Back to Journals » International Journal of Nanomedicine » Volume 12
Multifunctionalized polyethyleneimine-based nanocarriers for gene and chemotherapeutic drug combination therapy through one-step assembly strategy
Authors Jiang D, Wang M, Wang T, Zhang B, Liu C, Zhang N
Received 31 May 2017
Accepted for publication 25 September 2017
Published 6 December 2017 Volume 2017:12 Pages 8681—8698
DOI https://doi.org/10.2147/IJN.S142966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Dandan Jiang,1,* Mingfang Wang,1,* Tianqi Wang,1 Bo Zhang,1 Chunxi Liu,2 Na Zhang1
1Department of Pharmaceutics, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, Jinan, China; 2Pharmaceutical Department, Qilu Hospital of Shandong University, Jinan, China
*These authors contributed equally to this work
Abstract: Gene therapy combined with chemotherapy to achieve synergistic therapeutic effects has been a hot topic in recent years. In this project, the human tumor necrosis factor-related apoptosis-inducing ligand-encoding plasmid gene (TRAIL) and doxorubicin (Dox)-coloaded multifunctional nanocarrier was constructed based on the theory of circulation, accumulation, internalization, and release. Briefly, polyethyleneimine (PEI) was selected as skeleton material to synthesize PEI–polyethylene glycol (PEG)–TAT (PPT). Dox was conjugated to PEI using C6-succinimidyl 6-hydrazinonicotinate acetone hydrazone (C6-SANH), and a pH-sensitive Dox-PEI (DP) conjugate was obtained. Then, intracellular cationic pH-sensitive cellular assistant PPT and DP were mixed to condense TRAIL, and TRAIL–Dox coloaded PPT/DP/TRAIL (PDT) nanocarriers were obtained by one-step assembly. TRAIL was completely condensed by DP or PPT when mass ratios (DP/PPT to TRAIL) were up to 100:64, which indicated that DP and PPT could be mixed at any ratio for TRAIL condensation. The intracellular uptake rate of PDT was enhanced (P<0.05) when the contents of PPT in PPT+DP increased from 0 to 30%. Free Dox and TRAIL-loaded nanocarriers (PPT/C6-SANH-PEI/TRAIL [PCT]) were selected as controls to verify the synergistic antitumor effects of PDT. Compared with free TRAIL, TRAIL-protein expression was upregulated by PDT and PCT on Western blotting assays. The in vitro cytotoxicity of PDT was significantly enhanced compared to free Dox and PCT (P<0.01). Furthermore, murine PDT nanocarriers showed higher in vivo antitumor ability than both the Dox group (P<0.05) and the murine PCT group (P<0.05). These results indicated that the TRAIL + Dox synergistic antitumor effect could be achieved by PDT, which paves the way to gene–drug combination therapy for cancer.
Keywords: multifunctional, gene therapy, chemotherapy, TRAIL, one-step assembly strategy, CAIR theory
Introduction
Mutation and inactivation of tumor-suppressor genes are important pathogeneses during the development of tumors.1,2 Advances in understanding and manipulating genes have set the stage to prevent or treat cancer by altering human genetic material.3,4 Even though gene therapy has been increasingly applied in clinical trials for cancer treatment over the past decade, its clinical applications have not been growing as expected.5,6 Till now, only two cancer-gene therapies have been approved in China, and none has been approved by the US Food and Drug Administration. The limited clinical outcome of gene therapy might be due to tumor heterogeneity and drug resistance. Combination therapies using multiple therapeutic strategies can synergistically elevate antitumor effects with lowering doses of each agent, hence reducing side effects.7 Recently, the combination of chemotherapy and gene therapy has attracted a lot of attention with enhanced therapeutic effects of drugs and transfection efficiency of genes.1,8
The human tumor necrosis factor-related apoptosis-inducing ligand-encoding plasmid gene (TRAIL) is in the TNF superfamily of related proteins and can induce apoptosis in cancer cells instead of most normal cells.9 TRAIL has shown impressive anticancer activity in preclinical models, and this promise has promoted the application of TRAIL in clinical trials.10,11 To enhance TRAIL-induced apoptotic potency, combination therapies of TRAIL and chemotherapeutics have been being studied in recent years.12,13 Doxorubicin (Dox), which has been used clinically to treat several kinds of tumors in clinical settings, was selected as the model chemotherapeutic in this work.14 There are several advantages to the Dox and TRAIL combination. TRAIL-induced apoptosis requires clustering of DR5 within ceramide-enriched membrane platforms.10 DR5 clustering can facilitate recruitment of the adaptor protein FADD and pro-caspase 8 and transactivation of caspase 8, leading to induction of apoptosis.15,16 Triggered release of ceramide in plasma membrane or addition of exogenous ceramide can both mediate the formation of ceramide-enriched membrane platforms and enhance apoptosis in cancer cells.17 The potential of Dox to promote ceramide production has been identified by Vitovski et al.18 Dox enhances TRAIL-induced apoptosis in cancer cells via ceramide-enriched membrane platforms. Dox has been identified to have the greatest potential for sensitization of tumor cells to TRAIL through upregulation and activation of DR4 and DR5.19 Combining short-term TRAIL treatment with Dox prevents the development of TRAIL-resistant cells.20,21
The rate-limiting step in successful gene–drug combination therapy is the preparation of highly efficient nanocarriers.22,23 Nanocarrier-based gene–drug combination therapy can be classified into three groups: general chemotherapy combined with gene-carrying nanocarrier, use of separate nanocarriers for chemotherapy and gene therapy, and codelivery of chemical and gene therapy within a single nanocarrier.22 Among these three strategies, gene–drug combination therapy using a single nanocarrier is highly effective, due to its ability to affect multiple disease pathways in the same tumor cell.23 For example, Wang et al25 prepared a hyaluronic acid-decorated polyethylenimine (PEI)–poly(lactide-co-glycolic acid) nanoparticle system for targeted codelivery of Dox and miR5423p. The nanoparticles increased cytotoxicity in MDA-MB231 cells and further promoted triple-negative breast cancer-cell apoptosis via activating p53 and inhibiting surviving expression. Xu et al26 prepared double-walled microspheres consisting of a poly(lactide-co-glycolic acid) core surrounded by a polylactic acid shell to achieve the codelivery of “chi-p53” and Dox, resulting in a combined antitumor effect.
This codelivery of gene and chemotherapeutic drug was achieved by developing complex nanocarriers with various materials. Nowadays, one of the main challenges for preparation of gene–drug coloaded nanocarriers is how to make the preparation process more simplified and controllable.26 In this project, one-step assembly nanocarriers were designed by conjugating a chemotherapeutic drug to condensed gene material. For gene delivery, PEI has attracted much attention for its high transfection efficiency and multifunctional modification ability.27,28 For example, Yang et al30 prepared nanohydrogels comprising carboxymethylcellulose complexed with cationic branched PEI (bPEI) as gene-delivery vehicles. The gene-delivery efficacy showed that the carboxymethylcellulose nanogel complexed with bPEI showed high uptake and gene-transfection ability. Therefore, PEI, which can both achieve gene condensation and carriage of chemotherapeutic drugs, was selected as the skeleton material in this project.
A gene–drug coloaded nanocarrier delivering its cargo efficiently to the cytoplasm of cancer cells in solid tumors must go through a cascade of four steps: long circulation in the blood (C), accumulation in the tumor via the enhanced permeability and retention effect (A), internalization by tumor cells (I), and intracellular drug release (R), collectively referred to as the CAIR theory. As such, a nanocarrier achieves high delivery and therapeutic efficacy only if it efficiently meets all conditions of the CAIR theory.30 Therefore, to develop PEI-based nanocarriers in view of the CAIR theory, multiple elements should be modified in the skeleton material (Scheme 1): in blood circulation, polyethylene glycolation (PEGylation) can be used to make nanocarriers able to escape extracellular nucleases, evade the reticuloendothelial system, and avoid aspecific interaction with blood cells;31–34 to increase accumulation at tumor sites, suitable particle sizes are necessary30 (nanocarriers around 100 nm have longer blood-circulation times and better tumor accumulation than smaller particles); and cell-penetrating peptides, which can improve translocation across the plasma membrane and increase intracellular drug concentration, are often used to increase internalization of payloads by tumor cells.32 TAT peptide, a classical cationic cell-penetrating peptide, is known to facilitate and enhance cellular internalization efficiently in a large variety of electrostatically or covalently bound cargoes in a nontoxic fashion.34,35 For example, Koren et al37 prepared pH-sensitive PEGylated liposomes modified with TAT peptide, which showed increased cytotoxicity with exposed TAT (lower pH values) than shielded TAT (higher pH values). Peng et al38 synthesized TAT peptide conjugated novel cationic metal nanoparticles as highly efficient carriers for gene delivery to stem cells. Therefore, the TAT peptide was selected to improve gene and drug translocation across the plasma membrane in this project. Finally, effective intracellular gene and drug release can be achieved by pH-sensitive chemical bonds38,39 and endosomal escape materials.40,41 To achieve the pH-responsive release of the chemotherapeutic drug Dox, Dox-PEI (DP) conjugates were prepared through hydrazone bonds based on our group’s previous work.43 The pH-buffering ability of PEI can be used to achieve endosomal escape of cargoes through its “proton sponge” effect. As shown in Scheme 1, there are only free amino groups on PEI, and endosomal escape is achieved. The programmable release of Dox and gene via an endosomal pH-triggered mechanism has been verified in previous work.42 In short, Dox can be released from a Dox–gene coloaded nanocarrier rapidly, while the gene is protected from degradation under acidic endosomal conditions. Furthermore, due to the proton-sponge effect of free PEI, the gene and Dox are released into the cytoplasm.
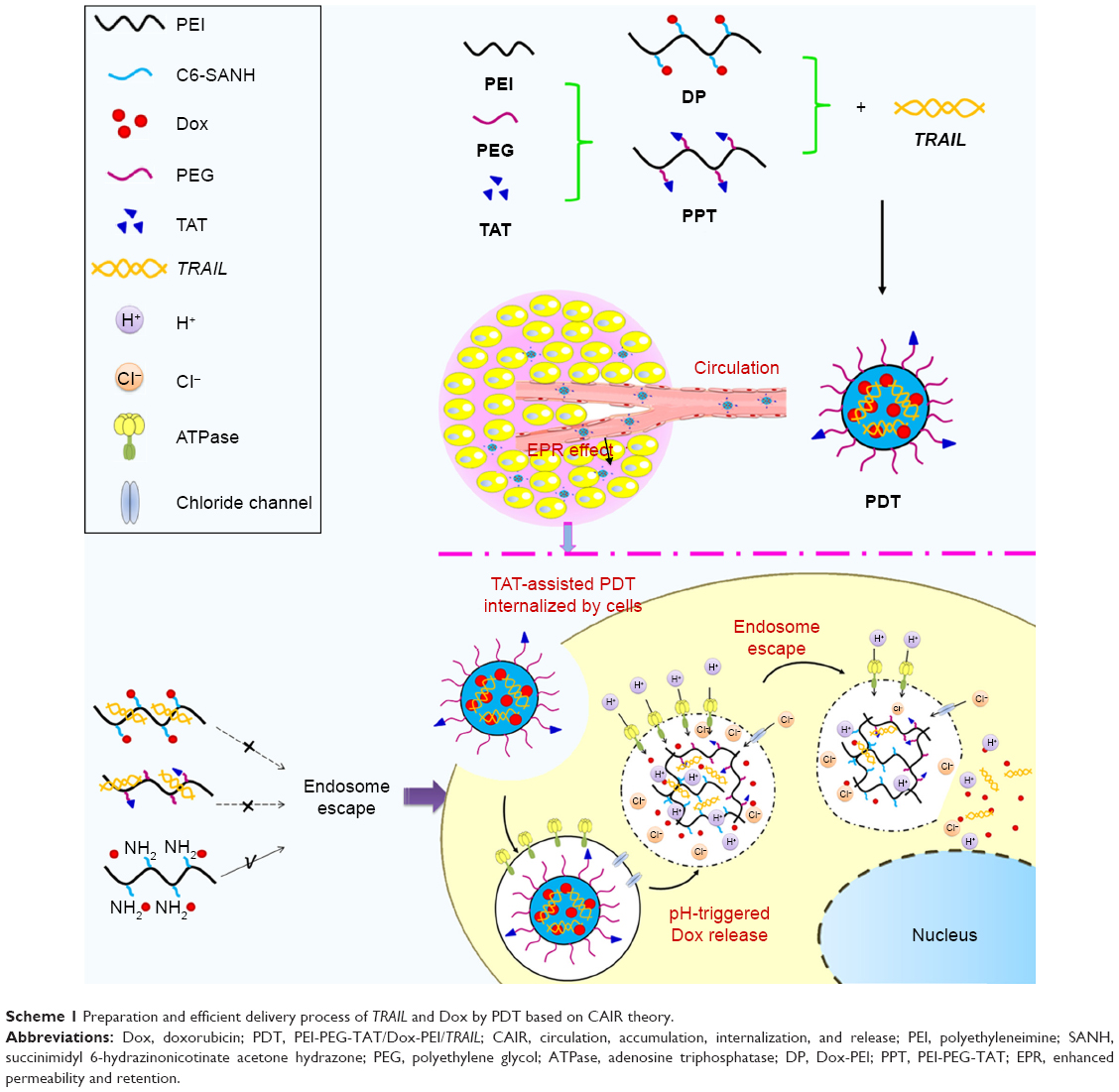 | Scheme 1 Preparation and efficient delivery process of TRAIL and Dox by PDT based on CAIR theory. |
In this project, PEI-based intracellular assistant material (PEI-PEG-TAT [PPT]) and pH-sensitive material (DP) were synthesized and characterized by 1H nuclear magnetic resonance (NMR) spectroscopy. Then, PPT and DP of ideal properties were used to condense TRAIL to form Dox–TRAIL coloaded PPT-DP-TRAIL nanocarriers (PDT). Morphology, particle-size distribution, and ζ-potential of PDT were investigated. Cell-uptake testing was performed in HEPG2 and SKOV3 cells to verify the intracellular uptake-assistance ability of PPT. The expression of the TRAIL protein of PDT was investigated by Western blot assay. Finally, the synergistic antitumor effect of TRAIL–Dox coloaded PDT was confirmed by in vitro cytotoxicity testing and in vivo antitumor evaluation.
Materials and methods
Materials
Branched PEI (molecular weight [MW] 25 kDa) was purchased from Sigma-Aldrich (St Louis, MO, USA). C6-S-HyNic (C6-succinimidyl 6-hydrazinonicotinate acetone hydrazone [C6-SANH]) was purchased from Solulink (San Diego, CA, USA). Dox was purchased from Meilun Biology Technology (Dalian, China). Bifunctional PEG derivatives with a maleimide and N-hydroxysuccinimide ester group (Mal-PEG-NHS; MW 2 kDa) were purchased from Kaizheng Biotech Development (Beijing, China). TAT with a cysteine on the C-terminal (sequence YGRKKRRQRRRC) was purchased from and synthesized by Shanghai Apeptide. MTT was purchased from Sigma-Aldrich. GoldView was purchased from Saibaisheng Biological Engineering (Beijing, China). Lipofectamine 2000 was purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents were of commercial special grade and used without further purification. Human TRAIL under an hEF1α promoter and EGFP under a CMV promoter plasmid were purchased from Yingrun Biotechnology (Changsha, China). Murine TRAIL under a CMV promoter plasmid was purchased from Vigene Biosciences (Rockville, MD, USA).
Cell culture
HEPG2 and SKOV3 cells, both purchased from the Chinese Academy of Sciences (Shanghai, China), were kindly provided by the Institute of Immunopharmacology and Immunotherapy of Shandong University (Jinan, China). They were both cultured in DMEM supplemented with 10% FBS at 37°C with 5% CO2. The sensitivity of TRAIL on HEPG2 and SKOV3 cells has been identified before.44–47
Synthesis of multifunctional materials
Intracellular pH-sensitive cationic DP was synthesized and characterized by 1H NMR spectroscopy (Avance DPX-300; Bruker, Billerica, MA, USA) as per our previous work.29 C6-SANH-PEI was the intermediate product in the DP-synthesis process, which was also characterized by 1H NMR spectroscopy in our previous work. Synthesis of the cellular internalization-assistant material PPT is depicted in Figure 1. First, PEI-PEG was synthesized through amide reaction between the NHS ester group of PEG and the amino groups of PEI. Briefly, PEI and NHS-PEG-Mal (2 kDa) were dissolved in dimethyl sulfoxide 20 mL in a 50 mL glass flask under stirring. Then, 20 μL TEA was added to the reaction solution and stirred for 48 hours at room temperature. The product obtained was purified by dialysis against distilled water (MW cutoff [MWCO] 8,000–14,000 Da) for 48 hours and lyophilized. The chemical structure of the PEI-PEG was confirmed by 1H NMR spectroscopy.
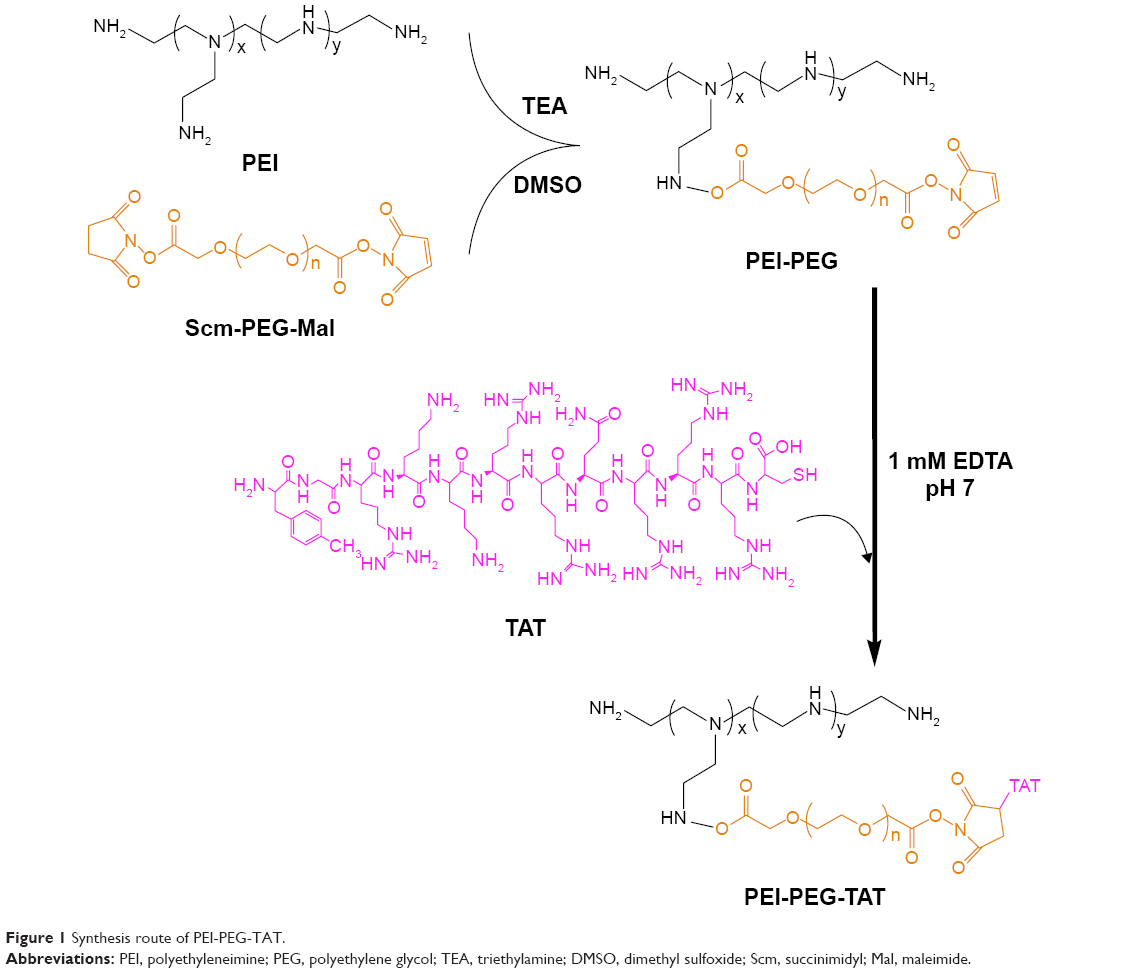 | Figure 1 Synthesis route of PEI-PEG-TAT. |
Then, PPT was synthesized. Briefly, TAT and PEI-PEG were dissolved in 1 mM EDTA containing PBS buffer (20 mL) and reacted for 48 hours in a nitrogen atmosphere. TAT was coupled to the end of PEI-PEG via the reaction between the Mal residues of the PEG and sulfhydryl groups of TAT. The target product was purified by dialysis against distilled water (MWCO 8,000–14,000 Da) and then lyophilized. The chemical structure of PPT was also confirmed by 1H NMR.
Agarose gel-electrophoresis assay
The condensation abilities of DP-TRAIL (DT), C6-SANH-PEI/TRAIL (CT), and PPT-TRAIL were analyzed with 0.8% agarose gel. To obtain specified weight ratios of multifunctional materials, TRAIL was added to different concentrations of DP, C6-SANH-PEI, and PPT solutions at equal volumes under vortexing (Vortex 5; Kylin–Bell Lab Instruments, Jiangsu, China). The mixed solutions were incubated at room temperature for 30 minutes to form DP-TRAIL, CT, and PPT-TRAIL complexes.
Briefly, the concentration of TRAIL was fixed at 80 μg/mL, and DT, CT, and PPT-TRAIL solutions were all prepared at different mass ratios (25:128, 25:64, 25:32, 25:16, 25:8, 25:4, and 25:2). The obtained DT, CT, and PPT-TRAIL complexes with different mass ratios were mixed with an appropriate amount of 6× loading buffer and electrophoresed on a 0.8% (w:v) agarose gel containing an appropriate amount of GoldView (Tris-acetate-EDTA buffer, 90 V, 20 minutes). The electrophoretic mobility of DT, CT, and PPT-TRAIL was visualized by an ultraviolet transilluminator and a digital imaging system (IS-2200; Alpha Innotech, San Leandro, CA, USA).
In vitro transfection test of DT
Since DP and PPT have similar condense ability, the first step of prescription optimization was the determination of mass ratio between DP and TRAIL. DT particle sizes with mass ratios of 25:16, 25:8, 25:4, and 25:2 were measured with a nanoparticle analyzer (DelsaNano S; Beckman Coulter, Brea, CA, USA). The transfection efficiency of DT with suitable particle sizes was further evaluated to determine the optimal preparation.
In the in vitro transfection test, pEGFP-N1 plasmid (pDNA), which contains the gene encoding EGFP, expressed from a CMV promoter, was used as the reporter gene. Briefly, HEPG2 and SKOV3 cells were seeded in 24-well culture plates at a density of 5×104 cells/well. Culture media were removed after cells had reached 80% confluence, and cells were treated with fresh media (without FBS) containing DP-pDNA with different mass ratios (2 μg pDNA/well) for 4 hours at 37°C in an incubator. Then, the culture media containing DP-pDNA were removed and washed twice with cold PBS and all the cells cultured for another 48 hours with fresh media. At the end of the incubation, cells were washed with PBS three times and cell nuclei labeled by incubating the samples with Hoechst 33342 for 10 minutes. Finally, the cells were washed with PBS three times and 1 mL PBS added to each cell. Dox and GFP expression in cells was observed using inverted fluorescence microscopy (BX40; Olympus, Tokyo, Japan). Fluorescent images were obtained with a DP70 digital imaging system (Olympus), and the results were analyzed by Olysia BioReport imaging software (version 3.2; Olympus).
Optimization of PPT content in PPT+DP by intracellular uptake test
The second step of prescription optimization was the determination of PPT content in PPT+DP. The optimal mass ratio of DT was fixed at 25:4 according to the in vitro transfection test. The mass ratio of PPT+DP:TRAIL was also fixed at 25:4 to keep consistent condensation ability. To select the optimal PPT content in the mixed materials of PPT+DP, PPT+DP mixtures with different PPT content (0, 1%, 5%, 10%, 20%, 30%, 40%, and 50%) were used to condense TRAIL (80 μg/mL). Particle sizes of PDT with different PPT contents were characterized with the DelsaNano S.
The internalization abilities of PDT with different PPT contents in HEPG2 and SKOV3 cells were visualized and quantified by fluorescence microscopy (BX40) and flow cytometry, respectively. Briefly, HEPG2 and SKOV3 cells were seeded in 24-well culture plates at a density of 5×104 cells per well. Culture media were removed after cells had reached 80% confluence, and cells were treated with fresh media containing PDT with different PPT contents for 4 hours at 37°C in an incubator. Then, culture media containing the tested PDT with different PPT contents were removed and washed twice with cold PBS. Cells were incubated with Hoechst 33342 and washed twice with cold PBS 10 minutes later. Then, cellular uptake of PDT was observed using inverted fluorescence microscopy with the DP70 digital imaging system and the results analyzed with Olysia BioReport. After that, all cells were harvested and washed three times with cold PBS. The fluorescence intensity of the cells was measured using flow cytometry equipped with a 488 nm argon laser for excitation. For each sample, 10,000 events were collected and fluorescence detected. The percentage of positive events was calculated from the events within the gate divided by the total number of events, excluding cell debris. Finally, the transfection efficiency of PDT with optimal PPT content was further evaluated by the method in the “In vitro transfection test of DT” section.
Characterization of DT, PDT, PCT, and PCD
To establish the synergistic effect of TRAIL and Dox on PDT, DT, PPT-CT (PCT), and PPT/C6-SANH-PEI/pDNA (PCD) were prepared and used as controls in the subsequent experiments. Particle sizes and ζ-potential of DT, PDT, PCT, and PCD were analyzed with the DelsaNano S. The morphology of DT, PDT, PCT, and PCD was examined by transmission electron microscopy (JEM-1200EX; JEOL, Tokyo, Japan).
Western blotting assay
The successful expression of the TRAIL protein of PDT was the precondition for the exertion of TRAIL–Dox combined/synergistic effects, and expression of the TRAIL protein of PDT was determined by Western blotting assay. Briefly, HEPG2 cells were seeded in six-well plates at a density of 4×105 cells/well. Culture media were removed after cells had reached 80% confluence, and cells were treated with fresh media containing PDT, PCT, PCD, and Dox for 4 hours at 37°C in an incubator, with the Lipofectamine 2000/TRAIL as positive control and PCD as negative control (equivalent to 5 μg of TRAIL or 1.75 μg of Dox each well). Then, media were removed and supplemented with DMEM with 10% FBS. After incubation for another 48 hours, media were discarded and washed twice with cold PBS. Then, the protein was extracted from the cells in each group. The sodium dodecyl sulfate polyacrylamide-gel electrophoresis was applied to separate the specified proteins, which were transferred to polyvinylidene fluoride membranes. After that, membranes were blocked by 5% skimmed milk and incubated with the primary antibodies anti-TRAIL (Bioss, Beijing, China) and anti-β-actin (Sigma-Aldrich) overnight at 4°C. HRP-conjugated antibodies were incubated with the membranes after washing. An enhanced chemiluminescence kit was used to visualize the bands in darkness, and bands were quantified using the IS-2200 and AlphaEaseFC 4.0 (Alpha Innotech).
In vitro cytotoxicity assays
To evaluate the synergistic effects of Dox and TRAIL in PDT, the MTT assay was used to evaluate the in vitro antitumor effects of PDT in HEPG2 and SKOV3 cells. Briefly, HEPG2 and A549 cells were seeded in 96-well plates at a density of 8,000 cells per well and incubated for 24 hours at 37°C. Then, different concentrations of Dox, PCT, PDT, and PCD (equivalent Dox concentrations were 0.01, 0.05, 0.1, 0.5, 1, 2, 4, 6, 8, and 10 μg/mL and TRAIL concentrations 0.029, 0.14, 0.29, 1.4, 2.9, 5.8, 11.6, 17.4, 23.2, and 29 μg/mL) were added to five wells each and incubated for 48 hours. Following that, 20 μL of MTT reagents (5 mg/mL) was added to each well and the cells incubated at 37°C for another 4 hours. The media were removed, and formazan crystals formed by the living cells were dissolved in 200 μL dimethyl sulfoxide per well. Absorbance at 570 nm of the solution was measured on a microplate reader (model 680; Bio-Rad, Laboratories, Hercules, CA, USA), and all experiments were repeated three times.
In vivo antitumor evaluation
The in vivo synergistic antitumor effect of PDT was proved by using murine TRAIL and evaluating the synergistic antitumor effect of murine TRAIL–Dox coloaded PPT–DP–murine TRAIL nanocarriers (mPDT) on H22 tumor-bearing BALB/c mice. Briefly, H22 tumor-bearing BALB/c female mice were prepared by subcutaneous injection at the right axillary space with 0.1 mL cell suspension containing 106 H22 tumor cells. Ten days later, when tumor volumes were approximately 100 mm3, mice were randomly assigned to four treatment groups (n=3 for each group). Then, mice were treated every 2 days with normal saline (NS; intravenous [IV]), Dox solution (Dox 1 mg/kg, IV), mPCT (mouse TRAIL 1 mg/kg, IV) and mPDT (Dox 1 mg/kg, mouse TRAIL 1 mg/kg, IV). Body weights and tumor volumes were recorded every other day. Tumor volumes were measured with a Vernier caliper every other day using the formula (length × width2)/2, where length is the longest dimension and width the widest dimension. At day 12, the mice were killed and tumors from each group surgically excised, rinsed with NS, wiped, and photographed. Then, tumor sections (5 mm) were cut from 10% neutral buffered formalin-fixed and paraffin-embedded tissue blocks. Apoptotic cell death in tumor tissue was evaluated histologically by TUNEL staining (in situ cell-death-detection kit; Hoffman-La Roche, Basel, Switzerland) and imaged by fluorescence microscopy (BX4).
Hemolysis assessment
Whole blood from rabbits was collected in heparinized tubes and centrifuged at 3,000 rpm for 4 minutes at 4°C. Then, hemocyte suspensions were washed with NS (5–10 mL) three times and diluted to 2% erythrocyte suspension. The erythrocyte suspension was always freshly prepared and used within 24 hours after collection of DT, PDT, PCT, and PCD (0.1 mL) with the same concentration of in vivo administration were tested. NS and deionized water were used as negative (no hemolysis) and positive (100% hemolysis) controls, respectively. Specimens were incubated at 37°C±0.5°C for 3 hours and centrifuged at 3,000 rpm for 10 minutes before collecting the supernatant. The absorbance value of the hemoglobin released from the erythrocyte cells was measured at 576 nm. All trials were performed three times. The hemolysis rate was calculated thus:
|
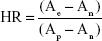
where Ae, An, and Ap are the absorbance value of the experimental group, the negative-control group, and the positive-control group, respectively.
Statistical analysis
All experiments were repeated a minimum of three times and measured at least in triplicate. Results are reported as means ± SD. Statistical significance was analyzed using Student’s t-test. Differences between the experimental groups were considered significant when P<0.05.
Results and discussion
Synthesis of multifunctional materials
Successful synthesis of functionalized materials paved the way to construct desirable nanocarriers. DP and C6-SANH-PEI were synthesized and characterized by 1H NMR in our previous work.34 In addition, the intracellular pH-sensitivity of DP was confirmed in 0.1 M PBS buffer at pH 7.4 and 5. Dox was released from DP completely after 96 hours at pH 5 and only 27.84% of Dox was released from DP at pH 7.4.
1H NMR spectra of PEI, succinimidyl (Scm)–PEG-Mal, TAT, and PPT are shown in Figure 2. The peaks (Figure 2D) at 2.2–3.2 ppm and 3.2–4 ppm were related to PEI and SCM-PEG-Mal, respectively, which indicated that SCM-PEG-Mal was successfully conjugated to PEI. Moreover, the successful conjugation of TAT to PEI-PEG was also confirmed by the chemical shifts belonging to TAT (−CH2−, 1.6–1.9 ppm; aromatic protons of TAT, 6.8–7.3 ppm) in Figure 2D. These results indicated that the cellular internalization-assistant material PPT was successfully synthesized. The integration ratio of PEI (3.2–4 ppm), PEG (2.2–3.2 ppm), and TAT (6.8–7.3 ppm) indicated that the molar ratio among PEI, PEG, and TAT was approximately 1:95:2. The modification ratio of PEG:PEI was enough for long circulation of PDT in blood.
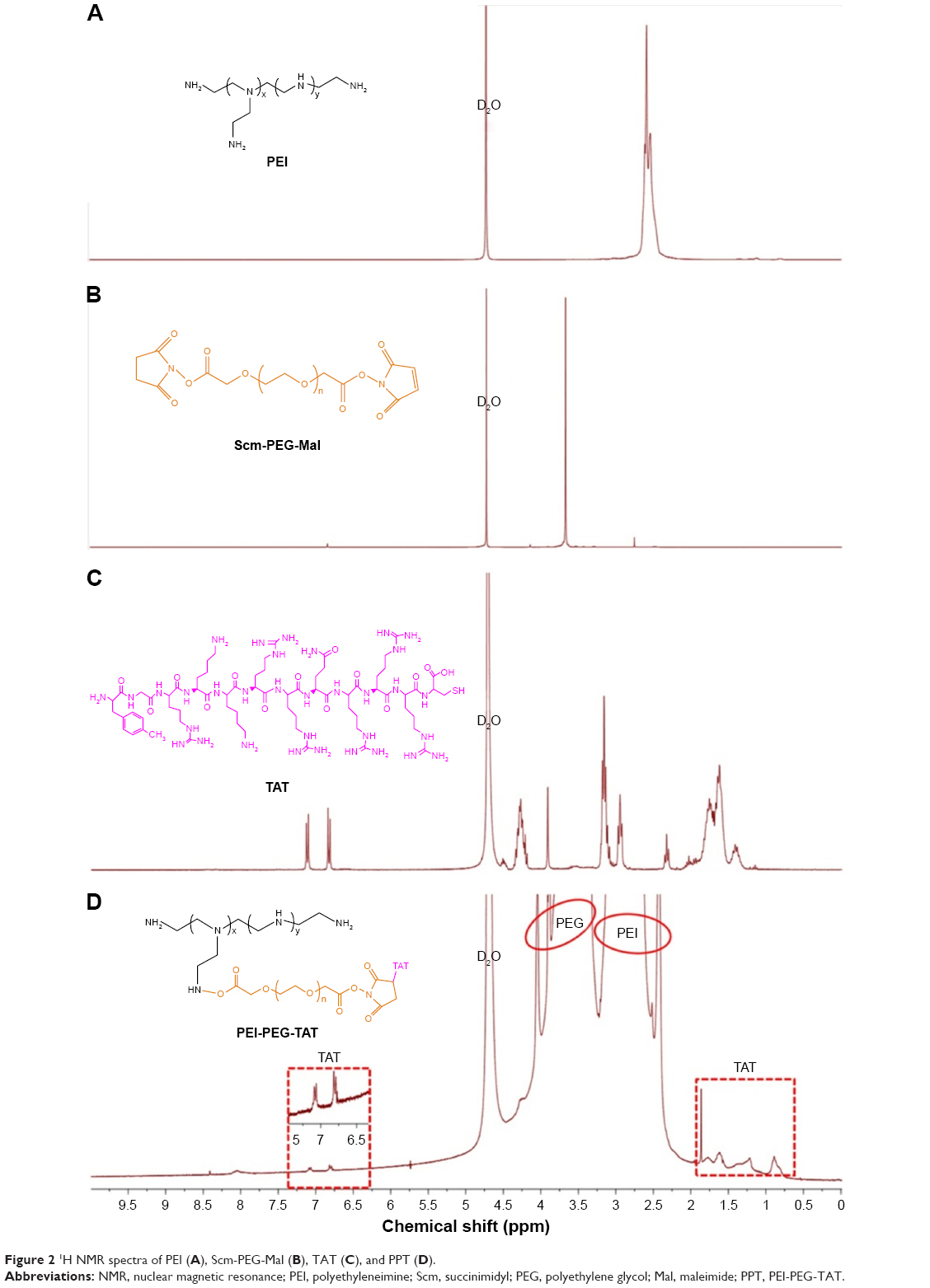 | Figure 2 1H NMR spectra of PEI (A), Scm-PEG-Mal (B), TAT (C), and PPT (D). |
Agarose gel-electrophoresis assay
The condensation ability of DP, C6-SANH-PEI, and PPT was analyzed by 0.8% agarose gel. As shown in Figure 3, TRAIL was completely condensed by DP, C6-SANH-PEI, and PPT when mass ratios (TRAIL) were up to 100:64, which indicated the condensation abilities of DP, C6-SANH-PEI, and PPT to condense TRAIL were similar. Therefore, DP, C6-SANH-PEI, and PPT could be mixed at any ratio to condense TRAIL.
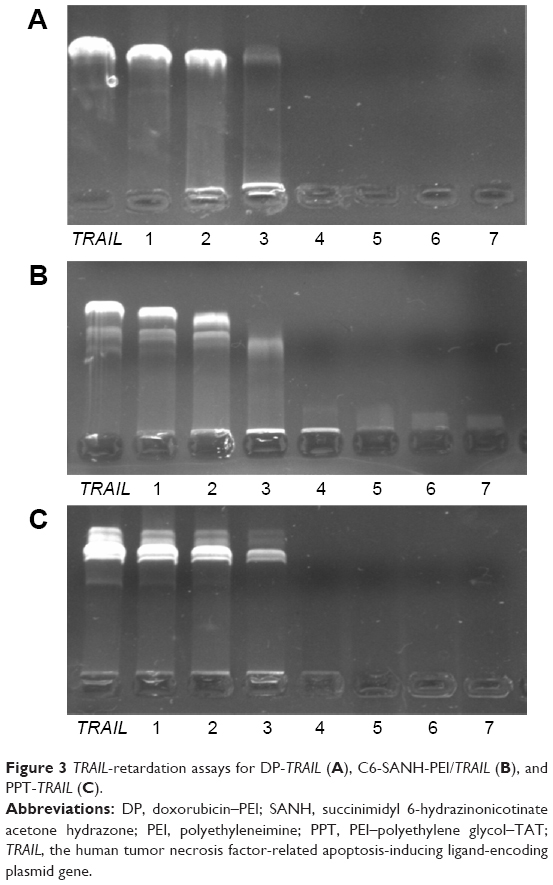 | Figure 3 TRAIL-retardation assays for DP-TRAIL (A), C6-SANH-PEI/TRAIL (B), and PPT-TRAIL (C). |
In vitro transfection test of DT
As shown in Table 1, particle sizes of DT were 184.9±5 nm, 85.8±3.9 nm, 81±3.5 nm, and 103.5±2.1 nm when mass ratios of DT were 100:64, 100:32, 100:16, and 100:8, respectively. In view of the toxicity of PEI and the multimodal particle-size distribution of DT (100:8), DP:pDNA with mass ratios of 25:16–25:4 were used to perform the in vitro transfection test.
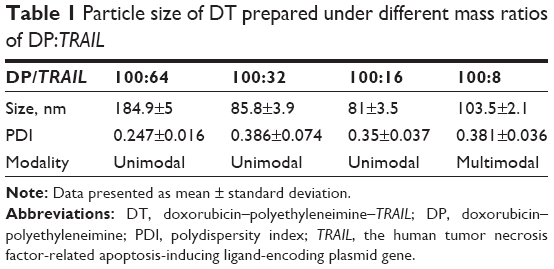 | Table 1 Particle size of DT prepared under different mass ratios of DP:TRAIL |
As shown in Figure 4, cells were incubated with Hoechst 33342 at 37°C for 1 hour to label cell nuclei with blue. The transfection efficiency of DP-pDNA enhanced with mass ratios of DP:pDNA increased from 100:64 to 100:16 in both HEPG2 and SKOV3 cells. Meanwhile, the intensity of Dox was also enhanced with mass ratios of DP/pDNA increased in both HEPG2 and SKOV3 cells, which might have been due to the increased concentration of DP in DP-pDNA. When Dox and pDNA were merged with the nucleus, the white color (merge of red, green, and blue) meant both Dox and transcribed pDNA were in the nucleus, which means both were released from the DP-pDNA after 48-hour incubation.
 | Figure 4 Fluorescence micrography of DP-pDNA complexes in HEPG2 cells (A) and SKOV3 cells (B) following 48-hour incubation at 37°C. |
Optimization of PPT in PPT+DP by cell-uptake test
Particle size is an important factor that affects the in vivo distribution of nanocarriers. With mass ratio of PPT+DP:TRAIL fixed at 25:4, PDT with different PPT contents was analyzed with the DelsaNano S. As shown in Figure 5, PDT particle sizes were about 80 nm, with PPT content of 0%–30%, and there were no significant differences. When the PPT contents in PPT+DP were 40% and 50%, particle sizes were around 100 nm. To confirm the optimal PPT contents in PDT, further investigation would be needed.
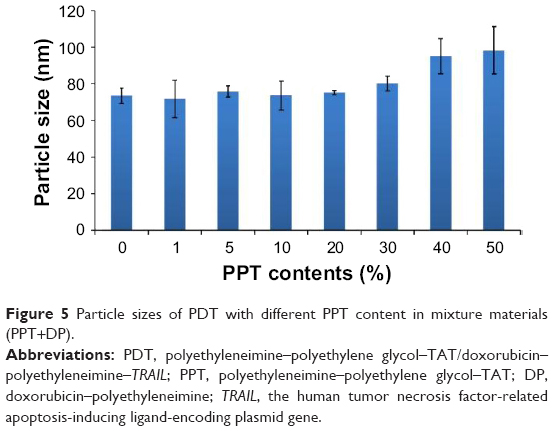 | Figure 5 Particle sizes of PDT with different PPT content in mixture materials (PPT+DP). |
HEPG2 and SKOV3 cells were chosen to determine the cellular uptake of PDT with different PPT contents and select the optimal PPT contents in PDT. As shown in Figure 6 (HEPG2 cells) and Figure 7 (SKOV3 cells), cellular uptake of PDT was enhanced, with PPT contents in PDT increased from 0% to 30% (P<0.05). When the PPT contents in PDT increased from 40% to 50%, cellular uptake dropped compared to PDT with 30% PPT contents (P<0.05 vs 30% PPT contents). This result could have been due to the relatively large particles of PDT. For this reason, PDT with 30% PPT content was selected as the optimal formulation.
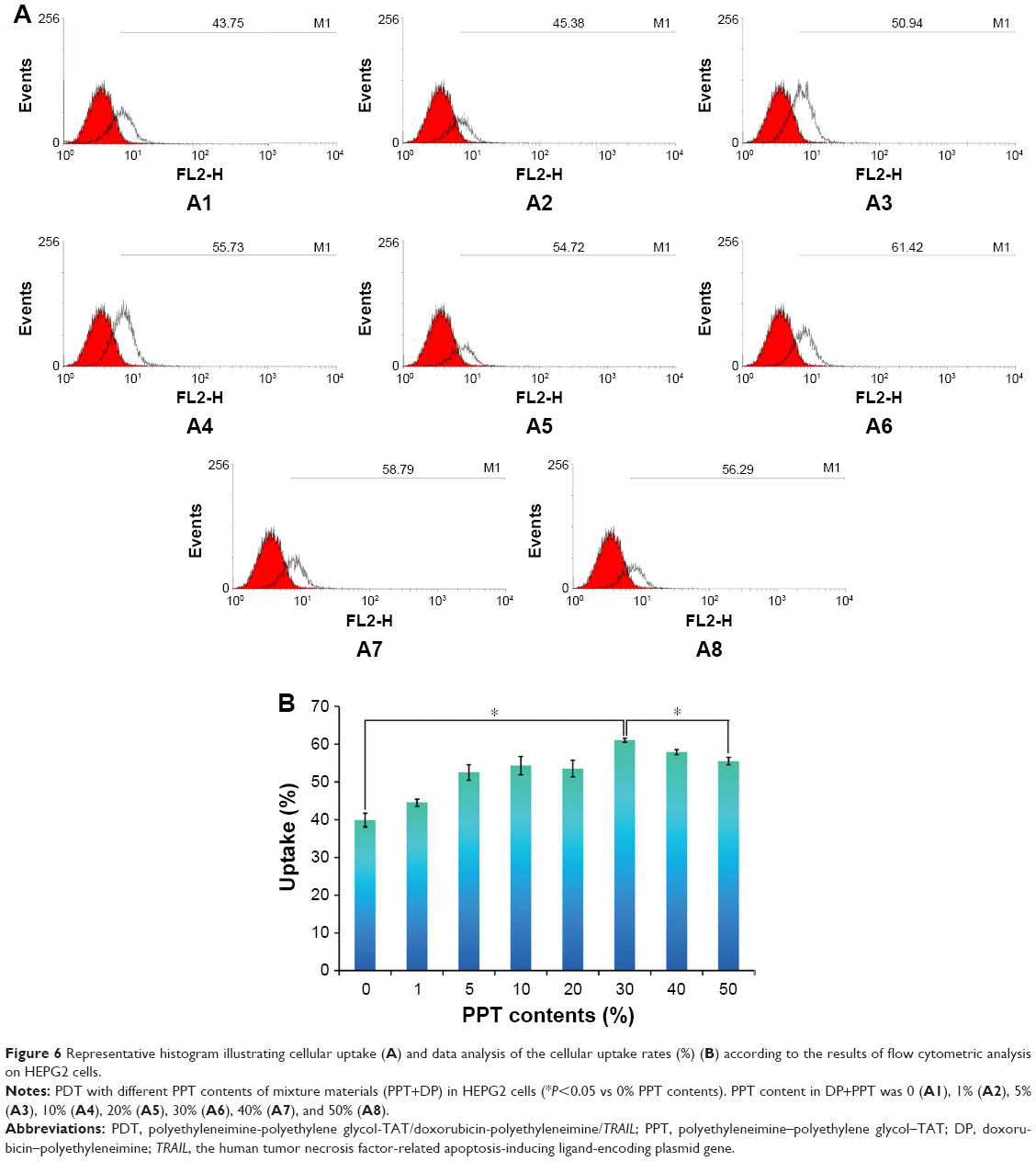 | Figure 6 Representative histogram illustrating cellular uptake (A) and data analysis of the cellular uptake rates (%) (B) according to the results of flow cytometric analysis on HEPG2 cells. |
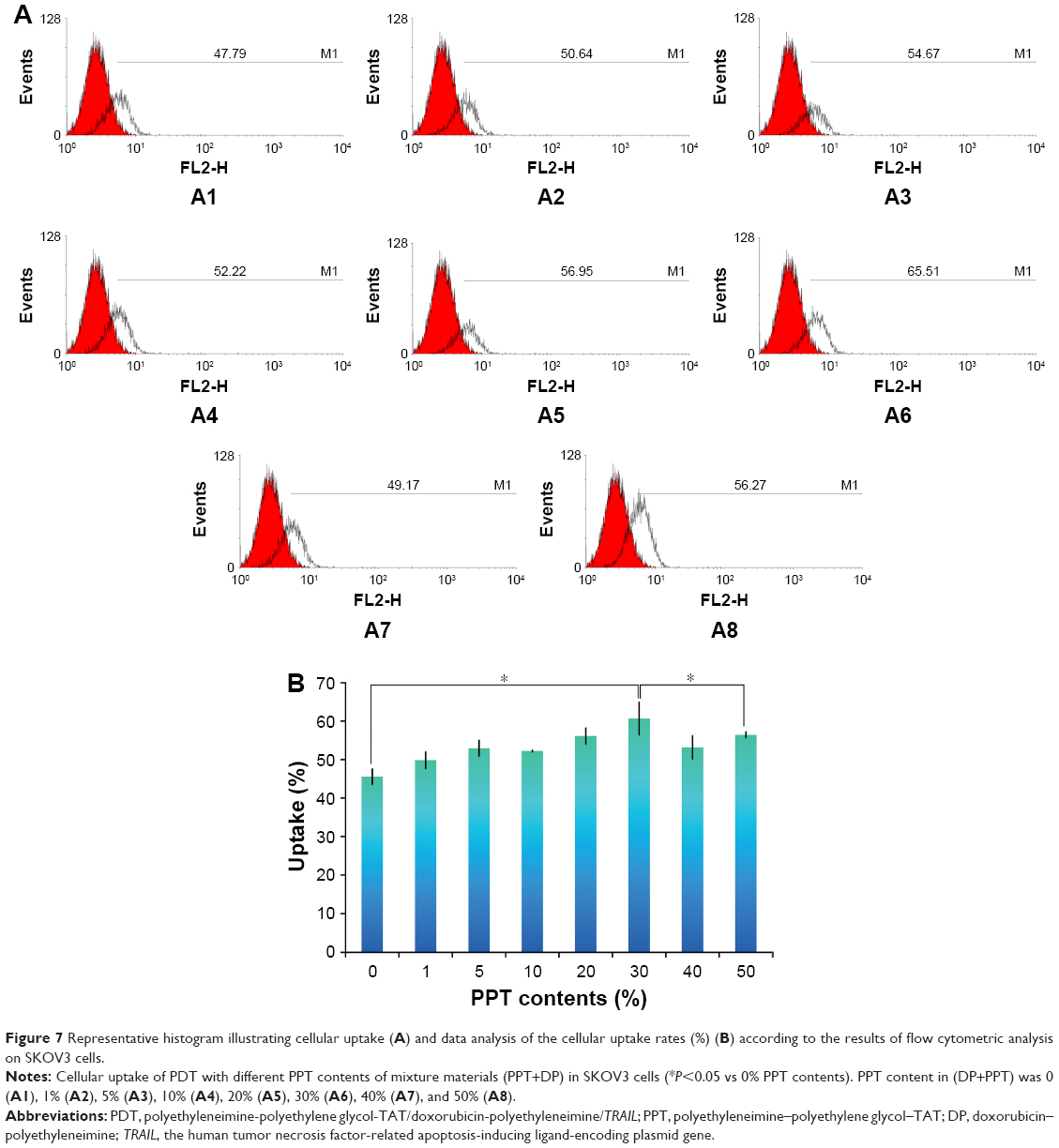 | Figure 7 Representative histogram illustrating cellular uptake (A) and data analysis of the cellular uptake rates (%) (B) according to the results of flow cytometric analysis on SKOV3 cells. |
As shown in Figure 8, inverted fluorescence microscopy was consistent with cell-uptake analysis through flow cytometry: the intensity of Dox enhanced with PPT contents in PDT increased from 0 to 30%. When PPT contents in PDT increased from 40% to 50%, the intensity of Dox dropped compared to PDT with 30% PPT contents. As shown in Figure 9, the results showed that pDNA was able to be transcribed well with DP-PPT-pDNA.
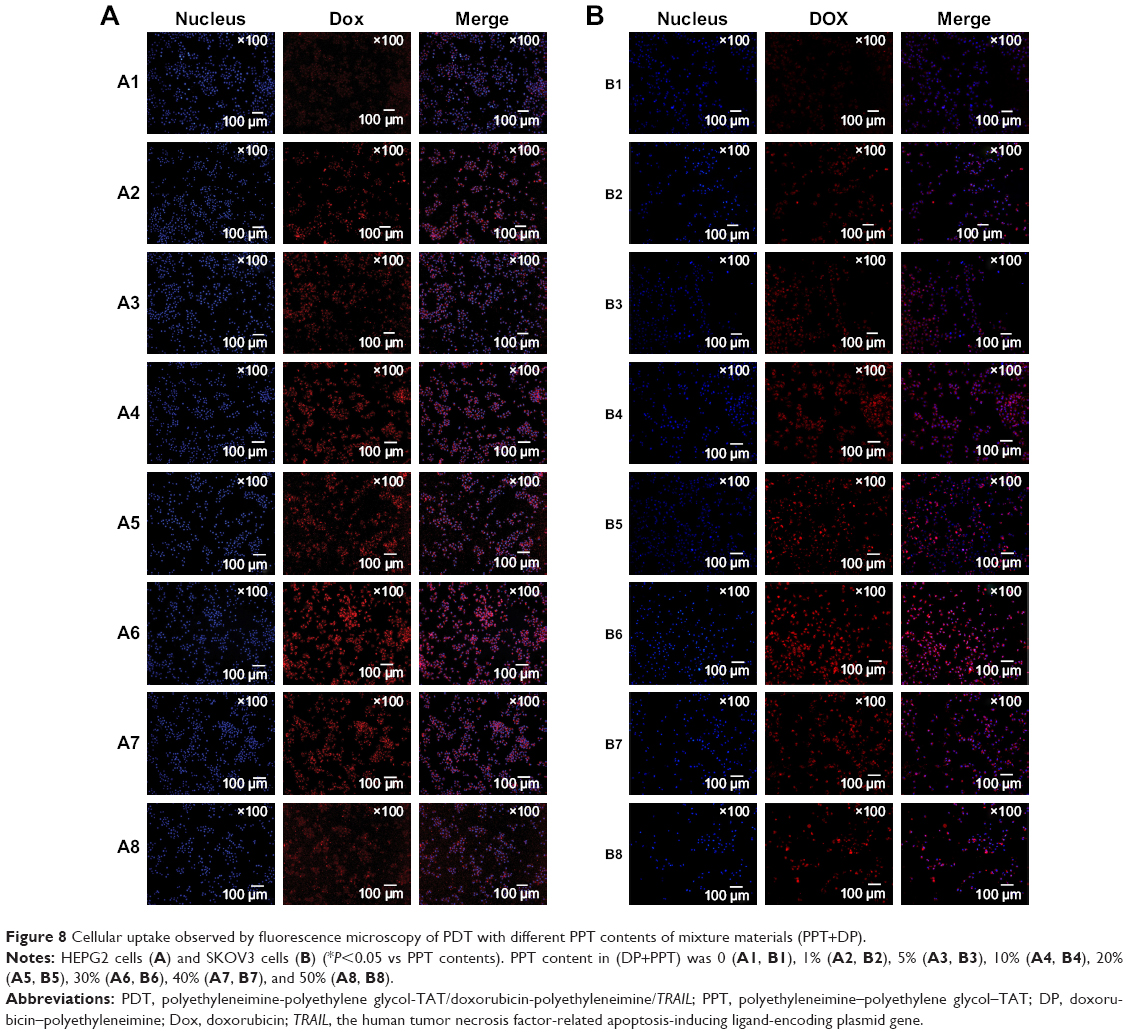 | Figure 8 Cellular uptake observed by fluorescence microscopy of PDT with different PPT contents of mixture materials (PPT+DP). |
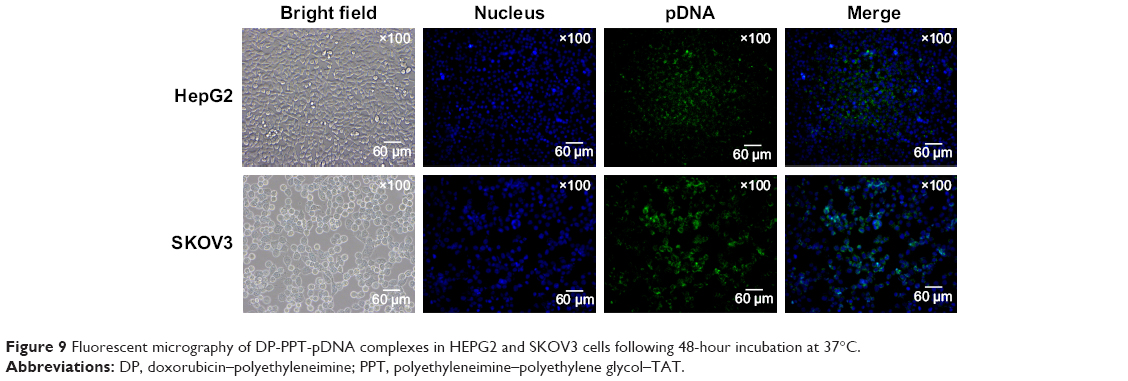 | Figure 9 Fluorescent micrography of DP-PPT-pDNA complexes in HEPG2 and SKOV3 cells following 48-hour incubation at 37°C. |
Characterization of DP, PDT, PCT, and PCD
The therapeutic effect of the nanocarriers was significantly affected by the physicochemical characteristics. Particle sizes of DT, PDT, PCT, and PCD were analyzed. As shown in Table 2, DT, PDT, PCT, and PCD had similar average sizes, and average particle size was about 80 nm. The ζ-potentials of DT, PDT, PCT, and PCD were 12.3±1.971 mV, 12.6±0.289 mV, 11±2.11 mV, and 9.33±1.44 mV, respectively. DT, PDT, PCT, and PCD were spherical under optimal conditions, as shown in Figure 10. Therefore, the subsequent evaluations (in vitro Western blot assay and in vitro cytotoxicity assay) would not have been affected by the similar physicochemical characteristics of the nanocarriers.
 | Table 2 Particle size and PDI of DT, PDT, PCT, and PCD |
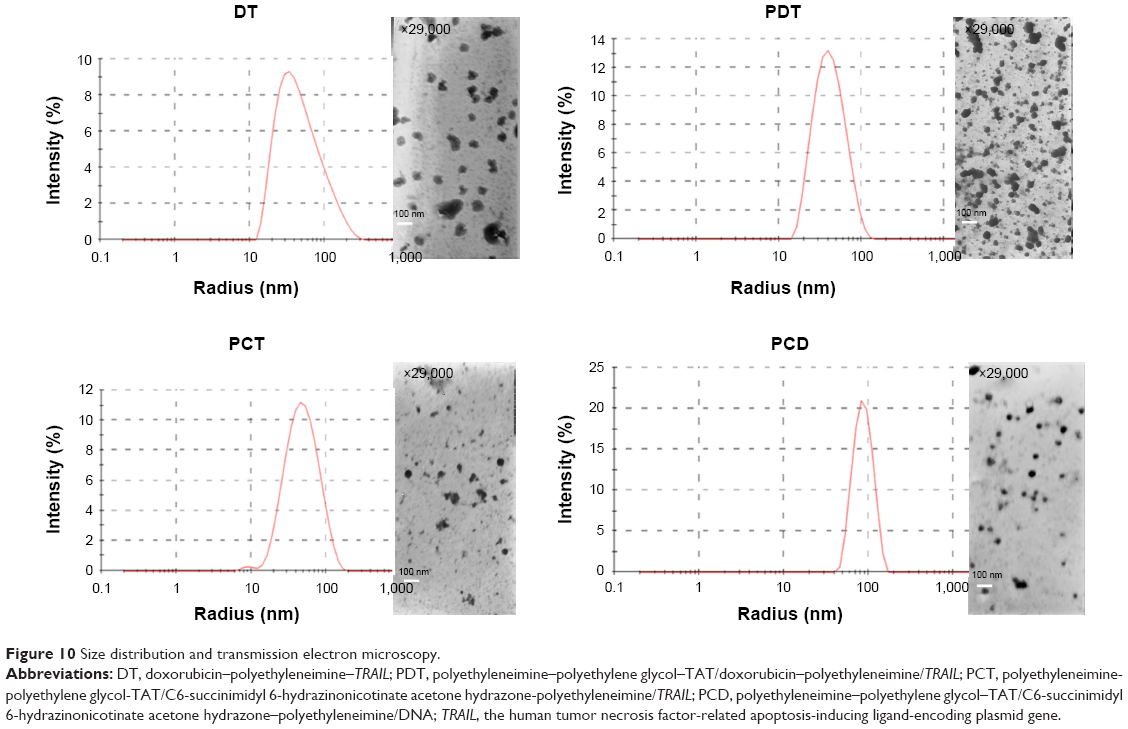 | Figure 10 Size distribution and transmission electron microscopy. |
Western blot assay of TRAIL
The Western blot assay was used to evaluate TRAIL-protein expression after HEPG2 cells had been transfected by PDT, which was the precondition of drug–gene synergistic effects. As shown in Figure 11, TRAIL expression in HEPG2 cells after treatment with PDT, PCT, PCD, and Dox was evaluated by Western blot assay. Among the four groups, PCD represented PPT/C6-SANH-PEI/pDNA and was a blank nanocarrier without Dox/TRAIL loaded. There was no TRAIL transcription in HEPG2 cells incubated with PCD. PCD was selected as the negative control and Lipofectamine 2000/TRAIL selected as positive control. Compared with PCD, TRAIL-protein expression was upregulated by PDT and PCT compared with PCD, while TRAIL expression in HEPG2 cells was not upregulated by free Dox or TRAIL. These results indicated that the PDT can be successfully transfected in HEPG2 cells and express TRAIL protein. In this case, the internalized PDT would effectively exert gene-therapy effects. However, the TRAIL expression of Lipofectamine 2000/TRAIL in HEPG2 cells was higher than PDT. For this reason, more efforts should be made to enhance the transfection efficiency of PDT in future research.
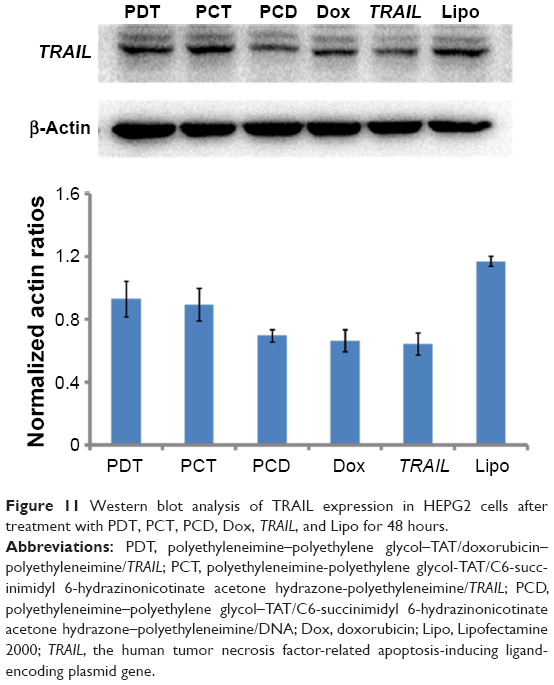 | Figure 11 Western blot analysis of TRAIL expression in HEPG2 cells after treatment with PDT, PCT, PCD, Dox, TRAIL, and Lipo for 48 hours. |
In vitro cytotoxicity assay
The in vitro cytotoxicity assay was used to evaluate the in vitro synergistic antitumor effects of PDT. As shown in Figure 12 and Table 3, the antitumor effect of PDT was enhanced compared to free Dox and PCT in both HEPG2 and SKOV3 cells (P<0.05). These results indicated that enhanced antitumor effects were achieved by PDT. After Dox and TRAIL had been delivered to tumor cells, the cells would be killed by Dox and the expression of TRAIL. Therefore, synergistic antitumor effects of PDT were achieved. Furthermore, after HEPG2 and SKOV3 cells had been incubated with PCD for 48 hours, cell viability was above 80% at different concentrations. This result indicated that the blank nanocarriers PCD did not have obvious cytotoxicity in HEPG2 or SKOV3 cells.
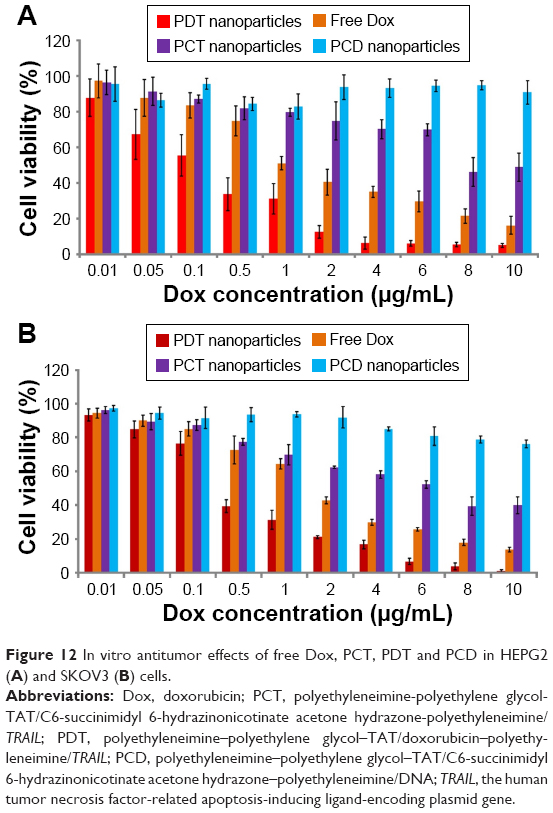 | Figure 12 In vitro antitumor effects of free Dox, PCT, PDT and PCD in HEPG2 (A) and SKOV3 (B) cells. |
 | Table 3 IC50 of HEPG2 and SKOV3 cells incubated with free Dox, PCT, PDT, and PCD at 48 hours |
In vivo antitumor evaluation
The antitumor efficacy of mPDT was assessed on H22 tumor-bearing BALB/c mice. Changes in body weight and tumor volumes in the mice are shown in Figure 13. After 12 days of treatment, the average tumor volume of mice treated with NS reached about 550 mm3 (Figure 13B), which was larger than that for the Dox (P<0.05), mPCT (P<0.05), and mPDT groups (P<0.01). For the mPDT group, average tumor volume was about 200 mm3, which was smaller than both the Dox group (P<0.05) and the mPCT group (P<0.05). Moreover, Figure 13C shows a direct visual representation of the tumor-suppression effect, in accordance with Figure 13B. These results indicated that mPDT had a stronger tumor-inhibition effect than Dox and mPCT, and a synergistic antitumor effect of Dox and TRAIL was achieved by mPDT.
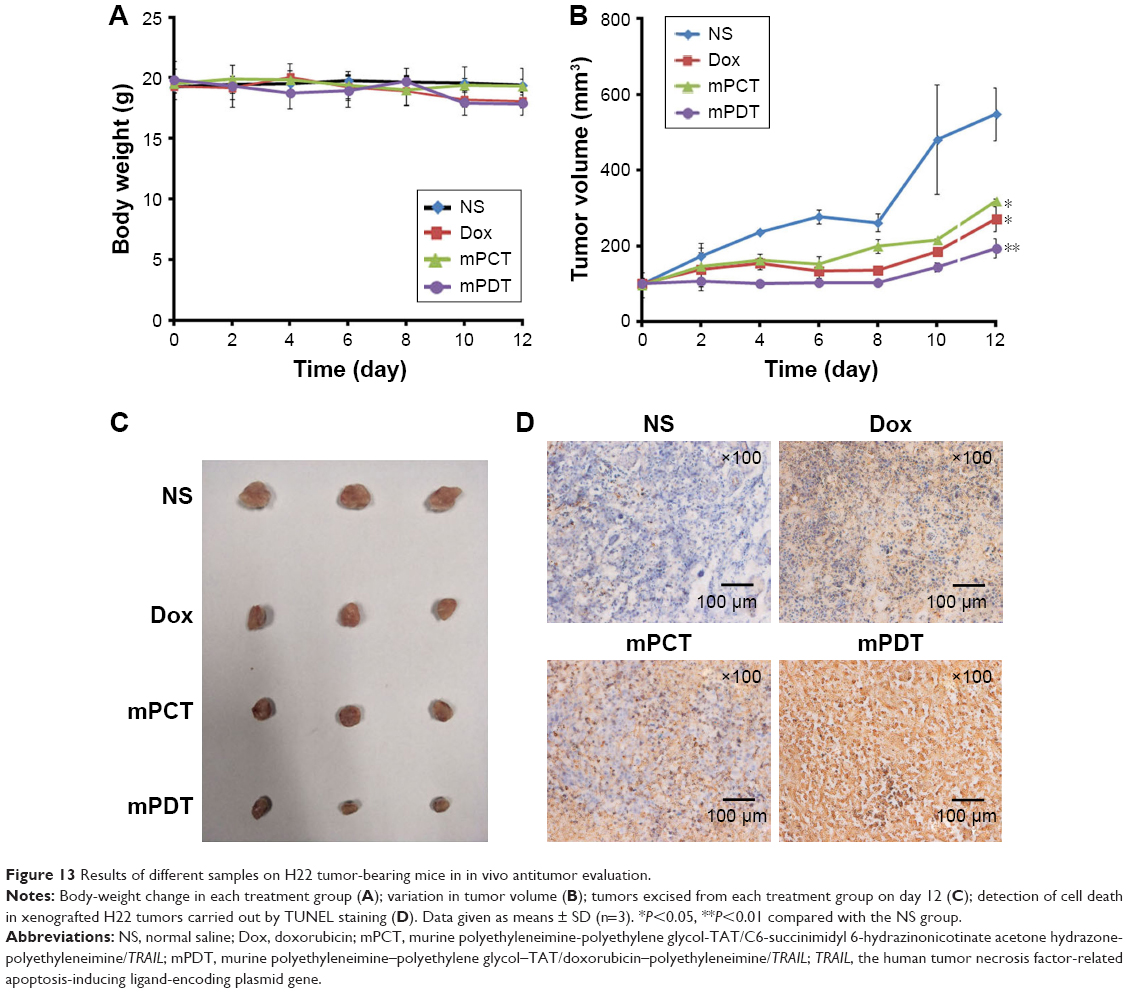 | Figure 13 Results of different samples on H22 tumor-bearing mice in in vivo antitumor evaluation. |
Body-weight variations were monitored to monitor the adverse effects of various formulations. As shown in Figure 13A, no obvious body-weight loss induced by Dox, mPCT, or mPDT was observed in comparison with the NS group (P>0.05), which indicated that body-weight loss caused by systemic toxicities was not revealed in this test.
We next investigated the effect of treatments on apoptosis in vivo by TUNEL staining of paraffin-embedded sections of the H22-xenografted tumors. As shown in Figure 13D, there were greater degrees of apoptosis in the mPDT, Dox, and mPCT groups compared with the NS group. Either Dox or mPCT caused a modest increase in the number of TUNEL-positive cells (brown color) compared with the NS group. However, mPDT dramatically increased the number of TUNEL-positive cells compared with the Dox and mPCT groups. These results indicated a significant increase in cell death in the tumors in the mPDT group compared with the Dox and mPCT groups. Taken together, these data further identify that a synergistic antitumor effect of Dox and TRAIL can be achieved by mPDT.
Hemolysis assessment
To determine whether the gene–drug codelivery nanocarriers were safe for IV injection, hemolytic properties were evaluated. No hemolysis or aggregation was observed for DT, PDT, PCT, or PDT after centrifugation. As shown in Table 4, hemolysis rates of DT, PDT, PCT, and PDT were less than 5%. Generally, a hemolysis percentage less than 5% is regarded as nontoxic and safe, so the hemolytic activities of DT, PDT, PCT, and PDT were negligible (<5%). These results indicated that the prepared DT, PDT, PCT, and PDT had good hemocompatibility during the preliminary safety evaluation, and were suitable for IV administration.
 | Table 4 Hemolysis rates of DT, PDT, PCT and PCD |
Conclusion
In conclusion, Dox–TRAIL coloaded PDT based on one-step assembly was developed to achieve synergistic antitumor effects. The intracellular cationic pH-sensitive cellular assistant PPT and DP, with similar consistency condensation ability, were mixed along to condense the TRAIL by one-step assembly, resulting in the PDT obtained. The cellular uptake of PDT was enhanced with increased PPT contents in PDT (P<0.05), which verified the intracellular uptake-assistant ability of PPT. TRAIL-protein expression was upregulated by PDT when compared with free TRAIL in Western blot assays. Furthermore, the in vitro antitumor effect of PDT was significantly enhanced compared to free Dox and PCT in both HEPG2 and SKOV3 cells (P<0.01). mPDT had stronger tumor-inhibition effects than Dox (P<0.05) and mPCT (P<0.05) in vivo, which showed that the synergistic antitumor effect of Dox and TRAIL was achieved by mPDT. Therefore, the in vivo synergistic antitumor effect of PDT was identified indirectly. It can thus be said that Dox–TRAIL coloaded PDT holds promising potential as a multifunctional drug–gene codelivery nanocarrier for combination tumor therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81402867) and Joint Research Fund for Overseas Chinese, Hong Kong, and Macao Young Scholars of National Natural Science Foundation of China (81628014). The authors are thankful for English grammar review by Livesey David Olerile.
Disclosure
The authors report no conflicts of interest in this work.
References
Nastiuk KL, Krolewski JJ. Opportunities and challenges in combination gene cancer therapy. Adv Drug Deliv Rev. 2016;98:35–40. | ||
Morris LG, Chan TA. Therapeutic targeting of tumor suppressor genes. Cancer. 2015;121:1357–1368. | ||
Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016;240:287–301. | ||
Yan CY, Gu JW, Hou DP, et al. Synthesis of Tat tagged and folate modified N-succinyl-chitosan self-assembly nanoparticles as a novel gene vector. Int J Biol Macromol. 2015;72:751–756. | ||
Ambattu LA, Rekha MR. Betaine conjugated cationic pullulan as effective gene carrier. Int J Biol Macromol. 2015;72:819–826. | ||
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. | ||
Kemp JA, Shim MS, Heo CY, Kwon YJ. “Combo” nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Deliv Rev. 2016;98:3–18. | ||
Shi X, Li C, Gao S, et al. Combination of doxorubicin-based chemotherapy and polyethylenimine/p53 gene therapy for the treatment of lung cancer using porous PLGA microparticles. Colloids Surf B Biointerfaces. 2014;122:498–504. | ||
Guo L, Fan L, Pang Z, et al. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J Control Release. 2011;154:93–102. | ||
He CL, Tang ZH, Tian HY, Chen X. Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev. 2015;98:64–76. | ||
Jang B, Kwon H, Katila P, Lee SJ, Lee H. Dual delivery of biological therapeutics for multimodal and synergistic cancer therapies. Adv Drug Deliv Rev. 2016;98:113–133. | ||
Guo L, Fan L, Ren J, et al. A novel combination of TRAIL and doxorubicin enhances antitumor effect based on passive tumor-targeting of liposomes. Nanotechnology. 2011;22:265105. | ||
Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–5400. | ||
Yu X, Zhang B, Wang T, et al. Two-stage pH-sensitive doxorubicin hydrochloride loaded core-shell nanoparticles with dual drug-loading strategies for the potential anti-tumor treatment. RSC Adv. 2016;6:104049–104066. | ||
Modrak DE, Cardillo TM, Newsome GA, Goldenberg DM, Gold DV. Synergistic interaction between sphingomyelin and gemcitabine potentiates ceramide-mediated apoptosis in pancreatic cancer. Cancer Res. 2004;64:8405–8410. | ||
Dumitru CA, Gulbins E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene. 2006;25:5612–5625. | ||
Dumitru CA, Carpinteiro A, Trarbach T, Hengge UR, Gulbins E. Doxorubicin enhances TRAIL-induced cell death via ceramide-enriched membrane platforms. Apoptosis. 2007;12:1533–1541. | ||
Vitovski S, Chantry AD, Lawson MA, Croucher PI. Targeting tumour-initiating cells with TRAIL based combination therapy ensures complete and lasting eradication of multiple myeloma tumours in vivo. PLoS One. 2012;7:e35830. | ||
Jiang TY, Mo R, Bellotti A, Zhou JP, Gu Z. Gel-liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24:2295–2304. | ||
Hu CL, Gu FF, Tai ZG, et al. Synergistic effect of reduced polypeptide micelle for co-delivery of doxorubicin and TRAIL against drug-resistance in breast cancer. Oncotarget. 2016;7:61832–61844. | ||
Jiang HH, Kim TH, Lee S, Chen X, Youn YS, Lee KC. PEGylated TNF-related apoptosis-inducing ligand (TRAIL) for effective tumor combination therapy. Biomaterials. 2011;32:8529–8537. | ||
Huang W, Chen LQ, Kang L, Jin MJ, Sun P, Xin X, Gao ZG, Bae HY. Nanomedicine-based combination anticancer therapy between nucleic acids and small-molecular drugs. Adv Drug Deliv Rev. 2017;115:82–97. | ||
Yhee JY, Son S, Lee H, Kim K. Nanoparticle-based combination therapy for cancer treatment. Curr Pharm Des. 2015;21:3158–3166. | ||
Oh B, Han J, Choi E, Tan X, Lee M. Peptide micelle-mediated delivery of tissue-specific suicide gene and combined therapy with Avastin in a glioblastoma model. J Pharm Sci. 2015;104:1461–1469. | ||
Wang SP, Zhang JM, Wang YT, Chen MW. Hyaluronic acid-coated PEI-PLGA nanoparticles mediated co-delivery of doxorubicin and miR-542-3p for triple negative breast cancer therapy. Nanomedicine. 2016;12:411–420. | ||
Xu Q, Xia Y, Wang CH, Pack DW. Monodisperse double-walled microspheres loaded with chitosan-p53 nanoparticles and doxorubicin for combined gene therapy and chemotherapy. J Control Release. 2012;163:130–135. | ||
Liu C, Liu F, Feng L, Li M, Zhang J, Zhang N. The targeted co-delivery of DNA and doxorubicin to tumor cells via multifunctional PEI-PEG based nanoparticles. Biomaterials. 2013;34:2547–2564. | ||
Lim HJ, Kim JK, Park JS. Complexation of apoptotic genes with polyethyleneimine (PEI)-coated poly-(DL)-lactic-co-glycolic acid nanoparticles for cancer cell apoptosis. J Biomed Nanotechnol. 2015;11:211–225. | ||
Yang X, Kim JC. β-Cyclodextrin grafted polyethyleneimine hydrogel immobilizing hydrophobically modified glucose oxidase. Int J Biol Macromol. 2011;48:661–666. | ||
Yang HN, Park JS, Jeon SY, Park KH. Carboxymethylcellulose (CMC) formed nanogels with branched poly(ethyleneimine) (bPEI) for inhibition of cytotoxicity in human MSCs as a gene delivery vehicles [sic]. Carbohydr Polym. 2015;122:265–275. | ||
Sun Q, Sun X, Ma X, et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26:7615–7621. | ||
Han X, Li Z, Sun J, et al. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin delivery: probing the effect of uncovalent PEGylation degree on cellular uptake and blood long circulation. J Control Release. 2015;197:29–40. | ||
Li Y, Xu B, Bai T, Liu W. Co-delivery of doxorubicin and tumor-suppressing p53 gene using a POSS-based star-shaped polymer for cancer therapy. Biomaterials. 2015;55:12–23. | ||
Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov. 2015;14:239–247. | ||
Han SS, Li ZY, Zhu JY, et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small. 2015;11:2543–2554. | ||
Boisguérin P, Deshayes S, Gait MJ, et al. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv Drug Deliv Rev. 2015;87:52–67. | ||
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273. | ||
Peng LH, Niu J, Zhang CZ, et al. TAT conjugated cationic noble metal nanoparticles for gene delivery to epidermal stem cells. Biomaterials. 2014;35:5605–5618. | ||
Yilmaz MD, Xue M, Ambrogio MW, et al. Sugar and pH dual-responsive mesoporous silica nanocontainers based on competitive binding mechanisms. Nanoscale. 2015;7:1067–1072. | ||
Wang C, Zhao T, Li Y, Huang G, White MA, Gao J. Investigation of endosome and lysosome biology by ultra pH-sensitive nanoprobes. Adv Drug Deliv Rev. 2017;113:87–96. | ||
Kim J, Kim H, Kim WJ. Single-layered MoS2-PEI-PEG nanocomposite-mediated gene delivery controlled by photo and redox stimuli. Small. 2016;12:1184–1192. | ||
Kim H, Okamoto H, Felber AE, et al. Polymer-coated pH-responsive high-density lipoproteins. J Control Release. 2016;228:132–140. | ||
Wang MF, Liu TX, Han LQ, Gao WW, Yang SM, Zhang N. Functionalized O-carboxymethyl-chitosan/polyethylenimine based novel dual pH-responsive nanocarriers for controlled co-delivery of DOX and genes. Polym Chem. 2015;6:3324–3335. | ||
Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–2014. | ||
Charette N, Saeger CD, Horsmans Y, Leclercq I, Stärkel P. Salirasib sensitizes hepatocarcinoma cells to TRAIL-induced apoptosis through DR5 and survivin-dependent mechanisms. Cell Death Dis. 2013;4:e471. | ||
Fan H, Hu QD, Xu FJ, Liang WQ, Tang GP, Yang WT. In vivo treatment of tumors using host-guest conjugated nanoparticles functionalized with doxorubicin and therapeutic gene pTRAIL. Biomaterials. 2012;33:1428–1436. | ||
Lu JZ, Zhang L, Xie F, et al. Mild oxidative stress induced by a low dose of cisplatin contributes to the escape of TRAIL-mediated apoptosis in the ovarian cancer SKOV3 cell line. Oncol Rep. 2016;35:3427–3434. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.