Back to Archived Journals » Biosimilars » Volume 4
Multidisciplinary approach to evaluating immunogenicity of biosimilars: lessons learnt and open questions based on 10 years' experience of the European Union regulatory pathway
Authors Chamberlain P
Received 18 February 2014
Accepted for publication 3 April 2014
Published 25 June 2014 Volume 2014:4 Pages 23—43
DOI https://doi.org/10.2147/BS.S50012
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Paul D Chamberlain
NDA Advisory Board, NDA Advisory Services Ltd, Surrey, UK
Abstract: Clinical evaluation of comparative immunogenicity represents an important component of the European Union regulatory review process for candidate biosimilar products. The clinical evaluation is part of a multidisciplinary review that cross-refers to product quality attributes as well as preclinical and ongoing risk management considerations. Results from the monitoring of anti-drug antibody formation in relevant populations treated for an adequate period of time are interpreted in relation to clinically relevant endpoints, including pharmacokinetics, pharmacodynamics, efficacy, and safety parameters. The European Union regulatory standard for designation of biosimilarity requires a suitable weight of evidence, determined on a product-specific basis, to demonstrate that the immunogenicity associated with the biosimilar product does not lead to a higher negative impact on clinically relevant outcomes compared with the reference product. The experience gained during the 10-year period following the implementation of the European Union biosimilars pathway indicates that a suitably cautious approach was applied, insofar as no immunogenicity-related issues have emerged for the approved applications of the different biosimilar products. In some cases, product quality-related issues were identified in the preauthorization setting as being potentially relevant for heightened risk of immunogenicity and were duly taken into account for the biosimilarity decision. Some unresolved issues remain, most notably concerning the limitation of noninterventional post-marketing surveillance measures to monitor the potential for changes in immunogenicity over the longer term, eg, following introduction of changes in manufacture, formulation, or primary product container. Lack of standardization of bioanalytical methods precludes comparison of anti-drug antibody formation for different products that are evaluated in noncomparative clinical studies, and correlation with relevant clinical parameters is also lacking.
Keywords: immunogenicity, biosimilar, regulatory, clinical, preclinical, bioanalytical
Introduction
The purpose of this paper is to review the experience gained in the European Union (EU) regulatory system for evaluation of undesirable immunogenicity of candidate biosimilar products, within the context of the overall scientific assessment required for registration on the basis of Article 10(4) of Directive 2001/83/EC (“Similar Biological Medicinal Products”). This legal pathway was established in 2004, enabling the authorization between 2006 and December 2013 of biosimilar versions of five different reference products, ie, human growth hormone, filgrastim, erythropoietin-alfa, infliximab, and follitropin-alfa. In addition to the approved products, applications for three candidate biosimilar insulin products were withdrawn during the review procedure, and one negative opinion was given in respect of a candidate biosimilar interferon-alfa product.1 Table 1 summarizes the candidate and approved biosimilar products considered in this paper.
 | Table 1 Biosimilar candidates reviewed for marketing authorization in the European Union to the end of 2013 |
The effectiveness of the applied regulatory standards for managing the potential unfavorable effects associated with undesirable immunogenicity of these products is reviewed in a multidisciplinary manner, taking into account product quality, bioanalytical, preclinical, clinical, and post-authorization risk management aspects.
The impact of immunogenicity-related risks on overall benefit and risk is also considered in relation to extrapolation to therapeutic indications not studied in the preauthorization phase, as well as interchangeability of biosimilar and innovator versions.
Regulatory guidance
The most relevant reference points for guiding the design of the immunogenicity evaluation of candidate biosimilar products for registration in the EU are summarized in Table 2. In addition to the primary (often referred to as “overarching”) guidelines for biosimilar products, product-specific guidelines contain additional recommendations for evaluation of immunogenicity; these should be consulted for advice on the extent of the clinical immunogenicity evaluation required for different product types.
 | Table 2 European Union regulatory guidelines relevant for immunogenicity risk assessment of candidate biosimilar products* |
It is important to emphasize that these guidelines are not legally binding; an applicant may elect to apply alternative approaches, subject to adequate scientific justification. This is particularly the case for selection of bioanalytical methodology, ie, the choice of technical format for pharmacokinetic and anti-drug antibody (ADA) assays is the responsibility of the applicant, although the validation of suitability should comply with prevailing regulatory standards. Regulatory guidelines are dynamic, ie, are updated on a regular basis to reflect the experience gained. For example, as discussed later in the section Nonclinical immunogenicity evaluation, the role of immunogenicity evaluation in nonclinical studies has been moderated in the more recent EU biosimilar guidelines.2
Product quality-related issues
General considerations
Biosimilar candidates for registration in the EU should always have 100% identical amino acid sequence to the reference product.3 In general, the post-translational modification profile should be within the range demonstrated for the reference product, although there may be exceptions in the case of identified safety risks for the originator (eg, for cetuximab). Designation of biosimilarity is based on a highly rigorous demonstration of comparability of product attributes, taking into account variables that could influence undesirable immunogenicity. From the immunogenicity perspective, the requirement for 100% amino acid sequence identity implies that the biosimilar candidates would be expected to contain the same T-cell epitopes as the reference product, but that differences in immunogenic potential could be related to differences in:
- conformational structures, eg, multimers or aggregates of the therapeutic protein that alter recognition by B-cell receptors and/or uptake by antigen-presenting cells, or are able to stimulate immune effector cells directly
- post-translational glycosylation or PEGylation, involving structures that can be recognized by pre-existing antibodies in sensitized individuals or might alter antigen processing, binding to major histocompatibility complex, or recognition by B-lymphocytes or T-lymphocytes
- process-related impurities, eg, Escherichia coli-derived proteins or yeast beta-glucans or endotoxins, which could stimulate innate immune cells to modify the adaptive immune response to the therapeutic protein.
Aggregates and subvisible particles
Recent work has revealed that aggregation of therapeutic proteins influences antigen uptake, processing, and presentation via a mass action mechanism in addition to activating antigen-presenting cells.4
The presence of tungsten residues in prefilled syringes has been cited as a risk factor for enhanced immunogenicity of therapeutic proteins, being associated with detection of increased levels of product aggregates.5 This was implicated as a causal factor for induction of neutralizing antibodies in two patients with chronic kidney disease following subcutaneous administration of a biosimilar erythropoietin product.6,7
Issues for erythropoietin
In preauthorization studies of Binocrit® and Silapo® using intravenous administration, there was no difference detected in clinical immunogenicity of the biosimilar candidates in direct comparison with the reference product. Since it was not possible to perform a directly comparative clinical comparison of subcutaneously administered Binocrit or Silapo versus the reference product, Erypo®, at the time of the initial Phase III nephrology studies (because administration of the reference product by the subcutaneous route had been temporarily contraindicated due to an elevated incidence of antibody-mediated pure red cell aplasia consequent to a change in product formulation), the initial marketing authorizations for Binocrit and Silapo in patients with chronic kidney disease were restricted to intravenous administration.
Phase III studies comparing Binocrit or Silapo versus the reference product were then performed at a later date. Therapeutic equivalence and comparable safety and immunogenicity were demonstrated for Silapo in direct comparison with Erypo, resulting in the approval of Silapo for subcutaneous as well as intravenous administration in the chronic kidney disease population. However, the sponsor voluntarily halted the first Phase III study comparing subcutaneous administration of Binocrit versus Erypo following the detection of neutralizing anti-erythropoietin antibodies in two subjects.6 Pure red cell aplasia was diagnosed in one of these subjects; this subject was an 86-year-old male who had a number of comorbidities, a history of food allergy, and an interrupted treatment pattern in the Phase III study. The sponsor performed a thorough follow-up investigation that revealed a potential risk factor for a heightened immune response to the therapeutic protein, ie, an association with residual tungsten in the prefilled syringes was suggested as a possible causal factor for induction of the detected neutralizing antibodies.7 A follow-up Phase III study using low-tungsten prefilled syringes is ongoing.
It is relevant to note that cases of pure red cell aplasia, featuring the development of antibodies that react with, and neutralize the biological activity of, the endogenous cytokine are detected at very low frequency in subjects treated with different recombinant erythropoietin products.8
Differences in excipient composition could influence immunogenicity indirectly by modification of the solubility of a therapeutic protein, potentially to increase levels of subvisible particles. For example, trehalose may reduce the propensity of certain therapeutic monoclonal antibodies to form aggregates in solution: replacement of trehalose with an alternative sugar, eg, sucrose, might result in an increased risk of aggregation that enhances immunogenicity, making it advisable to generate comparative stability data using worst-case storage conditions of the drug product formulated with different excipients.9
Post-translational glycosylation
The EU regulatory pathway for biosimilars permitted the use of a different host cell line for expression of a nonglycosylated therapeutic protein, namely growth hormone (Valtropin® [BioPartners GmbH, Reutlingen, Germany]), but acceptability for glycosylated proteins is equivocal.10 As a general rule, for a glycoprotein biosimilar candidate, the host cell substrate should be as similar as possible to that used for expression of the reference product, reflecting the influence of the host cell on post-translational glycosylation profile. For cetuximab, where the particular nonhuman glycosylation profile associated with the SP2/0 host cell line used for the originator was implicated in the induction of serious systemic hypersensitivity reaction,11 there would be a justifiable case to use an alternative cell line (eg, CHO) that enables expression of cetuximab bearing negligible levels of the problematic galactose-α-1,3-galactose carbohydrate in the Fab domain.12
Even in the case that a similar host cell line were used, it is possible that there could be detectable differences in levels of the potentially nonhuman antigenic glycan, N-glycolyl neuraminic acid (Neu5Gc), that could result in an altered propensity for binding to pre-existing antibodies in human subjects.13 This issue was raised during the review of the marketing authorization application for a human follitropin-alfa expressed in CHO cells (Ovaleap® [Teva Pharma B.V., Utrecht, the Netherlands]), which was found to have slightly higher levels of Neu5Gc compared with the reference product.14 The risk was mitigated by quantitative analysis of Neu5Gc by high-performance anion-exchange chromatography with pulsed amperometric detection, measurement of pre-existing Neu5Gc-reactive antibodies in the clinical trial population (incidence found to be 18%), and demonstration that baseline status for Neu5Gc-reactive antibodies did not impact treatment outcomes in direct comparison with the reference product.
Process-related impurities
Elevated levels of host cell-derived protein impurities were associated with detection of treatment-emergent antibodies reactive with these impurities, in association with increased levels of antibodies to the therapeutic protein, in the case of an investigational formulation of Omnitrope® (Sandoz GmbH, Kundl, Austria).15
Issues for growth hormone
A biosimilar growth hormone candidate manufactured using an early version of the manufacturing process induced antibodies that reacted to the Escherichia coli-derived protein impurity in association with a reported increase in antibodies reactive with the human growth hormone product.15 The purification process was modified in addition to improving the performance of the assay used to measure the levels of the contaminating host cell protein. Anti-growth hormone antibody levels were then found to decline in subjects who started treatment on the earlier version of the product but who continued therapy with the drug product from the improved process (Table 3). Most notably, for subjects receiving the commercial grade product (EP2K-02-PhIII-Lyo study), none developed anti-growth hormone antibodies and only one subject had a positive result for anti-E. coli protein antibodies at the 12-month treatment time point.
 | Table 3 Preauthorization clinical studies providing immunogenicity data* |
Clinical evaluation
General considerations
Regulatory guidelines in the EU require evidence that the immunogenicity of the biosimilar candidate does not have a higher negative clinical impact in direct comparison with the reference product. It is important to note that the arbiter is clinical impact, and not the incidence or magnitude of detected levels of ADA per se. Also, it is acceptable for a biosimilar candidate to be less immunogenic than the reference provided, provided that the two product versions are demonstrated to have equivalent efficacy and posology.2
Because safety may not extrapolate across different therapeutic indications in the same manner as efficacy, additional safety data might be required in particular therapeutic indications/patient populations that were not evaluated in the main therapeutic equivalence studies. This consideration may be relevant from the immunogenicity perspective as discussed below.
The clinical studies should be designed for an adequate duration to demonstrate differences, if these were to occur, in clinically significant immunogenicity. Clearly, the experience gained for the reference product would be most relevant. In general, for products administered in a chronic basis, it is advisable to provide 12-month continuous exposure data to support registration.2 For products administered in a cyclical manner, samples for ADA testing should be collected prior to the first treatment cycle, then prior to one or more (depending on identified immunogenicity risk level for reference product) subsequent cycles, and finally at 4–6 weeks following the last treatment.
In cases where it may be difficult to detect differences in low incidence adverse effects, heightened monitoring in the post-authorization setting may be required to compensate for the uncertainty at the time of registration.2 Indeed, as discussed below, immunogenicity has featured in the risk management plan for all of the biosimilar products approved to date in the EU.
Weight of evidence
The extent of the clinical data submitted in support of approval, as reported in the European public assessment reports1 and other information in the public domain, is summarized in Table 3. Since the assessment reports do not provide a fully comprehensive picture of the data submitted, Table 3 is not complete with respect to the full weight of evidence actually submitted to the agency.
The biosimilarity assessment for the products reviewed to date by EU regulatory agencies has always included directly comparative clinical evidence of immunogenicity of the biosimilar candidate versus the reference product in at least one therapeutic indication. Notable features for the clinical immunogenicity evaluation of these biosimilar candidates are summarized in the next paragraph.
Main features of immunogenicity review for biosimilars in the European Union
- Immunogenicity was assessed in a descriptive manner by interpreting results of anti-drug antibody testing relative to clinical outcomes, including pharmacokinetics/pharmacodynamics (filgrastim, epoetin, infliximab), efficacy (all products), and safety (all products)
- The clinical indication used for demonstration of therapeutic equivalence also provided the main evidence for assessment of comparative immunogenicity
- Where the product was to be used in settings with a potentially different immunogenicity risk profile (subcutaneous administration of epoetin in renal disease or oncology subjects), it was necessary to provide additional preauthorization immunogenicity data to rule out heightened risk
- For chronic administration products, immunogenicity monitoring for a 12-month period of continuous treatment (no switching) was required
- Anti-drug antibody sampling enabled characterization of the timing of antibody formation, as well as magnitude and neutralizing capacity of anti-drug antibody
- A study design involving immunogenicity monitoring in subjects switched between candidate biosimilar and reference products was not required in the preauthorization setting
- Follow-up, post-treatment monitoring of subjects treated in pivotal comparative clinical studies was required for products with identified immunogenicity-related risks (epoetin and growth hormone)
- Product quality differences were influential in the interpretation of anti-drug antibody test results (erythropoietin, growth hormone, follitropin-alfa)
- Preauthorization immunogenicity assessment of the candidate biosimilar was based on the drug product formulation/primary container combination intended for commercialization.
In the case of some therapeutic monoclonal antibodies, supportive data on relative immunogenicity may be obtained from a single-dose comparative pharmacokinetic study in healthy volunteers. Accordingly, it is recommended to include ADA monitoring at appropriate time points in the Phase I study.16
The agency raised a number of concerns relating to the marketing authorization applications for Alpheon® (BioPartner GmbH) and the insulins marketed by Marvel LifeSciences Ltd (Harrow, United Kingdom) (Solumarv®, Isomarv Medium®, and Combimarv®), as summarized in the following two paragraphs.
Issues for interferon-alfa
The refusal assessment report for Alpheon®17 referred to major concerns relating to uncertainties about product quality and clinical safety and efficacy. Specific concerns about the extent of immunogenicity evaluation were cited as contributing to the overall negative opinion. These included an incomplete validation of the specificity and dilutional linearity of the double-antigen enzyme-linked immunosorbent assay method used to monitor anti-drug antibodies to Alpheon compared with Roferon-A®. The commercial kit method, whilst acceptable in principle, had not been fully validated prior to the clinical sample analysis; and the retrospective validation was considered incomplete. The agency noted that false negatives were not adequately excluded, there was no reporting of antibody titers and there was no assay of neutralizing capacity of the detected anti-drug antibody. Thus, although the incidence of anti-drug antibody formation in the respective treatment groups was similar, the agency considered that bioanalytical limitations precluded a definitive conclusion on relative immunogenicity of Alpheon compared with Roferon-A.
Issues for insulin
Following an initial application in 2007, which was subsequently withdrawn, Marvel Life Sciences resubmitted Marketing Authorisation Applications for three biosimilar candidate insulin products in December 2011: Solumarv® (soluble rapid-acting insulin); Isomarv Medium® (intermediate-acting isophane insulin); Combimarv® (a mixture of the other two products, 70% long-acting isophane insulin + 30% soluble rapid-acting insulin). As a result of major concerns expressed by the agency, the applicant withdrew these applications in November 2012. The respective withdrawal assessment reports many observations and deficiencies for the three Marvel insulins18 catalog, including lack of demonstration of comparability of product attributes and pharmacodynamics. The analysis of clinical safety revealed a higher incidence of hypersensitivity reactions in subjects receiving the biosimilar insulin candidates; in addition, the Marvel insulins had lower efficacy during the 12–24-week treatment period compared with the reference product versions. Unfortunately, insufficient data were available to exclude a role for heightened immunogenicity in these clinical safety and efficacy observations.
Although the anti-drug antibody incidence was similar for the treatment groups, patients treated with Marvel insulins had higher mean anti-insulin antibody titer at week 28. The agency noted that differences in the product process could have contributed to higher levels of potentially immunogenic impurities. However, the anti-drug antibody assay specificity had not been validated for detection of the specific impurity. Furthermore, clinical samples were not tested for antibodies reactive with host-cell derived proteins or for product-specific immunoglobulin E antibodies. Although a neutralizing antibody assay was applied, the agency questioned the suitability of this method, on the basis of equivocal relevance to measure inhibition of the biological activity of insulin.
The submission did not include immunogenicity results from the 6-month open-label extension period of the main safety study. The risk management plan was not adequately specific regarding the proposed post-authorization activities to address the uncertain impact of immunogenicity on treatment outcome.
Data correlation
The example of infliximab represents a case in which there was a relatively high, but comparable, incidence of subjects with rheumatoid arthritis and ADA during treatment with Remsima® (Celltrion Healthcare Hungary Kft, Budapest, Hungary) or Remicade® (Janssen Biologics B.V., Leiden, the Netherlands). There was a high level of concordance both in timing and relative magnitude of neutralizing antibodies to the two products.19 Although detection of ADA was associated with reduced peak and trough drug concentrations, the magnitude of this impact was the same for Remsima when compared with Remicade. In addition, the incidence of infusion-related reactions was similar for the two treatment groups. This case illustrates the critical importance of being able to correlate ADA signals with relevant clinical parameters in a temporal manner to assess whether there could be a heightened risk of clinically significant immunogenicity associated with a candidate biosimilar product.
Predefined acceptance criteria
To date, EU regulators have not required application of predefined acceptance margins of detected ADA incidence for the biosimilar candidate versus the reference product. This reflects the rather different sensitivities of the bioanalytical assays used, allied to lack of knowledge of what level of difference in detected ADA might have a negative influence on clinical parameters. Thus, it is clinical outcome, rather than detected ADA per se, that is the main arbiter for assessing relative immunogenicity for the biosimilarity decision.
Extrapolation of therapeutic indications
The approval of biosimilar versions of filgrastim, growth hormone, and infliximab represent examples in which the biosimilar product was granted all of the therapeutic indications that had been authorized for the reference product. The extrapolation was based on extensive evidence of commonality of the principal mechanisms of action of the product across the different indications, allied to lack of evidence that there could be an increased risk of clinically relevant immunogenicity in the therapeutic indications that were not studied in preauthorization studies. Residual uncertainty at the time of approval was addressed by provisions included in the risk management plan for monitoring of immunogenicity in the post-marketing setting.
For filgrastim, a potential concern could be administration to healthy volunteers for stem cell transplantation, since these subjects would not derive any benefit from the procedure. In regard to the biosimilar filgrastim products approved to date, the post-approval experience of use in hematopoietic stem cell mobilization, comprising a total of 904 subjects, including 156 sibling or unrelated volunteer donors, has not revealed a different safety profile from that of the reference product, Neupogen® (Amgen Europe B.V.).20 However, data on ADA formation are lacking.
The approval of Omnitrope was based on demonstration of equivalent efficacy and safety, including immunogenicity, in the most sensitive population, namely treatment-naïve growth hormone-deficient children. This enabled a valid extrapolation to all of the indications approved for the reference product.21 In accordance with the provisions of the risk management plan, the sponsor has initiated noninterventional post-marketing studies in children and in adults that will include monitoring for anti-growth hormone antibodies and the potential clinical effects of such antibodies.22,23
Given the known association between anti-infliximab antibodies, reduced efficacy, and increased incidence of infusion-related reactions, it is pertinent to question whether there could be a differential risk for a biosimilar product relative to the reference product in alternative therapeutic settings. For example, controlled clinical studies performed using patients who receive less immunosuppressive medication might reveal differences in immunogenicity that are not apparent in a patient population receiving a more intensive concomitant immunosuppressive regimen.24
However, selection of the most sensitive population for an objective comparison of the anti-infliximab antibody response must take account of the potential interference of residual drug in different bioanalytical assay formats.25,26 This represents a critical variable because a lower dose level (3 mg/kg) is used in patients with rheumatoid arthritis than in those with other indications (5 mg/kg in Crohn’s disease, psoriasis, ulcerative colitis, and ankylosing spondylitis), thereby potentially enhancing sensitivity to detect differences in patients with rheumatoid arthritis that could be obscured by higher circulating drug levels in, for example, patients with ankylosing spondylitis.27 Higher drug interference is a possible reason why the reported incidence of anti-infliximab antibodies was lower in the ankylosing spondylitis population (not receiving methotrexate, but possibly receiving other immunosuppressive medications) compared with rheumatoid arthritis patients receiving methotrexate in the studies comparing Remsima with Remicade. Most importantly, the preauthorization studies for Remsima demonstrated that there was no difference in clinically impactful immunogenicity across the rheumatoid and ankylosing spondylitis populations when directly compared with the reference product. This evidence supports the extrapolation of a conclusion of no difference in immunogenic potential from a study performed in the rheumatoid arthritis population to the other authorized indications.
Nonclinical immunogenicity evaluation
EU experience
The hierarchical nature of the biosimilar approach applied in the EU requires a substantial weight of evidence of similarity at the product quality level.3 Accordingly, the role of nonclinical studies is to assess the impact of any detected differences, or of residual uncertainties, for the benefit-risk to proceed into comparative clinical studies.2
Product-specific guidelines (see Table 2) in the EU for candidate biosimilars containing recombinant growth hormone, erythropoietin, filgrastim, insulins, or interferon-alfa recommend that immunogenicity should be evaluated during nonclinical, repeat-dose toxicology studies. Three (for recombinant growth hormone, filgrastim and insulins) of five of these guidelines advise that “special emphasis” should be placed on evaluation of the immune response, while the other two (recombinant erythropoietin and interferon-alfa) simply recommend that antibody determination should be performed. In addition, the currently effective version of the overarching guideline on nonclinical and clinical requirements for biosimilar products (CHMP/BMWP/42832/20005) endorses a role for nonclinical evaluation of immunogenicity in recommending the inclusion of antibody measurements, encompassing both cross-reactivity and neutralizing capacity, as part of the comparative toxicokinetics data package.
As a consequence of this regulatory guidance, the initial wave of biosimilar submissions in the EU incorporated data on the comparative immunogenicity of the respective candidate biosimilar versus reference products as measured during nonclinical toxicology/toxicokinetics studies. Table 4 provides a summary of the extent of the nonclinical immunogenicity evaluations and the results reported in the various assessment reports available on the European Medicines Agency website.1
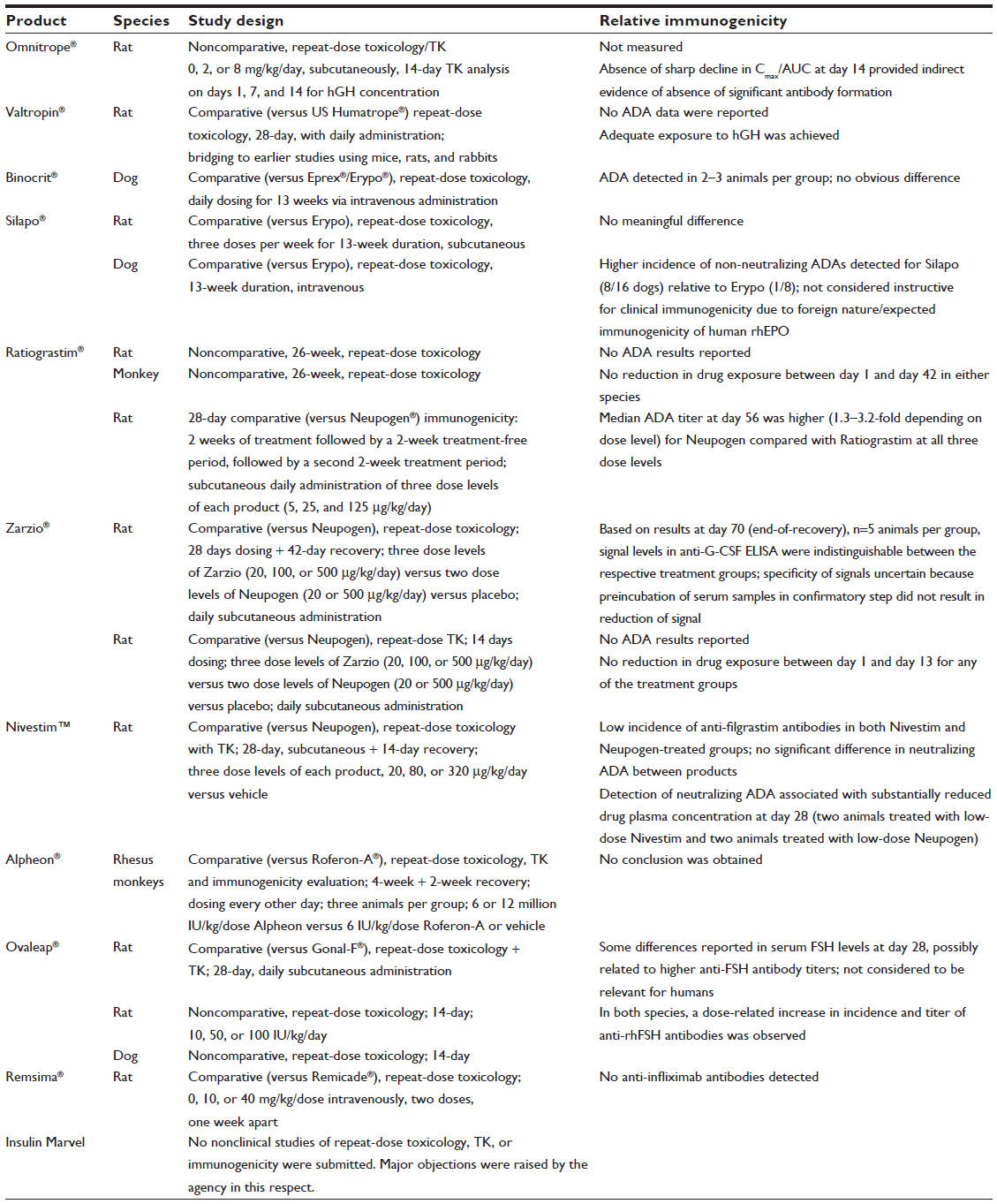 | Table 4 Summary of nonclinical studies contributing to immunogenicity assessment for biosimilar candidates reviewed in the European Union |
Table 4 indicates that the EU regulatory agencies applied a cautious approach in seeking to maximize the potential value of nonclinical data to inform the benefit-risk decision to proceed into clinical studies of these biosimilar candidates. Although some differences were detected in the apparent magnitude of the treatment-emergent antibody response to particular products, overall the results from the nonclinical immunogenicity evaluation were deemed not to have been instructive for the biosimilarity assessment: aside from the equivocal validity of extrapolating from an irrelevant immunological context to humans, imprecision associated with high biological variability and small group sizes makes data interpretation extremely difficult; in addition, differences in ADA assay sensitivity preclude a predefinition of meaningful differences based on historical data for the product class.
Bearing in mind the limitations of these studies with regard to providing a reliably instructive index of relative immunogenicity for the biosimilarity assessment, and building on this initial experience, the more recently issued EU regulatory guidelines reflect a transition from an “obligatory” to an “as needed” approach. Thus, both the draft revision to the overarching guidance on nonclinical and clinical considerations for biosimilar products2 and the product-specific guidance on biosimilar monoclonal antibodies13 recommend: “blood samples should be taken and stored for future evaluations if then needed.” This less extensive approach, if confirmed in the final effective version of the revised overarching guideline, would then supersede the recommendations in the earlier product-specific guidelines, enabling “as needed” ADA evaluation.
The trigger for performing ADA evaluation of samples collected from nonclinical in vivo studies of biosimilar candidates is not specifically described in the draft revision to the overarching guideline. However, it would be logical to cross-refer to the recommendations given in the International Conference on Harmonisation S6R1 guideline,28 whereby ADA testing samples from comparative (biosimilar versus reference product) in vivo nonclinical studies should be triggered in three situations: differences in pharmacodynamic activity between the biosimilar and reference groups; increase or decrease in exposure in the absence of a pharmacodynamic marker; or increase in incidence or severity of immune-mediated reactions. Accordingly, a difference in area under the curve for the biosimilar candidate relative to the reference product could be a trigger for ADA testing if there were no pharmacodynamic marker available to assess impact. If a nonclinical immunogenicity evaluation were performed, the primary purpose would be to enable a valid interpretation of the nonclinical findings, rather than to predict immunogenic potential in humans. The clinical immunogenicity data, not the nonclinical data, will always be the most relevant arbiter for the biosimilarity assessment regarding the decision to grant approval for marketing.
Supportive preclinical evidence for risk mitigation of identified differences
Although not an obligatory or standard part of the biosimilarity exercise, regulatory guidance29 continues to acknowledge the potential of nonclinical data to provide, in the case of molecules having substantial primary amino sequence homology between species, supportive data to detect differences in immunogenic potential associated with identified differences in product-related factors. This reflects the availability of data from comparative studies performed in wild-type animals30,31 and transgenic animal models32–34 to detect differences in the ADA response between structurally related molecules. Accordingly, it would be unwise to dismiss a role for comparative in vivo immunogenicity evaluation, eg, as part of the preclinical risk minimization exercise for a biosimilar candidate formulated differently to the reference product. Such studies have been used to contribute supportive data to justify suitability of a new formulation of human interferon-beta.35,36
Likewise, in principle, there might be a potential role for in vitro technologies to contribute to the immunogenicity risk mitigation exercise for a biosimilar development program, in the case that there are identified differences in product properties that have a potential or an uncertain impact on treatment-emergent immune responses in humans. In this respect, in vitro evaluation of stimulation of CD86 expression on human dendritic cells was useful in moderating the potential risk associated with degradation products of polysorbate in a formulation of recombinant human erythropoietin.37 In addition, a combination of in vitro and ex vivo analyses was used to assess the potential of aggregates of a human monoclonal antibody to influence the interaction between innate and adaptive immune responses.38
Presently, there is a lack of published data to demonstrate the discriminatory sensitivity of comparative measures of endocytosis, antigen presentation, and CD4+ T-helper cell stimulation for biosimilar candidates that meet the EU standard of structural comparability, ie, 100% identical primary amino acid sequence and a highly similar post-translational modification profile. While differences in post-translational glycosylation could affect uptake by and endocytotic processing in antigen-presenting cells,39,40 bioinformatics approaches suggest a rather low level (less than 4%) of glycosylation of T-cell epitopes in general.41 The latter could explain the limited sensitivity of in vitro major histocompatibility complex binding and T-cell activation assays to reveal differences between highly glycosylated factor VIII variants.42
Overall, it is still unclear whether in vitro immunogenicity risk mitigation methods could contribute incremental value for early screening of biosimilar candidates, ie, to detect differences that are not within the sensitivity range of state-of-the-art physicochemical methods. Nevertheless, it could be interesting to compare the relative in vitro activation of CD4+ T-cells in response to biosimilar candidates versus the reference product using blood from subjects who have been previously treated with the reference product, since this would be expected to increase sensitivity to detect differences.
Bioanalytical considerations
Assay sensitivity versus clinical relevance
EU regulatory practice has been to review the bioanalytical signals detected in comparative clinical studies of biosimilar candidates versus reference products relative to clinically relevant endpoints to assess whether a detected difference in the incidence or magnitude of ADA could have a negative influence on overall clinical benefit and risk. The evidence is discussed in more detail below.
From the perspective of the design of the bioanalytical strategy, the implication is that the methodology to detect ADA should be sufficiently sensitive, taking into account confounding factors such as interference by residual drug, which can be a particularly influential factor for therapeutic monoclonal antibodies,43 to detect ADA that could have a negative clinical impact. Since there may be limited historical data to guide the decision, the most pragmatic solution would be to apply methods that are least confounded by drug interference and to correlate ADA assay results with clinically relevant endpoints.
This conundrum is best illustrated by the work of van Schouwenberg et al,44 who applied alternative ADA assay methods to measure treatment-emergent ADA to adalimumab. Although the two methods had widely differing sensitivities, the additional sensitivity to detect ADA gained by the pH-shift anti-idiotype method compared with the antigen-binding test did not reveal an incremental negative impact on the measured drug trough concentration. This is explained by the apparent need to induce relatively high levels of ADA in order to reduce the circulating level of functional adalimumab to an extent that has an impact on the therapeutic response. This observation underlines the importance of interpreting measures of ADA in relation to relevant clinical parameters.
Hierarchical testing scheme
Samples from clinical studies should be evaluated in a blinded manner using a hierarchical testing scheme, following the recommendations applicable in the regulatory guidance for biopharmaceutical products.29,45 Thus, positive samples detected in the screening assay should be subjected to a confirmatory test in the presence of an excess of competing antigen (unlabeled biosimilar candidate versus unlabeled reference product). Samples confirmed as positive should then be further qualified for antibody titer and neutralizing capacity.
Testing for ADA of immunoglobulin (Ig)E class is necessary only where there are clinical observations of potential immunoglobulin IgE-mediated hypersensitivity in clinical subjects.46 Ideally, the screening assay(s) should be designed to detect IgG and IgM class antibodies. Testing for antibodies reactive with host cell proteins is not usually required, but might be merited for proteins manufactured using an E. coli cell substrate.
Screening and confirmatory assay cut-points
Assay cut-points should be established using samples from treatment-naïve subjects, according to recommendations for ADA testing for biopharmaceutical products.29,45 In the author’s experience, application of a confirmatory cut-point with a false-positive rate of 0.1% was effective in classifying a number of “gray zone” signals as confirmed negatives for different product classes associated with a relatively low incidence of treatment-emergent ADA.
Choice of assay format
The sponsor has the flexibility to select from a wide range of different assay formats, and then to validate the suitability of the methodology for the intended purpose, ie, to detect ADA that could impact on clinically relevant parameters. Review of the assessment reports for the biosimilar products reviewed to date in the EU indicates the diversity of methods applied (summarized in Table 5).
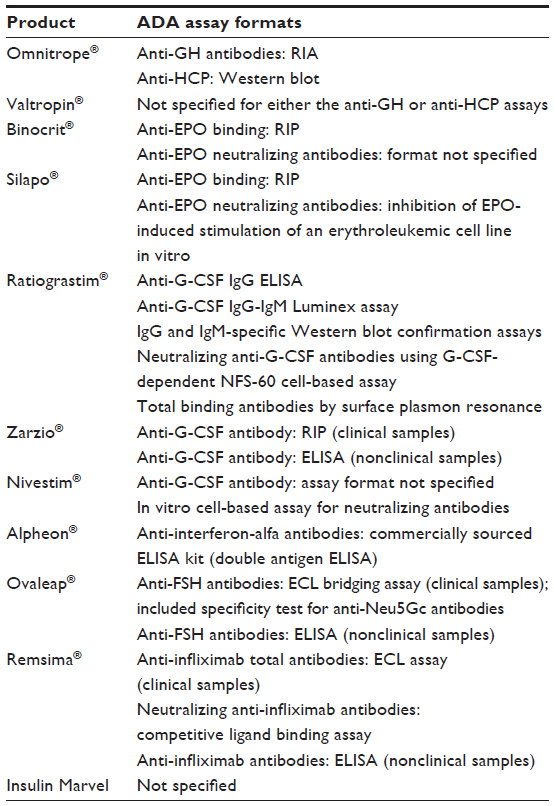 | Table 5 Anti-drug antibody assay formats reported in assessment reports for biosimilar product candidates reviewed in the European Union* |
For a PEGylated therapeutic protein, it is important to compare the sensitivity of different assay formats, particularly direct binding versus bridging assay formats, because the relatively large hydrodynamic volume of the poly(ethylene glycol) moiety may alter antigen labeling efficiency and/or capacity for bivalent binding of the ADA (author’s observations).
Generally, sponsors should develop a neutralizing assay format to provide an additional level of analysis of all confirmed positive samples detected in the screening assay. Depending on the product, this may be an in vitro cell-based assay or a competitive ligand-binding assay. Thus, in the case of Remsima, a competitive ligand-binding assay was used, reflecting the direct blocking effect of anti-infliximab antibodies on binding of the target antigen by infliximab. On the other hand, cell-based assays were used for measuring the neutralizing capacity of anti-erythropoietin or anti-filgrastim antibodies.
One versus two screening assays
The draft revision to the overarching guideline on nonclinical and clinical considerations for biosimilar products2 recommends that: “Assays should be performed with both the reference and biosimilar molecule in parallel (in a blinded fashion) to measure the immune response against the product that was received by each patient.”
However, even for a highly complex and relatively immunogenic molecule such as infliximab, duplicative testing in parallel assays (one assay using labeled versions of the biosimilar candidate and the other assay using labeled versions of the reference product) did not reveal a difference.19 Moreover, the risk that labeling of the respective test products could modify antigenicity to an unpredictable extent47 implies a higher burden for reagent quality control. The use of separate assays/reagents might be expected to decrease assay precision; gain in specificity by using separate assays is equivocal, given the possibility that biotinylation and/or ruthenylation could alter affinity for binding to epitopes proximal to labeling sites.
Therefore, to minimize any bioanalytical bias associated with assays that use different labeled antigens, it would be preferable to use a single assay; this should measure binding of ADAs to the candidate biosimilar in the screening assay, with a test of inhibition of the binding in the confirmatory step using both the unlabeled candidate biosimilar and reference products as competing antigens. The operator should be blinded to the identity of clinical samples. This approach should detect ADAs that recognize novel epitopes on the candidate biosimilar, whilst avoiding the additional level of assay variability associated with use of two different assays/labeled antigens. The alternative approaches are illustrated schematically in Figure 1.
 | Figure 1 Comparison of one-assay versus two separate screening and confirmatory assays. |
In the case of PEGylated proteins, it would still be acceptable to use a single screening assay, allied to confirmatory testing of signal specificity with the PEGylated and non-PEGylated versions to distinguish between antibodies reactive with the respective [protein versus PEGylated protein versus poly(ethylene glycol)] moieties. The same approach would be applicable in the case of fusion proteins.
Positive control
Based on the author’s experience gained during the EU regulatory approval of the biosimilar candidates reviewed to date, it is not essential for each sponsor to prepare in-house positive control reagents using either the biosimilar candidate or the reference product as an immunogen. A commercially sourced antibody reagent specific for the given protein is perfectly acceptable, if qualified appropriately. This reflects the role of the positive control antibody to provide a benchmark of relative assay sensitivity in a nonquantitative assay to detect a heterogeneous population of ADA, rather than as a calibrator for accurate measurement of the same molecule present in a test sample.
Suitability of a candidate positive control reagent should be demonstrated by confirming equivalent reactivity in a ligand-binding assay with the biosimilar and reference products: this could be achieved by titrating the positive control antibody signal in the screening assay format with increasing amounts of competing antigen (biosimilar versus reference product); the inhibition curves for the biosimilar and reference product competing antigens should be essentially overlapping. If not, the reason for the apparent difference in antigenicity would require further investigation.
Drug interference
The drug tolerance limit should be validated using the selected positive control antibody reagent according to the recommendations given in United States Pharmacopeia Chapter 1106.45 Drug interference is a particularly important limiting factor for detection of ADAs to monoclonal antibodies.43 Inclusion of acid-dissociation and partitioning steps provides an opportunity to overcome this interference.48
Baseline positives
In the case of some products, eg, insulin, it may not be feasible to recruit treatment-naïve subjects for comparative clinical studies of biosimilarity. Since a relatively high proportion (up to 80%) of subjects develop antibodies to insulin,49 it is preferable to establish assay cut-points using samples from treatment-naïve subjects, to avoid a confounding influence of earlier treatment.
Relationship to historical data
In the author’s experience, application of state-of-the-art bioanalytical methods has often revealed a substantially higher incidence of ADA compared with the results reported for the originator products. This situation is analogous to the markedly different results obtained using ADA assays of differing sensitivities for detecting anti-adalimumab antibodies in clinical samples.50
Therefore, historical data obtained using different ADA assays are not relevant for comparative purposes, or for setting predefined margins for acceptable differences in ADA incidence or median titer within the statistical analysis plan for bioequivalence or therapeutic equivalence studies of a biosimilar product versus a reference product.
Reporting of results
ADA (neutralizing and non-neutralizing) results for respective treatment groups at each sample time point should be reported in terms of percent confirmed ADA positives/confirmed ADA positive + confirmed ADA negatives and median antibody titer. These data outputs should then be correlated with relevant clinical endpoints.
Risk management plan
The immunogenicity-related provisions associated with the EU approvals of biosimilar human growth hormone, erythropoietin, and filgrastim have been clearly summarized in a separate review.51 Therefore, only the provisions relating to the more recently approved biosimilar infliximab, Remsima, are summarized in Figure 2.
 | Figure 2 Immunogenicity-related provisions in the European Union’s risk management plan for Remsima® (Celltrion Healthcare Hungary Kft, Budapest, Hungary). |
Items 3 and 4 are the most significant items on this list in terms of potential for providing data on immunogenicity in a different population, ie, Crohn’s disease, and on the potential influence of immunogenicity on longer-term treatment outcomes in the rheumatoid arthritis population. Although no specific reference is given to immunogenicity monitoring, it would seem implausible for these studies to omit monitoring of ADA and drug levels relative to efficacy and safety indices.
Limitations of post-marketing data
The Achilles heel for the argument that there is no higher immunogenicity-related risk for biosimilar products approved according to the EU regulatory standards is the limited availability of directly comparative post-marketing data from monitoring of ADA levels relative to clinically relevant parameters of efficacy and safety.
Even if preauthorization studies were sufficiently sensitive and of adequate duration to detect clinically relevant differences in immunogenicity that might manifest in the longer term, the regulatory process does not impose an obligation on biosimilar product manufacturers to reconfirm equivalent efficacy, safety, and immunogenicity by direct comparison with the reference product to support implementation of changes in the post-marketing lifecycle. In this respect, it is important to remember that manufacturing changes, changes in the formulation or primary container, or even new therapeutic indications, could be authorized in the post-marketing phase for either the reference or biosimilar products. Authorization of such changes would be based on a demonstration of comparability of the pre-change versus post-change material,10,52 ie, a direct comparison of the post-change biosimilar versus the reference product (or other biosimilar products) is not mandatory. Theoretically, this creates a risk for manufacturing drift to manifest as changes in clinically relevant immunogenicity.
The absence of standardized ADA assays, or common positive control antibody reagents to benchmark relative assay sensitivity, precludes reliance on noncomparative clinical data to monitor the longer-term immunogenic potential of the different product versions.
One potential solution would be for patient registries to include ADA monitoring, using a standardized assay, for both reference and biosimilar products. This might require prospectively planned, interventional, observational cohort studies, rather than noninterventional studies, if additional blood sampling were required above the level recognized as normal clinical practice. Measurement of drug levels and recording of potential immune-mediated adverse events (eg, infusion reactions) should also be included.
Immunogenicity risk associated with switching medications
It is evident from Table 3 that the preauthorization clinical experience for different biosimilar candidates included open-label extension periods in which patients who had been treated with the reference product were then switched to receive treatment with the biosimilar candidate. Immunogenicity was monitored during open-label periods, thereby providing some evidence of the lack of potential for induction of ADA by the biosimilar candidate in the same patients who had been treated earlier with the reference product.
As discussed in a separate review,53 there are limited additional data from other studies that have evaluated switching from the reference product to the biosimilar product and, less commonly, from the biosimilar to the reference product.
In the author’s experience, scientific advice from the US Food and Drug Administration has encouraged the design of pivotal studies for certain biosimilar candidates to include re-randomization at the end of the controlled double-blind study period to continue subjects on either the biosimilar candidate or the reference product during an open-label extension period that also includes immunogenicity monitoring. This strategy would provide comparative data for a single switch in either direction.
However, one issue that is not addressed is the longer-term impact of switching between multiple products. Since there could be important ethical and feasibility challenges for designing a multiple-switching study, it would again impose upon on the patient registry/observational cohort scenario to assume the role for longer-term monitoring of treatment outcomes using relevant measures, including ADA formation, maintenance of drug levels and efficacy, and the incidence/severity of immune-mediated adverse events.
Interchangeability and substitution
Since the EU regulatory decision to grant a marketing authorization for a biosimilar product has no implication for the substitutability of medicines at the pharmacy level, which is the jurisdiction of individual member states, there are no additional regulatory provisions in the EU governing “interchangeability”.
On the other hand, it is conceivable that national decisions on pricing and reimbursement or product selection, and/or acceptance by clinicians, could be influenced by the relative immunogenicity of the authorized products. Although it is too early to comment on how actual or perceived differences in immunogenicity might play out in the longer term, there would seem to be an essential role for post-marketing immunogenicity data that are comparable in nature, ie, using standardized methodology for monitoring patient populations exposed to different product versions.
Such data might provide objective information regarding the potential for manufacturing drift to modify relative immunogenicity of different product versions. Possibly, organizations such as the World Health Organization could provide standardized assays and reagents for ADA assays, and the risk management plans for approved biosimilars would be linked to appropriate patient registries/observational cohort studies that include different product versions.
Arguably, an open-ended designation of “interchangeability” (in a regulatory jurisdiction such as the USA where this implied “substitutability”) would carry an unsustainably heavy burden to confirm continuing biosimilar status,54 including no difference in clinically relevant immunogenicity following implementation of changes (eg, manufacturing process, formulation, primary container). It would then be logical to consider “switchability”, guided by the appropriately informed judgment of the prescribing physician, as the major factor to address.
Lessons learnt
To date, the only immunogenicity-related issues that have been identified for the biosimilar candidates reviewed in the EU registration process have all occurred in the preapproval setting (summarized in Boxes 1, 2, 4, and 5). A common theme across the four cited examples was the detection of differences in product quality attributes that might have contributed to a heightened risk of clinically relevant immunogenicity.
In the case of growth hormone, an elevated level of a process-related impurity that was not detected by the original analytical method was associated with an increased level of treatment-emergent antibodies in clinical samples from patients treated with an investigational formulation; process improvements implemented in the pre-authorization setting resolved this issue, such that the authorized product (Omnitrope®) had an immunogenicity profile that was indistinguishable from that of the reference product.15
In the case of erythropoietin, use of prefilled syringes containing a tungsten impurity is the most likely explanation for the detection of neutralizing ADA in two subjects with chronic kidney disease receiving subcutaneous administration of Binocrit®.7
Differences in product-related substances and/or process-related impurities between other biosimilar candidates (Alpheon® and the insulins marketed by Marvel LifeSciences Ltd) and their respective reference products were cited as reasons for noncomparability.18,19 Directly related to these differences in product quality attributes, inadequate validation of the specificity and sensitivity of ADA assays allied to clinical signals of reduced efficacy and/or increased incidence/severity of immune-mediated adverse events, contributed to the failure of these candidates to achieve designation as biosimilar.
Comparative immunogenicity evaluation in nonclinical toxicology studies was not instructive for the biosimilarity decision to enable marketing authorization. Nevertheless, non-clinical studies might provide helpful data to justify decisions on formulation of biosimilar candidates, or for investigation of unexpected findings arising from changes in the manufacturing process or in primary container or storage conditions.
Overall, based on the experience gained during a 10-year period since implementation of the EU regulatory pathway for biosimilar medicinal products, the regulatory standard for approval has been validated as suitably cautious by the absence of observed differences in clinically relevant immunogenicity between the approved biosimilar and originator products following authorization. Although, as in the case of registration of any medicinal product, some degree of uncertainty has been accepted at the time of marketing authorization, a combination of post-marketing data and a heightened level of pharmacovigilance has not revealed any incremental immunogenicity-related risks in respect of the authorized use of biosimilars in the EU.
Open questions
How to optimize post-marketing monitoring to detect longer-term differences in clinically relevant immunogenicity?
The absence of standardized assays and control reagents, allied to the lack of correlation of results of ADA assays to clinically relevant measures of efficacy and safety, is a major impediment to our understanding of whether there is any unrecognized incremental risk of immunogenicity for biosimilar products that have been approved to the EU regulatory standard.
As a result, the clinical uptake of EU-approved biosimilar products could be compromised by an unfounded perception that immunogenicity-related risks had not been adequately evaluated or controlled.
One solution could be for patient registries comprising patients treated with different product versions to incorporate longer-term periodic monitoring of clinically relevant parameters. This might still not be a feasible solution for detection of very rare events such as erythropoietin-induced antibody-mediated pure red cell aplasia, but could yield an objective basis for understanding whether there is a potential for post-approval manufacturing drift to affect other immunogenicity-influenced endpoints, including drug concentration, efficacy, and tolerability.
Is there a negative impact of switching between different product versions?
Inclusion of two-way switching, eg, by re-randomization of patients treated in the blinded treatment phase of a Phase III study, could provide some assurance that switching between the biosimilar and reference versions is not associated with clinically relevant differences in immunogenicity. Duration of exposure to the different product versions would need to reflect the dynamics of the treatment-emergent immune response in addition to effective washout of the effects of the previous treatment.
On the other hand, it would not be feasible to compare switching between different biosimilar product versions in preapproval studies. More realistically, adequate monitoring of patients treated with different product versions within a patient registry or observational cohort study, with access to a centralized laboratory able to apply standardized assays, could represent the most practical option for comparing longer-term outcomes associated with elective switching between the respective versions of the product.
What is an acceptable threshold for differences?
The lack of standardized bioanalytical assays or control reagents for ADA assays will continue to impose a waste of resources, because results using different assays can never be validly compared. For the regulator, the substantially different results obtained by assays with lower or higher sensitivity forces a reliance on clinical evidence of lack of impact of differences detected (or not detected) in bioanalytical assays. Most importantly, it means that it is rarely possible to interpret the clinical relevance of bioanalytical indices of ADA formation; equally, it is not possible to define an equivalence margin for incidence or titer of ADA formation that is relevant for designation of biosimilarity.
Even more challenging for the physician is the questionable validity of reporting arbitrary measures of ADA formation in the prescribing information of the reference and the biosimilar products – what should count is the comparative impact on clinically relevant parameters, such as drug concentration, efficacy and tolerability.
Disclosure
The valuable critical comments of Dr Matthew Baker (Antitope UK Limited) regarding the immunological considerations for product quality and preclinical sections of this article are acknowledged. The final text reflects the views of the author only. PC reports no other conflicts of interest in this work.
References
European Medicines Agency. European Public Assessment Reports. Omnitrope®, Valtropin®, Binocrit®, Silapo®, Ratiograstim®, Zarzio®, Nivestim®, Alpheon®, Ovaleap®, Remsima® and Marvel insulins. Available from: http://www.ema.europa.eu. Accessed April 26, 2014. | |
European Medicines Agency. Similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues, CHMP/BMWP/42832/2005 rev1, Draft Revision, June 3, 2013. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500144124&mid=WC0b01ac058009a3dc. Accessed April 26, 2014. | |
European Medicines Agency. Similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues, CHMP/BMWP/247713/2012, Draft Revision, May 24, 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf. | |
Rombach-Riegraf V, Karle AC, Wolf B, et al. Aggregation of recombinant human monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS One. 2014;9:e86322. | |
Liu W, Swift R, Torraca G, et al. Root cause analysis of tungsten-induced protein aggregation in pre-filled syringes. PDA J Pharm Sci Technol. 2010;64:11–19. | |
Haag-Weber M, Eckhardt KU, Horl WH, Roger SD, Vetter A, Rother HK. Safety, immunogenicity and efficacy of subcutaneous biosimilar epoetin-α (HX575) in non-dialysis patients with renal anemia: a multi-center, randomized, double-blind study. Clin Nephrol. 2012;77:8–17. | |
Seidl A, Hainzl O, Richter M, et al. Tungsten-induced denaturation and aggregation of epoetin-alfa during primary packaging as a cause of immunogenicity. Pharm Res. 2012;29:1454–1467. | |
MacDougall IC, Rossert J, Casadevall N, et al. A peptide-based erythropoietin-receptor agonist for pure red-cell aplasia. N Engl J Med. 2009;361:1848–1855. | |
Onitsuka M, Tatsuzawa M, Asano R, et al. Trehalose suppresses antibody aggregation during the culture of Chinese hamster ovary cells. J Biosci Bioeng. 2013;117:632–638. | |
Schneider CK, Vleminkx C, Gravanis I, et al. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30:1179–1185. | |
Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1,3-galactose. N Engl J Med. 2008;358:1109–1117. | |
Lammerts van Beuren JJ, Rispens T, Verploegen S, et al. Anti-galactose-α-1,3-galactose IgE from allergic patients does not bind α-galactosylated glycans on intact therapeutic antibody Fc domains. Nat Biotechnol. 2011;29:574–576. | |
Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 2010;28:863–867. | |
European Medicines Agency. European public assessment report for Ovaleap®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002608/human_med_001689.jsp&mid=WC0b01ac058001d124. Accessed April 26, 2014. | |
European Medicines Agency. European public assessment report for Omnitrope®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000607/human_med_000946.jsp&mid=WC0b01ac058001d125. Accessed April 26, 2014. | |
European Medicines Agency. Similar biological medicinal products containing monoclonal antibodies: non-clinical and clinical issues, CHMP/BMWP/403543/2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf. Accessed April 26, 2014. | |
European Medicines Agency. Refusal assessment report for Alpheon®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000585/human_med_000643.jsp&mid=WC0b01ac058001d125. Accessed April 26, 2014. | |
European Medicines Agency. Withdrawal assessment reports for Solumarv®, Isomarv Medium® and Combimarv®. Available from: http://www.ema.europa.eu. Accessed April 26, 2014. | |
European Medicines Agency. European public assessment report for Remsima®. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002576/human_med_ 001682.jsp&mid=WC0b01ac058001d124. Accessed April 26, 2014. | |
Schmitt M, Publicover A, Orchard KH, et al. Biosimilar G-CSF based mobilization of peripheral blood hematopoietic stem cells for autologous and allogeneic stem cell transplantation. Theranostics. 2014;4:280–289. | |
Pavlovic M, Girardin E, Kapetanovic L, Ho K, Trouvin J-H. Similar biological medicinal products containing recombinant human growth hormone: European regulation. Horm Res. 2008;69:14–21. | |
Pfaffle R, Schwab KO, Marginean O, et al. Design of, and first data from, PATRO Children, a multicentre, noninterventional study of the long-term efficacy and safety of Omnitrope® in children requiring growth hormone treatment. Ther Adv Endocrinol Metab. 2013;4:3–11. | |
Beck-Peccoz P, Minuto F, Leal-Cerro, Zabransky M, Stalla G. Rationale and design of PATRO Adults, a multicentre, noninterventional study of the long-term efficacy and safety of Omnitrope® for the treatment of adult patients with growth hormone deficiency. Ther Adv Endocrinol Metab. 2012;3:85–91. | |
Lee H. Is extrapolation of the safety and efficacy data in one indication to another appropriate for biosimilars? AAPS J. 2014;16:22–26. | |
Steenholdt S, Bendtzen K, Brynskov J, Thomsen OO, Ainsworth MA. Cut-off levels and diagnostic accuracy of infliximab trough levels and anti-infliximab antibodies in Crohn’s disease. Scand J Gastroenterol. 2011;46:310–318. | |
Krintel SB, Grunert VP, Hetland ML, et al. The frequency of anti-infliximab antibodies in patients with rheumatoid arthritis treated in routine care and the associations with adverse drug reactions and treatment failure. Rheumatology. 2013;52:1245–1253. | |
Ducourau E, Mulleman D, Paintaud G, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13:R105. | |
European Medicines Agency. ICH S6 (R1) guideline. Pre-clinical safety evaluation of biotechnology-derived pharmaceuticals. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf. Accessed April 26, 2014. | |
European Medicines Agency. Immunogenicity assessment of biotechnology-derived therapeutic proteins, CHMP/BMWP/14327/2006. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf. Accessed April 26, 2014. | |
Fradkin AH, Carpenter JF, Randolph TW. Immunogenicity of aggregates of recombinant growth hormone in mouse models. J Pharm Sci. 2009;98:3247–3264. | |
Jeandidier N, Boullu S, Busch-Brafin M-S, et al. Comparison of antigenicity of Hoechst 21PH insulin using either implantable intraperitoneal pump or subcutaneous external pump infusion in type 1 diabetic patients. Diabetes Care. 2002;25:84–88. | |
Braun A, Kwee L, Labow MA, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-alpha) in normal and transgenic mice. Pharm Res. 1997;14:1472–1478. | |
Hermeling S, Aranha L, Damen JM, et al. Structural characterization and immunogenicity in wild-type and immune tolerant mice of degraded recombinant human interferon alpha2b. Pharm Res. 2005;22:1997–2006. | |
van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Aggregated recombinant human interferon beta induces antibodies but no memory in immune-tolerant transgenic mice. Pharm Res. 2010;27:1812–1824. | |
Jaber A, Driebergen R, Giovannoni G, Schellekens H, Simsarian J, Antonelli M. The Rebif new formulation story: it’s not trials and error. Drugs R D. 2007;8:335–348. | |
Bellomi F, Muto A, Palmieri G, et al. Immunogenicity comparison of interferon beta-1a preparations using the BALB/c mouse model: assessment of a new formulation for use in multiple sclerosis. New Microbiol. 2007;30:241–246. | |
Mueller R, Karle A, Vogt A, et al. Evaluation of the immuno-stimulatory potential of stopper extractables and leachables by using dendritic cells as readout. J Pharm Sci. 2009;98:3548–3561. | |
Joubert MK, Hokom M, Eakin C, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem. 2012;287:25266–25279. | |
Dong X, Storkus WJ, Salter RD. Binding and uptake of agalactosyl IgG by mannose receptor on macrophages and dendritic cells. J Immunol. 1999;163:5427–5434. | |
Werdelin O, Meldal M, Jensen T. Processing of glycans on glycoprotein and glycopeptides antigens in antigen-presenting cells. Proc Natl Acad Sci U S A. 2002;99:9611–9613. | |
Szabó TG, Palotai R, Antal P, et al. Critical role of glycosylation in determining the length and structure of T-cell epitopes. Immunome Res. 2009;5:1–12. | |
Delignat S, Repessé Y, Gilardin L, et al. Predictive immunogenicity of Refacto® AF. Haemophilia. December 30, 2013. [Epub ahead of print.] | |
Wang Y-M, Fang L, Zhou L, Wang J, Ahn H-Y. A survey of applications of biological products for drug interference of immunogenicity assays. Pharm Res. 2012;29:3384–3392. | |
Van Schouwenberg PA, Kriekaert CL, Rispens T, Aarden L, Wolbink GJ, Wouters D. Long-term measurement of anti-adalimumab using pH-shift-anti-idiotype antigen binding test shows predictive value and transient antibody formation. Ann Rheum Dis. 2013;72:1680–1686. | |
United States Pharmacopeia. Chapter 1106. Immunogenicity assays – design and validation of immunoassays to detect anti-drug antibodies. Available from: http://www.usp.org. Accessed May 27, 2014. | |
Clinical and Laboratory Standards Institute. Design and validation of immunoassays for assessment of human allergenicity of new biotherapeutic drugs: approved guideline. Clinical and Laboratory Standards Institute document I/LA34-A, 2011. Available from http://www.clsi.org. Accessed May 27, 2014. | |
Hoyer-Hansen G, Hamers MJ, Pedersen AN, et al. Loss of ELISA specificity due to biotinylation of monoclonal antibodies. J Immunol Methods. 2000;235:91–99. | |
Smith HW, Butterfield A, Sun D. Detection of antibodies against therapeutic proteins in the presence of residual therapeutic protein using a solid-phase extraction with acid dissociation (SPEAD) sample treatment prior to ELISA. Regul Toxicol Pharmacol. 2007;49:230–237. | |
Mianowska B, Szadkowska A, Pietrzak I, et al. Immunogenicity of different brands of human insulin and rapid-acting insulin analogs in insulin-naïve children with type 1 diabetes. Pediatr Diabetes. 2011;12:78–84. | |
Van Scouwenberg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D. A novel method for the detection of antibodies to adalimumab in the presence of drug levels reveals “hidden” immunogenicity in rheumatoid arthritis patients. J Immunol Methods. 2010;362:82–88. | |
Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012;30:1186–1190. | |
European Medicines Agency. ICH Q5. Note for guidance on biotechnological/biological products subject to changes in their manufacturing process. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002805.pdf. Accessed April 26, 2014. | |
Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12:1473–1485. | |
Ebbers HC, Chamberlain P. Interchangeability: an insurmountable 5th hurdle? Generics and Biosimilars Initiative Journal. 2014;3(2). Available from http://www.gabi-journal.net/interchangeability-an-insurmountable-5th-hurdle.html. Accessed May 21, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.