Back to Journals » International Medical Case Reports Journal » Volume 19
Multiple Red Flags of Transthyretin Amyloid Cardiomyopathy in a Single Patient: A Case Report on Diagnostic Challenge
Authors Yao L, Chen K, Zhang F, Qin J
Received 29 December 2025
Accepted for publication 3 March 2026
Published 13 March 2026 Volume 2026:19 589746
DOI https://doi.org/10.2147/IMCRJ.S589746
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Lingling Yao,1,* Kexin Chen,1,* Fusheng Zhang,1 Jun Qin2
1Cardiac Care Unit, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China; 2Department of Hematopathology, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Qin, Department of Hematopathology, Renmin Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China, Email [email protected]
Background: Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, infiltrative cardiomyopathy caused by misfolded transthyretin proteins forming amyloid fibers that deposit in the myocardial interstitium. It may present with multi-systems involvement. Diagnostic delay is common due to non-specific symptoms.
Case Summary: We present a case of ATTR-CM with autonomic neuropathy as initial symptom, accompanied by multi-system manifestations. After being shuffled between multiple hospitals and departments, it was eventually found that his symptoms matched several “red flags” of ATTR-CM. Following the diagnostic algorithm, he underwent a 99Tcm-PYP imaging and was finally diagnosed with ATTR-CM. In our manuscript, we outline the diagnostic pathway recommended by the latest guidelines and review treatment strategies that are available and emerging.
Discussion: Since early diagnosis is crucial for improving clinical outcomes for patients, we would like to highlight the pitfalls in diagnosing this disease and emphasize the need to be aware of its variable clinical presentation and red flags.
Keywords: amyloidosis, cardiomyopathy, transthyretin, autonomic neuropathy, multi-organ damage
Introduction
Cardiac amyloidosis (CA) refers to the deposition of insoluble amyloid fibrils formed by misfolded proteins in the interstitial spaces of myocardial cells, leading to a restrictive cardiomyopathy characterized primarily by diastolic dysfunction, often manifesting as refractory heart failure and arrhythmias.1 The precursor proteins that trigger amyloidosis are mainly monoclonal immunoglobulin light chains (L) and transthyretin (TTR), accounting for over 98% of all CA cases. These amyloid proteins deposit in tissues and organs, causing structural damage and organ dysfunction, often resulting in multi-system damage. Thus, amyloidosis is a systemic disease that can involve the autonomic nervous system, kidneys, gastrointestinal tract, skin, liver, and other organs.2 Due to the lack of specificity in clinical manifestations, it often leads to under-recognition and delayed diagnosis. For clinical doctors, being familiar with the cardiac and extra-cardiac warning signs (“red flags”) is particularly important for the diagnosis of the disease. In this article, we report a case with ATTR-CM in which the diagnosis was delayed by six years after the onset of symptoms. This case highlights the challenges in recognizing CA early and underscores the importance of timely diagnosis to prevent irreversible organ damage and improve patient outcomes. Our findings aim to enhance clinical awareness and reduce diagnosis delays in similar cases.
Case Presentation
A 70-year-old male retired teacher was admitted to the cardiology department of our hospital in June 2024 with a 1-week history of palpitations, dyspnea, and general fatigue. A detailed history delineated a six-year, insidious progression of disease, commencing with non-specific constitutional symptoms before manifesting as severe cardiac and neurologic dysfunction. His first consultation in 2018 was due to diarrhea, with a colonoscopy showing no abnormalities. Since then, he has had chronic diarrhea (3–5 times/day) with long-term upper abdominal distension, early satiety, poor appetite, and progressive weight loss. Since 2019, he developed progressive limb weakness and numbness. He was diagnosed as peripheral neuropathy by neurologist. He also has experienced recurrent dizziness and syncope for 5 years, mostly triggered by positional changes, with documented chronic hypotension (lowest 70/40 mmHg). Each syncope episode lasted 3–5 minutes before regaining consciousness. No specific discomfort was reported upon waking. And repeated cranial Magnetic resonance imaging (MRI) showed bilateral frontal lobe lacunar infarcts. Mildly abnormal renal function was reported for 3 years, showing creatinine (Cr) 128–162 umol/L and urine protein ± to +. He had experienced palpitations, chest tightness, shortness of breath, and intermittent lower limb edema for 3 years, and was previously diagnosed with chronic heart failure, atrial flutter, and valvular heart disease during hospitalization. Cardiac troponin T (cTnT) levels had been mildly elevated since 2022, with persistently increased NT-proBNP. A Computed Tomography Angiography (CTA) suggested coronary atherosclerosis, showing mild stenosis at the opening of the left anterior descending artery. Hypothyroidism for 5 years, with cold intolerance and little sweating, he was treated with levothyroxine 50 μg daily with partial symptom improvement, although thyroid function was followed irregularly. Other manifestations included hypothyroidism, intermittent hoarseness, long-standing voiding difficulties, anxiety, and low mood. He had no history of hypertension, diabetes, or relevant family history. The patient had consulted multiple hospitals and departments due to the above symptoms, with some symptoms improving after symptomatic treatment but easily recurring.
On admission, he was conscious but lethargic. Vital signs were: afebrile, BP 78/54 mmHg, HR 46 bpm, RR 18 breaths/min and SpO2 98%. He was cachectic with body mass index (BMI) of 16 kg/m2. Respiratory examination revealed decrease air entry at left lower lung, no rales bilaterally. Cardiovascular examination revealed bradycardia, irregular rhythm, and a soft systolic murmur 2/6. Neurological examination revealed severe muscle atrophy, absent tendon reflexes in the lower limbs, and globally reduced muscle strength (4+ in upper limbs, 4- in lower limbs).
Investigations
After admission, he underwent relevant auxiliary examinations. Full blood Count showed hemoglobin concentration 87 g/L. Biochemical tests showed cTnT of 0.137 ng/mL, NT-proBNP of 10207 pg/mL, BUN of 8.6 mmol/L, Cr of 117.3 umol/L, and TSH of 11.215 uIU/mL. Stool culture and identification were negative. An electrocardiogram (ECG) examination demonstrated atrial flutter with slow ventricular response, complete right bundle branch block and left anterior fascicular block. Additionally, there was poor R wave progression in the anterior wall, and low voltage in limb leads (Figure 1A). Echocardiography showed slightly dilated ascending aorta, enlarged atria, granular echogenicity in the myocardium, thickening of the interventricular septum and left ventricular wall, thickening of the right ventricular anterior wall, moderate regurgitation of the tricuspid and mitral valves, mildly elevated pulmonary artery systolic pressure, small pericardial effusion, and left ventricular ejection fraction (LVEF) of 62%. Left ventricular global longitudinal strain (LV GLS) was reduced at −12.5% with an apical sparing pattern, and E/e′ ratio was elevated at 18 (Figure 1B). Musculoskeletal ultrasound revealed thickening of the right median nerve at the carpal tunnel, with a cross-sectional area of 17 mm2, whereas of 9 mm2 on the left side. Based on this finding, carpal tunnel syndrome was considered, which is a recognized extracardiac red flag for ATTR amyloidosis (Figure 1C). Electromyography showed peripheral nerve injury in the upper and lower limbs. Ultrasound of both kidneys and prostate showed no structural abnormalities. Chest CT suggested a small nodule in the left upper lobe, bilateral mild to moderate pleural effusion, and atelectasis in both lower lungs. Contrast-enhanced abdominal CT indicated gastric wall edema, multiple small lymph nodes in the hepatogastric space, possible cholecystitis, as well as abdominal and pelvic effusion, mesenteric haziness, and subcutaneous edema in the abdominal and pelvic wall. Laryngoscopy performed for intermittent hoarseness showed pharyngitis and benign nasal polyps in the right nasal cavity, tongue base, and epiglottis. The antinuclear antibodies, anti-ENA antibody, anti-neutrophil cytoplasmic antibody were negative. α-galactosidase A activity was 6.74 umol/L/h (2.20~17.65), thus Fabry disease is excluded. Laboratory tests revealed that the serum immunoglobulin free light chain (FLC) kappa and lambda levels were normal, and the FLC ratio was 1.058 (0.26–1.65). Furthermore, no M protein was detected in either serum or urine, thereby excluding AL amyloidosis. Myocardial scintigraphy with 99mTc-pyrophosphate (99mTc-PYP) findings is as follows: a 1-hour H/CL ratio of 1.55 and 3-hour myocardial uptake greater than rib uptake, with a Perugini (semiquantitative) grade of 2–3 (Figure 2).
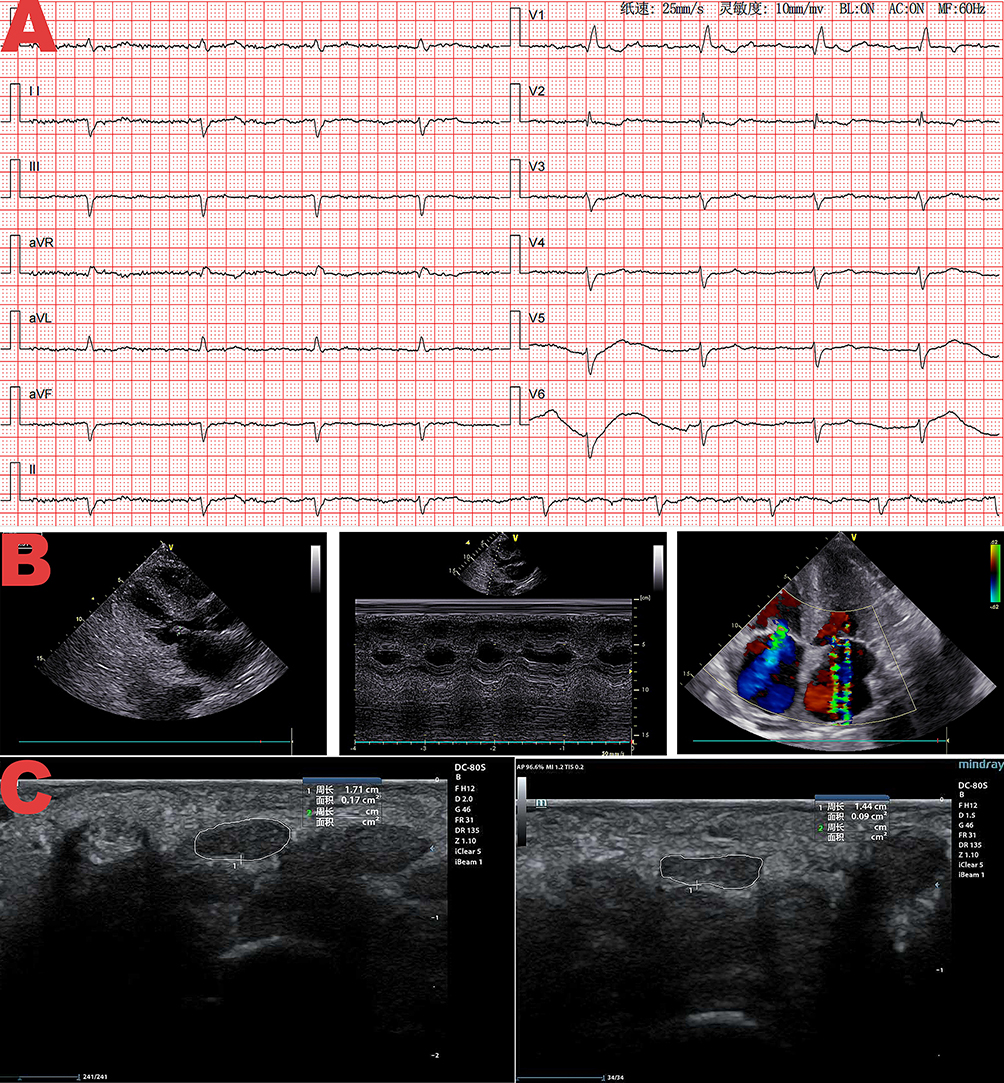 |
Figure 1 (A) Electrocardiogram showing atrial flutter with slow ventricular response, complete right bundle branch block, left anterior fascicular block, low voltage in limb leads (a typical electrocardiographic feature of transthyretin amyloid cardiomyopathy [ATTR-CM]), and poor R wave progression in the anterior wall. (B) Echocardiographic images: (left) Four-chamber view demonstrating biatrial dilation; (middle) M-mode echocardiography revealing concentric ventricular hypertrophy (interventricular septum: 14 mm, posterior left ventricular wall: 13 mm, right ventricular anterior wall: 6.6 mm at diastole); (right) Color Doppler imaging showing moderate tricuspid and mitral valves regurgitation. Left ventricular global longitudinal strain (LV GLS) of −15% and E/e′ ratio of 18 (key functional indices for ATTR-CM evaluation, consistent with diastolic dysfunction in ATTR-CM) were also measured in this examination. (C) Musculoskeletal ultrasound images of the carpal tunnel: (left) Right median nerve with a cross-sectional area of 17 mm2; (right) Left median nerve with a cross-sectional area of 9 mm2, indicating right median nerve thickening. This finding suggests carpal tunnel syndrome—a well-recognized extracardiac red flag for transthyretin amyloidosis. |
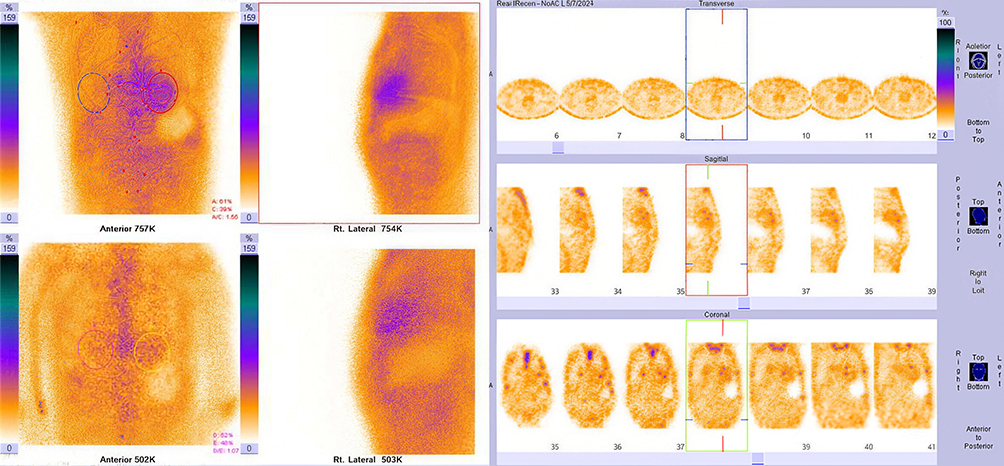 |
Figure 2 99mTc-PYP scintigraphy images: (Top panels) Anterior and right lateral planar acquisitions (2-hour and 3-hour) showing myocardial radiotracer uptake; (Bottom panels) Transaxial, sagittal, and coronal tomographic images (centered on the heart) demonstrating persistent myocardial radiotracer retention. Key quantitative/qualitative findings: 1-hour heart-to-contralateral lung (H/CL) ratio = 1.55; 3-hour myocardial PYP uptake was greater than rib uptake, with a Perugini (SQA) rating of 2–3. SPECT/CT was not performed in this examination to exclude blood pool activity; however, the above findings (≥2-grade myocardial uptake + exclusion of AL amyloidosis) are consistent with the diagnostic criteria for transthyretin amyloid cardiomyopathy (ATTR-CM). |
Diagnosis and Outcome
Taken together, the patient presented with heart failure with preserved ejection fraction, complicated with the typical ECG features, exclusion of monoclonal gammopathy, and grade 2–3 myocardial uptake on 99mTc-PYP scintigraphy established the non-invasive diagnosis of ATTR-CM, without the need of endomyocardial biopsy, consistent with current diagnosed criteria. The patient’s neurological features were consistent with a mixed polyneuropathy, including autonomic manifestations (chronic diarrhoea, gastroparesis, dysuria, orthostatic intolerance, and syncope) and sensorimotor manifestations (carpal tunnel syndrome, muscle weakness, and atrophy). The comprehensive neurological assessment led to the neuro diagnosis of concomitant transthyretin amyloidosis polyneuropathy (ATTR-PN). Additionally, the patient exhibited multiple organs damage including the kidneys, vocal cords, and thyroid glands. If inferred monistically, these could all be related to amyloidosis.
The patient and his family declined genetic testing, tissue biopsy, and disease-modifying therapy due to economic constraints and concerns about potential adverse effects of targeted drugs, despite detailed counseling on their benefits and risks. Management was limited to supportive care (anticoagulation, diuretics, nutritional support). The patient’s condition progressively declined, and he passed away suddenly at home 7 months post-discharge. Key time points in his diagnostic and treatment course are summarized in Table 1.
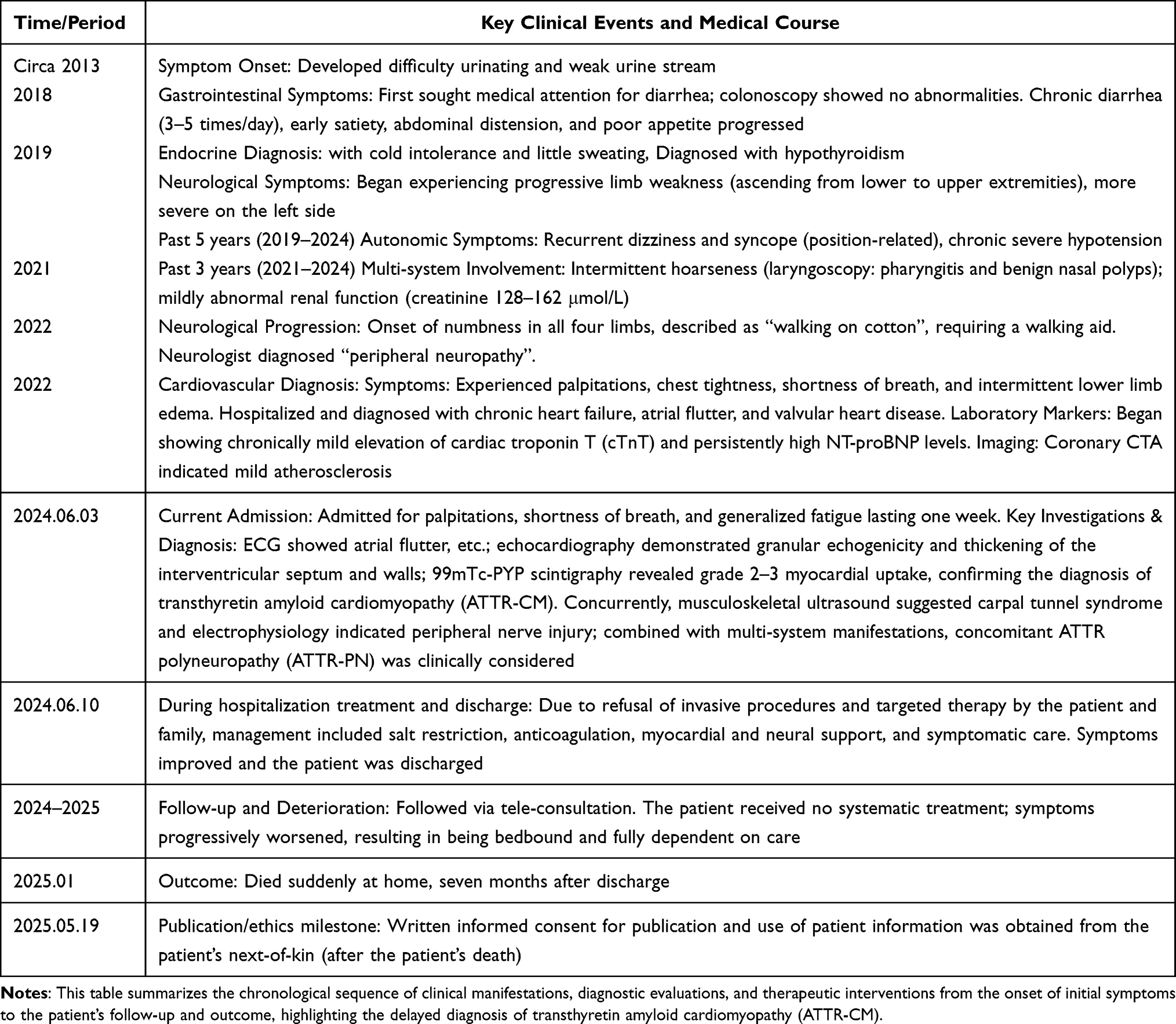 |
Table 1 Timeline of Key Clinical Events and Medical Course of the Patient |
Discussion
ATTR-CM is an underdiagnosed chronic disease, characterized by its insidious onset and lack of specific clinical manifestations. Due to the limitations of non-invasive diagnostic methods in the past, early diagnosis of this disease is difficult, resulting in a significant disease burden. Diagnosis delay is an important challenge in disease management of ATTR-CM, and studies have found that patients with ATTR-CM take an average of 6–8 years from onset to diagnosis.3 While patients with ATTR-CM historically had poor prognosis with median survival time of only 2 to 6 years after diagnosis, early diagnosis and treatment combined with evolving therapeutic has substantially improved outcomes. However, those with diagnostic delays and advanced disease continue to have a survival period of less than 2 years.4 Most patients die from cardiovascular causes, including sudden death and heart failure. This patient in the case was diagnosed 6 years after the onset of symptoms and died less than a year later. The entire diagnostic and treatment process was full of difficulties. Therefore, it is crucial to enhance clinicians’ awareness of ATTR-CM.
Many countries and institutions have proposed some clinical warning signs, also known as red flags, for cardiac amyloidosis: (1) Elderly heart failure (LVEF ≥ 40%) with unexplained LVH; (2) Echocardiographic LVH without electrocardiographic evidence of QRS high voltage; (3) Persistently elevated low-level troponin; (4) Low-gradient, low-velocity aortic stenosis in the elderly with right ventricular hypertrophy; (5) Hypotension intolerance to angiotensin system inhibitors and/or beta-blockers; (6) Polyneuropathy (PN), particularly with autonomic dysfunction (unexplained diarrhea and constipation, orthostatic hypotension, urinary retention, incontinence, etc); (7) Familial PN; (8) Bilateral carpal tunnel syndrome and/or lumbar spinal stenosis in the elderly; (9) Recurrent bilateral cataracts.5,6 This patient exhibited 7 of the above red flag signs. According to the ATTR-CM diagnostic pathway, the monoclonal immunoglobulin test was performed, and no M protein was found. Both blood and urine free light chain tests were negative, ruling out AL-CM. Further, 99mTc-PYP scintigraphy showed an H/CL ratio of 1.55 at 1 hour, with myocardial uptake higher than bilateral ribs at 3 hours, SQA rating of 2–3, suggesting ATTR-CM.
The gene encoding TTR is located on chromosome 18 and is classified into hereditary/mutant (ATTRm) and wild-type (ATTRwt) based on the presence or absence of gene mutations. ATTRm is an autosomal dominant genetic disease, and more than 130 gene mutations are known to cause ATTRm. A study involving 202 Chinese ATTRm patients showed that the most common gene mutation is Val30Met (19.8%), followed by Ala97Ser (15.8%).7 The Val30Met mutation gene often leads to ATTR-related neuropathy, also known as familial transthyretin amyloid polyneuropathy (ATTR-PN).8 ATTRwt is believed to be caused by age-related misfolding of TTR proteins, and studies have shown that the clinical symptoms usually appear after the age of 70, also known as senile systemic amyloidosis, and are more prevalent in males than females. The pathogenesis may be related to abnormal protein oxidation during the aging process. When ATTR deposits in tissues and organs other than the heart, multi-system manifestations can occur. Common extra-cardiac manifestations of ATTRm include multiple peripheral sensory-motor neuropathies, presenting as pain and sensory abnormalities starting from the lower limbs, followed by muscle weakness and motor function impairment, and in severe cases, inability to walk. Additionally, autonomic nerve involvement, such as sweating disorders, alternating constipation and diarrhea, weight loss leading to malnutrition, postural hypotension, and urinary retention are also common. However, symptoms related to neuropathy are relatively rare in ATTRwt patients.9 Of note, the prominent polyneuropathy and autonomic neuropathy with extensive multi-system involvement observed in this patient are unusual in wild-type ATTR-CM, but are frequently seen in hereditary/variant forms. This clinical phenotype strongly suggests an underlying TTR gene mutation with potential familial implications. Unfortunately, the patient and his family declined genetic testing due to concerns about economic constraints.
99mTc-PYP scintigraphy is a non-invasive test that identifies the presence or absence of myocardial amyloid protein deposition at an early stage, making early diagnosis of ATTR-CM possible. After excluding AL-CM, the specificity and positive predictive value of 99mTc-PYP imaging for diagnosing ATTR-CM can reach 100%.10 The visual scoring based on delayed planar imaging (1–3 hours post-injection) can grade by comparing the degree of radiotracer uptake in cardiac regions to that in ribs. No myocardial uptake is graded as 0, uptake lower than that of bone tissue is graded as 1, equal to bone tissue as 2, and higher than bone tissue as 3. Additionally, semi-quantitative analysis (SQA) as an image interpretation method can also be used to diagnose CA. It quantifies the uptake levels in the heart and the contralateral lung region separately, thereby obtaining the ratio of the two, namely H/CL value, which can serve as an evaluation parameter for scintigraphy results, with H/CL ≥1.5 at 1 hour or ≥1.3 in the 3-hour indicating a positive result. Studies have found that 2–3 grade myocardial uptake, with showing H/CL ≥1.5, indicates a high likelihood of ATTR-CM Patients with cardiac amyloidosis who are negative for blood and urine light chains can be diagnosed with ATTR-CM if their myocardial scintigraphy score is ≥2, without the need for endomyocardial biopsy.11,12 In this case, the patient’s 99mTc-PYP scintigraphy results showed an H/CL ratio >1.5, with myocardial uptake graded at 2–3, meeting international standards, and negative serum monoclonal immunoglobulin tests, thus confirming the diagnosis of ATTR-CM.
Current therapeutic strategies for ATTR-CM address symptomatic manifestation and disease pathogenesis. While conventional heart failure therapies manage symptoms and arrhythmias, novel agents focus on targeting amyloid cascade. Conventional heart failure medications, such as diuretics, neuroendocrine inhibitors, are poorly tolerated in patients with CA due to hypotension and preload dependence. The use of digoxin should be cautiously and closely monitored rather than absolutely contraindicated, as it is more likely to bind with amyloid fibril deposits, increasing its blood concentration and the risk of digoxin toxicity, even sudden death. The most common arrhythmia in CA patients is atrial fibrillation, with studies indicating that early radiofrequency ablation may be chosen for such patients, while late-stage patients are not recommended due to a high risk of recurrence. In CA patients with severe conduction block, those meeting the criteria for pacemaker implantation may undergo pacemaker therapy.13
The disease-modifying therapies for ATTR-CM mainly target different points in the TTR amyloid protein cascade reaction, including stabilizers, inhibitors, and degraders, which have been developed or are currently under investigations.14,15 Tafamidis, as the first approved drug for ATTR-CM, can specifically bind to the TTR tetramer, preventing the dissociation of the TTR tetramer into unstable monomers, which are prone to aggregate into amyloid proteins. If Tafamidis is unavailable or unaffordable, the non-steroidal anti-inflammatory drug (NSAID) diflunisal can be used as an alternative, stabilizing the TTR tetramer in vitro. However, its clinical application is limited by significant safety concerns, including gastrointestinal and renal adverse effects, and it is not a straightforward alternative to Tafamidis. Additionally, the novel stabilizer acoramidis (AG10) has a slightly higher stabilizing efficacy than Tafamidis, as well as significantly higher efficacy than diflunisal. It has been approved in December 2024 as the second drug globally for ATTR-CM. TTR inhibitors include small interfering RNA drugs (Patisiran and Vutrisiran) and antisense oligonucleotide drugs (Inotersen and Eplontersen), which are only approved for peripheral neuropathy in ATTRm, recent clinical trials have demonstrated significant cardiac benefits of these drugs in ATTR-CM patients, and vutrisiran has been approved for the treatment of ATTR-CM in some countries. Nexiguran ziclumeran (Nex‑Z, also known as NTLA‑2001) is an innovative in vivo CRISPR‑Cas9 gene‑editing therapy that targets the TTR coding gene, reducing TTR synthesis at the source and achieving long‑term efficacy with a single administration. While this therapy has shown favorable safety and durable efficacy in early‑stage trials, it remains investigational and has not been approved for clinical use to date. ALXN-2220, a TTR-specific degrader, can target misfolded TTR and clear amyloid protein deposits, and it has received fast track designation from the FDA for the treatment of ATTR-CM, but it has not been approved for clinical use to date, and its Phase 3 clinical trial is still ongoing. Furthermore, since the liver produces most of the TTR, liver transplantation and combined heart-liver transplantation are also treatment options to inhibit TTR synthesis.16
Conclusion
ATTR-CM is a systemic disease with multi-system involvement, and early clinical manifestations are non-specific, leading to missed and misdiagnoses. While untreated or late-diagnosed patients may have poor outcomes, early diagnosis combined with emerging disease-modifying therapies has substantially improved prognosis. Therefore, it is essential to raise awareness among clinicians about this condition. Standardized identification and screening in patients with “warning signs” can help in early diagnosis and treatment processes, thereby improving patient prognosis.
Ethics Approval and Consent for Participate
Institutional approval was required for the publication of this case report. This study protocol was approved by the Ethics Committee of Renmin Hospital, Hubei University of Medicine (Approval SYRMYY-2024-136). Written informed consent for the use of patient information and the publication of case reports was obtained from the patient’s next-of-kin on May 19th, 2025.
Acknowledgments
The authors would like to extend their gratitude to the patient’s family for their support and cooperation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Kittleson MM, Ruberg FL, Ambardekar AV, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis. J Am Coll Cardiol. 2023;81(11):1076–8. doi:10.1016/j.jacc.2022.11.022
2. Muchtar E, Grogan M, Aus Dem Siepen F, et al. Supportive care for systemic amyloidosis: international Society of Amyloidosis (ISA) expert panel guidelines. Amyloid. 2025;32(2):93–116. doi:10.1080/13506129.2025.2463678
3. Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC. 2019;7(8):709–716. doi:10.1016/j.jchf.2019.04.010
4. Antonopoulos AS, Panagiotopoulos I, Kouroutzoglou A, et al. Prevalence and clinical outcomes of transthyretin amyloidosis: a systematic review and meta‐analysis. Eur J Heart Fail. 2022;24(9):1677–1696. doi:10.1002/ejhf.2589
5. Yun S, Casado J, Pérez-Silvestre J, et al. Clinical suspicion, diagnosis and management of cardiac amyloidosis: update document and executive summary. Revista Clínica Española. 2024;224(5):288–299. doi:10.1016/j.rceng.2024.04.009
6. Heart Failure Group of Chinese Society of Cardiology, Editorial Board of Chinese Journal of Cardiology. Chinese expert consensus on the diagnosis and treatment of transthyretin cardiac amyloidosis. Zhonghua Xin Xue Guan Bing Za Zhi. 2021;49(4):321–332.
7. Chu X, Kang J, Xu J, et al. A multicenter study of hereditary transthyretin amyloidosis in China. Ann Neurol. 2025;97(6):1158–1167. doi:10.1002/ana.27203
8. Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6. doi:10.1186/s13023-017-0726-x
9. Chompoopong P, Mauermann ML, Siddiqi H, Peltier A. Amyloid neuropathy: from pathophysiology to treatment in light‐chain amyloidosis and hereditary transthyretin amyloidosis. Ann Neurol. 2024;96(3):423–440. doi:10.1002/ana.26965
10. Hanna M, Ruberg FL, Maurer MS, et al. Cardiac Scintigraphy with Technetium-99m-Labeled bone-seeking tracers for suspected amyloidosis. J Am Coll Cardiol. 2020;75(22):2851–2862. doi:10.1016/j.jacc.2020.04.022
11. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16):1554–1568. doi:10.1093/eurheartj/ehab072
12. Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503–3626. doi:10.1093/eurheartj/ehad194
13. Giancaterino S, Urey MA, Darden D, Hsu JC. Management of Arrhythmias in Cardiac Amyloidosis. JACC Clin Electrophysiol. 2020;6(4):351–361. doi:10.1016/j.jacep.2020.01.004
14. Gonzalez-Lopez E, Maurer MS, Garcia-Pavia P. Transthyretin amyloid cardiomyopathy: a paradigm for advancing precision medicine. Eur Heart J. 2025;46(11):999–1013. doi:10.1093/eurheartj/ehae811
15. Griffin JM, Grodin JL, Ruberg FL, Masri A, Hanna M, Maurer MS. Current landscape of therapies for transthyretin amyloid cardiomyopathy. JACC Heart Fail. 2025;13(5):685–694. doi:10.1016/j.jchf.2025.03.017
16. Saito Y, Nakamura K, Ito H. Molecular mechanisms of cardiac amyloidosis. Int J Mol Sci. 2021;23(1):25. doi:10.3390/ijms23010025
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.