neurol. 1996;40(6):912–917. | ||
Matsubara-Tsutsui M, Yasuda M, Yamagata H, et al. Molecular evidence of presenilin 1 mutation in familial early onset dementia. Am J Med Genet. 2002;114:292–298. | ||
Gómez-Tortosa E, Barquero S, Barón M, et al. Clinical-genetic correlations in familial Alzheimer’s disease caused by presenilin 1 mutations. J Alzheimers Dis. 2010;19(3):873–884. | ||
Larner AJ, Ray PS, Doran M. The R269H mutation in presenilin-1 presenting as late-onset autosomal dominant Alzheimer’s disease. J Neurol Sci. 2007;252(2):173–176. | ||
Tanahashi H, Mitsunaga Y, Takahashi K, Tasaki H, Watanabe S, Tabira T. Missense mutation of S182 gene in Japanese familial Alzheimer’s disease. Lancet. 1995;346:440. | ||
Lendon CL, Martinez A, Behrens IM, et al. E280A PS-1 mutation causes Alzheimer’s disease but age of onset is not modified by ApoE alleles. Hum Mutat. 1997;10(3):186–195. | ||
Kaneko H, Kakita A, Kasuga K, et al. Enhanced accumulation of phosphorylated α-synuclein and elevated β-amyloid 42/40 ratio caused by expression of the presenilin-1 ΔT440 mutant associated with familial Lewy body disease and variant Alzheimer’s disease. J Neurosci. 2007;27:13092–13097. | ||
Shioi J, Georgakopoulos A, Mehta P, et al. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007;101(3):674–681. | ||
Hamaguchi T, Morinaga A, Tsukie T, Kuwano R, Yamada M. A novel presenilin 1 mutation (L282F) in familial Alzheimer’s disease. J Neurol. 2009;256:1575–1577. | ||
Tabira T, Chui de H, Nakayama H, Kuroda S, Shibuya M. Alzheimer’s disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70(3):367–372. | ||
Aoki M, Abe K, Oda N, et al. A presenilin-1 mutation in a Japanese family with Alzheimer’s disease and distinctive abnormalities on cranial MRI. Neurology. 1997;48(4):1118–1120. | ||
Page RM, Baumann K, Tomioka M, et al. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283(2):677–683. | ||
Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. | ||
Besançon R, Lorenzi A, Cruts M, et al. Missense mutation in exon 11 (Codon 378) of the presenilin-1 gene in a French family with early-onset Alzheimer’s disease and transmission study by mismatch enhanced allele specific amplification. Hum Mutat. 1998;11:481. | ||
Ikeuchi T, Kaneko H, Miyashita A, et al. Mutational analysis in early-onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement Geriatr Cogn Disord. 2008;26(1):43–49. | ||
Kumar-Singh S, Theuns J, Van Broeck B, et al. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27(7):686–695. | ||
Traykov DL, Mehrabian S, Van den Broeck M, et al. Novel PSEN1 mutation in a Bulgarian patient with very early-onset Alzheimer’s disease, spastic paraparesis, and extrapyramidal signs. J Alzheimers Dis Other Demen. 2009;24(5):404–407. | ||
Campion D, Brice A, Hannequin D, et al. A large pedigree with early-onset Alzheimer’s disease: clinical, neuropathologic, and genetic characterization. Neurology. 1995;45:80–85. | ||
Yasuda M, Maeda S, Kawamata T, et al. Novel presenilin-1 mutation with widespread cortical amyloid deposition but limited cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry. 2000;68(2):220–223. | ||
Matsushita S, Arai H, Okamura N, et al. Clinical and biomarker investigation of a patient with a novel presenilin-1 mutation (A431V) in the mild cognitive impairment stage of Alzheimer’s disease. Biol Psychiatry. 2002;52(9):907–910. | ||
Ishikawa A, Piao YS, Miyashita A, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol. 2005;7(3):429–434. | ||
Pasalar P, Najmabadi H, Noorian AR, et al. An Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala). Neurology. 2002;58:1574–1575. | ||
Zekanowski C, Styczyńska M, Pepłońska B, et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol. 2003;184(2):991–996. | ||
Ch’ng GS, An SS, Bae SO, Bagyinszky E, Kim S. Identification of two novel mutations, PSEN1 E280K and PRNP G127S, in a Malaysian family. Neuropsychiatr Dis Treat. 2015;11:2315–2322. | ||
Zou Z, Liu C, Che C, Huang H. Clinical genetics of Alzheimer’s disease. Biomed Res Int. 2014;2014:291862. | ||
Waring SC, Rosenberg RN. Genome-wide association studies in Alzheimer disease. Arch Neurol. 2008;65:329–334. | ||
Kim DH, Yeo SH, Park JM, et al. Genetic markers for diagnosis and pathogenesis of Alzheimer’s disease. Gene. 2014;545(2):185–193. | ||
Chung SJ, Lee JH, Kim SY, et al. Association of GWAS top hits with late-onset Alzheimer disease in Korean population. Alzheimer Dis Assoc Disord. 2013;27:250–257. | ||
Chung SJ, Kim MJ, Kim YJ, et al. CR1, ABCA7, and APOE genes affect the features of cognitive impairment in Alzheimer’s disease. J Neurol Sci. 2014;339(1–2):91–96. | ||
Jin C, Li W, Yuan J, Xu W, Cheng Z. Association of the CR1 polymorphism with late-onset Alzheimer’s disease in Chinese Han populations: a meta-analysis. Neurosci Lett. 2012;527(1):46–49. | ||
Liu QY, Yu JT, Miao D, et al. An exploratory study on STX6, MOBP, MAPT, and EIF2AK3 and late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34(5):.e13–.e17. | ||
Tan L, Yu JT, Zhang W, et al. Association of GWAS-linked loci with late-onset Alzheimer’s disease in a northern Han Chinese population. Alzheimers Dement. 2013;9:546–553. | ||
Liu G, Zhang S, Cai Z, et al. BIN1 gene rs744373 polymorphism contributes to Alzheimer’s disease in East Asian population. Neurosci Lett. 2013;544:547–551. | ||
Liu G, Zhang S, Cai Z, et al. PICALM gene rs3851179 polymorphism contributes to Alzheimer’s disease in an Asian population. Neuromolecular Med. 2013;15(2):384–388. | ||
Shang H, Fu J, Zhang XM, Song RR, Wang WZ. Association between EXOC3L2 rs597668 polymorphism and Alzheimer’s disease. CNS Neurosci Ther. 2013;19(10):834–839. | ||
Wang J, Yu JT, Tan L. PLD3 in Alzheimer’s Disease. Mol Neurobiol. 2015;51:480–486. | ||
Xu W, Xu J, Wang Y, et al. The genetic variation of SORCS1 is associated with late-onset Alzheimer’s disease in Chinese Han population. PLoS One. 2013;8:e63621. | ||
Tan MS, Yu JT, Jiang T, et al. NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. J Neuroimmunol. 2013;265(1–2):91–95. | ||
Jiang T, Yu JT, Wang YL, et al. NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. J Neuroimmunol. 2013;265(1–2):91–95. | ||
Ohara T, Ninomiya T, Hirakawa Y, et al. Association study of susceptibility genes for late-onset Alzheimer’s disease in the Japanese population. Psychiatr Genet. 2012;22(6):290–293. | ||
Miyashita A, Koike A, Jun G, et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One. 2013;8(4):e58618. | ||
Miyashita A, Wen Y, Kitamura N, et al. Lack of genetic association between TREM2 and late-onset Alzheimer’s disease in a Japanese population. J Alzheimers Dis. 2014;41(4):1031–1038. | ||
Miyashita A, Arai H, Asada T, et al. GAB2 is not associated with late-onset Alzheimer’s disease in Japanese. Eur J Hum Genet. 2009;17(5):682–686. | ||
Komatsu M, Shibata N, Kuerban B, Ohnuma T, Baba H, Arai H. Genetic association between clusterin polymorphisms and Alzheimer’s disease in a Japanese population. Psychogeriatrics. 2011;11(1):14–18. | ||
Ohnuma T, Nakamura T, Takebayashi Y, et al. No associations found between PGBD1 and the age of onset in Japanese patients diagnosed with sporadic Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2012;2(1):496–502. | ||
Vo Van G, An SSA, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11:89–143. | ||
Lin B, Wang J, Cheng Y. Recent patents and advances in the next-generation sequencing technologies. Recent Pat Biomed Eng. 2008;2008(1):60–67. | ||
Mitsui J, Tsuji S. Genomic aspects of sporadic neurodegenerative diseases. Biochem Biophys Res Commun. 2014;452(2):221–225. | ||
Lill CM, Bertram L. Towards unveiling the genetics of neurodegenerative diseases. Semin Neurol. 2011;31(5):531–541. | ||
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. | ||
Guerreiro R, Brás J, Hardy J, Singleton A. Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations. Hum Mol Genet. 2014;23(R1):R47–R53. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. | ||
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. | ||
Bagyinszky E, Bae S, Kim SY, An SS. In silico modeling of pathogenic point mutations in PSEN1 as studied in South-east Asia Toxicol. Environ Health Sci. 2016;8:135. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. | ||
Tosto G, Reitz C. Genome-wide association studies in Alzheimer’s disease: a review. Curr Neurol Neurosci Rep. 2013;13(10):381. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.