Back to Journals » International Journal of Nanomedicine » Volume 11
Multiple nanoemulsion system for an oral combinational delivery of oxaliplatin and 5-fluorouracil: preparation and in vivo evaluation
Authors Pangeni R ![]() , Choi SW, Jeon O, Byun Y, Park JW
, Choi SW, Jeon O, Byun Y, Park JW ![]()
Received 31 August 2016
Accepted for publication 4 November 2016
Published 30 November 2016 Volume 2016:11 Pages 6379—6399
DOI https://doi.org/10.2147/IJN.S121114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Rudra Pangeni,1,* Sang Won Choi,1,* Ok-Cheol Jeon,2 Youngro Byun,3 Jin Woo Park1
1Department of Pharmacy, College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Muan-gun, 2Pharosgen R&D Center, Asan Institute for Life Sciences, 3Department of Molecular Medicine and Biopharmaceutical Science, Graduate School of Convergence Science and Technology, College of Pharmacy, Seoul National University, Seoul, Republic of Korea
*These authors contributed equally to this work
Abstract: Oxaliplatin (OXA) is a third-generation cisplatin analog that has been approved as first-line chemotherapy in combination with 5-fluorouracil (5-FU) for the treatment of resectable and advanced colorectal cancer. However, the therapeutic efficacy of oral OXA and 5-FU is limited by their low bioavailability due to poor membrane permeability. The aim of the present study was to develop an oral delivery system for OXA and 5-FU. We constructed an ion-pairing complex of OXA with a deoxycholic acid derivative (Nα-deoxycholyl-L-lysyl-methylester, DCK) (OXA/DCK) as a permeation enhancer. Next, we prepared multiple water-in-oil-in-water nanoemulsions incorporating OXA/DCK and 5-FU to enhance their oral absorption. To evaluate their membrane permeability, we assessed in vitro permeabilities of OXA/DCK and 5-FU through an artificial intestinal membrane and Caco-2 cell monolayer. Finally, oral bioavailability in rats and tumor growth inhibition in the colorectal adenocarcinoma cell (CT26)-bearing mouse model were investigated after oral administration of nanoemulsion containing OXA/DCK and 5-FU. The droplet size of the optimized nanoemulsion was 20.3±0.22 nm with a zeta potential of -4.65±1.68 mV. In vitro permeabilities of OXA/DCK and 5-FU from the nanoemulsion through a Caco-2 cell monolayer were 4.80- and 4.30-fold greater than those of OXA and 5-FU, respectively. The oral absorption of OXA/DCK and 5-FU from the nanoemulsion also increased significantly, and the resulting oral bioavailability values of OXA/DCK and 5-FU in the nanoemulsive system were 9.19- and 1.39-fold higher than those of free OXA and 5-FU, respectively. Furthermore, tumor growth in CT26 tumor-bearing mice given the oral OXA/DCK- and 5-FU-loaded nanoemulsion was maximally inhibited by 73.9%, 48.5%, and 38.1%, compared with tumor volumes in the control group and the oral OXA and 5-FU groups, respectively. These findings demonstrate the therapeutic potential of a nanoemulsion incorporating OXA/DCK and 5-FU as an oral combination therapy for colorectal cancer.
Keywords: oxaliplatin, 5-fluorouracil, deoxycholic acid derivative, ion-pairing complex, nanoemulsion, oral delivery
Introduction
Oral chemotherapy is attracting increasing attention because it avoids the discomfort of injections after hospitalization and can be conducted at home, which can improve patient cooperation and quality of life. In addition, a prolonged exposure of anticancer drugs to cancerous cells can be implemented by maintaining an appropriate plasma drug concentration, which can increase drug efficacy and decrease adverse effects, as well as facilitate the use of anticancer drugs for chronic treatment regimens as prophylactics against recurrence and metastasis.1,2 However, oral delivery of anticancer drugs is a challenge because of their particular physicochemical properties and physiological barriers; these barriers include pre-systemic metabolism, gastrointestinal (GI) instability, low aqueous solubility, poor intestinal membrane permeability, and high levels of P-glycoprotein (P-gp) efflux.2–5
Oxaliplatin (cis-[(1R,2R)-1,2-cyclohexanediamine-N, N′] [oxalato(2)-O, O′] platinum, OXA) is a third-generation platinum antitumor compound, which exerts its effects by interfering with DNA replication and transcription machinery through nuclear DNA adduct formation. These Pt–DNA adducts typically take the form of Pt–guanine–guanine bonds. At the nucleotide level, Pt–DNA complexes will activate either DNA repair mechanisms or apoptotic pathways.6–9 Clinically, OXA is used as first-line chemotherapy in combination with 5-fluorouracil (5-FU) for treatment of advanced colorectal cancer. The US Food and Drug Administration has also approved OXA as first- and second-line therapy for the treatment of metastatic colorectal cancer in combination with 5-FU and leucovorin, known as FOLFOX, and other malignancies, such as pancreatic cancer.10,11 However, both OXA and 5-FU are currently administered by intravenous infusion and exhibit poor oral bioavailability because of their low intestinal membrane permeability; their log P-values are −0.47 and −0.9, respectively, limiting the use of oral formulations for long-term dosing.2,12,13 Therefore, various approaches have been proposed for oral delivery of OXA or 5-FU. Oral formulations for effective local delivery of OXA to treat colorectal cancer tissue have been developed by encapsulation of OXA in pH-sensitive, mucoadhesive chitosan-based alginate microspheres and hyaluronic acid-coupled chitosan nanoparticles, followed by coating with Eudragit S100.12,14 Colon-specific pectin/ethylcellulose film-coated 5-FU pellets and poly(lactic-co-glycolic acid) nanoparticles for the oral delivery of 5-FU have also been prepared.15,16 Nanocarriers composed of an inorganic hydroxyapatite core and alginate shell for pH-responsive drug release, nanoparticles comprising iron oxide and alginate linked with a cancer cell-targeting ligand for targeted hyperthermia therapy, and porous iron oxide microspheres showed great potential as drug delivery systems for synergistic anticancer therapy.17–19
In our previous studies, we prepared a positively charged bile acid derivative, Nα-deoxycholyl-L-lysyl-methylester (DCK), by chemically linking deoxycholic acid (DOCA) with L-lysine as a permeation enhancer, and demonstrated that intestinal membrane permeability and oral bioavailability of polar and hydrophilic drugs such as bisphosphonates and insulin were increased after the ion-pairing complex formation between drug molecule and DCK.20,21 An ion-pairing complex with a highly lipophilic cation, which can also be recognized by transporters in the GI lumen, can avoid dilution of enhancer in the GI fluid. Thus, unlike with other conventional penetration enhancers, our approach can effectively improve drug absorption with a minimal quantity of enhancer. An ion-pairing complex of OXA with DCK (OXA/DCK) significantly improved the intestinal membrane permeability and oral bioavailability of OXA in rats by increasing its lipophilicity. Moreover, the orally administered OXA/DCK complex significantly inhibited tumor growth in squamous cell carcinoma (SCC7)-bearing mice.22
The objective of the present study was to design an oral combinational delivery system for OXA and 5-FU based on multiple water-in-oil-in-water (w/o/w) nanoemulsions, incorporating an amphiphilic OXA/DCK complex and a hydrophilic 5-FU to enhance the oral absorption of OXA and 5-FU simultaneously. To achieve this goal, we constructed the OXA/DCK complex by ion pairing to confer a lipophilic property on OXA. Next, OXA/DCK- and 5-FU-loaded multiple w/o/w nanoemulsions were prepared using titration; we characterized the droplet size by transmission electron microscopy (TEM) and dynamic laser light scattering. We found an optimum formulation for a multiple-nanoemulsive system containing OXA/DCK and 5-FU by assessing in vitro permeability of OXA/DCK and 5-FU across an artificial intestinal membrane and Caco-2 cell monolayer. Finally, oral bioavailability in rats and tumor growth inhibition efficacy of the nanoemulsion for combinational therapy in the tumor-bearing mice were evaluated following oral administration.
Materials and methods
Materials
OXA was prepared by Laurus Labs (Hyderabad, India). 5-FU, DOCA, ethyl chloroformate, N-methylmorpholine, Nε-Boc-L-lysine methyl ester hydrochloride (H-Lys(Boc)-OMe·HCl), lithium aluminum hydride (LiAlH4), trimethylsilyl diazomethane (2 M solution in hexane), ammonium acetate, and formic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Caprylocaproyl macrogol-8-glycerides (Labrasol), diethylene glycol monoethyl ether (Transcutol HP), and propylene glycol monocaprylate (Capryol 90) were obtained from Gattefossé (St Priest, France). Polyethoxylated castor oil (Cremophor EL) was provided by BASF (Ludwigshafen, Germany). Tetrahydrofuran (THF), chloroform, methanol, acetyl chloride, and n-hexane (analytical grade) were obtained from Merck (Kenilworth, NJ, USA). Solvents for high-performance liquid chromatography (HPLC) and atomic absorption spectroscopy were obtained from Merck and Thermo Fisher Scientific (Waltham, MA, USA).
Animals
Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee (IACUC) of Mokpo National University (Jeonnam, Republic of Korea). All animal experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the guidelines of IACUC.
BALB/c mice (males, 20–25 g) and Sprague Dawley rats (males, 200–250 g) were purchased from Orient Co., Ltd. (Gyunggi-do, Republic of Korea). The animals were housed under standard housing conditions of temperature (23°C±2°C), relative humidity (55%±10%), and light (12/12-h light/dark cycle with no ultraviolet exposure). The animals had free access to a standard laboratory diet (Nestlé Purina, St Louis, MO, USA) and ion-sterilized tap water.
Preparation and characterization of OXA/DCK ionic complex
DCK as an oral enhancer was synthesized by conjugating DOCA with a positively charged lysine, as previously described.23 Briefly, H-Lys(Boc)-OMe·HCl in N-methylmorpholine (2.7%, w/v) was added to DOCA (26 g) in a solvent mixture of ethyl chloroformate (6.4 mL), N-methylmorpholine (7.4 mL), and THF (800 mL); the mixture was then refluxed for 2 h. After the reaction mixture was stirred overnight at room temperature, the precipitates were filtered and dried by removing solvent through evaporation. Lys(Boc)DOCA was obtained by column chromatography of the precipitates with a solvent mixture of chloroform and methanol. The Lys(Boc)DOCA was further dissolved in a mixture of acetyl chloride and methanol in an ice bath. After the complete removal of the solvent, the residue was dissolved in water and washed with chloroform. Finally, the aqueous layer was gathered and freeze-dried to obtain DCK in powder form.
The ion-pairing complex of OXA with DCK (OXA/DCK) was prepared to enhance the intestinal membrane permeability of OXA by increasing its lipophilic property. Briefly, an aqueous solution of DCK with a pH of 7.0 adjusted with sodium bicarbonate was added to an OXA aqueous solution (pH 7.0 adjusted with sodium bicarbonate) at a complexation molar ratio of 1:2 (OXA:DCK) under continuous stirring. The solution was then centrifuged and freeze-dried at −70°C to remove water until the loss of drying value of the complex at 105°C was less than 0.1%.
To confirm the complex formation between OXA and DCK, the characteristic crystalline features of pure OXA, DCK, physical mixtures of OXA and DCK, and the OXA/DCK complex were compared using powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). PXRD patterns were measured by a D8 advance diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) at 40 mA and 40 kV using Cu-Kα1 radiation (λ =1.5418 Å). The powder samples were deposited on an adhesive support with 0.5 mm thickness and then placed in the diffractometer. PXRD patterns were recorded in step-scan mode in the range of 3°≤2θ≤40° at a scanning rate of 0.02° per second. Furthermore, thermal analyses of the samples were performed using a DSC Q1000 V9.9 Build 303 (TA Instrument Inc., New Castle, DE, USA). Approximately 1.5 mg of each sample was weighed into a non-hermetically sealed aluminum pan and scanned at a heating rate of 5°C/min over a temperature range of 25°C–250°C. All DSC measurements were made under a nitrogen atmosphere at a flow rate of 100 mL/min.
Preparation of w/o/w nanoemulsion
A two-step spontaneous emulsification method was employed to prepare a w/o/w nanoemulsion. First, pseudo-ternary phase diagrams for the primary nanoemulsion were constructed by an oil titration method using Capryol 90, Labrasol, Transcutol HP, and deionized water as oil phase, surfactant, co-surfactant, and an aqueous phase, respectively. The primary w/o nanoemulsion was identified as the region in the pseudo-ternary diagram where clear and transparent droplets were obtained based on visual observation. The optimum w/o formulation with lowest droplet size and maximum amount of aqueous phase was selected as the following formulation: 21.4% aqueous phase, 50.0% surfactant and co-surfactant mixture (Smix,1; Labrasol:Transcutol HP, 1:2, w/w), and 28.6% oil phase.
Second, we further dispersed the w/o nanoemulsion containing 23.36% OXA or 67.52% OXA/DCK complex with 23.36% 5-FU in its aqueous phase for the w/o/w nanoemulsion as an oil phase by an aqueous phase titration method. In this system, Cremophor EL and Transcutol HP were applied as a surfactant and co-surfactant (Smix,2), respectively, with weight ratios of 2:1, 1:1, and 1:2. The oil phase (primary w/o nanoemulsion) and a fixed weight ratio of Smix,2 were mixed in different weight ratios (from 1:9 to 9:1), and each pseudo-ternary phase diagram was then constructed by the slow addition of water. The clear zone of w/o/w nanoemulsion was identified, and several formulations were selected from each pseudo-ternary phase diagram and subjected to further physicochemical characterization and in vitro intestinal membrane permeability studies.
Characterization of w/o/w nanoemulsion system
Average droplet size, polydispersity index (PDI), and zeta potential of the w/o/w nanoemulsions were determined by dynamic laser light scattering analyzer (Malvern Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). Each drug-loaded nanoemulsion was diluted with deionized water (1:20), followed by sonication for 1 min to minimize multiple scattering effects; the measurements were performed at 25°C. The morphological evaluation of the selected microemulsion was performed using TEM. The w/o/w nanoemulsion was diluted by 100 times with deionized water, and a drop of each microemulsion was placed on a copper grid. After removing the excess with filter paper, one drop of 2% aqueous solution of phosphotungstic acid was added onto the grid to allow negative staining. The excess was removed with filter paper, and the grid was observed by TEM (JEM-200; JEOL, Tokyo, Japan).
In vitro artificial intestinal membrane permeability
A parallel artificial membrane permeability assay (BD Biosciences, San Jose, CA, USA) for nanoemulsions containing OXA, OXA/DCK, or 5-FU was performed to evaluate their intestinal membrane permeability. Each drug or nanoemulsion was diluted with phosphate-buffered saline (PBS, pH 6.8) at the concentration of 125.8 μM OXA, 125.8 μM OXA/DCK complex, and 384.4 μM 5-FU. Then, a 200 μL drug solution or nanoemulsion was added to each donor well plate, and 300 μL of buffer (PBS, pH 6.8) was added to each well of the receptor plate. The plate assembly was set by coupling the donor plate with the receiver plate and incubated at room temperature for 5 h. After incubation, samples were withdrawn from both receptor and donor plates. The concentrations of OXA and OXA/DCK complex were measured by HPLC using a C18 column (4.6×250 mm, 5 μm, 100 Å; 20 μL sample injection) at 40°C. The mobile phase consisted of water (pH 3.0 adjusted with phosphoric acid)–acetonitrile (99:1, v/v) and was run at a flow rate of 0.8 mL/min. OXA or OXA/DCK was measured using a UV detector at 210 nm. For 5-FU, 20 μL aliquots from each sample were injected into an HPLC system equipped with a C18 column (4.6×250 mm, 5 μm, 100 Å), and the samples were chromatographed using an isocratic mobile phase of deionized water (pH 3.2 adjusted with phosphoric acid) at a flow rate of 0.8 mL/min. The quantification of 5-FU was carried out at 260 nm.
Effective permeability (Pe) of each drug was calculated using the following formula: Pe = −ln[1 − CA(t)/Cequilibrium]/[A × (1/VD + 1/VA) × t], where Pe is the permeability (cm/s), A is the effective filter area (ƒ ×0.3 cm2; ƒ means the apparent porosity of the filter, ƒ =0.76), VD is the volume of the donor well (0.2 mL), VA is the volume of the receptor well (0.3 mL), t is the total time of incubation in seconds, CA(t) denotes the concentration of drug in the receptor well at time t, and Cequilibrium = [CD(t) × VD + CA(t) × VA]/(VD + VA), where CD(t) denotes the concentration of drug in the donor well at time t.
In vitro Caco-2 cell monolayer permeability
The permeabilities of OXA, OXA/DCK complex, and 5-FU incorporated in the nanoemulsion were also investigated across a Caco-2 cell monolayer. Caco-2 cells were seeded onto each 12-well Transwell® filter insert (pore size 0.4 μm, surface area 1.12 cm2; Corning Incorporated, Corning, NY, USA) at a density of 3×105 cells/well. The culture medium, Dulbecco’s Modified Eagle’s Medium (DMEM; Lonza, Basel, Switzerland) containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific) and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific), was changed every 48 h for 21–29 days, and the cell monolayers with a transepithelial electrical resistance (TEER) of greater than 350 Ω cm2 were used for transport experiments. The culture medium was removed, and the monolayer was pre-incubated with 0.5 mL of Hanks’ balanced salt solution (HBSS) for 20 min at 37°C. After measuring the TEER, the HBSS was removed, and 0.5 mL of 125.8 μM OXA, 125.8 μM OXA/DCK, and 384.4 μM 5-FU in HBSS or nanoemulsion diluted with HBSS, and 1.5 mL of HBSS were added to the each apical and basolateral compartment, respectively. During incubation at 37°C, 100 μL samples were taken from the basolateral compartment at 0.5, 1, 2, 3, 4, and 5 h. The samples were filtered through a membrane filter (0.45 μm, polyvinylidene fluoride) and stored at 4°C for analysis. The concentration of OXA, OXA/DCK complex, or 5-FU permeated though the monolayer was determined using the HPLC system with a UV detector, as described earlier. The apparent permeability coefficient (Papp) of OXA, OXA/DCK complex, or 5-FU was calculated according to the following equation: Papp = dQ/dt × 1/(A × C0), where dQ/dt indicates the linear appearance rate of mass in the basolateral sides (μmoL/s), C0 is the initial concentration of OXA, OXA/DCK complex, or 5-FU on the apical side (μmoL/mL), and A is the surface area of the monolayer (cm2).
In vitro drug dissolution
Dissolution tests were performed in 500 mL of medium containing 0.1 N HCl solution (pH 1.2) or phosphate buffer (pH 6.8) at 37°C±0.2°C, using the USP type 1 apparatus (basket) rotating at 100 rpm. Ten milligrams of OXA or 38.8 mg of OXA/DCK complex (equivalent to 10 mg of OXA) with 10 mg of 5-FU powder or in 800 mg of w/o nanoemulsions with Smix,2 (formulations C, E, F, G, and H without secondary aqueous phase) was encased in a hard gelatin capsule size 00. Each capsule was subjected to a dissolution test, and 1 mL samples were withdrawn at 15, 30, 45, 60, 90, and 120 min. After filtration, the amount of OXA or OXA/DCK complex and 5-FU in the samples was quantified by HPLC using a UV detector, as described earlier.
In vitro cytotoxicity study
To evaluate the cytotoxic effects of free OXA, OXA/DCK, and 5-FU, as well as their nanoemulsions, a cell counting (CCK-8 kit; Dojindo Molecular Technologies, Rockville, MD, USA) assay was performed. CT26 (murine colon carcinoma) cells were seeded at a density of 5×103 cells/well in 100 μL DMEM containing 10% FBS and cultured at 37°C for 24 h. The cells were then treated with 157, 79, 39, 20, 10, and 5 μM of OXA or OXA/DCK and 480, 240, 120, 60, 30, and 15 μM of 5-FU in DMEM or nanoemulsion diluted with DMEM. The cells were cultured for an additional 24 h. To determine cell viability, a 10 μL WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt] solution was added and incubated for 2 h. The absorbance was then measured using a microplate reader (PerkinElmer multimode plate reader; PerkinElmer, Waltham, MA, USA) at 450 nm. The percentage of viable cells was calculated by comparing the values of treated cells with those of untreated cells.
In vivo pharmacokinetic study in rats
To evaluate the improvement in intestinal absorption of OXA and 5-FU by complex formation with bile acid derivatives and formulation as a nanoemulsion, OXA or OXA/DCK complex incorporated with 5-FU in the nanoemulsion (formulation E) was administered orally to rats. Each rat was orally administered 400 μL of aqueous solution consisting of OXA (10 mg/kg), OXA/DCK complex (equivalent to 10 mg/kg of OXA), or 5-FU (20 mg/kg), as well as a 400 μL nanoemulsion diluted with water to comprise OXA (10 mg/kg) or OXA/DCK complex (equivalent to 10 mg/kg of OXA) with 5-FU (20 mg/kg). To evaluate oral bioavailability, 150 μL of OXA (5 mg/kg) or 5-FU (5 mg/kg) in water was also prepared and injected via the tail vein. After administration, blood samples (200 μL) were collected from a capillary in the retro-orbital plexus at different time intervals and directly mixed with 50 μL of sodium citrate (3.8% solution). Blood samples were then immediately centrifuged (2,500× g, 15 min, 4°C). The plasma samples were isolated and kept frozen at −70°C until analysis.
The plasma concentration of OXA was estimated by the Pt concentration using atomic absorption spectroscopy (AAS) after dilution with 5 mL of concentrated nitric acid and heating for 6 h in a 100°C heating block for digestion. Before analysis, the samples were diluted with 10 mL of 3% nitric acid in ultrapure water and then injected into the furnace for AAS analysis. All measurements were carried out using a Thermo Scientific iCE 3500 AAS (Thermo Fisher Scientific) equipped with a GFS35 graphite furnace and a GFS35Z autosampler. Argon was used as the purging gas at a flow rate of 200 mL/min, and the injection volume of the sample was 20 μL. The spectrometer was provided with a Zeeman-based background corrector, and the selected wavelength was 266 nm.
To determine the plasma concentration of 5-FU, 100 μL of each standard and plasma samples were diluted with 50 μL of 20 μg/mL internal standard (chlorouracil) and 100 μL of saturated ammonium sulfate solution. After vortexing for 1 min, the diluted plasma sample was extracted with a 2 mL solvent mixture of propanol and diethylether (80:20, v/v). The mixture was then centrifuged at 2,500× g for 10 min, and the collected organic phase was evaporated under nitrogen at 37°C. The dried residues were reconstituted with 100 μL of mobile phase for HPLC analysis (ammonium phosphate buffer with pH 6.8/methanol, 98:2, v/v), followed by vortexing and centrifugation at 3,000× g for 1 min. The plasma concentration of 5-FU was then determined by HPLC with a C8 column (4.6×250 mm, 5 μm), and the mobile phase was run at a flow rate of 1 mL/min. A 50 μL sample was injected, and 5-FU was measured using a UV detector at 260 nm.
In vivo tumor growth inhibition by orally administered OXA or OXA/DCK with 5-FU nanoemulsion
The CT26 cells were subcutaneously grafted into the flanks of BALB/c mice at a concentration of 1×106 cells/100 μL of PBS (pH 7.4). On day 14, when each tumor became palpable (70–100 mm3), mice were randomly divided into five groups of 10 animals each: control (not treated), OXA-S (once-daily oral administration of 10 mg/kg OXA in water), OXA/DCK-S (once-daily oral administration of OXA/DCK complex as 10 mg/kg OXA in water), 5-FU-S (once-daily oral administration of 10 mg/kg 5-FU in water), and OXA/DCK-5-FU-NE (once-daily oral administration of nanoemulsion [formulation E] containing OXA/DCK complex as 10 mg/kg OXA and 10 mg/kg 5-FU) groups. During the 18-day treatment, mice were fasted for 4 h before administration and 2 h after administration. Tumors were measured every 3 days in two dimensions using a caliper, and tumor volumes were calculated as a2 × b ×0.52, where a is the width and b is the length. Body weight was also measured. At 18 days after treatment, the isolated tumor masses were measured, and tumor tissues were fixed in 10% formalin for histological evaluation. Staining with proliferating cell nuclear antigen (PCNA) for cell proliferation and fluorescent terminal deoxynucleotidyl transferase-mediated dUPT nick end labeling (TUNEL) for apoptosis were carried out.
Pharmacokinetic and statistical analyses
Pharmacokinetic parameters were obtained using a non-compartmental method in WinNonlin® software (ver. 5.3; Pharsight Corporation, Mountain View, CA, USA). All data are expressed as mean ± standard deviation. A P-value less than 0.05 was considered to indicate statistical significance using a t-test between two mean values for unpaired data or one-way analysis of variance followed by Tukey’s multiple-comparison test among more than three mean values for unpaired data.
Results
Preparation and characterization of OXA/DCK ionic complex
OXA has diaminocyclohexane as a carrier group for Pt and oxalato as a leaving group. It has been reported that a water molecule can attack the platinum atom, which leads to the opening of the oxalato ring.24,25 Therefore, OXA can reversibly exist as an oxalato monodentate intermediate, which has a carboxylic acid with negative charge in the aqueous solution. At neutral pH, OXA will be semi-stable, constantly opening and closing the oxalate ring; however, a positively charged DCK may form a connection with the ring-opened intermediate, resulting in a continuous conversion of OXA via the formation of OXA/DCK ion-pairing complex.
To identify the OXA in the solid state, the PXRD spectra of the OXA, DCK, and the physical mixtures were compared with that of the OXA/DCK complex (Figure 1A). Pure OXA showed diffraction peaks at 11.95°, 15.77°, 17.92°, 19.57°, 22.25°, 23.95°, 25.42°, 26.09°, 31.57°, 31.61°, 33.15°, and 36.76° over the 2θ range in the PXRD spectra, characteristic peaks of crystalline OXA. Characteristic peaks for OXA were also evident in the physical mixture, indicating that the drug still existed in a crystalline form. However, no sharp diffraction peaks for crystalline OXA were seen in the OXA/DCK complex. The disappearance of OXA crystallinity in the complex was also confirmed by DSC analysis (Figure 1B). The characteristic endothermic peak was observed at 292°C in the thermogram of the pure OXA, while it was absent from the DSC thermogram of the OXA/DCK complex. Based on the PXRD and DSC spectra, the OXA was molecularly dispersed with DCK and existed in an amorphous form after complex formation.
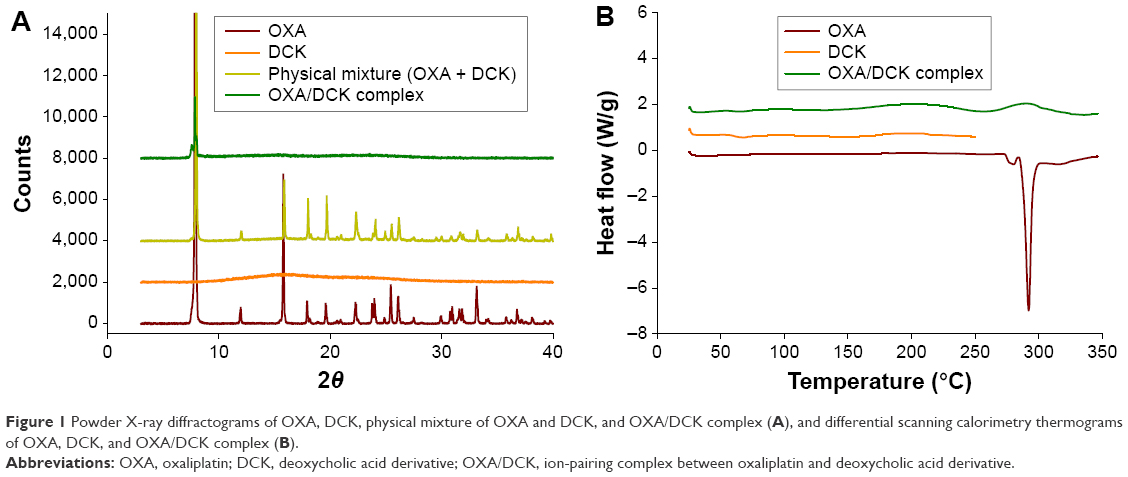 | Figure 1 Powder X-ray diffractograms of OXA, DCK, physical mixture of OXA and DCK, and OXA/DCK complex (A), and differential scanning calorimetry thermograms of OXA, DCK, and OXA/DCK complex (B). |
Pseudo-ternary phase diagrams and preparation of drug-loaded nanoemulsion
First, pseudo-ternary phase diagrams for primary w/o nanoemulsion were constructed to identify the nanoemulsifying region and optimum concentration ratio of oil, surfactant, and co-surfactant. The phase diagrams represented the w/o system containing Capryol 90 as oil, Labrasol as surfactant, and Transcutol HP as co-surfactant at different Smix,1 ratios (2:1, 1:1, and 1:2), and the area implying the isotropic nanoemulsion was varied with the Smix,1 ratio (Figure 2A–C). When the co-surfactant amount was lower than the surfactant amount, that is, Smix,1 2:1, the area for nanoemulsion was less than that of the Smix,1 1:1 ratio, indicating that the solubilization of the oil phase by the Smix,1 was lower. The maximum concentration of the aqueous phase that could be solubilized in the oil phase was found to be 13% by adding approximately 42% of the Smix,1 2:1. When the co-surfactant concentration was increased to the ratio of Smix,1 1:2, the nanoemulsion region was further increased by shifting the area toward a water-rich region compared to the Smix,1 1:1, and the maximum concentration of water that was solubilized in the phase diagram was 24% by incorporating around 50% Smix,1 1:2, which showed the domination of Transcutol HP in nano-sizing. However, on further increment of the co-surfactant concentration in the Smix,1 ratio, a significant decrease in the nanoemulsion region was observed. It is possible that this was a liquid crystalline phase formed by Transcutol HP and was not stabilized by the given amount of Labrasol. These results showed that the Gibbs free energy of nanoemulsion formulation may rely on the extent of which the surfactants and co-surfactant passively reduce the interfacial tension of the w/o interface and the variation in dispersion entropy.13,26,27 Therefore, several nanoemulsions, which appeared clear and transparent, were selected from the phase diagram constructed by the Smix,1 1:2, and each combination was further examined for its water content, particle size, PDI, and in vitro membrane permeability (data not shown). The optimum w/o primary emulsion was composed of 21.4% aqueous phase (23.36% OXA or 67.52% OXA/DCK complex with 23.36% 5-FU), 50% mixture of Labrasol and Transcutol HP (1:2, w/w), and 28.6% Capryol 90. For w/o/w nanoemulsion, the primary w/o emulsion was applied as an oil phase, and a mixture of Cremophor EL and Transcutol HP, and water was used for Smix,2 and aqueous phase, respectively. Figure 2D–F represents the pseudo-ternary phase diagrams for w/o/w nanoemulsion with different Smix,2. The area for nanoemulsion increased comparatively with an increase in the total concentration of surfactant. When surfactant and co-surfactant concentrations were kept equal (Smix,2 1:1), we observed that the double nanoemulsion region increased significantly compared to Smix,2 2:1. The maximum amount of w/o nanoemulsion that was found to be solubilized in this ratio was 35% with a lower concentration of Smix,2 1:1 (29%). When the concentration of co-surfactant was increased with respect to surfactant (Smix,2 1:2), we observed that the double nanoemulsion region decreased compared to Smix,2 1:1. The maximum amount of w/o primary nanoemulsion solubilized was 23% by 47% of Smix,2 1:2. When the co-surfactant concentration was further increased with respect to surfactant, the region for nanoemulsion was further decreased (data not shown). After construction of phase diagrams, eight formulations with composition of the primary emulsion and Smix,2 (in 1:1 to 1:6 ratios) forming the self-nanoemulsion under infinite dilution with water were selected for further evaluation from each phase diagram at different Smix,2.
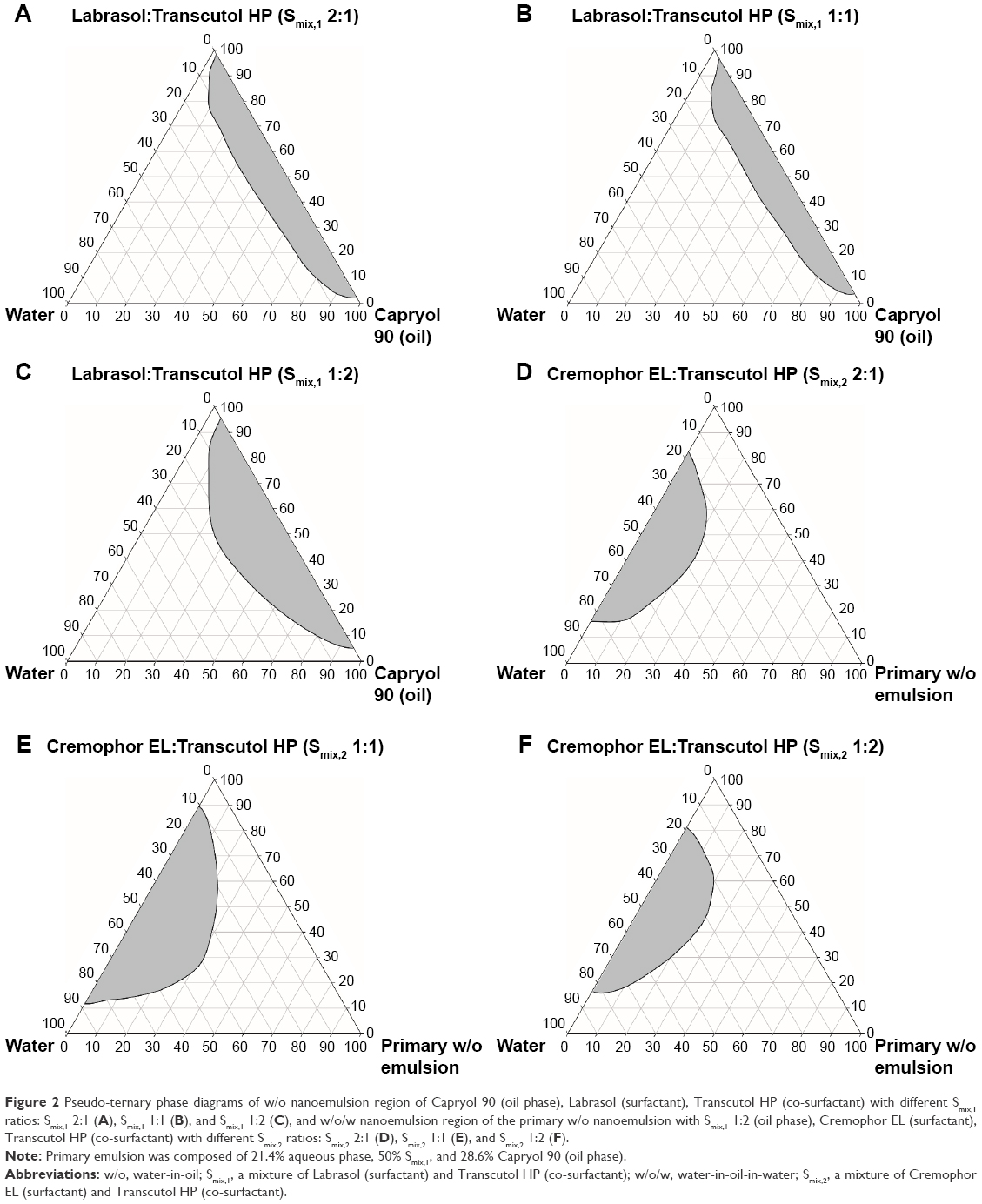 | Figure 2 Pseudo-ternary phase diagrams of w/o nanoemulsion region of Capryol 90 (oil phase), Labrasol (surfactant), Transcutol HP (co-surfactant) with different Smix,1 ratios: Smix,1 2:1 (A), Smix,1 1:1 (B), and Smix,1 1:2 (C), and w/o/w nanoemulsion region of the primary w/o nanoemulsion with Smix,1 1:2 (oil phase), Cremophor EL (surfactant), Transcutol HP (co-surfactant) with different Smix,2 ratios: Smix,2 2:1 (D), Smix,2 1:1 (E), and Smix,2 1:2 (F). |
Characterization of w/o/w nanoemulsion
Droplet size, PDI, and zeta potential of a series of transparent and monophasic w/o/w nanoemulsions produced by conventional low-energy emulsification technique are summarized in Table 1. All formulations representing nano-sized droplets ranged from 13.5 to 232 nm with a PDI lower than 0.59 indicating uniform size with narrow distribution. The largest droplet size was observed in formulation A with the highest percentage of oil phase (w/o primary emulsion) concentration. At each Smix,2, it was observed that the droplet size increased rapidly as the concentration of the oil phase was increased from 10% to 30%; droplet size of formulation A and C: 61.4±4.06 vs 18.1±0.31 nm at Smix,2 2:1; 146±1.17 vs 17.5±0.15 nm at Smix,2 1:1; 232±5.06 vs 19.0±1.06 nm at Smix,2 1:2. These results reveal that droplet size of w/o/w nanoemulsion is proportional to the concentration of oil phase in the formulations. When the concentration of the oil phase was constant at 20% or 30%, the droplet size decreased as the concentration of surfactant was increased. Therefore, formulation A with a lower concentration of surfactant (Smix,2 1:2) and 30% of the oil phase was found to generate the largest droplet size. However, the droplet size of formulations containing less than 10% of the oil phase was similar and smaller than 20 nm regardless of the Smix,2 ratio or its content. Among the nanoemulsions prepared with the Smix,2 ratio 1:1, formulation E produced 20.3±0.22 nm droplets with 0.18±0.01 PDI, and formulation H was found to produce the lowest mean droplet size compared to other formulations (13.5±0.14 nm). Zeta potential values of all nanoemulsions were observed in the range of −4.65 to 0.14 mV (neutral), which means that fatty acids and esters of the oil layer in the w/o primary emulsion were completely covered with surfactant and co-surfactant in the w/o/w double emulsion.
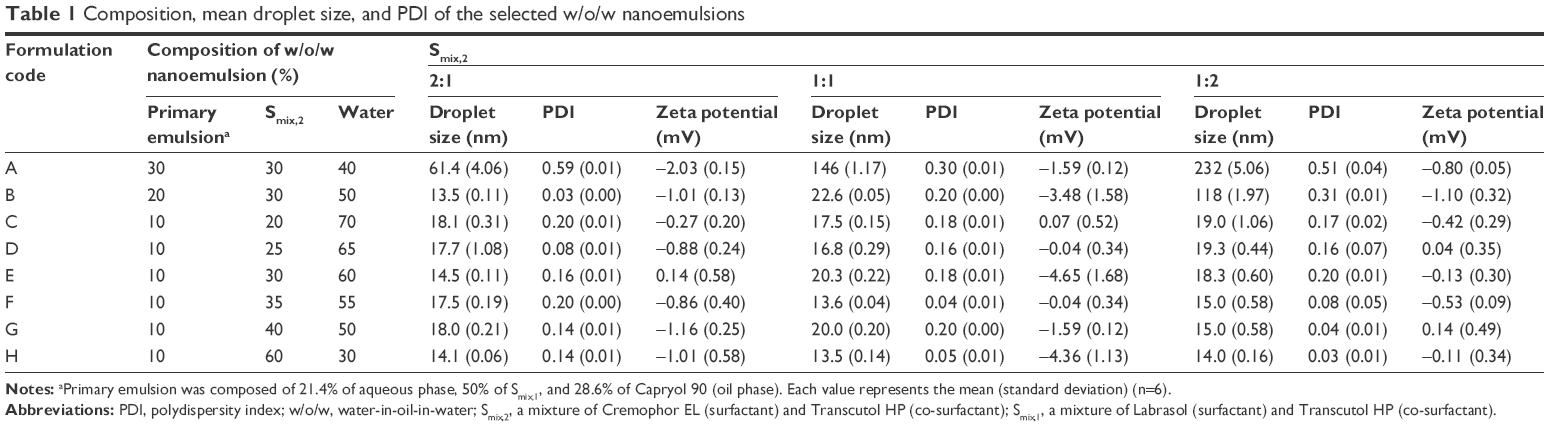 | Table 1 Composition, mean droplet size, and PDI of the selected w/o/w nanoemulsions |
Figure 3 shows TEM images for formulation E (Smix,2 1:1) containing OXA or OXA/DCK complex with 5-FU. The nano-sized globules were dispersed uniformly and had a narrow size distribution with a diameter less than 50 nm, in accordance with the findings measured by particle size analyzer.
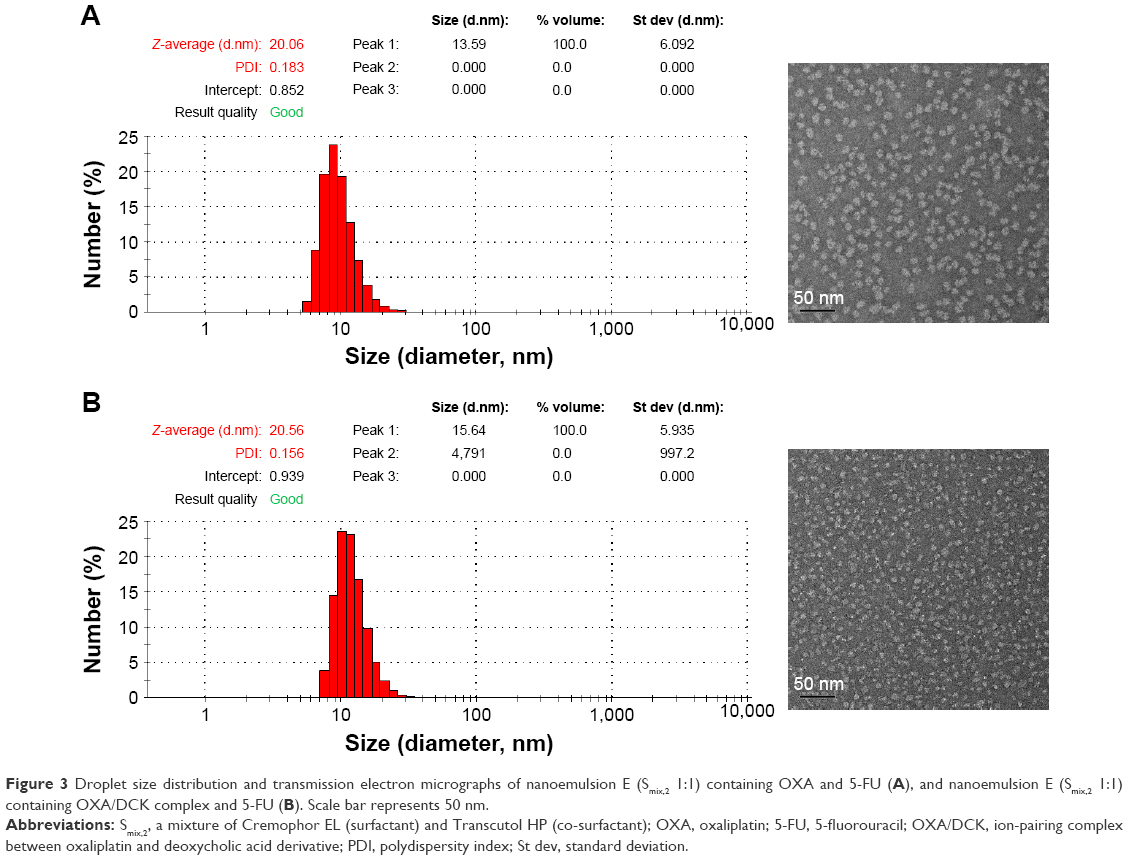 | Figure 3 Droplet size distribution and transmission electron micrographs of nanoemulsion E (Smix,2 1:1) containing OXA and 5-FU (A), and nanoemulsion E (Smix,2 1:1) containing OXA/DCK complex and 5-FU (B). Scale bar represents 50 nm. |
In vitro artificial intestinal membrane permeability
The overall artificial intestinal membrane permeability values for OXA, OXA/DCK complex, and 5-FU were significantly increased after formulation into the w/o/w nanoemulsion (Figure 4). In the case of OXA, the permeability was influenced by the surfactant-to-co-surfactant ratio (Smix,2) and Smix,2-to-oil phase (the primary w/o emulsion) ratio. Except for formulation H, the permeability value of OXA from the nanoemulsion prepared by Smix,2 1:1 was relatively higher than those from the formulations with Smix,2 2:1 and 1:2 at the same composition. When the Smix,2 1:1-to-oil phase ratio was increased from 2 to 6 at 10% of the oil phase (formulations C–H), the maximum membrane permeability of OXA was observed in the formulation E (Smix,2 1:1-to-oil phase ratio 1:3), where the permeability of OXA was 9.24-fold greater than that of OXA solution (4.99±0.59 vs 0.54±0.12 (×10−6 cm/s)). However, the permeability of OXA/DCK complex was still 37.4- and 4.02-fold greater than those of free OXA and OXA from formulation E (Smix,2 1:1), respectively. At a higher surfactant-to-co-surfactant ratio (Smix,2 2:1), the membrane permeability of the OXA/DCK complex was relatively higher than those from the nanoemulsions with a higher or lower content of the oil phase (the primary w/o emulsion), while the OXA/DCK permeability was not significantly improved by incorporating Smix,2 1:2 at the same composition. The OXA/DCK from nanoemulsion E (Smix,2 1:1) was also maximally permeated through the artificial intestinal membrane, and its permeability value was 2.85 and 1.46 times greater than those from the solution and nanoemulsion A (Smix,2 2:1). Moreover, the permeability of the OXA/DCK complex from formulation E (Smix,2 1:1) was 107- and 11.5-fold greater than those of free OXA and OXA from formulation E (Smix,2 1:1), respectively (57.2±5.23 vs 0.54±0.12 vs 4.99±0.59 [×10−6 cm/s]).
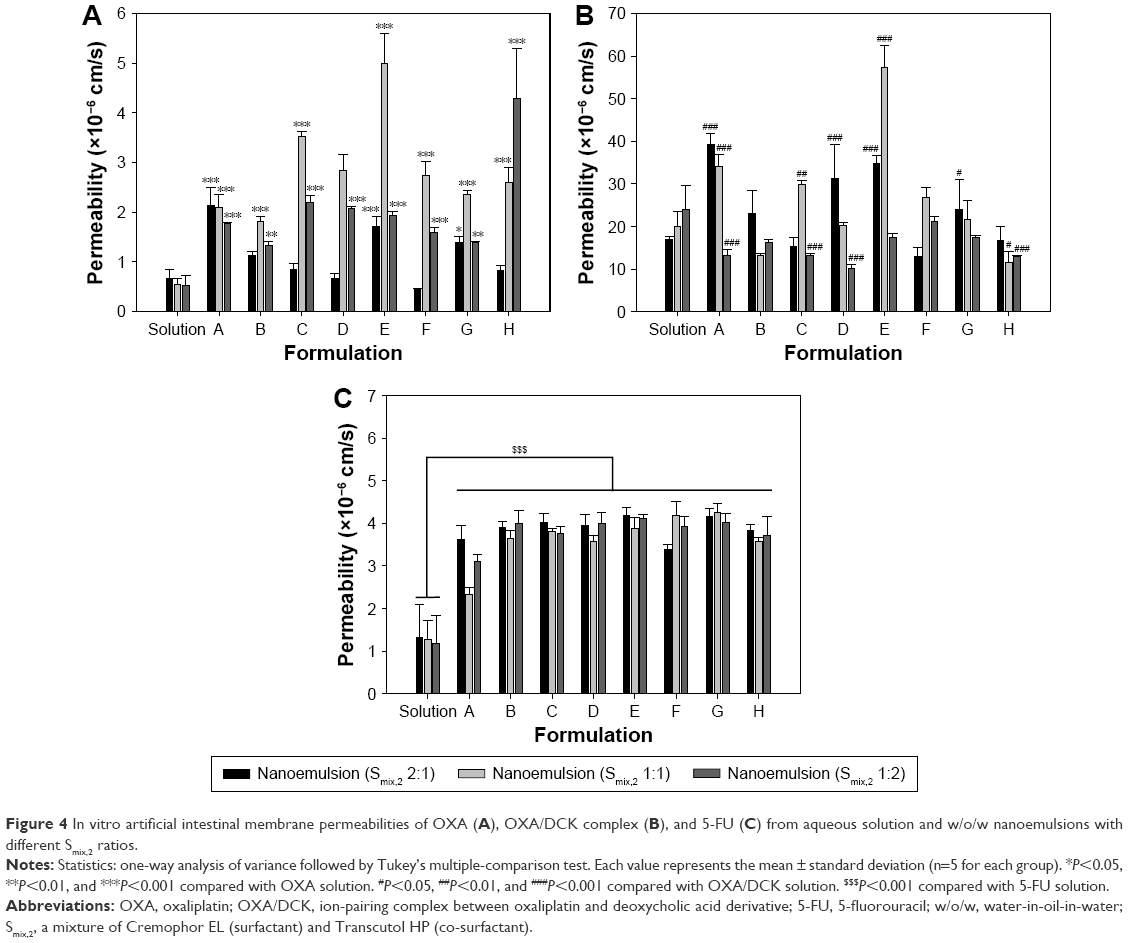 | Figure 4 In vitro artificial intestinal membrane permeabilities of OXA (A), OXA/DCK complex (B), and 5-FU (C) from aqueous solution and w/o/w nanoemulsions with different Smix,2 ratios. |
In the case of 5-FU, its membrane permeability increased as the amount of oil phase decreased at all Smix,2 ratios, while there were small differences in the permeability values for formulations C–H, which may have been due to their uniform content of oil phase and similar droplet size. The permeability of 5-FU from formulation E (Smix,2 1:1) was 3.05-fold greater compared to that of free 5-FU (3.78±0.26 vs 1.27±0.46 [×10−6 cm/s]). Based on the in vitro artificial membrane permeabilities of OXA, OXA/DCK, and 5-FU, we further performed Caco-2 cell monolayer permeability studies and dissolution tests using nanoemulsions prepared with 10% of oil phase (w/o nanoemulsion) and Smix,2 1:1 to find an optimum concentration ratio of the oil phase to Smix,2 for enhancement of the oral absorption of OXA or OXA/DCK complex and 5-FU simultaneously.
In vitro Caco-2 cell monolayer permeability
The in vitro permeabilities of OXA, OXA/DCK, and 5-FU nanoemulsions across a Caco-2 cell monolayer are presented in Table 2. When OXA was formulated in nanoemulsion E (Smix,2 1:1), its Papp increased from 2.50±1.96 to 7.08±1.42 (×10−6 cm/s). The permeability of OXA was also significantly improved compared to that of free OXA by ionic complex formation with DCK. After the OXA/DCK complex formulated in nanoemulsion E, the apical-to-basal Papp of the OXA/DCK complex was further enhanced 2.13- and 4.80-fold compared to those of the OXA/DCK complex and OXA alone, respectively (12.0±1.91 vs 5.63±3.03 vs 2.50±1.96 [×10−6 cm/s]). The permeability of 5-FU was also significantly increased by incorporation into the nanoemulsion, and the permeability of 5-FU in formulation E (Smix,2 1:1) was 4.30-fold greater than that of 5-FU solution (14.9±3.08 vs 3.46±1.22 [×10–6 cm/s]).
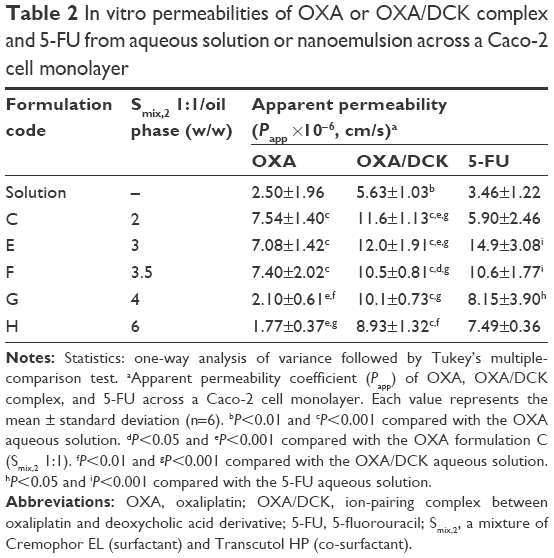 | Table 2 In vitro permeabilities of OXA or OXA/DCK complex and 5-FU from aqueous solution or nanoemulsion across a Caco-2 cell monolayer |
The permeability of OXA, OXA/DCK complex, or 5-FU nanoemulsion across a Caco-2 cell monolayer displayed a tendency to increase as the weight ratio of Smix,2 to oil increased up to 3:1, and then significantly decreased at the ratio of 6:1. Based on the results of the in vitro Caco-2 cell monolayer permeability analysis, we performed further tests by using a nanoemulsion E (Smix,2 1:1) containing OXA or OXA/DCK complex with 5-FU to assess in vivo intestinal absorption in rats and tumor growth inhibition efficacy in mice.
In vitro drug dissolution study
The prepared nanoemulsions were also tested for in vitro dissolution. At pH 1.2, more than 90% of OXA or OXA/DCK was released from the nanoemulsions within 120 min (Figure 5A and B). However, the dissolution rates of OXA or OXA/DCK from nanoemulsions G and H were relatively delayed compared with the dissolution rate of the powder form or the other formulations. In contrast, more than 85% of 5-FU was released from the nanoemulsions within 15 min, and the dissolution rate of 5-FU at pH 1.2 was not significantly influenced by incorporating it in the nanoemulsion (Figure 5C). In a medium with pH set to 6.8, more than 90% of OXA and 5-FU was completely released from the nanoemulsion within 15 min (Figure 5D and F). In the case of the OXA/DCK complex, its powder form also exhibited a delayed release compared with release from the nanoemulsions (except formulation C). More than 90% of OXA/DCK was released from the nanoemulsion within 15 min (Figure 5E). This may have resulted from aggregation of the conjugated DCK molecules in the complex, as well as a lower Smix,2-to-oil (the primary w/o emulsion) ratio.
 | Figure 5 In vitro cumulative percentage release profiles of OXA (A), OXA/DCK complex (B), and 5-FU (C) in powder form or from various w/o/w nanoemulsions prepared with Smix,2 1:1 in pH 1.2 media, and OXA (D), OXA/DCK complex (E), and 5-FU (F) in powder form or from various w/o/w nanoemulsions prepared with Smix,2 1:1 in pH 6.8 media. |
In vitro cytotoxicity studies
To evaluate in vitro therapeutic effects of OXA or OXA/DCK complex with 5-FU formulated in the optimized nanoemulsion, we investigated cell viability in CT26 cells after treatment with various concentrations of OXA or OXA/DCK with 5-FU in aqueous solution or nanoemulsion E (Smix,2 1:1) (Figure 6). At more than 10 μM of free OXA in DMEM, cell viability decreased to less than 80% and was 30.0%±2.81% at 157 μM of OXA. The cell viability of OXA in nanoemulsion E (Smix,2 1:1) also decreased as the OXA concentration increased, and decreased significantly compared with effects of free OXA when the drug concentration was higher than 79 μM. When cells were treated with more than 79 μM of OXA and 240 μM of 5-FU in the aqueous solution or nanoemulsion, the cell viability values synergistically decreased compared to those of cells treated only with OXA, and maximally decreased by 49.2% at 5 μM of OXA and 15 μM of 5-FU solution compared to those treated with 5 μM of OXA solution. In addition, the cells treated with nanoemulsion containing 10 μM of OXA and 30 μM of 5-FU maximally decreased by 53.2% compared to those treated with 10 μM of OXA nanoemulsion. The OXA/DCK complex also displayed dose-dependent cell viability; cell viability values were 7.67%±0.19% and 7.17%±0.05% at 157 μM of OXA/DCK complex in solution and nanoemulsion, respectively. At the same time, the cytotoxicity of OXA/DCK nanoemulsion was lower than that of free OXA/DCK whose concentration was lower than 39 μM. At the same molar concentration, the free OXA/DCK showed higher cytotoxicity than that of free OXA, and the cell viability values were 2.93- and 3.92-fold less than those of OXA solution at 79 and 157 μM. The OXA/DCK complex also provided a synergistic cytotoxic effect on the CT26 cells by combinational treatment with 5-FU; the cell viability values of 5 μM OXA/DCK in solution and 39 μM OXA/DCK nanoemulsion decreased by 43.8% and 70.6%, respectively, by incorporating 15 and 120 μM of 5-FU. Nanoemulsion E (Smix,2 1:1) without drug also displayed cell toxicity at higher concentrations, equivalent to nanoemulsion containing 240 μM of 5-FU. In contrast, cell viability was not significantly influenced by the concentration of 5-FU, and the values ranged from 43.6% to 57.1% at all concentration levels of 5-FU solution. At the same time, the minimum cell viability was observed after treatment with a nanoemulsion containing 480 μM of 5-FU (6.94%±0.02%), which was found to be 6.28 times more effective than free 5-FU. However, the cell viability of cells treated with 15 μM of 5-FU in nanoemulsion was significantly increased by 75.1%±3.91%. The dose-independent cytotoxic effect of free 5-FU may be caused by its limited cell membrane permeability, which could be improved by formulation into the nanoemulsion. Altogether, these results indicated that drug-loaded nanoemulsion can be successfully administered orally for the prevention of recurrence and metastasis of colon cancer, and may show synergistic therapeutic effects though combinational drug delivery.
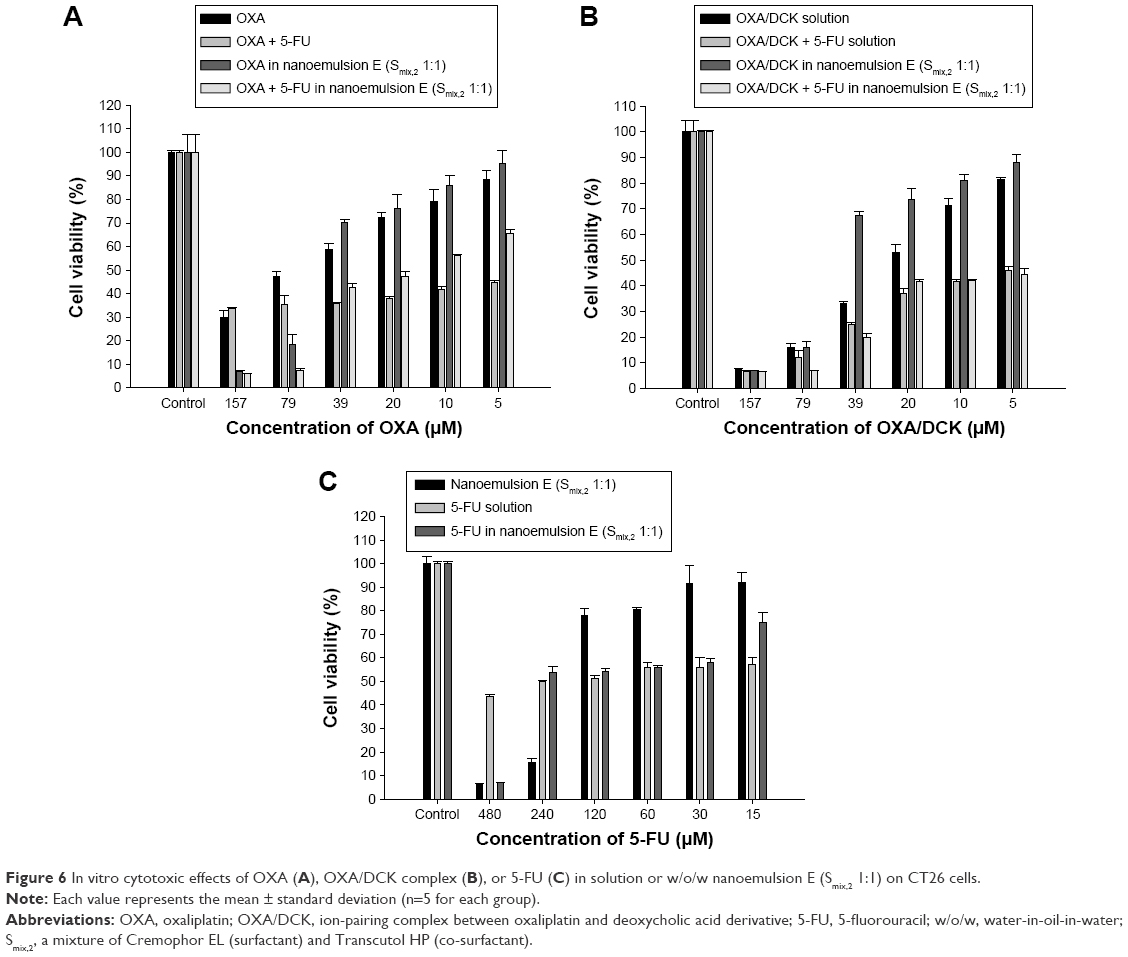 | Figure 6 In vitro cytotoxic effects of OXA (A), OXA/DCK complex (B), or 5-FU (C) in solution or w/o/w nanoemulsion E (Smix,2 1:1) on CT26 cells. |
In vivo oral absorption in rats
The plasma concentration–time profiles and the pharmacokinetic parameters of OXA and 5-FU resulting from the oral administration of aqueous solution of OXA or OXA/DCK with 5-FU or nanoemulsions (formulation E) containing OXA or OXA/DCK with 5-FU in rats are presented in Figure 7 and Tables 3 and 4, respectively. The intestinal absorption of OXA was improved significantly after formulation in the nanoemulsion, resulting in a 1.66- and 2.67-fold increase in the Cmax and AUClast, respectively. However, the oral absorption of OXA was more significantly enhanced by complex formation with DCK, and its Cmax and AUClast values were 306% and 540% improved, respectively. In addition, the Cmax and AUClast values for OXA/DCK nanoemulsion were 1.77- and 1.70-fold greater than those of free OXA/DCK, respectively, as well as 5.41- and 9.17-fold greater compared to those after administration of OXA solution (Figure 7B). The resulting oral bioavailability of OXA/DCK nanoemulsion was determined to be 9.19-fold higher than that of OXA solution due to the ionic complexed DCK molecules and nanoemulsification (Table 3). The Cmax value of 5-FU nanoemulsion, 0.164±0.044 μg/mL, was 1.78-fold higher than that of 5-FU solution (0.092±0.008 μg/mL). The AUClast value for the 5-FU nanoemulsion also increased by 1.39-fold compared to that of the 5-FU solution (0.242±0.058 μg·h/mL) (Figure 7C). Thus, the resulting oral bioavailability of the 5-FU nanoemulsion was evaluated as a 139% increase compared with that of free 5-FU (Table 4).
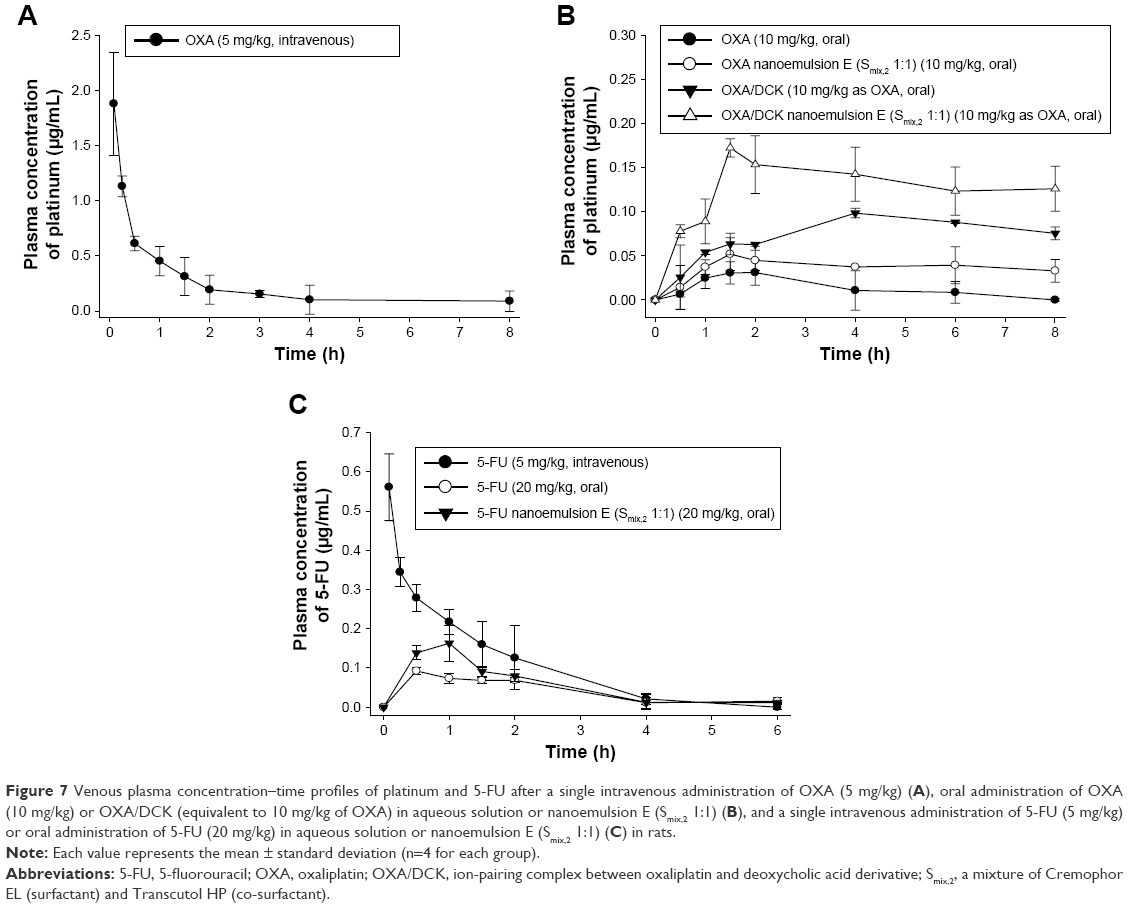 | Figure 7 Venous plasma concentration–time profiles of platinum and 5-FU after a single intravenous administration of OXA (5 mg/kg) (A), oral administration of OXA (10 mg/kg) or OXA/DCK (equivalent to 10 mg/kg of OXA) in aqueous solution or nanoemulsion E (Smix,2 1:1) (B), and a single intravenous administration of 5-FU (5 mg/kg) or oral administration of 5-FU (20 mg/kg) in aqueous solution or nanoemulsion E (Smix,2 1:1) (C) in rats. |
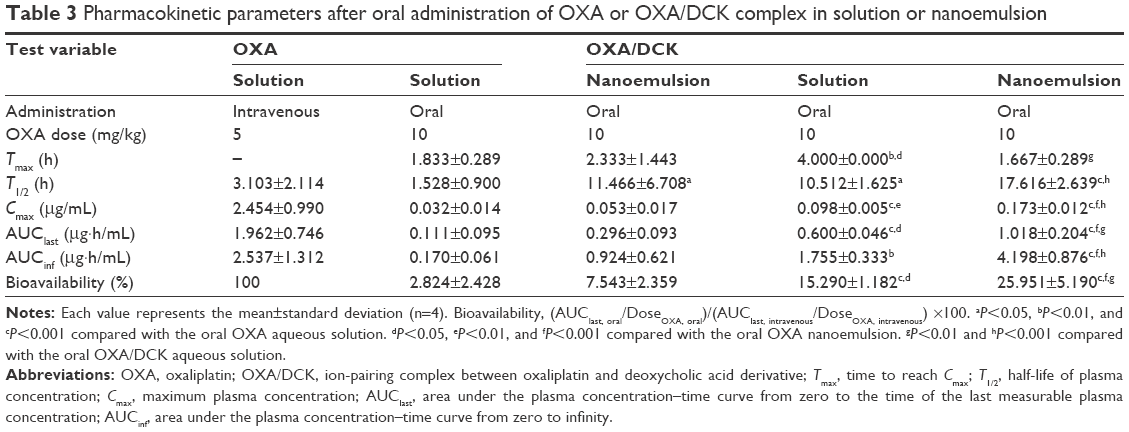 | Table 3 Pharmacokinetic parameters after oral administration of OXA or OXA/DCK complex in solution or nanoemulsion |
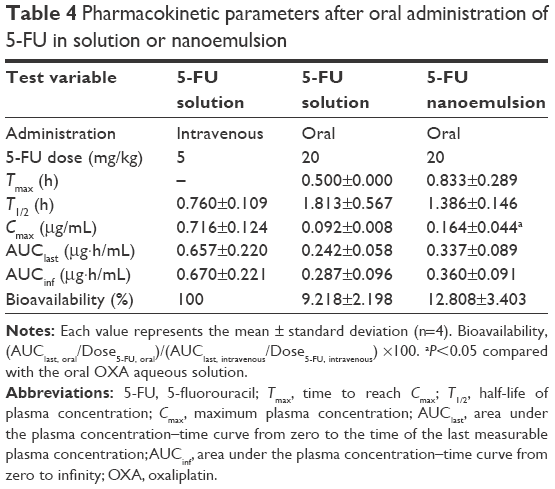 | Table 4 Pharmacokinetic parameters after oral administration of 5-FU in solution or nanoemulsion |
Inhibition effect of orally administered nanoemulsion comprising OXA/DCK and 5-FU on tumor growth
We then tested the anticancer effects of the oral combinational delivery system containing OXA/DCK and 5-FU as formulation with the w/o/w nanoemulsion E in an in vivo tumor model using CT26 cell lines. After a daily oral administration of OXA/DCK-S in mice-bearing murine tumors, the OXA/DCK complex demonstrated 60.3% and 21.4% inhibition of tumor growth compared to the control and once-daily oral administration of OXA-S, respectively (Figure 8A). 5-FU-S solution treatment also delayed tumor growth in mice by 57.9% compared to the control group; however, tumor growth after oral administration of pure 5-FU was not significantly different than in the OXA/DCK-S group. In contrast, the OXA/DCK-5-FU-NE group maximally inhibited the increase in tumor volume compared to the control (73.9%), OXA-S (48.5%), OXA/DCK-S (34.4%), and 5-FU-S (38.1%) groups. After 18 days of treatment, tumor masses in the OXA/DCK-5-FU-NE group were reduced by 43.0%, 26.0%, 11.6%, and 32.0% compared to the control, OXA-S, OXA/DCK-S, and 5-FU-S, respectively, and the oral combinational treatment using OXA/DCK and 5-FU did not affect body weight (Figure 8B–D). To determine the effect of oral combinational treatment using a nanoemulsion including OXA/DCK and 5-FU on tumor growth inhibition, the proliferating cell density and apoptosis in the isolated tumor tissues were investigated by staining with PCNA and TUNEL (Figure 9). Consistently, the OXA/DCK-5-FU-NE treatment exhibited a profound decrease in proliferating cell density, as well as increased tumor apoptosis compared with the control, OXA-S, OXA/DCK-S, and 5-FU-S groups. Based on these results, the oral administration of nanoemulsion comprising OXA/DCK and 5-FU produced a synergistic effect on tumor growth inhibition due to improvement of the oral bioavailability of OXA and 5-FU.
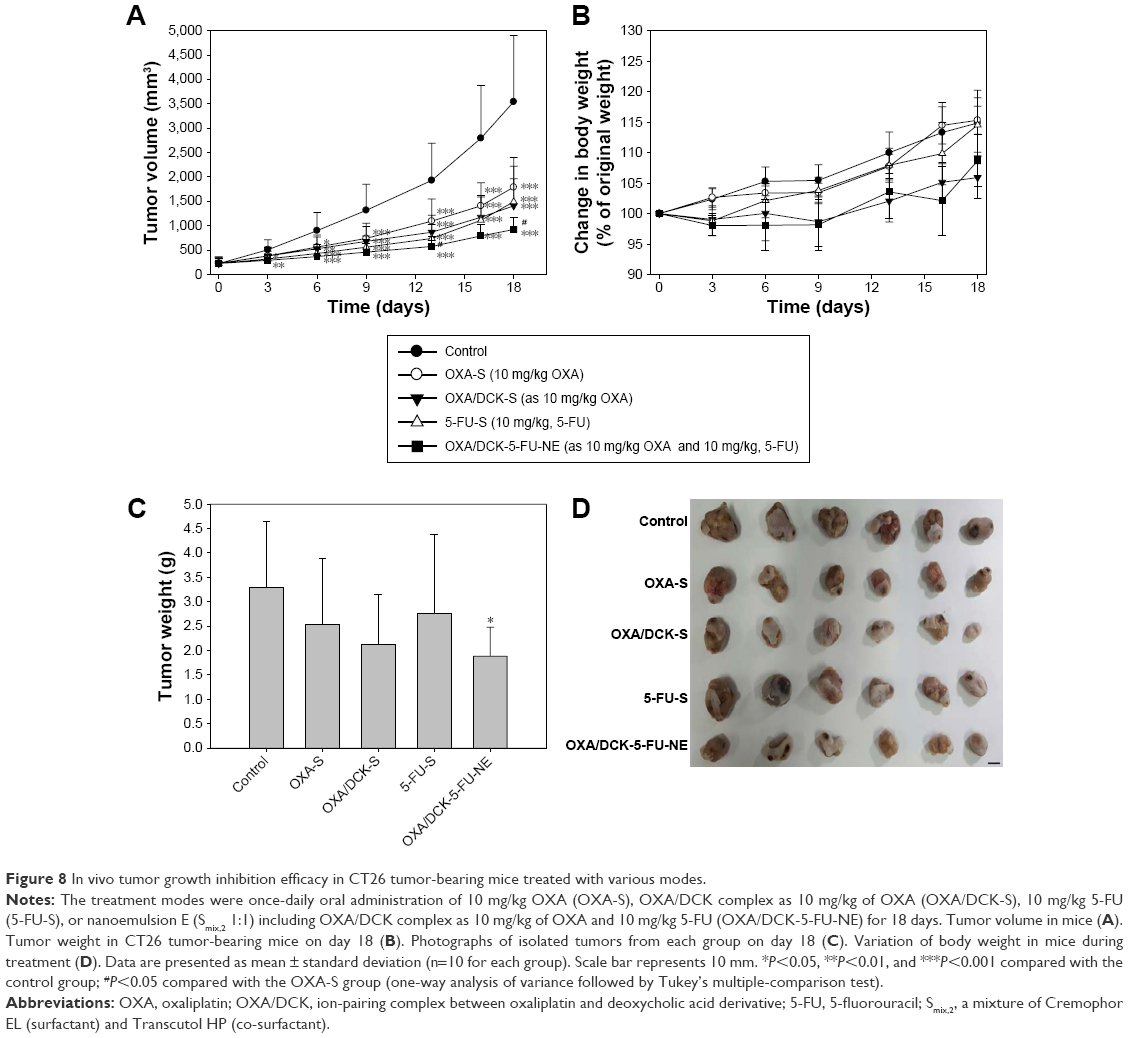 | Figure 8 In vivo tumor growth inhibition efficacy in CT26 tumor-bearing mice treated with various modes. |
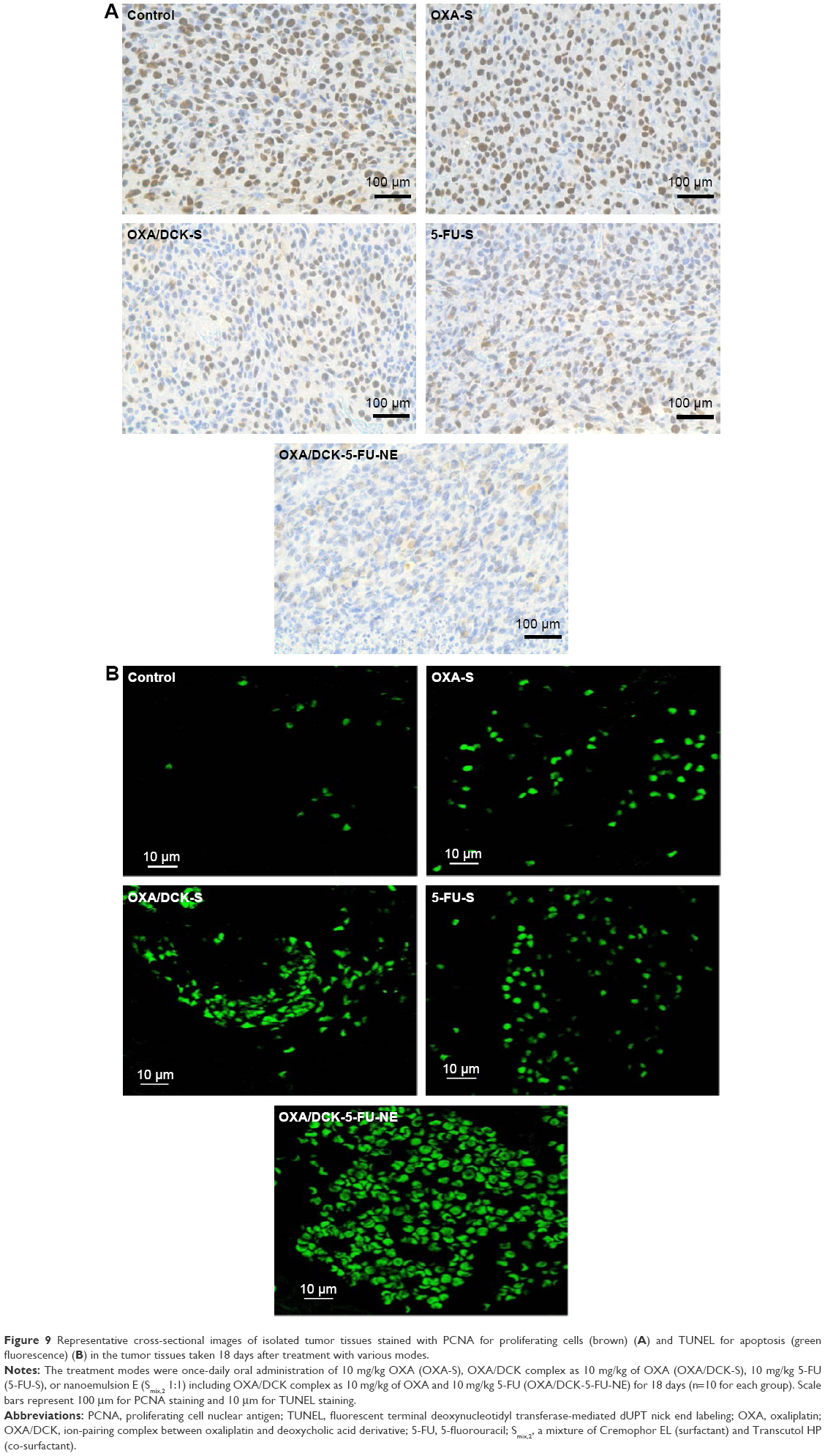 | Figure 9 Representative cross-sectional images of isolated tumor tissues stained with PCNA for proliferating cells (brown) (A) and TUNEL for apoptosis (green fluorescence) (B) in the tumor tissues taken 18 days after treatment with various modes. |
Discussion
In this study, we investigated a self-nanoemulsifying system for oral combinational delivery of OXA and 5-FU to enhance their membrane permeability and in vivo oral bioavailability. It is difficult to optimize oral delivery of a hydrophilic drug in the form of a self-nanoemulsifying system by w/o emulsion because phase separation of the external oil phase or fast precipitation of drug entrapped into the internal water phase can occur after exposure to the GI fluids.13,28 To overcome this problem, we adopted a w/o/w multiple self-nanoemulsifying system comprising a primary w/o emulsion of aqueous drug solution dispersed in the oil droplets, which can be easily dispersed in an external aqueous phase such as GI fluids using a secondary surfactant. In this study, Labrasol and Cremophor EL, which have a high hydrophilic–lipophilic balance value of 14, were used as surfactants to reduce interfacial energy required for the formation of nanoemulsion, because they are less affected by pH and ionic strength changes in the GI tract.28 However, Cremophor EL has been known to play a role in hypersensitivity reactions, including nephrotoxicity, neurotoxicity, and cardiotoxicity associated with intravenous paclitaxel administration.29,30 In addition, it can induce systemic and hematological toxicity through an oxidative stress-based mechanism, which is mainly responsible for the oxidative damage.31 Although it was reported that Cremophor EL was not detected after oral administration, and exhibited a half-maximal lethal dose (LD50) of greater than 6,400 mg/kg in rats, it did cause dose- and time-dependent damage in both endothelial and epithelial cells at concentrations higher than 0.1 and 5 mg/mL, respectively.28,30,32 Therefore, further toxicity studies are required, employing repeated oral administration of the nanoemulsion with various dose regimens. Transcutol HP was also used as a co-surfactant in both w/o and w/o/w nanoemulsions, to reduce interfacial tension and formation of mechanical barriers to coalescence. Therefore, surfactant and co-surfactant are preferentially adsorbed at the interface, reducing the interfacial energy and providing a mechanical barrier to coalescence.33,34 For an oral nanoemulsive system, the Smix ratio should be optimized to increase the dispersion entropy and interfacial area, as well as decrease the interfacial tension and free energy of the system, for a thermodynamically stable spontaneous dispersion at its minimum concentration value. Moreover, the oil phase-to-Smix ratio also plays an important role in the formation of nanoemulsions, because surfactants lower the interfacial tension, allowing a reduction in droplet size.34–36 Therefore, the droplet size decreased from 147 to 13 nm, as an increase in surfactant may be the result of more surfactant being available for adsorption and the formation of a more closely packed surfactant film at the oil–water interface, thereby providing stronger stabilization. In contrast, the droplet size was increased by increasing the co-surfactant concentration. The increased droplet size is probably due to the expansion of the interfacial film by the co-surfactant present.13,37
In this study, we incorporated an OXA/DCK complex with 5-FU into the multiple nanoemulsion. We demonstrated that the nanoemulsion resulted in a significantly increased drug permeability and higher oral bioavailability in rats. The underlying mechanisms for the improvement of oral bioavailability of OXA/DCK and 5-FU are not well understood yet. However, several modes of action could be proposed, as follows: First, membrane permeability enhancement of OXA by ion pairing with DCK was utilized to enhance the lipophilicity of the drug, which improved partitioning of OXA into the oil phase, as well as apparent liposolubility in the intestinal membrane.22 The reason is that DOCA is an amphiphilic molecule composed of a hydrophobic α-side and a hydrophilic β-side.38 It has also been reported that DOCA can incorporate perpendicularly into the surface of the bilayer membrane and perturb the acyl chain of the lipid matrix, thereby increasing the membrane flexibility and the solubility of lipophilic drugs in the membrane, resulting in improvement of the transcellular transport of OXA/DCK complex.38–41 Moreover, DOCA was shown to disrupt tight junctions in the epithelial lining by epithelial growth factor receptor phosphorylation in Caco-2 monolayers, resulting in increased paracellular transport of drug molecules via occluding dephosphorylation and cytoskeletal rearrangement at the tight junction level.42 Second, the selective reabsorption of DOCA via the bile acid transporter on the intestinal membrane can potentially improve the OXA/DCK permeability because DCK preserves the natural characteristics of DOCA, and can interact with the bile acid transporter after electrostatic interaction with OXA via the lysine part of DCK. In a previous study, the covalently or electrostatically conjugated DOCA could directly interact with the ileal brush-border membrane surface and be possibly recognized by the bile acid transporter, allowing high concentration gradients for both drug and enhancer at the mucosal surface, and an increase in their passive diffusion across the intestinal membrane.43,44 However, this mechanism should be verified in future studies. For these reasons, the OXA/DCK complex alone exhibited 37.4- and 2.25-fold higher permeability than free OXA through the artificial intestinal membrane and Caco-2 cell monolayer, respectively.
Another possible explanation for the promotion of oral absorption of OXA/DCK and 5-FU may be employment of the multiple w/o/w nanoemulsive forms as delivery carriers. It is thought that the oil droplets in this system may allow high dispersibility of the amphiphilic OXA/DCK complex in the nanoemulsive form, as well as protecting the complex from dissociation and minimizing the dilution effect of enhancers in the GI lumen.45 Multiple nanoemulsions have been known to facilitate drug absorption in the GI tract by changing the membrane structure and fluidity.46,47 In particular, the intestinal membrane permeation and the oral bioavailability of 5-FU could be enhanced by incorporating the drug into the multiple w/o/w nanoemulsive forms because we applied Labrasol and Cremophor EL as surfactants to disperse the inner water phase in the oil and the primary w/o nanodroplets in the outer aqueous phase, respectively. It has been reported that Labrasol can open the tight junction of the intestinal epithelium by interacting with filamentous actin and zonula occludens-1.48 Moreover, Cremophor EL has been shown to act as an enhancer by loosening the tight junction, as well as increasing cell membrane fluidity.49–51 Therefore, a surfactant-induced decrease in membrane integrity may promote the membrane permeation of 5-FU, a hydrophilic small molecule, by a paracellular pathway. Cremophor EL can also bind with the hydrophobic domain of P-gp, resulting in its conformation change and leading to reduced functionality for drug efflux.52 Thus, protection from P-gp-mediated 5-FU efflux may be attributed to its increased oral absorption.
For all these reasons, the permeability of the OXA/DCK complex from nanoemulsion E (Smix,2 1:1) through the artificial intestinal membrane and Caco-2 cell monolayer was 107 and 4.80 times greater than that of free OXA, respectively. At the same time, the permeability of 5-FU through the artificial intestinal membrane and Caco-2 cell monolayer was 3.05 and 4.31 times greater, respectively, than that of free 5-FU after entrapping it inside the nanodroplet of nanoemulsion E (S mix,2 1:1).
Consistently, the ion-pairing complex formation between OXA and DCK showed a 541% increase in the oral bioavailability of free OXA. At the same time, the oral absorption of OXA/DCK and 5-FU was notably improved after formulation in the multiple nanoemulsion; their oral bioavailability values increased by 919% and 139%, respectively, compared with those of free OXA and 5-FU. Moreover, the nanoemulsion system incorporating OXA/DCK and 5-FU demonstrated a prolonged half-life for OXA and 5-FU, which was extended 5.68- and 1.82-fold, respectively, than the half-life after intravenously administration of OXA and 5-FU, suggesting prolonged therapeutic activity of the drug.53,54
Like other platinum-based compounds, OXA exerts its cytotoxic effect mostly through DNA damage. Apoptosis of cancer cells can be caused by formation of DNA lesions, arrest of DNA synthesis, inhibition of RNA synthesis, and triggering of immunologic reactions.55,56 However, OXA alone displays only low activity in many tumors, and therefore, it is often combined with other chemotherapeutic agents, commonly 5-FU. The exact mechanism of synergism of combinational treatment of OXA and 5-FU is complex, but experimental observations suggest that OXA can downregulate or inhibit dihydropyrimidine dehydrogenase, slowing the catabolism of 5-FU.57–59 In our study, the in vitro cytotoxic effect of combining OXA (39 μM) and 5-FU (120 μM) was 1.65 and 1.44 times higher than that of the same amounts of OXA and 5-FU alone, respectively. Moreover, the cytotoxicity of a combination of free OXA (39 μM) and 5-FU (120 μM) was 1.80 times enhanced by treatment with a nanoemulsive form containing OXA/DCK (39 μM) and 5-FU (120 μM). For all of the above reasons, tumor growth in CT26 tumor-bearing mice was significantly suppressed after an oral administration of OXA/DCK- and 5-FU-loaded nanoemulsion, and tumor volume was maximally 3.84-, 1.94-, and 1.62-fold suppressed compared with tumor volume in the control group and the oral OXA-S and 5-FU-S groups, respectively. Although further studies are required to identify an optimum dosing regimen for the nanoemulsion of OXA/DCK and 5-FU, as well as to characterize the synergistic tumor growth inhibition efficacy of the nanoemulsion containing OXA/DCK and 5-FU in comparison with the nanoemulsion comprising OXA/DCK or 5-FU alone, these findings demonstrate that this formulation improves the oral absorption of OXA and 5-FU. We have shown the formulation’s therapeutic potential as an oral combination therapy for colorectal cancer, one which may improve patient compliance and expand the therapeutic applications of these drugs to the prevention of cancer recurrence and metastasis.
Conclusion
This study demonstrates that the nanoemulsion of OXA/DCK and 5-FU increases membrane permeability of OXA and 5-FU due to synergistic effects of enhancing the lipophilic property of OXA by amphiphilic ion-pairing complex formation with DCK and formulation in the multiple w/o/w nanoemulsive form. An orally administered nanoemulsion containing OXA/DCK and 5-FU showed a 9.19- and 1.39-fold increase in bioavailability compared with that of free OXA and 5-FU, respectively, and improved the pharmacokinetic effect. Moreover, the increase of GI absorption of OXA/DCK and 5-FU in the oral nanoemulsion contributed to inhibition of tumor growth in the colorectal cancer-bearing mice, resulting in a maximal decrease in tumor volume by 73.9%, 48.5%, and 38.1% compared with those of the control group and the oral OXA and 5-FU groups, respectively. Therefore, this nanoemulsion incorporating OXA/DCK and 5-FU for an oral combinational delivery system is expected to improve patient compliance by replacing the conventional intravenous infusion, and expand applications in metronomic cancer treatment, as well as prophylaxis of recurrence.
Acknowledgment
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (NRF-2014R1A1A1003122).
Rudra Pangeni and Sang Won Choi contributed equally as first authors to this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Banna GL, Collovà E, Gebbia V, et al. Anticancer oral therapy: emerging related issues. Cancer Treat Rev. 2010;36(8):595–605. | ||
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. | ||
Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm Res. 2003;20(8):1210–1224. | ||
Kuppens IE, Bosch TM, van Maanen MJ, et al. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother Pharmacol. 2005;55(1):72–78. | ||
Shin SC, Choi JS, Li X. Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm. 2006;313(1–2):144–149. | ||
Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol. 1998;25(2 Suppl 5):4–12. | ||
Woynarowski JM, Chapman WG, Napier C, Herzig MC, Juniewicz P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol. 1998;54(5):770–777. | ||
Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1(3):227–235. | ||
Alian OM, Azmi AS, Mohammad RM. Network insights on oxaliplatin anti-cancer mechanisms. Clin Transl Med. 2012;1(1):26. | ||
Giacchetti S, Perpoint B, Zidani R, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18(1):136–147. | ||
André T, Boni C, Mounedji-Boudiaf L, et al; Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. | ||
Jain A, Jain SK, Ganesh N, Barve J, Beg AM. Design and development of ligand-appended polysaccharidic nanoparticles for the delivery of oxaliplatin in colorectal cancer. Nanomedicine. 2010;6(1):179–190. | ||
Shakeel F, Haq N, Al-Dhfyan A, Alanazi FK, Alsarra IA. Double w/o/w nanoemulsion of 5-fluorouracil for self-nanoemulsifying drug delivery system. J Mol Liq. 2014;200(Part B):183–190. | ||
Urbanska AM, Karagiannis ED, Guajardo G, Langer RS, Anderson DG. Therapeutic effect of orally administered microencapsulated oxaliplatin for colorectal cancer. Biomaterials. 2012;33(18):4752–4761. | ||
He W, Du Q, Cao DY, Xiang B, Fan LF. Study on colon-specific pectin/ethylcellulose film-coated 5-fluorouracil pellets in rats. Int J Pharm. 2008;348(1–2):35–45. | ||
Li X, Xu Y, Chen G, Wei P, Ping Q. PLGA nanoparticles for the oral delivery of 5-fluorouracil using high pressure homogenization-emulsification as the preparation method and in vitro/in vivo studies. Drug Dev Ind Pharm. 2008;34(1):107–115. | ||
Liang YH, Liu CH, Liao SH, et al. Cosynthesis of cargo-loaded hydroxyapatite/alginate core-shell nanoparticles (HAP@Alg) as pH-responsive nanovehicles by a pre-gel method. ACS Appl Mater Interfaces. 2012;4(12):6720–6727. | ||
Bastakoti BP, Sukegawa H, Wu KC, Yamauchi Y. Synthesis of porous iron oxide microspheres by a double hydrophilic block copolymer. RSC Adv. 2014;4(20):9986–9989. | ||
Liao SH, Liu CH, Bastakoti BP, et al. Functionalized magnetic iron oxide/alginate core-shell nanoparticles for targeting hyperthermia. Int J Nanomedicine. 2015;10:3315–3327. | ||
Jeon OC, Seo DH, Kim HS, Byun Y, Park JW. Oral delivery of zoledronic acid by non-covalent conjugation with lysine-deoxycholic acid: in vitro characterization and in vivo anti-osteoporotic efficacy in ovariectomized rats. Eur J Pharm Sci. 2016;82:1–10. | ||
Mahmud F, Jeon OC, Al-Hilal TA, et al. Absorption mechanism of a physical complex of monomeric insulin and deoxycholyl-l-lysyl-methylester in the small intestine. Mol Pharm. 2015;12(6):1911–1920. | ||
Jeon OC, Byun Y, Park JW. Preparation of oxaliplatin-deoxycholic acid derivative nanocomplexes and in vivo evaluation of their oral absorption and tumor growth suppression. J Nanosci Nanotechnol. 2016;16(2):2061–2064. | ||
Park JW, Byun Y. Ionic complex of risedronate with positively charged deoxycholic acid derivative: evaluation of physicochemical properties and enhancement of intestinal absorption in rats. Arch Pharm Res. 2014;37(12):1560–1569. | ||
Jerremalm E, Eksborg S, Ehrsson H. Hydrolysis of oxaliplatin-evaluation of the acid dissociation constant for the oxalato monodentate complex. J Pharm Sci. 2003;92(2):436–438. | ||
Lucas MF, Pavelka M, Alberto ME, Russo N. Neutral and acidic hydrolysis reactions of the third generation anticancer drug oxaliplatin. J Phys Chem B. 2009;113(3):831–838. | ||
Gupta RR, Jain SK, Varshney M. AOT water-in-oil microemulsions as a penetration enhancer in transdermal drug delivery of 5-fluorouracil. Colloids Surf B Biointerfaces. 2005;41(1):25–32. | ||
Shishu, Kamalpreet, Maheshwari M. Development and evaluation of novel microemulsion based oral formulations of 5-fluorouracil using non-everted rat intestine sac model. Drug Dev Ind Pharm. 2012;38(3):294–300. | ||
Sigward E, Mignet N, Rat P, et al. Formulation and cytotoxicity evaluation of new self-emulsifying multiple W/O/W nanoemulsions. Int J Nanomedicine. 2013;8:611–625. | ||
Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. | ||
Kiss L, Walter FR, Bocsik A, et al. Kinetic analysis of the toxicity of pharmaceutical excipients Cremophor EL and RH40 on endothelial and epithelial cells. J Pharm Sci. 2013;102(4):1173–1181. | ||
Gutiérrez MB, Miguel BS, Villares C, Gallego JG, Tuñón MJ. Oxidative stress induced by Cremophor EL is not accompanied by changes in NF-kappaB activation or iNOS expression. Toxicology. 2006;222(1–2):125–131. | ||
Malingré MM, Terwogt JM, Beijnen JH, et al. Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol. 2000;18(12):2468–2475. | ||
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9(3):E344–E352. | ||
Bali V, Ali M, Ali J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids Surf B Biointerfaces. 2010;76(2):410–420. | ||
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. | ||
Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45(1):89–121. | ||
Parmar N, Singla N, Amin S, Kohli K. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B Biointerfaces. 2011;86(2):327–338. | ||
Gangadhar KN, Adhikari K, Srichana T. Synthesis and evaluation of sodium deoxycholate sulfate as a lipid drug carrier to enhance the solubility, stability and safety of an amphotericin B inhalation formulation. Int J Pharm. 2014;471(1–2):430–438. | ||
Elorza MA, Elorza B, Chantres JR. Stability of liposomal formulations: action of amphiphilic molecules. Int J Pharm. 1997;158(2):173–183. | ||
Guan P, Lu Y, Qi J, et al. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomedicine. 2011;6:965–974. | ||
Forsgård RA, Korpela R, Stenman LK, Osterlund P, Holma R. Deoxycholic acid induced changes in electrophysiological parameters and macromolecular permeability in murine small intestine with and without functional enteric nervous system plexuses. Neurogastroenterol Motil. 2014;26(8):1179–1187. | ||
Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G906–G913. | ||
Jeon OC, Hwang SR, Al-Hilal TA, et al. Oral delivery of ionic complex of ceftriaxone with bile acid derivative in non-human primates. Pharm Res. 2013;30(4):959–967. | ||
Al-Hilal TA, Park J, Alam F, et al. Oligomeric bile acid-mediated oral delivery of low molecular weight heparin. J Control Release. 2014;175:17–24. | ||
Hu J, Sun L, Zhao D, et al. Supermolecular evodiamine loaded water-in-oil nanoemulsions: enhanced physicochemical and biological characteristics. Eur J Pharm Biopharm. 2014;88(2):556–564. | ||
Cheng MB, Wang JC, Li YH, et al. Characterization of water-in-oil microemulsion for oral delivery of earthworm fibrinolytic enzyme. J Control Release. 2008;129(1):41–48. | ||
Fofaria NM, Qhattal HS, Liu X, Srivastava SK. Nanoemulsion formulations for anti-cancer agent piplartine – characterization, toxicological, pharmacokinetics and efficacy studies. Int J Pharm. 2016;498(1–2):12–22. | ||
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–486. | ||
Udata C, Patel J, Pal D, Hejchman E, Cushman M, Mitra AK. Enhanced transport of a novel anti-HIV agent – cosalane and its congeners across human intestinal epithelial (Caco-2) cell monolayers. Int J Pharm. 2003;250(1):157–168. | ||
Buyukozturk F, Benneyan JC, Carrier RL. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J Control Release. 2010;142(1):22–30. | ||
Lu Y, Qi J, Wu W. Absorption, disposition and pharmacokinetics of nanoemulsions. Curr Drug Metab. 2012;13(4):396–417. | ||
Yin YM, Cui FD, Mu CF, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. | ||
Jacobs SS, Fox E, Dennie C, Morgan LB, McCully CL, Balis FM. Plasma and cerebrospinal fluid pharmacokinetics of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin Cancer Res. 2005;11(4):1669–1674. | ||
Zhang J, Li M, Liu Z, Wang L, Liu Y, Zhang N. Preclinical studies of N3-O-toluyl-fluorouracil-loaded lipid-based nanosuspensions in H22-bearing mice. Int J Nanomedicine. 2014;9:2741–2751. | ||
Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. 2011;18(1):18–25. | ||
Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat Rev. 2005;31(2):90–105. | ||
Fischel JL, Formento P, Ciccolini J, et al. Impact of the oxaliplatin-5 fluorouracil-folinic acid combination on respective intracellular determinants of drug activity. Br J Cancer. 2002;86(7):1162–1168. | ||
Stoehlmacher J, Park DJ, Zhang W, et al. A multivariate analysis of genomic polymorphisms: prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br J Cancer. 2004;91(2):344–354. | ||
Aschele C, Friso ML, Pucciarelli S, et al. A phase I–II study of weekly oxaliplatin, 5-fluorouracil continuous infusion and preoperative radiotherapy in locally advanced rectal cancer. Ann Oncol. 2005;16(7):1140–1146. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.