Back to Journals » Drug Design, Development and Therapy » Volume 20
Multidimensional Regulatory Mechanisms and Targeted Therapeutic Strategies for Inhibited Keratinocyte Proliferation in Diabetic Wounds
Authors Liu C ![]() , Niu X, Xu Y, Liu W
, Niu X, Xu Y, Liu W
Received 23 January 2026
Accepted for publication 17 March 2026
Published 15 April 2026 Volume 2026:20 598433
DOI https://doi.org/10.2147/DDDT.S598433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Chengcheng Liu,1,* Xueli Niu,2,3,* Yunfa Xu,4 Wei Liu5
1Department of Rehabilitation, Shengjing Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Department of Dermatology, The First Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 3Key Laboratory of Immunodermatology, Ministry of Education and NHC; National Joint Engineering Research Center for Theranostics of Immunological Skin Diseases, Shenyang, Liaoning, People’s Republic of China; 4Department of Radiology, Shenyang Second Hospital of Traditional Chinese Medicine, Shenyang, Liaoning, People’s Republic of China; 5Department of Neurology, Shenyang Second Hospital of Traditional Chinese Medicine, Shenyang, Liaoning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Liu; Yunfa Xu, Email [email protected]; [email protected]
Abstract: Diabetic wound healing impairment represents a pressing clinical challenge worldwide, with its high disability rate and recurrence rate imposing a heavy burden on patients and healthcare systems. Keratinocytes are the core effector cells that drive re-epithelialization during wound healing, and their impaired proliferative capacity is a core pathological mechanism underlying healing arrest and chronic wound development. The functional status of other repair cell populations, including fibroblasts, endothelial cells and immune cells, also exerts direct or indirect regulatory effects on the entire wound healing process. Under diabetic conditions, multifaceted pathological changes triggered by hyperglycemia not only induce comprehensive functional impairment of keratinocytes, but also disrupt the synergistic interaction between various repair cells. This ultimately stalls the physiological wound healing cascade, leading to the development of chronic, non-healing wounds. Building upon this, this review further summarizes novel targeted therapeutic strategies addressing these mechanisms, encompassing cutting-edge approaches such as engineered exosome delivery systems, photobiomodulation therapy, metabolic enzyme small-molecule inhibitors, peptide agonists, and plant-derived nanovesicles. This review aims to delineate the crosstalk between core regulatory modules and identify key druggable targets, a theoretical framework for the development of precision combination therapies with multi-target synergistic effects. Existing evidence demonstrates that the synergistic dysfunction of multiple core molecular hubs represents the core pathological basis underlying suppression in diabetic wounds. Future therapeutic strategies should focus on the synergistic benefits of spatiotemporally controlled dynamic intervention and wound microenvironment reprogramming.
Keywords: diabetic wounds, keratinocytes, proliferation inhibition, targeted therapy, metabolic reprogramming, epigenetic regulation, extracellular matrix, exosomes
Introduction
Diabetes is one of the most prevalent chronic metabolic diseases worldwide. According to the International Diabetes Federation, the global diabetic population exceeded 500 million by 2025. Diabetic wounds, among its most devastating complications, occur in 15–25% of patients, with approximately 15–20% of chronic wounds ultimately progressing to lower limb amputation. This significantly diminishes patients’ quality of life and exacerbates healthcare resource consumption.1 Wound healing is a highly coordinated dynamic process involving sequential stages such as inflammatory response, cell proliferation, matrix deposition, and tissue remodeling. Re-epithelialization, a core component of wound healing, primarily relies on the proliferation, migration, and differentiation of keratinocytes to form a continuous epidermal barrier covering the wound surface.2 Under healthy physiological conditions, keratinocytes rapidly respond to injury signals, initiating proliferation programs and migrating toward the wound center to complete epidermal regeneration. However, in diabetic states, a series of pathological alterations induced by the hyperglycemic microenvironment severely impairs keratinocyte function, leading to stalled re-epithelialization and ultimately forming chronic wounds that are difficult to heal.3
In recent years, significant progress has been made in elucidating the mechanisms underlying keratinocyte dysfunction in diabetic wounds. Existing studies confirm that hyperglycemia-induced metabolic toxicity serves as one of the initiating factors for keratinocyte proliferation inhibition. Under hyperglycemic conditions, abnormal upregulation of fructose-1,6-bisphosphatase 1 inhibits glycolytic flux, reduces ATP production, and directly impairs keratinocyte proliferation and migration capacity.4 Concurrently, the accumulation of advanced glycation end products (AGEs) induced by high glucose activates the RAGE signaling pathway, exacerbating oxidative stress and inflammatory responses, thereby further damaging keratinocyte function.5,6 Epigenetic dysregulation has also been identified as a core mechanism regulating keratinocyte function. Imbalances in the expression of epigenetic regulators such as the long non-coding RNAs MALAT1 and UCA1, and the m6A demethylase FTO, indirectly suppress keratinocyte proliferation by modulating epithelial-mesenchymal transition, inflammatory pathways, and autophagy flux.7,8 Pathological remodeling of the extracellular matrix (ECM) is equally significant. In diabetic wounds, ECM stiffening and structural disorganization disrupt keratinocyte mechanosensing and adhesion/migration capabilities via the YAP/TAZ mechanosensing axis.9 Among these, hyperglycemia-mediated abnormal O-GlcNAc glycosylation modifications directly target core transcription factors, serving as a pivotal bridge linking metabolic disorders to proliferation suppression. Hypoxia-inducible factor 1α (HIF-1α) and stimulator of interferon genes (STING) emerge as two central molecular hubs mediating this pathological process. Specifically, in the hyperglycemic microenvironment of diabetic wounds, STING is abnormally activated by mitochondrial DNA leakage induced by oxidative stress and mitochondrial dysfunction. Once activated, STING triggers the TBK1-IRF3 signaling pathway to induce the production of type I interferons and pro-inflammatory factors, which in turn upregulate the expression of cell cycle inhibitors such as p21 and p27 in keratinocytes. This leads to the arrest of keratinocyte cell cycle progression, directly inhibiting their proliferative capacity. Additionally, STING activation further exacerbates keratinocyte dysfunction by promoting inflammatory senescence and disrupting autophagy balance, thereby forming a vicious cycle that perpetuates proliferation suppression. Microbiome dysbiosis and biofilm formation at wound sites disrupt keratinocyte function through dual mechanisms, namely virulence factor release and immune dysregulation.10 Pathological effects mediated by persistent hypoxia fundamentally rely on dysfunction of the HIF-1α signaling axis. This abnormal regulation not only indirectly impacts keratinocyte proliferation by inhibiting angiogenesis but also directly affects keratinocytes themselves. The hyperglycemic environment of diabetes impairs HIF-1α function, preventing effective initiation of transcription for downstream proliferation-related, metabolic regulatory, and anti-apoptotic genes. Concurrently, abnormal HIF-1α activity directly disrupts keratinocyte cell cycle progression, metabolic reprogramming, and autophagy balance, constituting a direct cause of impaired keratinocyte proliferation capacity.11 Persistent hypoxia, a hallmark of diabetic wounds, abnormally activates the HIF-1α signaling axis spatiotemporally. This not only inhibits angiogenesis, leading to inadequate nutrient supply, but also directly regulates keratinocyte metabolic reprogramming and autophagy balance, exacerbating proliferation suppression.12 The disintegration of neuropeptide networks further exacerbates this pathological process. Decreased levels of pro-reparative neuropeptides such as substance P and calcitonin gene-related peptide directly weaken keratinocyte proliferation drive and microenvironmental regulatory capacity.13
Although existing research has revealed multiple independent mechanisms underlying the suppression of keratinocyte proliferation in diabetic wounds, these mechanisms do not operate in isolation. Instead, they form an interwoven regulatory network through shared molecular hubs. For instance, HIF-1α dysfunction not only exacerbates hypoxia stress but also activates the STING inflammatory pathway by regulating ROS release. Conversely, STING-driven IFN-β inhibits HIF-1α transcriptional activity, creating a vicious cycle.12 Current research is limited by insufficient analysis of the crosstalk logic between these mechanisms and a lack of systematic network-level understanding. This knowledge gap confines clinical interventions to single-target approaches, such as growth factor supplementation and debridement, which yield limited efficacy and fail to reverse complex pathological homeostasis.14 Therefore, systematically deciphering the multidimensional regulatory network underlying keratinocyte proliferation suppression in diabetic wounds, identifying crosstalk mechanisms between modules and key druggable targets is the core scientific prerequisite for developing highly effective, precision treatment strategies.
This review focuses on a core pathological hallmark of impaired diabetic wound healing: the suppressed proliferation of keratinocytes. It systematically dissects this pathological process from the perspectives of metabolic toxicity, epigenetic dysregulation, extracellular matrix pathological remodeling, wound microenvironment imbalance, hypoxic stress, cutaneous microbiota dysbiosis, and neuropeptide signaling disorder. We comprehensively characterize the molecular mechanisms and core regulatory targets within each functional module, delineate the crosstalk pathways between different modules mediated by key molecular hubs, and clarify the core mechanism by which multiple pathological factors synergistically drive the impairment of keratinocyte proliferation. Building on this framework, we further summarize the mechanism of action and clinical translational potential of emerging targeted intervention strategies, including engineered exosome delivery systems, photobiomodulation therapy, metabolic enzyme inhibitors, and peptide agonists. Taken together, the overarching goal of this work is to provide a theoretical basis and novel translational insights for the precision treatment of diabetic chronic wounds.
Cell Proliferation
Cell proliferation is an orderly and programmed dynamic process, typically initiated when cells transition from a quiescent state into the preparatory phase for division. This occurs when proliferating cells detect signals from growth factors, cytokines, or sufficient nutrients. This process involves the coordinated action of core regulatory molecules, including cyclins,15,16 cyclin-dependent kinases (CDKs),16 retinoblastoma protein (Rb),17,18 and cell cycle inhibitors (CKIs),18 to establish a periodic division rhythm and form stringent checkpoint controls between phases. Upon signal reception in G0 phase cells, Cyclin D binds to CDK4/6 to form a complex, phosphorylating the Rb protein and releasing its inhibition of E2F transcription factors. This drives cells into the G1 phase and initiates the expression of genes associated with DNA synthesis.19 Upon entering S phase, the Cyclin E-CDK2 complex further propels the cell cycle by promoting the activation of DNA polymerases, ensuring complete genomic replication.20 During G2 phase, Cyclin B binds to CDK1 to prepare cells for mitotic entry, while the G2/M checkpoint monitors DNA damage and halts the cell cycle if unrepaired damage is present.20 During the M phase, the spindle attachment checkpoint ensures proper chromosome segregation. Subsequently, the cell completes mitotic and cytokinesis, producing two daughter cells. Some daughter cells may re-enter the G0 resting phase, while others continue participating in the proliferation cycle.21,22
Cell Proliferation in Normal Wounds
Cell proliferation is a central and ongoing biological process in wound healing, spanning the entire phases of inflammation, proliferation, and remodeling. Among these, the proliferation of keratinocytes is the key step in the reconstruction of the epidermal barrier, a process that is highly dependent on the synergistic actions of other cells in the wound microenvironment. Various cell types, including fibroblasts, endothelial cells, macrophages, and neutrophils, work in concert through their own finely regulated proliferation and functional activation to accomplish key processes such as pathogen clearance, angiogenesis, and extracellular matrix (ECM) deposition, thereby collectively establishing the microenvironment that regulates keratinocyte proliferation. The proliferative activity of these cells is finely regulated by growth factors, cytokines, and mechanical signals in the local microenvironment23 (Figure 1). Investigating the mechanisms of cell proliferation not only enhances our understanding of the physiological repair process of wounds but also provides a theoretical foundation for chronic wound treatment strategies.
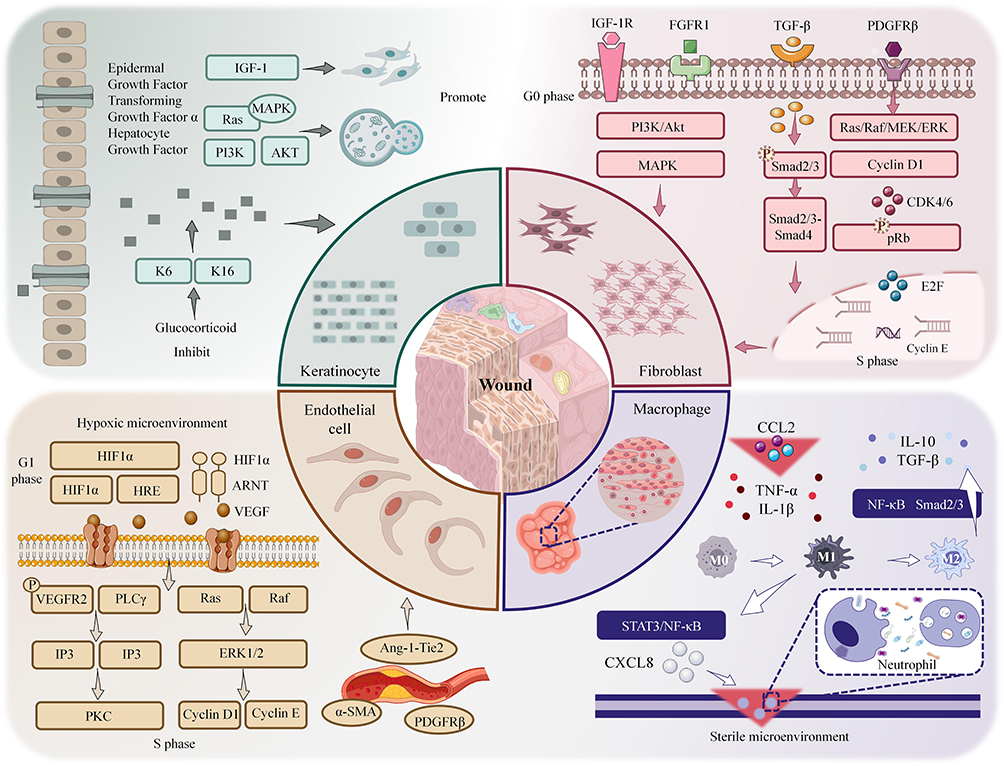 |
Figure 1 This diagram illustrates cell proliferation in normal wound healing. Keratinocytes activate via EGFR/MAPK/PI3K but are inhibited by glucocorticoids. Fibroblasts proliferate via PDGF-BB/TGF-β. Endothelial cells use HIF1α/VEGF for angiogenesis. Macrophages regulate proliferation via cytokines; neutrophils clear pathogens for repair. Upward arrows (↑) indicate upregulation or activation of relevant molecules and signaling pathways; downward arrows (T) indicate downregulation or inhibition of relevant molecules and signaling pathways. |
Keratinocyte Proliferation
The proliferation of keratinocytes is a core component of epidermal repair, directly determining whether the skin barrier can be promptly reestablished. Within hours of wound formation, basal keratinocytes near the wound margin exit quiescence and are activated to enter the cell cycle. During this activation, inflammatory mediators act on cell surface receptors via paracrine or autocrine mechanisms, triggering downstream signaling pathways to initiate DNA replication. Epidermal growth factor (EGF), transforming growth factor-alpha (TGF-α), and hepatocyte growth factor (HGF) play pivotal roles in this process (Table 1). They activate the Ras/MAPK and PI3K/Akt pathways by binding to EGFR or cMet receptors, respectively, propelling cells from G1 to S phase.24–27 Although insulin-like growth factor 1 (IGF-1) does not directly drive mitosis, it enhances the ability to form cell membrane protrusions, aiding cells in spreading and occupying space during migration. This indirectly supports the physical conditions required for subsequent proliferation. Once cells at the leading edge complete initial coverage, basal cells at the rear undergo extensive division to replenish the cellular reserve depleted by migration, ensuring the newly formed epithelium achieves normal thickness.28,29 Beyond the direct regulation by classical growth factors, the proliferation process of keratinocytes is profoundly influenced by stem cell fate regulation and epigenetic modifications. At the stem cell fate regulation level, the YAP/TAZ-Hippo mechanotransduction axis and the E2F/MYC-p21/p27 cell cycle regulatory pathway jointly participate in keratinocyte proliferation control. These two pathways coordinate the balance between stem cell self-renewal and differentiation, thereby determining epidermal regeneration efficiency.30 Cherkashina et al, using a human skin xenograft model, revealed that restoration of epidermal proliferation patterns depends on spatial rearrangement of basal layer stem cell subpopulations and mechanical coupling between stem cells and dermal papillae structures, with YAP nuclear localization playing a key regulatory role in this process.31 At the epigenetic level, processes such as DNMT1-UHRF1-mediated DNA methylation memory and JMJD3 histone demethylase activity have been demonstrated to profoundly regulate keratinocyte proliferation dynamics.32
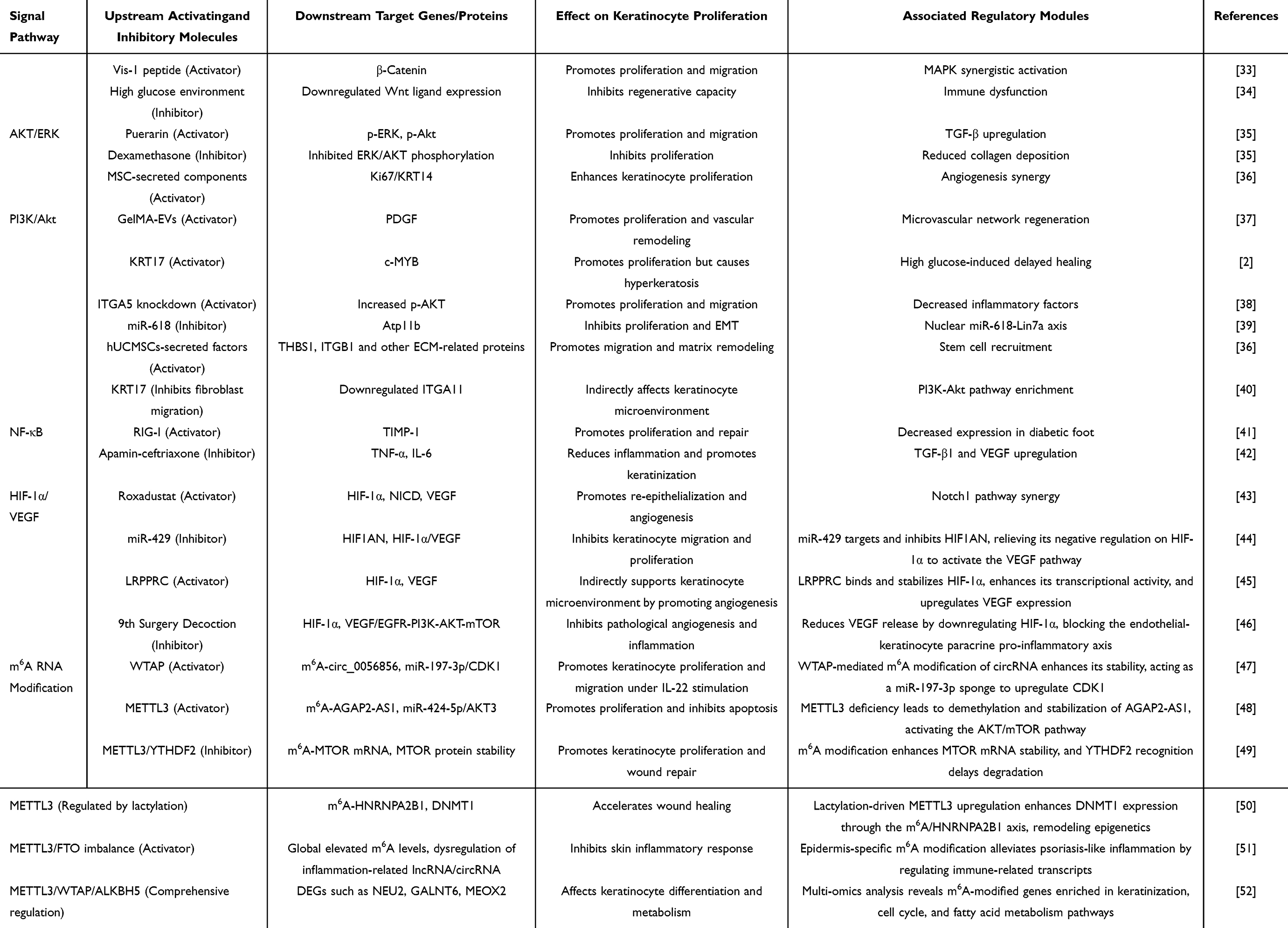 |
Table 1 Comprehensive Analysis of Signaling Pathways and m6A RNA Modification Regulating Keratinocyte Proliferation, Migration, and Wound Healing |
Keratinocyte proliferation is regulated by multiple negative signals, and the effects of pro-proliferative factors are not always synergistic. Studies have confirmed that transforming growth factor-β (TGF-β) can dose-dependently inhibit HGF- and KGF-induced keratinocyte proliferation and also exhibits inhibitory effects on EGF-induced proliferation.53 Furthermore, vasoactive intestinal peptide (VIP) itself lacks mitogenic activity. This neuropeptide significantly attenuates EGF-induced proliferation in HaCaT cells at 10−7 M by inhibiting EGFR tyrosine kinase activity, suggesting potential antagonistic interactions between neuropeptides and growth factors.54 Conversely, glucocorticoids significantly inhibit this repair process by downregulating K6/K16 keratin expression and blocking EGF-induced proliferation responses, indicating that systemic hormone levels influence local repair efficiency.55–57 Clinical studies further validate these mechanisms. In patients with chronic inflammation or prolonged use of exogenous hormones, the re-epithelialization process exhibits marked delays. This demonstrates that regulating keratinocyte proliferation not only accelerates wound healing but also serves as a critical target for intervening in pathological repair.58
Fibroblast Proliferation
The proliferation and activation of fibroblasts are fundamental to the formation of the dermal repair scaffold and the secretion of factors that promote keratinocyte proliferation; by reshaping the wound microenvironment, they indirectly determine the efficiency of keratinocyte proliferation. The proliferative activity of fibroblasts determines the quality and quantity of granulation tissue, serving as the fundamental force supporting wound filling and structural reconstruction. Following injury, platelets rapidly aggregate and release PDGF-BB, a potent chemokine that not only guides fibroblast migration to the wound site but also directly stimulates their entry into the mitotic cycle.59,60 Subsequently, TGFβ further amplifies this effect and induces some fibroblasts to express α-smooth muscle actin, endowing them with contractile capabilities that help reduce the wound area and create a stable wound matrix for the proliferation and migration of keratinocytes.61 Activated and proliferating fibroblasts are a major source of proliferative growth factors in the wound microenvironment; the various factors they secrete, such as IGF-1, VEGF, and bFGF, can act directly or indirectly on keratinocytes to drive their proliferation.22,59,60 Abnormal proliferation of fibroblasts directly leads to disruption of the dermal matrix scaffold structure, thereby inhibiting the normal proliferation of keratinocytes. The proliferative state of fibroblasts exhibits completely opposite phenotypes across different pathological wound conditions. In some diabetic wounds, despite high concentrations of growth factors, fibroblasts exhibit delayed or arrested proliferation. This indicates that intrinsic signaling pathway impairments and abnormal cell cycle regulation in fibroblasts are fundamental causes of impaired wound healing.62,63 Single-cell transcriptomic analysis reveals that distinct fibroblast subpopulations in aged wounds exhibit delayed regenerative capacity and downregulated expression of pro-reparative genes, alongside activated inflammatory and aging-related pathways.64,65 Conversely, in certain chronic venous ulcers, fibroblasts demonstrate abnormally hyperactive proliferation and migration capabilities.66
However, proliferation is not inherently beneficial; sustained or excessive cell expansion leads to uncontrolled collagen deposition, ultimately resulting in hypertrophic scarring.67 Studies indicate that M2 macrophages exert anti-fibrotic effects via IL-6, yet clinical observations reveal elevated proportions of M2 macrophages in hypertrophic scars. These conflicting conclusions indicate that IL-6’s role in fibrosis depends on local microenvironmental signaling integration.68 This further confirms that, proliferation must maintain a dynamic equilibrium with processes like apoptosis and differentiation to achieve functional rather than structural repair.59,60 Studies reveal that in chronic wounds associated with diabetes or aging, fibroblasts often exhibit delayed or arrested proliferation. Even in the presence of an abundant growth factor environment, their responsiveness remains significantly diminished, suggesting that intrinsic abnormalities in cell cycle regulation may be a fundamental cause of pathological repair.61,69 In the future, targeted modulation of cell cycle checkpoint proteins such as Cyclin D1 or CDK4/6 may emerge as an effective strategy for indirectly addressing keratinocyte proliferation defects and thereby restoring the homeostasis of the wound microenvironment.
Endothelial Cell Proliferation
Endothelial cell proliferation is a key prerequisite for wound angiogenesis. By forming a network of new blood vessels, it supplies the wound healing site with sufficient oxygen and nutrients, thereby providing a critical foundation for maintaining the normal proliferation and metabolism of keratinocytes. At the same time, proliferating and activated endothelial cells can also directly participate in the regulation of keratinocyte proliferation through paracrine signaling. Under hypoxic conditions, HIF1α accumulates stably and upregulates VEGF transcription. As the most potent angiogenic factor, VEGF activates PLCγ-PKC and Raf-MEK-ERK cascades by binding to VEGFR2, thereby promoting endothelial cells to exit G0 phase and enter active mitotic division.24,70,71 Concurrently, adjacent fibroblasts and macrophages secrete bFGF and angiopoietin-1, providing additional paracrine support that not only sustains endothelial cell survival but also promotes luminal structural stabilization.70,72–74 However, the results from the three-dimensional co-culture model diverge from the conventional understanding of endothelial cell-dominated vascular ingrowth. Within the simulated wound’s 3D matrix environment, fibroblasts migrate first and fill the defect, constructing a temporary matrix scaffold. In contrast, endothelial cells predominantly remain at the wound margins and do not actively sprout toward the central wound area.75 Chen et al demonstrated that locally applying concentrated mesenchymal stem cell conditioned medium to mouse wounds significantly increased CD34+ and Flk1+ endothelial progenitor cell numbers, accompanied by enhanced capillary density, confirming the central role of multifactorial synergy in vascular network reconstruction.76
Failure of angiogenesis due to impaired endothelial cell proliferation directly leads to local ischemia and hypoxia at the wound site; hypoxia and nutrient deprivation significantly inhibit the proliferative activity of keratinocytes and may even induce their apoptosis. Notably, neovascularization relies not solely on increased cell numbers; spatial arrangement and maturity are equally critical. Without adequate pericellular envelopment or basement membrane deposition, even extensively proliferating endothelium yields leaky, unstable vessels incapable of sustained blood supply.77 Clinical data further reveal that in ischemic ulcer patients, despite elevated local VEGF concentrations, impaired endothelial cell proliferation responses occur due to metabolic dysfunction or downregulated receptor expression, ultimately leading to failed vascular regeneration.78 Therefore, supplementation with a single factor alone cannot address the fundamental issue; comprehensive consideration of microenvironmental signal integration and intrinsic cellular state regulation is essential.
Macrophage and Immune Cell Proliferation
Macrophages and other immune cells form the core of the inflammatory microenvironment at the wound site; their proliferation, recruitment, and phenotypic polarization are key factors in regulating the initiation, maintenance, and termination of keratinocyte proliferation. Even with limited proliferative capacity, they can exert a decisive influence on the epidermal regeneration process through their secretory functions. Although macrophages lack significant self-renewal capacity, they play a pivotal role in regulating the proliferation of other cells. Monocytes chemotactically recruited to the wound site by CCL2 differentiate into macrophages, predominantly of the M1 phenotype in the early phase. As the repair process progresses, microenvironmental polarization signals prompt macrophages to differentiate into the M2 phenotype, leading them to secrete TGF-β and IL-10. On the one hand, this suppresses excessive inflammatory responses and prevents persistent inflammation from negatively inhibiting keratinocyte proliferation; on the other hand, it maintains moderate proliferative activity in fibroblasts, thereby indirectly supporting epidermal regeneration.79–81
However, the dichotomous classification of macrophages into M1/M2 exhibits significant limitations in applicability within the complex in vivo wound microenvironment. Studies using renal injury models confirm that the phenotypic classification of tissue macrophages should be based on their functional roles during different repair phases, rather than simplistically applying conclusions derived from in vitro polarization-induced models.82 The currently accepted linear perception of M1 macrophages as pro-inflammatory and repair-inhibiting, and M2 macrophages as anti-inflammatory and repair-promoting, oversimplifies the functional diversity of macrophages within the spatiotemporally dynamic wound microenvironment. Photobiomodulation studies further confirm the time-dependent nature of macrophage phenotype switching. Infrared laser exposure transiently upregulates TGFβ1 expression in M2 macrophages at 4 hours post-injury, followed by downregulation at 24 hours. This demonstrates that external interventions can modulate the temporal window for macrophage phenotype conversion, thereby influencing the proliferative behavior of downstream repair cells.83 Beyond macrophages, other immune-related cells also participate in regulating wound proliferation processes through self-proliferation or functional modulation. Circulating fibroblast precursors of bone marrow origin can also migrate to the wound bed under IL4/IL13 stimulation, undergoing limited proliferation and differentiation. Although their contribution is minor in healthy skin repair, they may undertake compensatory repair tasks in certain pathological states, such as extensive burns or radiation injury.68 Furthermore, although neutrophils lack sustained proliferative capacity, their early clearance of pathogens and necrotic debris creates the prerequisites for subsequent cellular proliferation. The overall coordination of immune cells determines whether proliferation can be initiated and terminated within an appropriate time window. Dysregulation at any stage may lead to repair stagnation or excessive proliferation. Lykov et al observed that in the pathological context of persistent infection or autoimmune disorders, the proportion of M1 macrophages at the wound site is abnormally increased, leading to sustained release of pro-inflammatory factors. This not only hinders the normal transition from the inflammatory phase to the proliferative phase but also directly inhibits the functional proliferation of keratinocytes, ultimately resulting in impaired wound healing.80,84
Microenvironment Regulation and Signal Integration
Cell proliferation is never an isolated event; it is always tightly coupled with other biological processes, including migration, differentiation, apoptosis, and even matrix remodeling. Matrix metalloproteinases not only degrade the extracellular matrix to clear pathways for cell movement but also release latent growth factors bound to the matrix, reactivating them to act on target cell receptors.67 Integrins and growth factor receptors often form complexes, amplifying pro-proliferative signals through the FAK-SRC pathway to heighten cellular responsiveness to external stimuli.57,72 Recent studies in single-cell and spatial omics have revealed significant cellular heterogeneity and localized signal gradient distributions within wound microenvironments. This finding suggests that traditional signaling pathway models, based on the assumption of a homogeneous environment, may obscure specific regulatory mechanisms within localized microenvironments. For instance, within a wound, hypoxic regions and vascular frontiers may exhibit fundamentally different responses to the same growth factor among cells of the same type.85,86 Beyond intercellular signaling and matrix microenvironment regulation, fine-tuned epigenetic control directly influences cellular proliferation processes, serving as a crucial intracellular relay for microenvironmental signals. Epigenetic regulation also plays a crucial role: demethylation of histone H3K27me3 lifts transcriptional repression on EGFR and cmys genes, thereby accelerating cell cycle progression.87,88 MicroRNAs provide more precise temporal control: miR-203 downregulation permits p63 expression, maintaining basal cell proliferative potential, while miR-21 overexpression suppresses abnormal hyperproliferation in chronic wounds by targeting EGR3, preventing excessive tissue overgrowth.89,90 These multi-layered regulatory mechanisms collectively ensure efficient proliferation within controlled parameters. Clinically, many refractory wounds lack not growth factors but impaired cellular signaling perception or execution, manifesting as impaired proliferation kinetics.67 In summary, extracellular matrix remodeling, signal pathway amplification, regional differences in the microenvironment, and epigenetic fine-tuning collectively regulate cell proliferation at the wound site and determine the efficiency of proliferative behavior. Impaired keratinocyte proliferation kinetics arise from compromised perception and execution of proliferative signals, along with an imbalance between inflammatory and proliferative processes, ultimately leading to difficult-to-heal wounds. Therefore, the treatment of chronic wounds must move beyond the traditional approach of simply supplementing growth factors and instead focus on restoring the normal regulatory mechanisms of keratinocyte proliferation by reestablishing the signaling homeostasis of the wound microenvironment.
Mechanisms and Regulatory Networks of Keratinocyte Proliferation Inhibition in Diabetic Wounds
One of the core pathological features of impaired wound healing in diabetes lies in the compromised proliferative capacity of keratinocytes. This impairment directly obstructs the re-epithelialization process, trapping the wound in a chronic, difficult-to-heal state. Keratinocyte proliferation suppression arises not from a single factor but from the interplay of multiple pathological mechanisms, hyperglycemia-mediated metabolic toxicity, epigenetic abnormalities, disrupted extracellular matrix remodeling, microenvironmental imbalance, hypoxia-ischemia, and dysbiosis of the microbiome (Figure 2). These factors engage in complex molecular cross-talk and synergistically disrupt keratinocyte proliferation pathways at multiple levels. These levels include intracellular metabolism, gene expression regulation and extracellular microenvironmental support, and this disruption ultimately exacerbates delayed wound healing.
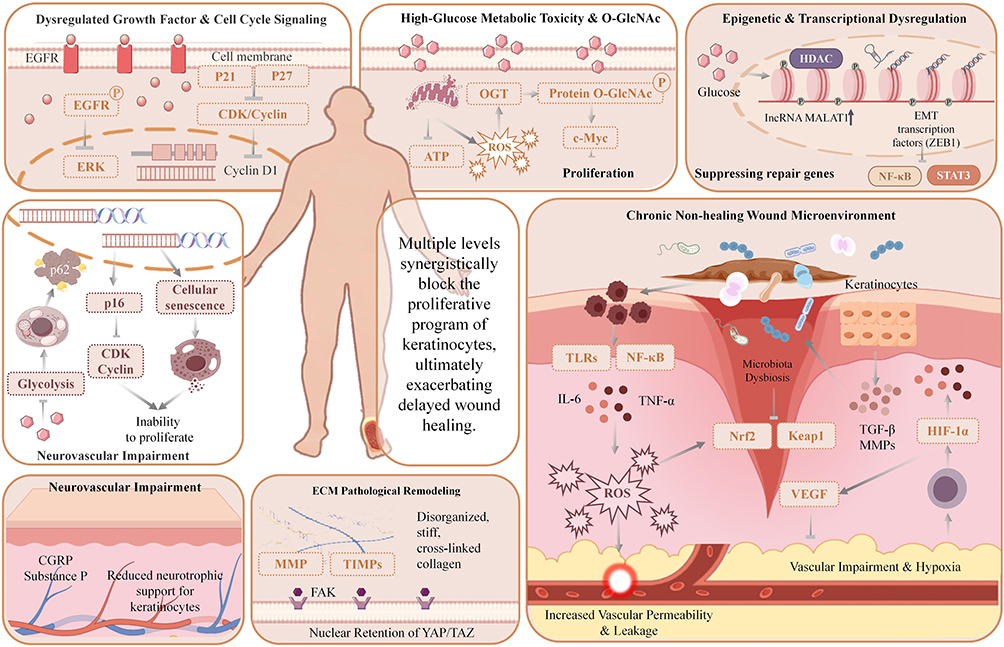 |
Figure 2 Schematic diagram of multiple synergistic mechanisms blocking keratinocyte proliferation in diabetic wounds. Disruption of EGR-1/EGFR signaling, IL-6/TNF-α-mediated chronic inflammation, dysregulated NF-κB pathway, high-glucose-induced imbalance of KLF5/TGF-β1, and dysfunction of O-GlcNAc-modified c-Myc collectively synergize to impede wound healing. Upward arrows (↑) indicate upregulation or activation of relevant molecules and signaling pathways; downward arrows (T) indicate downregulation or inhibition of relevant molecules and signaling pathways. |
High-Sugar Metabolic Toxicity
The effects of hyperglycemic metabolic toxicity include abnormalities in core molecular events such as post-translational modifications and metabolic enzyme activity, as well as imbalances in cellular physiological processes like autophagy flux and cellular senescence. Zhang et al discovered that abnormal elevation of O-GlcNAc glycosylation in high-sugar environments enhances c-Myc protein stability. This simultaneously activates downstream S100A6 molecules and directly traps cells in an abnormal proliferative state, preventing initiation of the migration repair program.91 Both metabolic reprogramming and post-translational modifications stem from early molecular disruptions induced by high glucose, ultimately impairing keratinocytes’ core repair capacity. Methylglyoxal generated under hyperglycemia damages keratinocytes and specifically induces significant upregulation of fructose-1,6-bisphosphatase 1 (FBP1). Further studies indicate that O-GlcNAc glycosylation modification can also directly arrest the G1/S transition in keratinocytes by regulating Cyclin D1 expression. This mechanism synergistically amplifies the proliferation-inhibitory effect through the c-Myc-mediated regulatory pathway. FBP1, a key negative regulator of glycolysis, becomes hyperactivated under hyperglycemia. This directly inhibits glycolytic flux, limiting intracellular ATP production which serves as the energy foundation for keratinocyte migration and proliferation. Insufficient energy supply directly leads to keratinocyte functional paralysis.92 Additionally, high glucose can induce downregulation of phosphofructokinase-2 (PFK-2) expression, further inhibiting glycolysis. This, combined with the abnormal activation of FBP1, exacerbates energy metabolism disorders. Simultaneously, high-sugar-mediated metabolic dysregulation extends beyond keratinocytes themselves, disrupting the energy homeostasis of fibroblasts and endothelial cells within the wound microenvironment. This leads to reduced secretion of pro-repair growth factors by these cells, indirectly impairing keratinocyte proliferation and creating a vicious cycle of multi-cell synergistic dysfunction. Asiatic acid and its H₂S donor derivative AA4 restore metabolic homeostasis by dual inhibition of FBP1, accelerating wound healing in diabetic mice. This demonstrates the high intervention feasibility of metabolic enzyme reprogramming mechanisms. Autophagy dysfunction represents a downstream cascade triggered by hyperglycemic metabolic toxicity, further amplifying keratinocyte functional impairment. Under high-glucose conditions, suppressed AMPK/ULK1 pathway activity reduces LC3II conversion and increases p62 accumulation, preventing keratinocytes from clearing damaged proteins and organelles via autophagy. This impairs proliferation and migration capabilities. Conversely, EGCG activates the AMPK/ULK1 pathway to restore the autophagy flux, not only directly improving keratinocyte function but also indirectly stimulating collagen synthesis in fibroblasts.93,94 In diabetic skin, downregulation of the RNA m6A demethylase FTO leads to elevated m6A modification levels on TRIB3 mRNA, which is subsequently recognized and degraded by YTHDF2. The resulting reduction in TRIB3 directly inhibits autophagy initiation and exacerbates keratinocyte apoptosis. Overexpression of FTO or TRIB3 partially reverses this phenotype, confirming the central role of m6A modification in autophagy regulation.7 Although these pathways target different molecules, both exacerbate cellular damage by blocking the autophagy flux. A high-sugar environment induces massive accumulation of reactive oxygen species (ROS) within keratinocytes while activating the receptor for advanced glycation end products (RAGE) pathway. These factors jointly upregulate p21 expression, triggering premature senescence that impairs keratinocyte proliferation. Concurrently, reduced synthesis of migration-related cytoskeletal proteins prevents participation in wound re-epithelialization.95 Conversely, human amniotic epithelial cell conditioned medium (hAECs-CM) effectively reverses high-glucose-induced premature senescence in keratinocytes by scavenging ROS and downregulating the RAGE/p21 pathway, thereby restoring their proliferation and migration functions. Additionally, ROS can oxidatively damage DNA, leading to activation of the p53 pathway, which further blocks the cell cycle and exacerbates the suppression of keratinocyte proliferation. Excessive ROS can diffuse into the extracellular space, mediating systemic oxidative stress imbalance within the wound microenvironment. This intensifies endothelial cell damage and inflammatory cascades, synergizing with pathological processes such as microbial colonization and matrix degradation to further deteriorate the repair microenvironment for keratinocytes.96,97 Hyperglycemia first induces early abnormalities in post-translational modifications and metabolic enzymes which directly disrupt the functional foundation of cells, triggering autophagy flow arrest and amplifying cellular damage. Ultimately, oxidative stress triggers premature senescence, leading to the complete loss of cellular survival and repair potential.
Epigenetic Modification Abnormalities: Multidimensional Mechanisms Synergistically Lock Keratinocyte Function in an Inhibited State
Epigenetic modification abnormalities serve as a core regulatory factor in suppressing keratinocyte function in diabetic wounds, with effects spanning histone modifications, DNA methylation, and non-coding RNA-mediated chromatin remodeling. High glucose-induced histone modification dysregulation directly silences repair-related genes, inhibiting core keratinocyte repair functions at the transcriptional level. Downregulation of IFNκ expression in wound-edge keratinocytes of type 2 diabetes (T2D) patients is closely associated with reduced H3K4me3 enrichment in the promoter region and silencing of the histone methyltransferase MLL1. Exogenous supplementation of IFNκ reverses these phenotypes, restoring early inflammatory responses, collagen deposition, and re-epithelialization.98 Long non-coding RNA MALAT1 is abnormally upregulated in high-glucose environments, driving TGF-β1-induced epithelial-mesenchymal transition (EMT) by activating ZEB1 and inhibiting keratinocyte migration. Silencing MALAT1 reverses EMT by restoring epithelial markers and releasing miR-205-mediated ZEB1 inhibition, thereby improving migration capacity.99 The lncRNA UCA1, abnormally upregulated in high-glucose conditions, activates the NF-κB inflammatory axis by binding METTL14 to stabilize HIF-1α protein. Simultaneously, it targets miR-140-5p to upregulate SOX9 expression, directly arresting the keratinocyte cell cycle and inhibiting proliferation. This dual pathway of inflammatory activation and cycle regulation exacerbates keratinocyte dysfunction.100
MicroRNAs (miRNAs), another crucial class of non-coding RNAs, extensively participate in suppressing keratinocyte proliferation by regulating target gene expression. Selective splicing of the JAM-A gene 3′-UTR releases miR-106b-5p, which targets and inhibits the PTEN/TIAM1 pathway, leading to hyperproliferation but migration arrest in keratinocytes.6 Under high-glucose conditions, miR-106b-5p expression further increases, not only inhibiting the PTEN/TIAM1 pathway but also directly suppressing Cyclin E1 expression to arrest the cell cycle and exacerbate proliferation suppression. CDK1-loaded extracellular vesicles (CDK1-sEVs) can restart the cell cycle by activating the AKT/ERK pathway, promoting re-epithelialization.101 DNA methylation and demethylation dynamics in macrophages, endothelial cells, and keratinocytes dynamically regulate wound healing-related signaling pathways; for instance, impaired TET3-mediated demethylation of the DSP gene impedes re-epithelialization.102,103 In the diabetic microenvironment, histone deacetylase activity (HDAC) inhibition by butyrate locally restores macrophage epigenetic programming, promoting inflammation resolution and tissue repair by suppressing STAT1 signaling through histone deacetylation.104
Pathological Remodeling of the Extracellular Matrix: Dysregulation of Keratinocyte Function Mediated by Mechanical Microenvironment Disturbances
In diabetic wounds, ECM structural disorganization and abnormal mechanical properties driven by high glucose and chronic inflammation can directly block keratinocyte repair programs by inhibiting the mechanosensitive YAP/TAZ signaling pathway. Existing animal studies confirm that the Agrin-MMP12 positive feedback loop can restore YAP/TAZ signaling, and treatment with its derivative sAgrin significantly accelerates wound healing.105 The YAP activator PY-60 also enhances YAP transcriptional activity by inhibiting its ubiquitination and degradation, thereby reversing keratinocyte functional suppression mediated by hyperglycemic ECM disruption.
Pathological ECM remodeling further disrupts keratinocyte-matrix interactions and functional homeostasis through additional molecular pathways. Dock5 deficiency leads to abnormal accumulation of ZEB1 protein, disrupting laminin-332/integrin axis stability. This not only causes defects in wound collagen deposition and exacerbates ECM structural disorganization but also directly impairs cell proliferation and migration by inhibiting Rac1 signaling-regulated keratinocyte cytoskeletal reorganization. Restoring Dock5 expression reverses all these pathological phenotypes.106 Concurrently, downregulated RIG-I expression in diabetic wounds reduces TIMP-1 secretion, disrupting the MMP/TIMP degradation-synthesis equilibrium and further exacerbating ECM structural damage. Beyond accelerating matrix degradation, diminished TIMP-1 directly inhibits keratinocyte proliferation by modulating the PI3K/AKT pathway, achieving synergistic effects in ECM remodeling and cellular function suppression.107
The pathological microenvironment of the ECM can also directly impede keratinocyte cell cycle progression and inhibit cell migration by interfering with downstream signaling pathways. In diabetic wounds, the weakened activity of the NO-cGMP signaling pathway not only blocks keratinocyte cell cycle progression and suppresses cell migration but also inhibits Cyclin D1-CDK4 binding by upregulating p27 expression, further obstructing cell cycle advancement and synergistically suppressing keratinocyte proliferation capacity. In contrast, the small-molecule agonist TOP-N53 can simultaneously reverse these proliferation and migration dysfunctions by elevating cGMP levels.108 In summary, pathological ECM remodeling synergistically suppresses keratinocyte function through multiple pathways, including mechanosensing, matrix interactions, and cell cycle regulation serving as a key driver of delayed re-epithelialization in diabetic wounds. This mechanism also identifies multidimensional potential targets for wound-specific therapeutic interventions.
Diabetic Wound Microenvironment
The biochemical and physical disturbances in the diabetic wound microenvironment systematically suppress keratinocyte proliferation and migration through multiple mechanisms. ECM homeostasis disruption and structural damage form a direct inhibitory barrier, while the hyperglycemic environment drives sustained overexpression of MMP-9 and MMP-8. These proteases not only degrade key ECM components like collagen and laminin but also disrupt the local homeostasis of pro-repair growth factors such as VEGF and bFGF.109
RIG-I deficiency further downregulates TIMP-1, thereby releasing the negative regulation on MMPs and exacerbating excessive degradation and structural disruption of the ECM. Concurrently, it fails to activate the PI3K/AKT pathway by binding to the CD63 receptor on keratinocyte surfaces, directly losing its pro-proliferative and anti-apoptotic effects on keratinocytes. This dual mechanism intensifies keratinocyte dysfunction.101 However, local delivery of recombinant TIMP-1 can simultaneously reverse these pathological alterations, restoring ECM homeostasis and keratinocyte-mediated re-epithelialization processes. Correlated with this loss of basement membrane structural integrity, Dock5 deficiency downregulates LAMA3 expression, disrupting integrin α6β4 signaling and severing desmosomal connections between keratinocytes and the basement membrane. This deprives keratinocytes of their physical anchorage, directly impeding migration.106 Disruption of vascular nourishment indirectly impairs keratinocyte function. High glucose abnormally activates fibroblasts to stimulate IL-7/IL-7R axis-mediated ANGPTL4 secretion, inhibiting endothelial lumen formation and severing vascular support. Blocking ANGPTL4 simultaneously enhances vascular density and keratinocyte proliferation.110 Concurrently, certain endogenous molecules retain repair-promoting potential but remain suppressed by the microenvironment, hindering their efficacy. The antimicrobial peptide AMP-IBP5 derived from IGFBP5 directly promotes keratinocyte migration and induces angiogenesis via the EGFR-STAT1/3-MAPK pathway.111 This effect depends on LRP1-mediated signaling, but in the disrupted wound microenvironment, such positive pathways are often overshadowed by the aforementioned negative mechanisms. In summary, diabetic wounds synergistically suppress keratinocyte proliferation potential through multiple mechanisms.
Chronic Hypoxia
The core pathological mechanism underlying impaired wound healing in diabetes revolves around chronic hypoxia, with dysregulation of hypoxia-inducible factor-1α (HIF-1α) playing a pivotal role. The hyperglycemic environment does not simply suppress or activate HIF-1α, but rather differentially regulates it according to cell type and repair stage, thereby suppressing the reparative functions at multiple levels.
During the early inflammatory phase, hyperglycemia drives abnormal HIF-1α activation in macrophages. This prolongs the proinflammatory state by activating pathways such as NF-κB and iNOS, hindering the transition to the repair phase.112 Manifestations include persistent inflammatory infiltration and heightened oxidative stress, leading to insufficient VEGF secretion and impaired angiogenesis.113 Concurrently, abnormally activated HIF-1α promotes macrophage secretion of proinflammatory factors such as TNF-α and IL-1β, thereby inhibiting keratinocyte proliferation. However, this pattern reverses upon entering the proliferative repair phase. In keratinocytes and endothelial cells, HIF-1α expression and stability are significantly suppressed. This leads to reduced transcriptional levels of downstream target genes such as vascular endothelial growth factor (VEGF), heme oxygenase-1 (HO-1), and inducible nitric oxide synthase (iNOS), ultimately resulting in insufficient angiogenesis and delayed epithelial regeneration.114 Further studies revealed that the inhibition of HIF-1α in proliferating keratinocytes is closely associated with increased ubiquitin-mediated degradation. A high-glucose environment induces upregulation of VHL protein expression, promoting HIF-1α ubiquitin-mediated degradation while simultaneously suppressing its transcriptional activation, thereby exerting a dual inhibitory effect.115,116 These mechanisms are primarily based on in vitro cellular experiments. Their specific regulatory roles within the complex microenvironment of diabetic wounds require further clarification.
Beyond HIF-1α, chronic hypoxia suppresses keratinocyte proliferation through additional pathways. Hypoxic conditions trigger ROS accumulation within keratinocytes, activating the p38 MAPK pathway to inhibit Cyclin D1 expression and arrest cell cycle progression.117 Hypoxia induces premature senescence in keratinocytes by upregulating p21 and p16 expression, thereby suppressing proliferation.9,11 Furthermore, hypoxia affects ECM synthesis and degradation, exacerbating ECM remodeling disorders and indirectly inhibiting keratinocyte proliferation; this is consistent with pan-cancer evidence showing hypoxia-driven ECM alterations via HIF-dependent gene regulation.118,119 In summary, the essence of impaired wound healing in diabetic conditions lies in the spatiotemporal dysfunction of HIF-1α signaling and the synergistic interaction between hypoxia and other pathological mechanisms.120 Future therapeutic approaches must shift toward multi-target synergistic interventions integrating metabolic regulation, immune reprogramming, and microenvironment engineering to effectively break the pathological cycle and achieve functional tissue regeneration.
Microbiome Dysbiosis
In the hyperglycemic, hypoxic microenvironment of diabetic wounds, dysbiosis of the microbiome is a key pathological factor that exacerbates keratinocyte proliferation inhibition and drives chronic wound progression. Chronic, difficult-to-heal wounds commonly exhibit reduced microbial diversity and dominant colonization by pathogens such as Staphylococcus aureus and Pseudomonas aeruginosa, which suppress keratinocyte proliferation through multiple direct mechanisms. First, pathogens can directly cause keratinocyte proliferation arrest and damage via virulence factors. Xanthine substances released by Staphylococcus aureus directly induce accumulation of pro-inflammatory mediators within keratinocytes while blocking cell cycle progression, thereby inhibiting proliferation at its source.121 Its secreted α-hemolysin directly disrupts keratinocyte membrane integrity, inducing apoptosis and reducing the number of proliferating keratinocytes. Second, pathogens can directly suppress keratinocyte proliferation by mediating immune amplification effects. Staphylococcus aureus superantigen SEB activates excessive T-cell proliferation through antigen presentation mediated by keratinocyte exosomes. The resulting secretion of cytokines such as IFN-γ and TNF-α directly inhibits keratinocyte cell cycle progression, forming an immune-mediated proliferation suppression.122 Additionally, dysregulation of the microbiome’s metabolism directly impairs keratinocyte proliferation capacity. Under normal physiological conditions, short-chain fatty acids (SCFAs) produced by skin commensal bacteria activate the GPR43 pathway to promote keratinocyte proliferation. However, dysbiosis in diabetic wounds leads to significantly reduced SCFA secretion, thereby diminishing this proliferative effect. Simultaneously, endotoxins produced by pathogenic bacteria activate the TLR4/NF-κB pathway, inducing pro-inflammatory factor secretion in keratinocytes, that directly inhibits cell proliferation and promotes apoptosis.
Neuropeptide Substances
Beyond overt pathological factors such as pathogen invasion and immune-metabolic imbalance, the neuropeptide dysregulation triggered by diabetic neuropathy represents a significant latent factor that inhibits keratinocyte proliferation and impedes wound healing. Neuropeptides serve as core signaling mediators regulating the local wound microenvironment within the nervous system. Their abnormal expression and function can suppress keratinocyte proliferation through multiple direct pathways. Substance P (SP) and calcitonin gene-related peptide (CGRP) represent the most pivotal pro-repair sensory neurotransmitters in skin tissue. Under healthy conditions, both directly activate the cAMP/PKA pathway within keratinocytes via their respective receptors. On one hand, this initiates the expression of cell cycle-related proteins, directly promoting keratinocyte proliferation. Concurrently, they inhibit the TLR4-MyD88 pathway to reduce production of proinflammatory factors like IL-6, thereby establishing a stable microenvironment conducive to keratinocyte proliferation.123 Clinical biopsy data reveal reduced PGP9.5-positive nerve fiber density in the dermis at wound margins of diabetic foot ulcer patients compared to healthy controls, directly diminishing neuropeptide-mediated proliferative effects.124 Furthermore, SP can bypass ligand-dependent receptor endocytosis through transactivation of EGFR, directly activating proliferative signaling pathways within keratinocytes. However, diabetic-induced nerve fiber degeneration reduces endogenous SP release below critical thresholds, rendering this compensatory pathway ineffective. Moreover, the absence of reparative neuropeptides impairs local wound angiogenesis and hinders inflammation resolution. By deteriorating the microenvironment, this indirectly weakens keratinocyte proliferation capacity. Compensatory increases in corticotropin-releasing factor (CRF) and α-melanocyte-stimulating hormone (α-MSH) further exacerbate the inhibitory effect on keratinocyte proliferation by promoting pro-inflammatory factor release and reducing paracrine growth factors125 (Table 2).
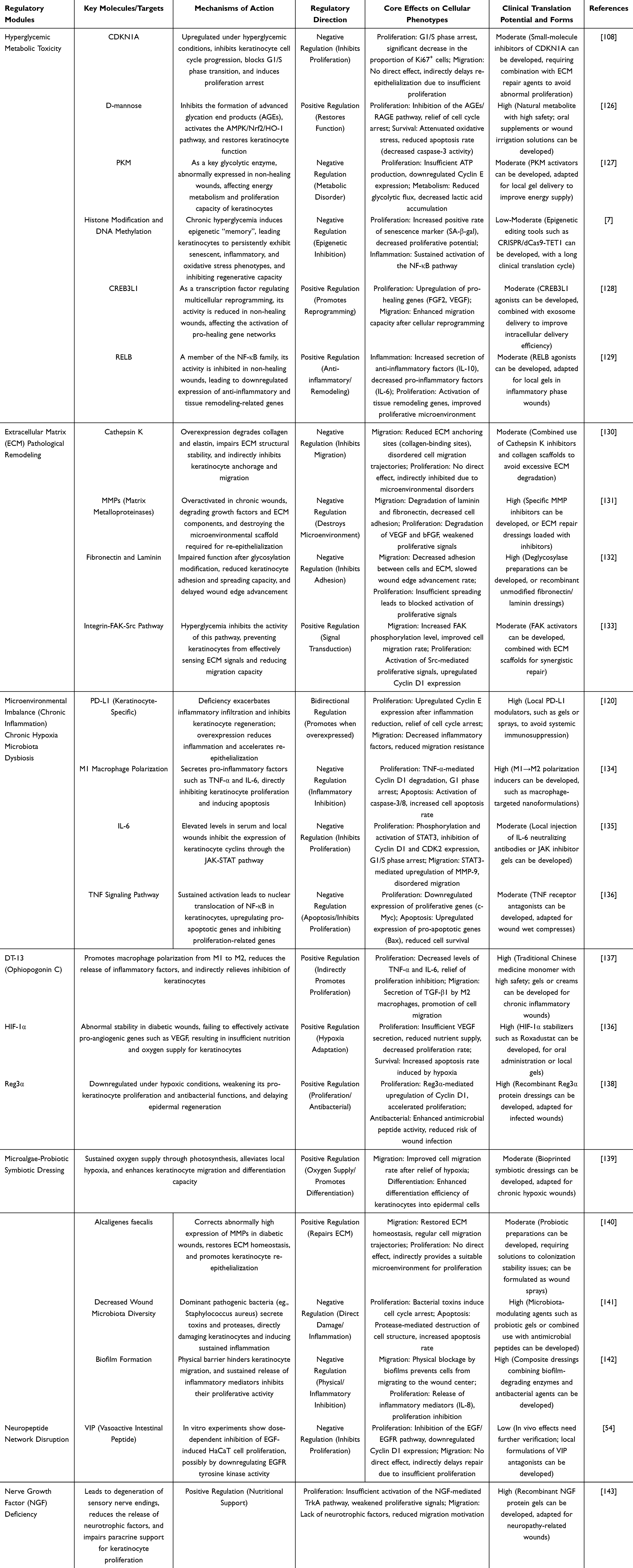 |
Table 2 Core Regulatory Modules and Key Targets for Inhibition of Keratinocyte Proliferation in Diabetic Wounds |
Imbalance of Wound Microbiome and Indirect Inhibitory Effects on Biofilm Formation
Pathogenicity Factors of Pathogens
Pathogenic bacteria disrupt host cell signaling pathways and structural integrity by secreting specific virulence factors. Even at low colonizing densities, pathogens can induce significant tissue damage, a process forming the initial pathological basis for chronic wound formation. Staphylococcus aureus and Streptococcus pyogenes utilize staphylococcal yellow pigment and SpeB protease as key virulence mediators, respectively. Although both target the extracellular matrix or inflammatory pathways, they exhibit highly specific and functionally differentiated characteristics at the molecular level. This feature of mechanistic differentiation with functional convergence reflects the pathogens’ evolutionary adaptation to maximize host microenvironment disruption efficiency at minimal metabolic cost. Staphylococcal yellow pigment secreted by Staphylococcus aureus confers an antioxidant barrier function to resist reactive oxygen species generated by neutrophil respiratory burst.144 Furthermore, staphylococcal yellow pigment activates the p38-MAPK pathway to induce excessive release of IL-6 and TNF-α, arresting keratinocytes in the G0/G1 phase.145 Unlike the virulence mechanism of Staphylococcus aureus, Streptococcus pyogenes disrupts the host microenvironment through SpeB protease-mediated protein cleavage. The SpeB protease secreted by Streptococcus targets and cleaves desmoglein-1, a core glycoprotein of desmosomes. This cleavage disrupts the integrity of intercellular junctions in epidermal cells while degrading fibronectin and laminin, causing migrating keratinocytes to lose their anchoring footholds. Proteases from clinical isolates also specifically cleave the extracellular domain of integrin β1. This cleavage blocks the FAK/Src signaling axis, inhibiting cytoskeletal reorganization and ultimately inducing significant epithelial regeneration impairment even under low bacterial colonization density.146 The synergistic degradation of staphylococcal yellow pigment and SpeB protease ultimately leads to the collapse of epithelial barrier function and epithelial regeneration impairment. This pathogenic mechanism, targeting critical regulatory nodes in the host, reflects the highly efficient pathogenicity developed by the pathogen through long-term coevolution and lays the pathological foundation for subsequent chronic wound progression.
Existing research on the pathogenic mechanisms of virulence factors presents contradictory findings. On one hand, the microenvironment of multi-species mixed infections significantly reshapes the expression of virulence and pathogenic effects in pathogens, disrupting the cognitive patterns established in single-species studies. For example, in mixed infection models, Staphylococcus aureus can secrete β-lactamases to protect coexisting Streptococcus pyogenes from penicillin-based antibiotics. This not only increases the risk of drug resistance but also alters the duration of Streptococcus pyogenes colonization and the kinetics of virulence output,147 indicating that single-species virulence mechanisms require reassessment in the context of multi-species interactions. Conversely, the pathogenic efficacy of virulence factors is highly dependent on the host’s microenvironmental state, with pathogen colonization and virulence release resulting from bidirectional interactions between microbe and host. Recent animal studies indicate that low-density bacterial biofilms can only induce chronic wound progression when accompanied by a concurrent increase in local oxidative stress levels at the wound site.148 Characterized by hyperglycemia, hypoxia, and immune dysfunction, the unique microenvironment of diabetic wounds not only amplifies the damaging effects of virulence factors but also reciprocally modulates the expression of pathogenic virulence genes. This forms a core barrier to the clinical translation of conclusions derived from in vitro experiments.
Immune-Metabolic Dysregulation
On the basis of initial damage caused by pathogens, the wound microenvironment in diabetic patients undergoes specific disruption. The synergistic imbalance between immune and metabolic processes becomes the core driver of wound healing failure, further exacerbating the pathological progression. The essence of impaired wound healing in diabetes lies in a multisystemic disorder triggered by immune homeostasis disruption. The core mechanism involves immune polarization-driven amplification of the inflammatory cascade, coupled with the dual synergistic effects of matrix remodeling and nutrient deprivation. In the pathological immune polarization process of diabetic wounds, pathogen-associated molecular patterns (PAMPs) maintain the stable presence of the M1 pro-inflammatory phenotype in macrophages through sustained activation of the TLR4/MyD88 pathway. This is driven by a positive feedback loop in the NF-κB pathway and persistent dysregulation of associated epigenetic modifications.149 The persistent accumulation of pro-inflammatory macrophages further triggers a series of downstream pathogenic effects. Diabetic wounds exhibit significantly elevated densities of CD86+ M1 macrophages with pathologically altered secretory profiles. The released IL-1β and TNF-α synergistically suppress keratinocyte cyclin D1 expression, directly arresting cell cycle progression. On the other hand, they disrupt the activity balance of matrix metalloproteinases by upregulating TIMP-1, jointly forming a dual inhibition of the epithelial regeneration program. However, recent studies confirm that this pro-inflammatory phenotype of macrophages is not entirely irreversible, with their functional abnormalities exhibiting an intervenable window of plasticity. Compounds such as apigenin and curcumin can effectively reverse the pro-inflammatory dominant phenotype of macrophages by upregulating miR-21 and promoting fatty acid oxidation metabolism, thereby accelerating wound closure in diabetic mice.150,151 This provides experimental evidence for targeted intervention in abnormal macrophage polarization.
These macrophages also lose their ability to transition to the M2 phenotype due to impaired STAT6 phosphorylation and defective PPARγ nuclear translocation.152 Functional loss of the TFAP2A-LIFR-Hippo-YAP axis, coupled with METTL16-mediated m6A modification abnormalities, further exacerbates the pro-inflammatory state. Concurrently, the coupled effects of matrix remodeling and nutrient deprivation synchronously deteriorate the wound microenvironment. Pathogenic bacteria-secreted proteases synergize with host-derived MMP-9 to form a degradative network, increasing wound matrix stiffness. Bacterial predatory consumption of nutrients induces local energy metabolism disruption. The hypoxic environment under multiple infections further induces mitochondrial autophagy, diminishing energy production efficiency. Neutrophil-released NETs exacerbate matrix degradation and amplify inflammatory responses, creating a vicious cycle.153 This synergistic imbalance between immunity and metabolism is not irreversible. Zhang et al demonstrate that nanoenzymes such as Zn-DHM can restore glucose homeostasis by downregulating hexokinase 2 (HK2) and the AGE-RAGE pathway, while simultaneously modulating the Th17/Treg balance, thereby breaking the aforementioned vicious cycle.154 This highlights the potential therapeutic value of metabolic reprogramming.
In summary, the synergistic effects of abnormal immune polarization, disordered matrix remodeling, and imbalanced metabolic reprogramming in diabetic wounds collectively construct a microenvironment that suppresses the repair process. However, existing research remains significantly limited. Most mechanistic studies are confined to the simplified M1/M2 binary model, with insufficient analysis of the functional roles of heterogeneous macrophage subpopulations and the core molecular hubs of immune-metabolic interactions. Clinical intervention strategies must transcend the traditional framework of simple anti-inflammatory approaches or growth factor supplementation, instead targeting the restoration of immune cell plasticity to reconstruct the metabolic microenvironment of the wound.
Biofilm-Structured Defense
When the wound microenvironment continues to deteriorate, it impedes the formation of the repair biofilm, directly leading to irreversible stagnation in wound healing and highlighting the complexity of chronic wound pathogenesis. During the pathological progression of chronic wounds, the biofilm establishes a self-sustaining, host-repair-resistant dynamic microecosystem through intricate biochemical and cellular mechanisms. The core of the biofilm’s defense system lies in structural heterogeneity and functional synergy, where diverse microbial populations form hierarchical protective networks through metabolic division of labor and spatial arrangement. Alginate secreted by Pseudomonas aeruginosa cross-links with PIA polysaccharides produced by Staphylococcus aureus, forming a dense hydrogel matrix that significantly inhibits the diffusion of antibiotic molecules.155,156 Candida albicans enhances matrix rigidity through β-glucan deposition while increasing resistance to host immune-mediated killing.157,158 Biofilms can also drive inflammatory homeostasis imbalance by pulsatile release of sublethal concentrations of pathogen-associated molecular patterns, inducing pathological remodeling of adhesion molecule expression profiles. This alteration leads to immune cell dysfunction and keratinocyte desensitization.159,160 Furthermore, nanomaterials such as silver nanoparticle-loaded hydrogels or DJ-K5 antimicrobial peptide combination therapies can significantly enhance antibiotic clearance efficiency against mixed biofilms by disrupting matrix structure or interfering with quorum sensing systems.161 Considering the pathophysiological characteristics of chronic wound healing, the pathological network formed by biofilms represents the core obstacle causing healing stagnation. This bottleneck substantially increases infection recurrence rates and the risk of major amputation.
The structural characteristics and pathogenic mechanisms of the aforementioned biofilms are largely derived from in vitro single- and dual-bacterial culture models and acute rodent infection models, raising questions about their applicability in real clinical settings. Although multiple meta-analyses confirm the widespread presence of biofilms in human chronic wounds,162 some studies suggest that biofilms represent secondary colonization following impaired wound healing capacity rather than the initiating factor driving chronicity. This controversy remains unresolved. Given this research landscape, an integrated research framework is urgently needed. This framework should not only target disrupting the synergistic pathological chain involving virulence factor invasion, immune-metabolic imbalance, and biofilm defense formation but also prioritize developing point-of-care diagnostic tools capable of simultaneously monitoring bacterial protease activity and host inflammatory markers. This approach would enable precise stratification of pathological stages and personalized interventions.163,164
The pathological mechanisms of wound microbiome dysbiosis and biofilm formation in diabetic wounds can be visually illustrated in Figure 3.
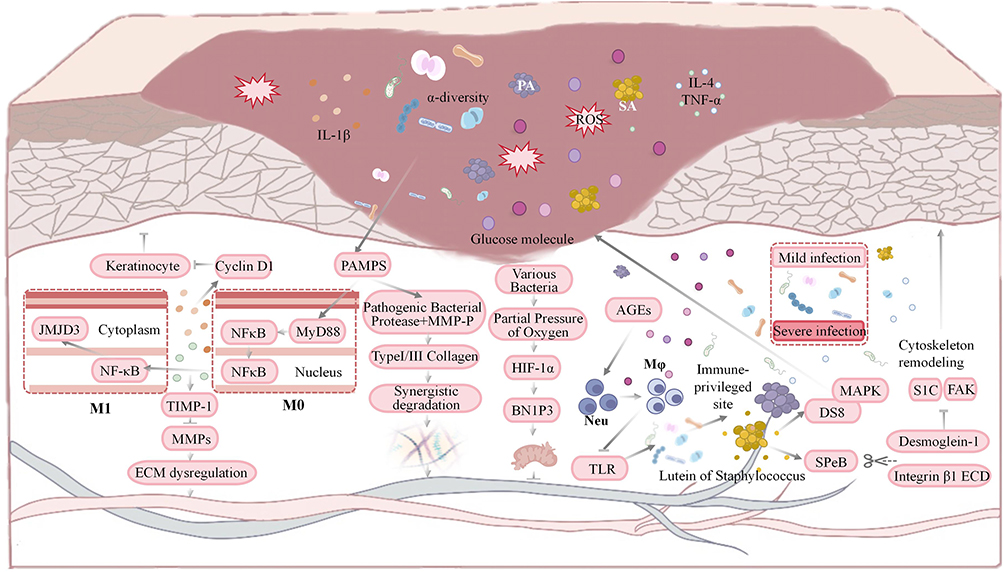 |
Figure 3 This diagram illustrates how microbiome imbalance in diabetic wounds inhibits healing: high glucose, pathogens release virulence factors, activating PAMPs/TLR4/NF-κB to boost pro-inflammatory factors/ROS; MMPs disrupt ECM, while biofilm impairs keratinocyte proliferation. Upward arrows (↑) indicate upregulation or activation of relevant molecules and signaling pathways; downward arrows (T) indicate downregulation or inhibition of relevant molecules and signaling pathways. |
Molecular Hub Links Damage Signals to Keratinocyte Proliferation Program
One of the core pathological features of impaired wound healing in diabetes is the significant suppression of epidermal keratinocyte proliferation capacity. This phenomenon is not caused by a single factor but is driven by a complex regulatory network formed by multiple molecular hubs at the levels of transcriptional regulation, metabolic stress, inflammatory response, neuromodulation, and extracellular vesicle-mediated intercellular communication. These pivotal molecules not only respond to local injury signals but also undergo functional reprogramming or expression dysregulation within the hyperglycemic microenvironment. This systematically disrupts keratinocytes’ ability to enter and sustain a proliferative state. Deepening our understanding of the mechanisms and interactions among these hubs will help uncover the fundamental causes of chronic diabetic wound healing failure and provide a theoretical basis for targeted interventions.
Zinc Finger Transcription Factors: The Dual Dilemma of Transcriptional Reprogramming and Competitive Inhibition
Zinc finger domain proteins are a class of transcription regulators that stabilize their three-dimensional structures through zinc ions and specifically bind to DNA or RNA. Their dysfunction manifests not merely as altered expression levels, but as multi-level regulatory failures including abnormal post-translational modifications, disrupted subcellular localization, and imbalanced recruitment of chromatin remodeling enzymes. This ultimately results in dual dysregulation: silencing of proliferative genes and sustained activation of inflammatory genes.
The microenvironment of diabetic wounds, characterized by hyperglycemia, chronic inflammation, and oxidative stress, directly disrupts upstream signaling pathways of transcription factors. The EGFR-ERK pathway exhibits reduced activity due to accelerated endocytosis, preventing the early-response factor Egr-1 from effectively inducing the expression of cell cycle genes such as Cyclin D1.165,166 However, Lukiw et al indicate that under specific inflammatory stimuli, glucocorticoids can selectively block the expression of inflammatory genes such as COX-2 by inhibiting the NF-κB pathway rather than the ERK pathway. This suggests that the role of Egr-1/Cyclin D1 in proliferation regulation within the complex microenvironment of diabetes remains to be explored.167 Concurrently, inflammatory mediators like TNF-α and IL-6 persistently activate NF-κB and JAK/STAT pathways. This causes RELA and STAT3 to chronically occupy promoters of proliferation-related genes, recruiting epigenetic modifiers to establish repressive marks. This phenomenon is termed transcriptional hijacking. This imbalance further exacerbates global gene silencing by competitively binding co-activators.168 Some in vitro studies indicate that while VIP inhibits EGF-induced proliferation in HaCaT cells, this effect does not occur through the canonical JAK/STAT or cAMP/PKA pathways. The pro-inflammatory and anti-proliferative roles of STAT3 in keratinocytes may be subject to cell type- or ligand-specific constraints.169 Krüppel-like factor 5 (KLF5) exhibits disease-course-dependent functional switching: during the acute phase, it promotes IL-8 secretion to clear necrotic tissue, while in chronic hyperglycemic environments, it triggers TGF-β1 autocrine circuits to induce premature keratinocyte differentiation. This switch correlates with the KLF family’s dual mechanisms promoting repair and fibrosis.170 c-Myc exhibits a glycosylation paradox of high expression with low function. Enhanced O-GlcNAc modification abnormally prolongs protein stability, paradoxically inhibiting timely E2F family activation. Although this mechanism has been extensively reported in tumor cells, no systematic comparative studies have yet been conducted to determine whether O-GlcNAc modification preferentially targets c-Myc, inhibits it, or affects other cell cycle regulators in diabetic keratinocytes.171 Furthermore, whether the degree of glycosylation exhibits a dose-response relationship with the amplitude or duration of blood glucose fluctuations remains poorly elucidated. Studies suggest this paradox may stem from post-translational modification-driven conformational changes.172 These dysfunctions in zinc finger transcription factors are driven by diabetic microenvironmental stress, forming a synergistic regulatory network through post-translational modifications and epigenetic regulation.
The abnormal regulatory mechanisms of zinc finger transcription factors on keratinocytes in diabetic wounds can be visually illustrated in Figure 4.
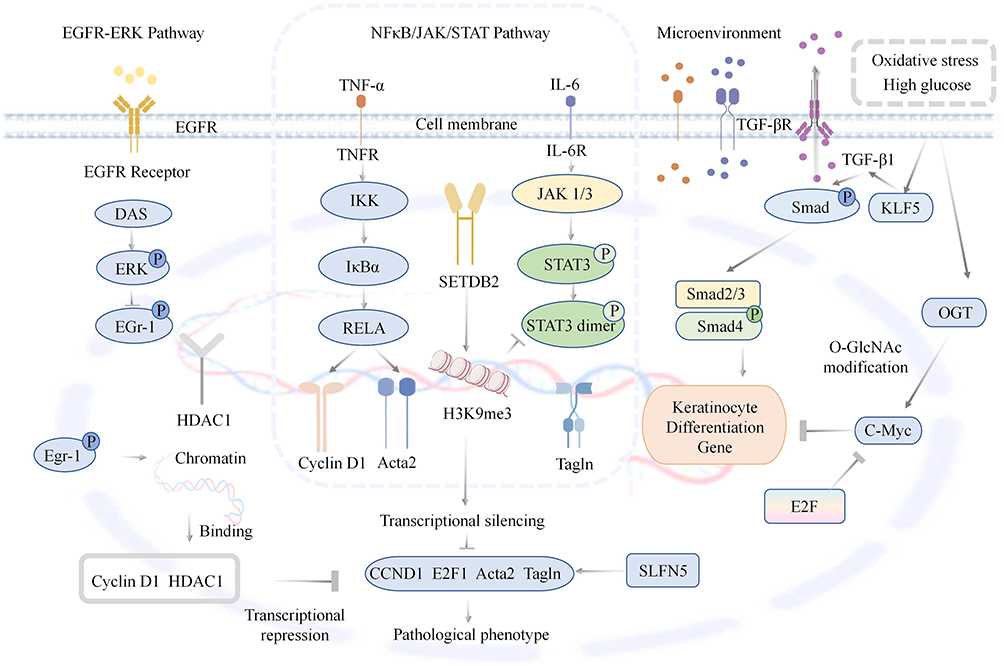 |
Figure 4 This diagram illustrates how zinc finger transcription factors dysregulate keratinocytes in diabetic wounds: EGFR-ERK pathway impairment blocks Egr-1/Cyclin D1 activation; NFκB/STAT3 drive transcriptional silencing of proliferative genes, high glucose-induced KLF5/TGF-β1 promote differentiation and O-GlcNAc-modified c-Myc inhibits proliferation. Upward arrows (↑) indicate upregulation or activation of relevant molecules and signaling pathways; downward arrows (T) indicate downregulation or inhibition of relevant molecules and signaling pathways. |
Hypoxia-Associated Molecules: Metabolic Sensing Dysfunction and Vascular-Epithelial Coupling Disruption
HIF-1α dysfunction exhibits marked tissue specificity and stage dependence. Studies confirm that in the early stage of diabetic foot ulcers, HIF-1α becomes excessively activated in inflammatory cells under hyperglycemic conditions, exacerbating NF-κB-mediated inflammatory amplification. By the late repair phase, HIF-1α activity is significantly suppressed in endothelial and keratinocytes.173 This dynamic shift indicates that therapeutic strategies solely targeting enhanced HIF-1α protein stability may produce counterproductive effects. Under diabetic conditions, O-GlcNAc glycosylation serves as a key mechanism mediating co-activator sequestration and transcriptional suppression. High glucose induces OGT activation, which directly modifies the transcription activation domain of HIF-1α. This modification creates steric hindrance, impeding the effective recruitment of HIF-1α to the transcription machinery.174 Recent studies have revealed that O-GlcNAc modification exerts bidirectional regulation on HIF-1α. Some research indicates that O-GlcNAc modification can suppress HIF-1α’s transcriptional activity by interfering with the binding of coactivators such as p300/CBP to HIF-1α.175,176 Conversely, other studies confirm that under specific cellular conditions or stress states, such as during placental development, an imbalance in the expression of O-GlcNAcylating enzymes OGT and OGA can instead promote angiogenesis by stabilizing HIF-1α protein expression.177 These conflicting findings suggest that the regulatory effects of O-GlcNAc modification on HIF-1α are highly dependent on the cellular microenvironment and substrate specificity of the modification.
Roxadustat intervention experiments have validated the reversibility of this mechanism. This PHD inhibitor not only restores HIF-1α protein levels in db/db mouse models but also reestablishes its transcriptional function, significantly increasing PCNA-positive cell density and wound re-epithelialization rates.178 Iridium metal complexes significantly upregulate downstream target gene expression in diabetic mouse models by blocking VHL-HIF-1α interactions, concurrently accelerating wound closure.179 These studies further confirm that restoring HIF-1α transcriptional activity holds greater therapeutic value than merely maintaining its protein stability.
It must also be clarified that some findings diverge significantly from conclusions drawn in the hyperglycemic environment of diabetes. Studies based on the EPEC effector NleB indicate that certain glycosylation modifications, such as arginine N-acetylglucosaminylation, actually enhance HIF-1α transcriptional activity and reprogram cellular glucose metabolism.180,181 This phenomenon stands in stark contrast to the HIF-1α transcriptional suppression observed in the hyperglycemic environment of diabetes, underscoring the intricate complexity of HIF-1α post-translational modification regulation. Therefore, in advancing the clinical translation of HIF-1α-targeted therapies, it is imperative to carefully distinguish the modification type, cellular localization, and spatiotemporal expression dynamics of HIF-1α across different stages of wound healing to avoid indiscriminate intervention approaches. Several critical questions remain unanswered in this field. Beyond the substrate preferences and model-specific variations in O-GlcNAc modification, precise regulation of HIF-1α activity during the inflammatory and reparative phases remains an unexplored area. Future research should establish more predictive molecular classification systems, propelling this field from descriptive summaries of phenomena toward precision medicine.
Inflammation Regulatory Molecules: The STING Pathway-Driven Immune-Metabolic Vicious Cycle
STING was initially defined as a downstream adaptor protein of the cytoplasmic DNA sensor cGAS, capable of responding to pathogen- or injury-associated DNA to activate the TBK1-IRF3 axis, thereby inducing type I interferon and proinflammatory factor expression.182 However, within the hyperglycemic microenvironment of diabetic wounds, the activation mechanism and pathological effects of STING undergo alteration. The hyperglycemic environment preferentially induces mitochondrial fission and oxidative stress, leading to mitochondrial DNA leakage into the cytoplasm. This DNA is then catalyzed by cGAS to form 2′3′-cGAMP, which binds to and activates STING, initiating pathological inflammatory signaling. In keratinocytes, STING exhibits distinct regulatory characteristics. On one hand, STING activation induces IFN-β production, which through autocrine activation of the JAK1-STAT1 pathway upregulates the cell cycle inhibitors p21 and p27. This subsequently suppresses CDK2/cyclin E complex activity, arresting the cell cycle.183,184 Conversely, STING activation reduces SETDB2 histone methyltransferase expression, diminishing the inhibitory H3K9me3 modification in the TNFα promoter region and releasing silencing of inflammatory genes. Notably, SETDB2 expression itself depends on the STING-mediated IFNβ/JAK/STAT1 pathway.185 This inflammation-signal-driven cell cycle arrest constitutes the unique inflammatory senescence phenotype of keratinocytes in diabetic wounds. Crucially, STING activation forms a vicious cycle with mitochondrial dysfunction, further amplifying pathological effects. Autophagy defects in diabetic keratinocytes cause abnormal STING protein accumulation, intensifying inflammatory activation. Persistent mitochondrial DNA leakage continuously supplies activation signals to the cGAS-STING pathway. Conversely, rapamycin-induced autophagy effectively reduces STING expression and accelerates wound healing.183,184 Strategies like nanozyme hydrogels also block abnormal activation of the cGAS-STING-NLRP3 axis by scavenging ROS and repairing mitochondrial DNA, confirming the pathological significance of this vicious cycle. The regulation of STING in macrophages is more complex. JMJD3 relieves STING gene silencing through epigenetic demethylation, forming the IL-6-JAK/STAT3-STING axis.186 Conversely, in another model, STING promotes macrophage chemotaxis and inflammatory resolution via STAT3.186 These seemingly contradictory outcomes reflect STING’s functional plasticity across distinct immune cell subsets and inflammatory phases. Collectively, STING has emerged as a central hub driving persistent inflammation through metabolic stress in diabetic wounds. Existing evidence supports STING as a therapeutic target, though its spatiotemporal activation patterns and tissue-specific downstream effectors require further clarification.
Neuropeptides: The Disintegration of the Neuro-Immune-Epithelial Triangular Regulatory Network
Diabetic neuropathy induces neuropeptide dysregulation, fundamentally representing the loss of the nervous system’s ability to regulate the local microenvironment. This imbalance acts on keratinocytes through three synergistic pathways: inhibition of angiogenesis, altered macrophage polarization, and direct interference with the cell cycle. Substance P (SP) and calcitonin gene-related peptide (CGRP) are primary sensory neurotransmitters. In healthy skin, both substances activate the cAMP/PKA pathway within keratinocytes via NK1R and CLR/RAMP1 receptors. This dual action promotes vascular endothelial growth factor (VEGF) and fibroblast growth factor-7 (FGF-7) secretion to support angiogenesis, while simultaneously inhibiting the TLR4-MyD88 signaling pathway to reduce interleukin-6 (IL-6) production.187,188
The absence of neuropeptides directly impairs vascular network remodeling. CGRP maintains microvascular vasodilation by inhibiting endothelin-1 synthesis; its deficiency reduces wound perfusion pressure, further exacerbating local hypoxia.189,190 More critically, it compromises immune regulatory function. SP induces macrophage expression of arginase-1 and secretion of interleukin-10 (IL-10), promoting macrophage polarization toward the M2 phenotype. This effect is completely absent in db/db mouse models.191
Under physiological conditions, SP can directly activate keratinocyte proliferation signals through the EGFR transactivation mechanism. However, diabetic-induced nerve fiber degeneration renders this compensatory pathway completely ineffective. Existing animal studies confirm that exogenous supplementation of NGF or CGRP can partially reverse the pathological phenotype of neuropeptide deficiency and accelerate wound healing,192,193 indicating that the wound site retains responsiveness to neuropeptides.
Exosomes
As nanocarriers for intercellular information exchange, the pathological restructuring of exosome contents creates molecular transmission barriers in diabetic wounds. This impairs the ability of diabetic wound keratinocytes to receive pro-proliferative signals from mesenchymal stem cells and fibroblasts, becoming a key factor hindering wound repair. A hyperglycemic environment first disrupts secretory cell function, causing mitochondrial dysfunction and autophagy flow blockage in bone marrow mesenchymal stem cells (BMSCs), resulting in the loss of proliferative activity in their exosomes. Healthy BMSC exosomes can increase the S-phase proportion in high-glucose HaCaT cells, whereas those derived from diabetic sources do not exhibit this effect.194 Mahheidari et al indicate that adipose-derived mesenchymal stem cells (ADSCs) and BMSCs produce functionally distinct exosomes. ADSC-derived exosomes are enriched in pro-angiogenic factors, primarily enhancing wound perfusion. Conversely, BMSC-derived exosomes concentrate cell cycle regulators such as CCND1 and CDK4 mRNA, more directly driving keratinocyte and fibroblast proliferation.195 This disparity necessitates selecting the appropriate exosome source based on the primary defect in the wound during clinical translation.
Abnormal exosomal contents stem not only from functional decline in secreting cells but also from uptake impairments in recipient cells. Downregulation of T-cell immunoglobulin mucin 4 (TIM4) and integrin αvβ5 on diabetic keratinocyte membranes reduces exosomal endocytosis efficiency. Even when internalized, abnormal lysosomal acidification accelerates degradation of contents due to reduced integrin αvβ5 expression.196 However, circ-Erbb2ip-engineered exosomes promote angiogenesis and antioxidant stress by regulating downstream target genes through a miRNA sponge mechanism.197 Furthermore, lncRNA H19 in ADSC-derived exosomes was demonstrated to drive macrophage M2 polarization via the miR-130b-3p/PPARγ/STAT3 axis, indirectly promoting fibroblast proliferation and angiogenesis,198 indicating the extensive regulatory role of non-coding RNA axes in wound healing.
Engineering strategies demonstrate significant advantages in overcoming these obstacles. For instance, circ-Erbb2ip-engineered exosomes target PTEN inhibition via a miRNA sponge mechanism, activating the PI3K/Akt pathway to promote angiogenesis and counteract oxidative stress, with therapeutic efficacy markedly superior to that of natural exosomes.199 Furthermore, lncRNA H19 in ADSC-derived exosomes was demonstrated to drive macrophage M2 polarization via the miR-130b-3p/PPARγ/STAT3 axis, indirectly promoting fibroblast proliferation and angiogenesis.200 Some circRNAs or lncRNAs may induce off-target effects or competitively inhibit endogenous regulatory networks when overexpressed, necessitating systematic safety evaluation in preclinical models.201,202
Treatment Methods
Photobiomodulation Therapy
The core pathological feature of impaired wound healing in diabetes lies in compromised keratinocyte proliferation and migration capacity. This phenomenon stems from the suppression of key signaling pathways by multiple factors, including hyperglycemia and chronic inflammation, presenting a major challenge in clinical treatment. Photobiomodulation therapy, a non-invasive physical intervention with minimal toxicity, precisely targets multiple molecular nodes of keratinocyte dysfunction through specific wavelengths of red and near-infrared light. By restoring function through multi-pathway synergistic effects, it has demonstrated the ability to enhance repair processes in preclinical studies.
The core regulatory effects of PBMT revolve around the activation of classical signaling pathways. It can upregulate epidermal growth factor secretion and activate p-EGFR, triggering the JAK2/STAT1/STAT5 signaling cascade to drive the expression of genes such as Cyclin D1 and promote proliferation. Simultaneously, it regulates migration-related genes to enhance cell motility.154 This therapy also amplifies proliferative signals by upregulating basic fibroblast growth factor and its receptor to activate the Ras-MEK-MAPK pathway.199 At the metabolic pathway level, PBMT activates the PI3K/AKT pathway,200,201 enhancing cell survival and ensuring energy supply by regulating targets like mTOR and GSK3β, while synergistically promoting regeneration through the Wnt pathway.203
PBMT also modulates cellular epigenetic states by acting on the microenvironment. It promotes Nrf2 nuclear translocation, upregulates antioxidant enzyme expression to scavenge reactive oxygen species, and simultaneously suppresses pro-inflammatory factors to improve the local microenvironment.204,205 Multispectral LED irradiation upregulates the expression of molecules such as phosphorylated focal adhesion kinase, enhances cell adhesion and spreading, and drives actin cytoskeleton reorganization to support migration.168 Collectively, PBMT targets the core pathological pathway of keratinocyte dysfunction in diabetic wounds. Current clinical treatments face limitations such as drug resistance and significant surgical trauma. PBMT’s multi-pathway synergistic advantages and non-invasive nature provide robust support for its clinical translation, offering promise as a novel therapeutic approach to improve chronic wound healing in diabetes.
Engineered Fat-Derived Exosomes
One of the core pathological features of impaired wound healing in diabetes is the compromised proliferation and migration capacity of keratinocytes, a phenomenon closely associated with hyperglycemia-induced signaling pathway dysregulation and an imbalanced inflammatory microenvironment. As natural carriers for intercellular signaling, exosomes possess excellent biocompatibility and efficient delivery capabilities for bioactive molecules, making them a research hotspot in this field. By loading components such as miRNAs and functional proteins, they can target and regulate keratinocyte functions, reconstructing key signaling pathways to promote re-epithelialization.
Exosomes synergistically regulate keratinocyte functions through multiple pathways. miR-21-5p-loaded adipose stem cell exosomes enhance local bioavailability by activating the Wnt/β-catenin pathway through PTEN downregulation and upregulating pro-proliferative genes such as Cyclin D1. Simultaneously, they target TGF-βI inhibition to enhance extracellular matrix remodeling, thereby promoting proliferation and migration through dual regulation.206–208 CDK1-loaded small extracellular vesicles directly deliver functional CDK1, achieving transient proliferation induction through dual-pathway synergistic activation of AKT and ERK.101 Adipose stem cell exosomes commonly activate the AKT/ERK pathway.209 AKT ensures energy supply, suppresses apoptosis, and upregulates HIF-1α, while ERK drives G1/S transition. Redundant regulatory mechanisms involving both proteins and non-coding RNAs exist.
Exosomes also indirectly support repair by improving the microenvironment and regulating migration pathways. Umbilical cord mesenchymal stem cell exosomes deliver HO-1 protein to inhibit NF-κB activation, reducing inflammatory cytokine release, thereby indirectly relieving proliferation suppression and synergistically promoting angiogenesis. Exosomes derived from HaCaT cells activate P38 MAPK and ERK1/2 pathways via ligands such as FGF2 and EGF, thereby regulating actin reorganization and focal adhesion turnover to mediate keratinocyte migration.210
In summary, exosomes have demonstrated significant reparative effects in animal models, yet clinical translation challenges remain, including large-scale preparation and optimization of targeted delivery. Future efforts should focus on the precise design of pathway-targeted exosomes, combined with the development of treatment strategies tailored to the dynamic pathological stages of wounds, to advance their transition from preclinical research to clinical application.
Small-Molecule Inhibitors Targeting Fructose
Abnormal metabolic reprogramming is a major contributor to keratinocyte dysfunction in diabetic wounds, with excessive activation of the gluconeogenesis pathway being particularly critical. Fructose-1,6-bisphosphatase 1 (FBP1), as the rate-limiting enzyme of gluconeogenesis, is abnormally upregulated under high-glucose conditions and directly inhibits keratinocyte proliferation and migration, making it a core therapeutic target.211 The novel small-molecule inhibitor AA4 exerts its effects through a dual mechanism: it directly competitively binds to the FBP1 active site to block gluconeogenic flux, and also functions as a hydrogen sulfide donor to induce FBP1 protein degradation. FBP1 deficiency leads to acetyl-CoA accumulation, promoting histone H3K9 acetylation to activate proliferative gene transcription. Simultaneously, it relieves glycolytic inhibition, enabling Warburg-like metabolic reprogramming to supply energy for cell cycle progression.212 Topical application of metabolic modulators in STZ-induced diabetic mouse models has been shown to accelerate wound closure, with efficacy independent of anti-inflammatory effects and attributable to repair driven by metabolic reprogramming.211
Metabolic abnormalities are frequently accompanied by post-translational modification dysregulation. Imbalanced O-GlcNAc glycosylation of c-Myc regulates the equilibrium between keratinocyte proliferation and migration. Under high-glucose conditions, enhanced O-GlcNAcyltransferase activity leads to excessive glycosylation at Thr58 of c-Myc.212 This modification stabilizes c-Myc protein and prolongs its half-life, driving abnormal cell proliferation while inhibiting migration. Topical application of O-GlcNAcyltransferase inhibitors or c-Myc-specific inhibitors reduces c-Myc stability, restores cell migration capacity, and promotes wound re-epithelialization.213 Additionally, FTO demethylates ULK1 mRNA, stabilizing its transcription and promoting autophagy, thereby mitigating endothelial cell damage.214,215 Nrf2 activation disrupts the oxidative stress-inflammation vicious cycle by upregulating antioxidant enzymes and suppressing the NF-κB pathway. RIG-I regulates TIMP-1 expression via an NF-κB-dependent mechanism, stabilizing the extracellular matrix to promote keratinocyte migration.41
In summary, targeted small-molecule inhibitors have established diversified intervention strategies targeting metabolic enzymes, post-translational modifications, epigenetic transcriptional regulation, and oxidative stress. These pathways cross-talk with each other, jointly regulating the balance between keratinocyte proliferation and migration. Future efforts should focus on developing tissue-selective formulations or multi-target synergistic approaches to overcome the limitations of single-target strategies and achieve efficient clinical translation.
Novel Peptide Agonists
Delayed re-epithelialization is a core clinical feature of diabetic wounds, with impaired keratinocyte function being the primary cause. Traditional exogenous growth factor therapies have shown limited clinical efficacy due to numerous constraints. Novel peptide agonists, characterized by high specificity and low toxicity, have emerged as a research focus. By precisely targeting receptors to activate downstream pathways, they multidimensionally remodel the proliferation and migration processes of keratinocytes.
FAP1, as an FGFR-specific agonist peptide, specifically binds to and phosphorylates FGFR1, activating the downstream MAPK pathway.216 This pathway promotes Cyclin D1 and c-Myc expression through ERK1/2 phosphorylation, driving keratinocyte G1/S transition and enhancing proliferative capacity.217 In diabetic mouse models, FAP1 increases keratinocyte Ki67 positivity and accelerates re-epithelialization.216 AMP-IBP5 synergistically counters hyperglycemic toxicity by simultaneously activating the EGFR, STAT1/3, and MAPK pathways, not only directly promoting keratinocyte migration but also inducing angiogenesis via paracrine signaling.218 Ghrelin activates the ERK1/2 pathway via GHSR1a to elevate keratinocyte mitotic index while downregulating inflammatory factor expression and stimulating collagen synthesis, addressing three key issues: proliferation deficiency, inflammatory dysregulation, and matrix defects.219 Frog-derived peptide OA-RD17 activates the MAPK pathway via TLR4 to initiate early proliferation. It subsequently induces miR-632 expression to inhibit GSK3β, stabilizing β-catenin to form a positive feedback loop in the Wnt pathway. Concurrently, it suppresses NF-κB nuclear translocation, facilitating the transition from the inflammatory phase to the proliferative phase.217 In summary, novel peptide agonists address core issues in diabetic wounds by multi-levelly intervening in keratinocyte signaling networks. Future efforts should optimize delivery systems to enhance bioavailability, conduct long-term safety assessments and clinical trials, and advance these agents from basic research to clinical application.
Plant-Derived Nanovesicles Synergistically Drive Precise Repair of Diabetic Wounds with Traditional Chinese Medicine Formulas
Chronic diabetic wounds exhibit limited responsiveness to conventional therapies due to keratinocyte dysfunction, persistent inflammation, and oxidative stress imbalance. In recent years, plant-derived nanovesicles combined with active components from traditional Chinese medicine via smart delivery systems have demonstrated therapeutic potential through multi-pathway synergistic regulation and targeted repair, offering new pathways for clinical translation (Table 3).
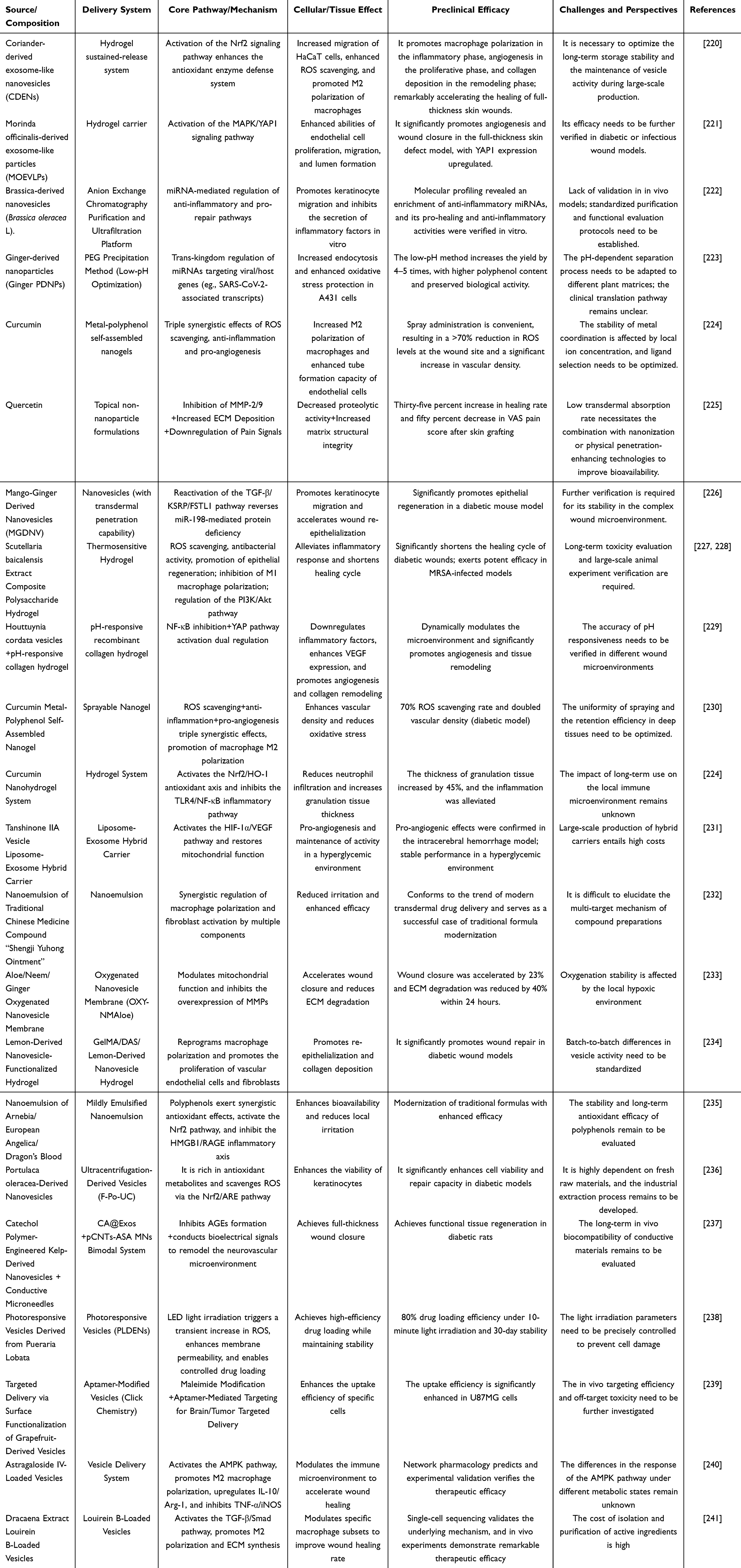 |
Table 3 Applications and Mechanisms of Synergistic Treatment with Plant Nanovesicles and Traditional Chinese Medicine for Diabetic Wounds |
Mango ginger-derived exosome-like nanovesicles (MGDNV) can specifically reactivate the TGF-β/KSRP/FSTL1 pathway, restoring FSTL1 protein levels depleted by miR-198 overexpression in diabetic wounds. This restoration promotes keratinocyte migration through phosphorylation modification at the S165 site of FSTL1.242 Wheatgrass juice-derived exosome-like nanovesicles (WDNVs) have been demonstrated to upregulate type I collagen expression and activate the Wnt/β-catenin pathway. These vesicles express typical exosomal markers including CD9, CD63, and HSP70 on their surface. WDNVs) have been demonstrated to enhance proliferation and migration of human epidermal keratinocytes (HaCaT) and dermal fibroblasts by upregulating type I collagen expression and activating the Wnt/β-catenin pathway. These vesicles express typical exosome markers such as CD9, CD63, and HSP70 on their surface. Furthermore, the bioactive components they carry exert anti-apoptotic effects without interfering with the cell cycle.243
In the field of traditional Chinese medicine, active components such as baicalin, quercetin, and curcumin accelerate repair through a triple mechanism of antioxidant, anti-inflammatory, and pro-angiogenic effects. For instance, composite polysaccharide hydrogels loaded with Scutellaria baicalensis extract scavenge reactive oxygen species, exhibit antibacterial activity, and promote epithelial regeneration.244 Houttuynia cordata vesicles combined with pH-responsive collagen hydrogels can simultaneously modulate NF-κB and YAP pathways, achieving dual synergistic effects of inflammation relief and angiogenesis.229 Studies show that topical quercetin significantly improves healing rates after skin grafting while reducing pain and MMP-2/9 levels, indicating its anti-proteolytic and ECM deposition-promoting effects.245 Despite promising prospects, standardized preparation, ingredient stability, and large-scale clinical validation remain bottlenecks. Future efforts should integrate network pharmacology to screen core targets and develop intelligent drug delivery systems, advancing precision medicine. The synergistic innovation of nanovesicles and traditional Chinese medicine formulations is progressively overcoming the challenges of diabetic wound healing, ushering in a new era of regenerative medicine.
Biological Dressing
The design of bio-dressings has shifted from passive coverage to active microenvironment regulation. Their core mechanism for promoting keratinocyte proliferation lies in mimicking the natural extracellular matrix and delivering signaling molecules. Recombinant hair keratin (eg., RK34, RK81) has been demonstrated to significantly enhance HaCaT cell migration rates by activating the PI3K-AKT pathway, upregulate KRT16/17 expression, and accelerate wound re-epithelialization in diabetic rats.246 Acetate- or butyrate-modified keratin fiber dressings remodel the inflammatory microenvironment through dual actions: inducing macrophage M2 polarization to reduce IL-1β levels while directly stimulating keratinocytes to express tight junction protein ZO-1 and adhesion molecule JAM, thereby enhancing epidermal barrier reconstruction.137 Furthermore, chiral hydrogels specifically adsorb type I collagen via their right-handed helical structure, activating keratinocyte proliferation through the integrin-YAP pathway. This bioinspired signaling mechanism overcomes limitations of traditional biochemical factor dependencies.247 In contrast, bioactive glass indirectly supports wound closure by enhancing the overall barrier function of the stratum corneum. While it does not directly accelerate single-cell migration, it achieves this by increasing trans-epithelial electrical resistance and improving the membrane localization of the tight junction protein claudin-1.248 These dressings share the commonality of establishing a local microenvironment conducive to keratinocyte functional recovery, yet their mechanisms of action differ markedly. Keratin-based dressings emphasize growth factor-like signaling, ion-modified dressings favor immune microenvironment reprogramming, while structurally biomimetic dressings utilize physical topological signals to trigger cellular responses.
Platelet-Rich Plasma
The application of platelet-rich plasma is evolving from crude extracts to standardized composite formulations, with its keratinocyte proliferation-promoting effects primarily dependent on the spatiotemporal release regulation of growth factor reservoirs. Traditional PRP exhibits inconsistent efficacy due to its short half-life and uncontrolled factor release. The novel zwitterionic hydrogel carrier PDGF-BB promotes keratinocyte proliferation by activating the MAPK/ERK or PI3K/AKT pathways. Its sustained-release formulation significantly enhances bioavailability and reduces required dosage.249 A more advanced strategy combines PRP with a chitosan-fucan polyelectrolyte complex, which substantially prolongs retention time, modulates inflammation, and promotes angiogenesis and re-epithelialization.250 PRP-derived exosomes demonstrate higher-order regulatory capabilities. The MALAT1 long non-coding RNA they carry binds miR-1914-3p, releasing its inhibition on MFGE8 and thereby activating the TGFB1/SMAD3 axis. This not only enhances keratinocytes’ self-repair capacity but also remodels macrophage phenotypes via paracrine signaling. Compared to monotherapy, PRP complexes offer advantages through multifactorial synergistic networks. However, their efficacy heavily depends on standardized preparation protocols, as platelet concentrations vary significantly with different centrifugation parameters. Current research focuses on engineered exosomes to overcome species-specific limitations and achieve targeted delivery.
Combination Therapy
Traditional single-target therapies for diabetic wound healing disorders struggle to meet the dynamic demands of each healing stage and fail to reverse the multifaceted signaling pathway dysregulation induced by hyperglycemia, resulting in limited clinical efficacy. With advancements in regenerative medicine and smart materials technology, combined treatment strategies have shifted from single-pathway interventions to precision models featuring synergistic target regulation. The core innovation lies in leveraging smart responsive biomaterials to dynamically regulate key signaling pathways. By focusing on the reconstruction of keratinocyte function, which is a core process suppressed by high-glucose environments, this approach simultaneously activates proliferation-related pathways such as Wnt/β-catenin, PI3K/Akt, and ERK while inhibiting NF-κB-mediated inflammatory responses. This achieves multi-level reprogramming of keratinocyte biological behavior, offering new possibilities for overcoming traditional treatment limitations (Figure 5).
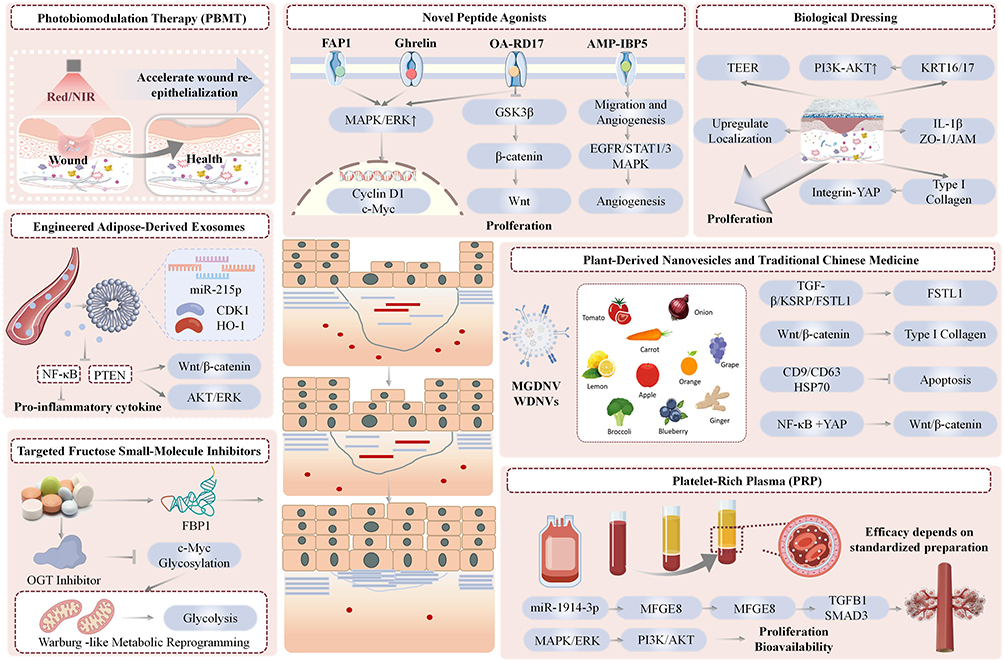 |
Figure 5 This diagram illustrates multiple therapies for diabetic wounds: Photobiomodulation Therapy accelerates re-epithelialization, while engineered adipose exosomes regulate key signaling pathways to support repair. Meanwhile, novel peptide agonists boost keratinocyte proliferation, and plant nanovesicles paired with traditional Chinese medicine target and repair disrupted molecular pathways. Additionally, small-molecule inhibitors reprogram abnormal metabolism linked to impaired healing. Finally, biological dressings and PRP activate coordinated repair cascades to enhance overall wound recovery. Upward arrows (↑) indicate upregulation or activation of relevant molecules and signaling pathways; downward arrows (T) indicate downregulation or inhibition of relevant molecules and signaling pathways. |
Keratinocytes, as the core cells responsible for wound re-epithelialization, exhibit impaired function that constitutes a critical bottleneck in delayed wound healing in diabetes. Under a high-glucose microenvironment, keratinocytes display pathological phenotypes including reduced proliferation and migration capacity, impaired autophagy, and abnormal differentiation, directly leading to stalled re-epithelialization. At the molecular level, hyperglycemia directly downregulates Wnt/β-catenin pathway activity, inhibiting β-catenin nuclear translocation and downstream proliferative gene expression. Concurrently, it activates the NF-κB/CCL20 axis, exacerbating local inflammation and obstructing M2 macrophage polarization, creating a vicious cycle. Recent studies further reveal that high glucose reduces expression of the m6A demethylase FTO, leading to decreased stability of TRIB3 mRNA, which in turn inhibits the autophagic flux and exacerbates apoptosis. Abnormal upregulation of KRT17 in keratinocytes also induces hyperkeratinization via the c-MYB/PI3K-Akt axis, disrupting normal repair processes. Collectively, these mechanisms form a complex molecular network underlying keratinocyte dysfunction.17,55 Addressing this core bottleneck, novel combination therapies achieve systematic regulation of key keratinocyte pathways through the synergistic action of spatiotemporally controlled delivery systems and precise molecular interventions. Smart hydrogels leverage wound microenvironment responsiveness for on-demand release of therapeutic factors. MMP-9-responsive hydrogels loaded with M2 macrophage exosomes promote M2 polarization during the inflammatory phase, mitigating NF-κB pathway activation. Upon transition to the proliferative phase, these hydrogels reactivate keratinocyte PI3K/Akt and Wnt/β-catenin pathways to accelerate re-epithelialization.8 GelMA hydrogels loaded with keratinocyte-derived exosomes improve wound blood supply by upregulating PDGF, indirectly enhancing keratinocyte migration.30 Dual-responsive hydrogels loaded with H₂S donors specifically mitigate hyperglycemic and hyperoxidative microenvironments, protecting keratinocyte function by inhibiting NF-κB nuclear translocation and activating the Nrf2 pathway.59 Combining physical stimuli with bioactive factors overcomes limitations of traditional drug delivery. Near-infrared-responsive hydrogels activate the YAP/TAZ pathway through mechanical force while synergistically releasing linagliptin to enhance Wnt signaling, dual-driving keratinocyte activation.58 Photothermal effects of gold nanorod-composite hydrogels improve the infected microenvironment, simultaneously synergizing with exosomes to activate the Akt/eNOS pathway for concurrent promotion of angiogenesis and epithelial regeneration.9 Small-molecule agonists/inhibitors enable precise reprogramming of signaling networks. The PPARβ/FFA1 dual-target agonist Y8 balances antioxidant and pro-migration effects, while the Wnt pathway activator KY19382 significantly enhances keratinocyte proliferation. The BRD9 degradation agent-loaded silk fibroin microneedle system achieves stage-specific regulation during inflammatory and proliferative phases.22,23,66 The SMART-EXO microgel system activates the integrin-FAK-RhoA pathway through mechanical self-contraction while releasing exosomal contents on demand, enabling synergistic mechanical-biochemical signal transduction.6 Transcriptomic evidence indicates that such combined therapies simultaneously enrich extracellular matrix-receptor interactions, focal adhesions, and Wnt signaling pathway-related genes, fundamentally reversing the pathological microenvironment of diabetic wounds.
In summary, combined therapy for diabetic wounds has entered a new phase. Compared to traditional passive interventions targeting single pathological pathways, novel combined strategies employ spatiotemporally precise interventions mediated by smart materials. These approaches target core proliferation pathways in keratinocytes while simultaneously regulating the immune microenvironment. Looking ahead, as the cross-regulatory mechanisms of these pathways are further elucidated and the clinical translation of smart delivery systems advances, such strategies will provide more efficient and precise clinical solutions for diabetic non-healing wounds, significantly improving patient outcomes.
Discussion
The chronicity of diabetic wounds has become a formidable challenge in global public health, with one of its core pathological features being the arrest of re-epithelialization triggered by keratinocyte proliferation dysfunction. Research on this pathological state has long focused on isolated modules such as metabolic toxicity and epigenetic modifications, overlooking that the core mechanism underlying impaired diabetic wound healing lies in the pathological interactions formed by multiple molecular hubs within the hyperglycemic microenvironment. The proliferation arrest of keratinocytes stems from cross-linking among five major modules: zinc finger transcription factors, HIF-1α, STING, neuropeptides, and exosomal communication. These pathways do not operate in parallel isolation but rather activate a cellular inhibitory state through bidirectional negative feedback, cascade amplification, and spatially heterogeneous activation.173,251
The most prominent issue in current basic research is the contradiction between the bidirectional effects of core regulatory pathways and research conclusions. The essence of these conflicting conclusions lies in the fact that most existing studies are confined to static observations of single cell types and single repair stages, overlooking that wound healing is a multicellular cooperative, temporally dynamic process. The overwhelming majority of preclinical studies employ a whole-process intervention model, which fails to account for the functional shifts of targets across different healing stages and cell types. This ultimately leads to effect biases when single-target interventions are applied to complex clinical wounds—a core reason why numerous single-target strategies demonstrate efficacy in animal models yet fail during clinical translation. This complexity further manifests in the cross-integration of mechanical signals and metabolic reprogramming. Pathological modules do not operate in parallel isolation but form bidirectional negative feedback loops and cascading amplification through core molecular hubs, creating pathological cascade effects. Single-target interventions are easily counteracted by compensatory mechanisms within the pathological network, explaining why traditional single-target therapies struggle to reverse chronic wound pathology.
Beyond limitations in target identification, the severe disconnect between preclinical research systems and real-world clinical settings represents a critical external factor contributing to translational failure, a pervasive challenge across the field. Most existing studies on diabetic wound mechanisms and interventions rely on acute wound models in rodents, which fundamentally differ from the pathophysiology of human diabetic foot ulcers. This discrepancy directly leads to discrepancies between preclinical efficacy and actual clinical outcomes. Research by Kato et al has confirmed that only plantar wound models can partially mimic the re-epithelialization process of human wounds. The contraction-dominant healing patterns observed in traditional dorsal and thigh wound models completely fail to reflect the true impact of intervention strategies on keratinocyte function. The modeling approaches and disease progression designs of existing animal models are severely misaligned with the clinical characteristics of diabetic foot ulcers. Most existing models employ a research design where diabetes is induced first, followed by the creation of acute wounds, with overall observation periods generally being short. The foot wound model established by Yu et al in type 2 diabetic rats had an observation period of only 8 days, far shorter than the natural disease course of human diabetic foot ulcers, which spans weeks to months.252 Such short-term models cannot reproduce the pathological steady state of human chronic wounds nor evaluate the long-term efficacy and safety of intervention strategies. Even when studies validate target efficacy, findings often stem from single, short-term interventions without evaluating long-term safety and tolerability of repeated dosing in clinical settings. A significant gap in pathological complexity also exists between clinical diabetic foot ulcers and experimental models. Clinical ulcers often involve multiple pathological factors such as peripheral neuropathy, peripheral arterial disease, and wound infection.253 In contrast, basic research predominantly employs models induced by a single hyperglycemic factor, failing to replicate the complex microenvironment of clinically relevant wounds where multiple factors overlap.254 This discrepancy in pathological scenarios directly leads to interventions effective in ideal models becoming completely ineffective in complex clinical settings. Beyond the inherent pathological bias in models, existing research also exhibits significant deviation from core clinical treatment endpoints in efficacy evaluation systems. Most preclinical studies prioritize short-term wound closure rates as primary metrics, whereas clinical treatment fundamentally requires achieving complete wound healing and reducing long-term recurrence and amputation rates.255 The accelerated short-term closure observed in animal models does not equate to functional healing of chronic wounds in clinical settings. This discrepancy in evaluation systems further widens the gap between basic research and clinical needs.
The disconnect between existing research and clinical practice is further evident in the severe neglect of patient heterogeneity, which represents a core challenge in clinical treatment. Type 1 and Type 2 diabetes differ in their fundamental pathogenesis, and their wound pathologies also exhibit significant distinctions. Type 1 diabetic wounds are primarily characterized by neuropathy and dysregulation of the TGF-β/SMAD pathway.256 In contrast, type 2 diabetic patients more frequently present with metabolic syndrome and peripheral vascular disease, manifesting as more pronounced suppression of the PI3K/AKT-FASN axis, metabolic disorders, and inflammatory imbalance.257 However, the vast majority of current preclinical studies fail to distinguish between diabetes subtypes, and clinical trials rarely employ stratified designs based on diabetes classification. This directly leads to a severe underestimation of overall treatment response rates. Clinically, diabetic wounds progress through distinct phases, which include infection, inflammation, proliferation, and remodeling,116 each characterized by fundamentally different core pathological contradictions. The infection phase centers on controlling infection and clearing necrotic tissue, while the proliferation phase focuses on promoting keratinocyte proliferation and angiogenesis. Existing intervention studies predominantly employ uniform protocols throughout all phases, failing to address stage-specific therapeutic needs and potentially exacerbating pathological damage during inappropriate phases. Clinically, diabetic foot ulcers are classified into neuropathic, ischemic, and neuro-ischemic mixed types, with distinct core pathological mechanisms across these subtypes. Neuropathic ulcers primarily involve neuropeptide deficiency and epithelial dysfunction, while ischemic ulcers center on microcirculatory impairment and hypoxia.258 Existing research rarely designs differentiated intervention strategies for distinct clinical phenotypes and lacks stratified validation based on wound phenotypes.
Concurrently, emerging therapeutic approaches themselves face substantial translational barriers, further limiting clinical adoption. Taking the most extensively studied exosome therapy as an example, while numerous studies confirm that engineered exosomes can promote keratinocyte proliferation and accelerate wound healing through multi-pathway regulation, their clinical translation remains constrained by multiple bottlenecks.259–261 High product heterogeneity among exosomes derived from different cell sources, isolation processes, and culture conditions hinders scalable, standardized production. Exosomes exhibit low in vivo targeting efficiency and insufficient storage stability, while potential immunogenicity and long-term safety concerns remain inadequately validated. For small-molecule inhibitors, peptide agonists, and plant-derived active ingredients, existing studies have mostly validated efficacy through topical application in animal models.262 However, systematic clinical data is lacking regarding transdermal absorption efficiency in human skin, metabolic stability within wound microenvironments, systemic exposure safety, and optimal dosing frequencies and regimens. Even photobiomodulation therapy, which has seen preliminary clinical application, lacks standardized parameters for optimal wavelength, energy density, and irradiation frequency, with severe shortages of multicenter, large-sample clinical validation data.263,264 More critically, nearly all novel intervention strategies lack large-scale, multicenter randomized controlled trials centered on clinical hard endpoints. The impact on long-term wound recurrence rates and amputation risks remains entirely unknown, hindering their integration into routine clinical practice.
Despite advancing understanding of the mechanisms underlying keratinocyte dysfunction in diabetic non-healing wounds, existing research exhibits multiple core shortcomings and limitations. Most existing data rely on static snapshot analyses, lacking continuous tracking of the activation sequence and feedback intensity changes of key molecules throughout the entire progression from acute to chronic wounds. Even studies reporting that JAM-A/miR-106b is significantly upregulated only in the late stages of wound healing, indicating a time-specific intervention window, face the technical challenge of precisely capturing such dynamic inflection points.6,265 Concurrently, existing research lacks systematic resolution of widespread contradictory conclusions within the field. Most studies focus solely on the role of individual regulatory axes in specific scenarios, failing to elucidate the determinants and boundary conditions of bidirectional effects within core pathways.266,267 More critically, the overall design of existing studies exhibits significant disconnect from clinical practice. Whether in experimental model selection, efficacy evaluation system establishment, or intervention protocol design, these approaches fail to adequately account for the complexity and heterogeneity of real-world clinical scenarios.268,269 This disconnect represents a major obstacle hindering the effective translation of basic research findings into clinical applications.
Given the current state of research, core bottlenecks, and clinical demands in this field, future studies must move beyond the traditional single-molecule, single-pathway research paradigm. Systematic breakthroughs are urgently needed across three dimensions, namely fundamental mechanisms, translational systems, and clinical research, to truly bridge the gap from basic discoveries to clinical applications. At the basic research level, the first step is to resolve contradictory conclusions regarding core pathways and establish a spatio-temporal dynamic regulatory network framework. Utilizing technologies such as single-cell spatio-temporal transcriptomics and in vivo dynamic imaging, researchers should map the dynamic activation profiles of key molecular hubs throughout the entire wound healing cycle. This will clarify the temporal windows and cell-specificity of target functional transitions while elucidating crosstalk logic between pathological modules and pathological cascade nodes.61 Second, establish standardized experimental models aligned with clinical pathological features. Prioritize plantar wound models simulating human re-epithelialization, develop composite chronic wound models incorporating neuropathy, ischemia, and infection, and extend observation periods to match chronic wound progression. This minimizes preclinical research bias at its source.270 At the translational research level, breakthroughs must focus on overcoming bottlenecks in targeted intervention and delivery systems. Based on the spatiotemporal dynamics of targets, develop phase-controlled, cell-specific intervention strategies. Utilize wound microenvironment-responsive delivery systems to achieve sequential precision dosing, mitigating potential risks from bidirectional target effects.271 Simultaneously, establish standardized production and quality control systems for emerging therapies, systematically complete preclinical safety evaluations, and lay the foundation for clinical trials.272 At the clinical research level, there is an urgent need to establish a standardized molecular classification system for diabetic wounds based on multicenter cohorts. This system should identify biomarkers and core intervention targets for wounds of different subtypes, stages, and phenotypes, driving the implementation of personalized precision treatment.273 Concurrently, rigorously designed stratified randomized controlled trials should be conducted, using clinical hard endpoints as core evaluation metrics to systematically validate the efficacy and long-term safety of intervention strategies, thereby filling critical gaps in clinical research.
Conclusions
Diabetic wounds remain a major clinical challenge worldwide, with impaired keratinocyte proliferation being a key contributor to failed re-epithelialization and chronic non-healing. In this review, we have summarized the wide range of pathological changes that disrupt keratinocyte proliferation in the diabetic setting, showing that these impairments do not stem from a single abnormal pathway, but arise from the combined dysfunction of multiple interrelated biological processes: metabolic toxicity driven by sustained hyperglycemia, dysregulated epigenetic control, pathological remodeling of the extracellular matrix, disrupted wound microenvironment homeostasis, hypoxic stress, dysbiosis of the cutaneous microbiota, and aberrant neuropeptide signaling. We have also outlined how signal crosstalk between these different processes shapes the progression of impaired wound healing, rather than each pathway acting in isolation. Going forward, further work is needed to fully unravel the regulatory mechanisms governing interactions between these pathological events across the full wound healing course, addressing critical gaps in our current understanding of this condition. There is also a need to refine the performance of emerging intervention approaches, including engineered exosome-based delivery systems and photobiomodulation therapy, to improve their targeted action, in vivo stability and overall safety profile, overcoming key barriers that currently limit their clinical application. In addition, well-designed, multicenter clinical studies that account for the different subtypes, stages and clinical presentations of diabetic wounds will be essential to verify the effectiveness and long-term safety of these treatment strategies in real-world patient populations. Taken together, the findings summarized here build a comprehensive understanding of the pathological changes driving impaired keratinocyte proliferation in diabetic wounds, and offer useful guidance for the development of new treatment approaches for this condition. Beyond diabetic wounds, these insights may also inform research into the mechanisms and treatment of other chronic, hard-to-heal wounds; ultimately, advances in this field hold the potential to reduce rates of disability, amputation and wound recurrence in affected patients, and ease the significant burden that diabetic wounds place on individuals and healthcare systems around the world.
Acknowledgments
Thanks for Figdraw.
Funding
National Natural Science Foundation of China (NSFC) Youth Program, Grant No. 82404163 (Xueli Niu).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fang W, Lan CE. The Epidermal Keratinocyte as a Therapeutic Target for Management of Diabetic Wounds. Int J Mol Sci. 2023;24:4290. doi:10.3390/ijms24054290
2. Zhou P, Feng H, Qin W, et al. KRT17 from skin cells with high glucose stimulation promotes keratinocytes proliferation and migration. Front Endocrinol. 2023;14:1237048. doi:10.3389/fendo.2023.1237048
3. Xie Z, Zhou S, Tang S, et al. High glucose combined with lipopolysaccharide stimulation inhibits cell proliferation and migration of human HaCaT keratinocytes by impacting redox homeostasis and activating the polyol pathway. Mol Biol Rep. 2024;51(1):1098. doi:10.1007/s11033-024-10042-5
4. Lu W, Huang G, Yu Y, et al. Fructose 1,6-bisphosphatase 1 is a potential biomarker affecting the malignant phenotype and aerobic glycolysis in glioblastoma. PeerJ. 2025;13:18926. doi:10.7717/peerj.18926
5. Lin H, Yang Y, Wang X, et al. Targeting the AGEs-RAGE axis: pathogenic mechanisms and therapeutic interventions in diabetic wound healing. Front Med. 2025;12:2296–45. doi:10.3389/fmed.2025.1667620
6. Gong T, Fan X, Wu M, et al. Post-transcriptional regulation of diabetic wound healing by junctional adhesion molecule A/miR-106b axis. Burns. 2025;51(6):107527. doi:10.1016/j.burns.2025.107527
7. Dong Z, Li S, Huang Y, et al. RNA N(6)-methyladenosine demethylase FTO promotes diabetic wound healing through TRIB3-mediated autophagy in an m(6)A-YTHDF2-dependent manner. Cell Death Dis. 2025;16:222. doi:10.1038/s41419-025-07494-3
8. Zhang L, Hung GC, Meng S, et al. LncRNA MALAT1 Regulates Hyperglycemia Induced EMT in Keratinocyte via miR-205. Non-Cod RNA. 2023;9:14. doi:10.3390/ncrna9010014
9. Schmidt A, von Woedtke T, Weltmann K, et al. YAP/TAZ, beta-catenin, and TGFb pathway activation in medical plasma-induced wound healing in diabetic mice. J Adv Res. 2025;72:387–400. doi:10.1016/j.jare.2024.07.004
10. Banerjee A, Singh P, Sheikh PA, et al. Simultaneous regulation of AGE/RAGE signaling and MMP-9 expression by an immunomodulating hydrogel accelerates healing in diabetic wounds. Biomat advanc. 2024;163:213937. doi:10.1016/j.bioadv.2024.213937
11. Shen Z, Du L, Fang X, et al. Nanozyme Cryogel Accelerates Diabetic Wound Healing by Targeting Biofilms and Inflammations of the Wound Bed. ACS nano. 2025;19:35081–35101. doi:10.1021/acsnano.5c12513
12. Cheng P, Xie X, Hu L, et al. Hypoxia endothelial cells-derived exosomes facilitate diabetic wound healing through improving endothelial cell function and promoting M2 macrophages polarization. Bioact Mater. 2024;33:157–173. doi:10.1016/j.bioactmat.2023.10.020
13. Maugeri G, D’Amico AG, Magrì B, et al. Protective effect of pituitary adenylate cyclase activating polypeptide in diabetic keratopathy. Peptides. 2023;170:171107. doi:10.1016/j.peptides.2023.171107
14. Jin W, Li Y, Yu M, et al. Advances of exosomes in diabetic wound healing. Burns Trauma. 2025;13:tkae078. doi:10.1093/burnst/tkae078
15. Ghafouri-Fard S, Khoshbakht T, Hussen BM, et al. A review on the role of cyclin dependent kinases in cancers. Can Cell Inter. 2022;22:325. doi:10.1186/s12935-022-02747-z
16. Khalaf N, El-Serag HB, Abrams HR, et al. Burden of Pancreatic Cancer: from Epidemiology to Practice. Clin Gastroenterol Hepatol. 2021;19:876–884. doi:10.1016/j.cgh.2020.02.054
17. Chen XQ, Zhao XL, Zhu XY, et al. Research Progress of Traditional Chinese Medicine Regulating Metabolic Reprogramming to Intervene in Hepatocellular Carcinoma. Traditional Chin Drug Res Clin Pharmacol. 2025;36:1. doi:10.3969/j.issn.1002-2600.2022.01.040
18. Jerka D, Bonowicz-Kozłowska K, Bai Y, et al. Reviving p18INK4c: harnessing a tumor suppressor for cancer treatment. Biochem Biophys Res Commun. 2025;784:152674. doi:10.1016/j.bbrc.2025.152674
19. Gettinger SN, Wurtz A, Goldberg SB, et al. Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non-Small Cell Lung Cancer. J Thorac Oncol. 2018;13:831–839. doi:10.1016/j.jtho.2018.03.008
20. Maiese K. The impact of aging and oxidative stress in metabolic and nervous system disorders: programmed cell death and molecular signal transduction crosstalk. Front Immunol. 2023;14:1273570. doi:10.3389/fimmu.2023.1273570
21. Charoensuk C, Thamtarana PJ, Chanprasert C, et al. Autosomal dominant diabetes associated with a novel ZYG11A mutation resulting in cell cycle arrest in beta-cells. Mol Cell Endocrinol. 2021;522:111126. doi:10.1016/j.mce.2020.111126
22. Bhalla K, Liu W, Thompson K, et al. Cyclin D1 represses gluconeogenesis via inhibition of the transcriptional coactivator PGC1α. Diabetes. 2014;63:3266–3278. doi:10.2337/db13-1283
23. Liu M, Zhang W, Chen Z, et al. Mechanisms of magnesium oxide-incorporated electrospun membrane modulating inflammation and accelerating wound healing. J Biomed Mater Res Part A. 2023;111:132–151. doi:10.1002/jbm.a.37453
24. Kobayashi R, Hoshikawa E, Saito T, et al. The EGF/EGFR axis and its downstream signaling pathways regulate the motility and proliferation of cultured oral keratinocytes. FEBS Open Bio. 2023;13:1469–1484. doi:10.1002/2211-5463.13653
25. Ademi H, Michalak-Micka K, Moehrlen U, et al. Effects of an Adipose Mesenchymal Stem Cell-Derived Conditioned medium and TGF-β1 on Human Keratinocytes In Vitro. Int J Mol Sci. 2023;24(19):14726. doi:10.3390/ijms241914726
26. Ling X, Dong S, Zhang L. Low dose TGF-β1 can improve vohwinkel syndrome by promoting the proliferation of keratinocytes. Acta Histochem. 2023;125:152010. doi:10.1016/j.acthis.2023.152010
27. Wang Y, Nguyen T, He Q, et al. Cytoneme-mediated intercellular signaling in keratinocytes is essential for epidermal remodeling in zebrafish. eLife. 2025;13. doi:10.7554/eLife.97400.3
28. Shahin H, Steinvall I, Sjöberg F, et al. Towards propagation of epidermal cells for wound repair: glass, as cell culture substrate, enhances proliferation and migration of human keratinocytes. Front Bioeng Biotechnol. 2025;13:1547044. doi:10.3389/fbioe.2025.1547044
29. Snowball JM, Jarrold BB, Deangelis Y, et al. Integration of transcriptomics and spatial biology analyses reveals Galactomyces ferment filtrate promotes epidermal interconnectivity via induction of keratinocyte differentiation, proliferation and cellular bioenergetics. Int J Cosmet Sci. 2024;46:927–940. doi:10.1111/ics.12991
30. Uddin A, Rahmani M, Sajini A. From basic biology to engineered therapies: the keratinocyte stem cell playbook. Front Med Technol. 2026;8. doi:10.3389/fmedt.2026.1763067
31. Cherkashina O, Tsitrina A, Abolin D, et al. The Recovery of Epidermal Proliferation Pattern in Human Skin Xenograft. Cells. 2025;14(6):448. doi:10.3390/cells14060448
32. Vlasova TI, Arsent’eva EV, Marzug BA. Intracellular mechanisms regulating the behavior of epidermal stem cells during skin regeneration (a review of literature). Univer Proceed Volga Region Med Sci. 2021;(3). doi:10.21685/2072-3032-2021-3-14
33. Joo BS, Baek J, Park MJ, et al. Effects of peptides derived from active sites of visfatin on wound healing. Sci Rep. 2025;15(1):22169. doi:10.1038/s41598-025-06751-x
34. Hegazi S, Aly R, Mesilhy R, et al. Diabetic foot ulcer wound healing and tissue regeneration: signaling pathways and mechanisms. In: Diabetic Foot Ulcers-Pathogenesis, Innovative Treatments and AI Applications. IntechOpen; 2024.
35. Nguyen LTH, Ahn S, Choi M, et al. Puerarin improves dexamethasone-impaired wound healing in vitro and in vivo by enhancing keratinocyte proliferation and migration. Appl Sci. 2021;11:9343. doi:10.3390/app11209343
36. Huang J, Deng Q, Tsang LL, et al. Mesenchymal stem cells from perinatal tissues promote diabetic wound healing via pi3k/akt activation. Stem Cell Res Ther. 2025;16:59. doi:10.1186/s13287-025-04141-8
37. Li Q, Zhou L, Li W, et al. Gelma hydrogel-loaded extracellular vesicles derived from keratinocytes promote skin microvasculature regeneration and wound healing in diabetic mice through activation of the pdgf-induced pi3k/akt pathway. Cell Biol Toxicol. 2025;41:103. doi:10.1007/s10565-025-10062-2
38. Wang X, Zhang W, Mao L, et al. Inflammation-related gene itga5 affects the healing of diabetic foot ulcers through pi3k-akt signaling pathway. Eur J Pharmacol. 2025;1002:177865. doi:10.1016/j.ejphar.2025.177865
39. Wu L, Fu W, Cao Y, et al. Inhibiting mir-618 promotes keratinocytes proliferation and migration to enhance wound healing in mice. Int J Mol Sci. 2024;25:7617. doi:10.3390/ijms25147617
40. Zhou P, Li Y, Zhang S, et al. Krt17 from keratinocytes with high glucose stimulation inhibit dermal fibroblasts migration through integrin α11. J Endocr Soc. 2024;8:bvad176. doi:10.1210/jendso/bvad176
41. Zhu H, Li Q, Huang Q, et al. Rig-i contributes to keratinocyte proliferation and wound repair by inducing timp-1 expression through nf-κb signaling pathway. J Cell Physiol. 2023;238:1876–1890. doi:10.1002/jcp.31049
42. Alamoudi AA, Alharbi AS, Abdel-Naim AB, et al. Novel nanoconjugate of apamin and ceftriaxone for management of diabetic wounds. Life. 2022;12(7):1096. doi:10.3390/life12071096
43. Tang D, Lin Q, Xu K, et al. Roxadustat: a catalyst for diabetic wound healing through re-epithelialization and angiogenesis. Cytojournal. 2025;22:90. doi:10.25259/Cytojournal_235_2024
44. Zheng R, Liu Y, Lei Y, et al. Upregulated microrna-429 confers endometrial stromal cell dysfunction by targeting hif1an and regulating the hif1a/vegf pathway. Open Med. 2023;18(1):20230775. doi:10.1515/med-2023-0775
45. Mo Y, Wu S, Liu Y, et al. Lrpprc promotes the progression of colorectal cancer via hif-1α/vegf-meditated angiogenesis. Pathol Res Pract. 2025;275:156223. doi:10.1016/j.prp.2025.156223
46. Zhu S, Feng X, Yan G, et al. 9th surgery decoction alleviates psoriasis-like inflammation through the downregulation of angiogenesis induced by hif-1α and the egfr-pi3k-akt-mtor pathways. Phytomedicine Plus;2025. 100817. doi:10.1016/j.phyplu.2025.100817
47. Du X, Shi L, Wang B, et al. Wtap mediated m6a-modified circ_0056856 contributes to the proliferation, migration, and invasion of il-22-stimulated human keratinocyte by mir-197-3p/cdk1 axis. Arch Dermatol Res. 2024;316(6):208. doi:10.1007/s00403-024-03097-8
48. Xian J, Shang M, Dai Y, et al. N(6)-methyladenosine-modified long non-coding rna agap2-as1 promotes psoriasis pathogenesis via mir-424-5p/akt3 axis. J Dermatol Sci. 2022;105:27–36. doi:10.1016/j.jdermsci.2021.11.007
49. Cui L, Chen Q, Cai J, et al. 0843 n6-methyladenosine rna modification promotes keratinocyte proliferation and cutaneous wound repair through regulation of mtor stability. J Invest Dermatol. 2025;145(8):S147. doi:10.1016/j.jid.2025.06.858
50. Hu X, Sui L, Pu C, et al. Lactylation-driven mettl3 regulates wound healing by enhancing m6a/hnrnpa2b1/dnmt1 signaling in keratinocytes. Genes Dis. 2025;13:101787. doi:10.1016/j.gendis.2025.101787
51. Cui L, Li B, Chen Z, et al. 993 epidermis-intrinsic n6-methyladenosine modification dampens skin inflammation. J Invest Dermatol. 2023;143(5):S170. doi:10.1016/j.jid.2023.03.1004
52. Gan L, Wu X, Song J. Comprehensive analysis of crucial m(6)a-related differentially expressed genes in psoriasis. Front Biosci. 2024;29:311. doi:10.31083/j.fbl2909311
53. Honma Y, Nishida K, Sotozono C, et al. Effect of Transforming Growth Factor-β1 and -β2 on Rabbit Corneal Epithelial Cell Proliferation Promoted by Epidermal Growth Factor, Keratinocyte Growth Factor, or Hepatocyte Growth Factor. Exp Eye Res. 1997;65(3):391–396. doi:10.1006/exer.1997.0338
54. Wollina U, Prochnau D, Hoffmann A, et al. Vasoactive intestinal peptide and epidermal growth factor: co-mitogens or inhibitors of keratinocyte proliferation in vitro? IntJ Mol Med. 1998;2(6):725–730. doi:10.3892/ijmm.2.6.725
55. Wang C, He D, Shi C. SIRT5 reduces the inflammatory response and barrier dysfunction in IL-17A-induced epidermal keratinocytes. Allergologia et immunopathologia. 2023;51:30–36. doi:10.15586/aei.v51i1.675
56. Xiao X, Qiu T, Cheng Q, et al. Uridine phosphorylase-1 promotes cell viability and cell-cycle progression in human epidermal keratinocytes via the glycolytic pathway. Clin Exp Pharmacol Physiol. 2024;51(7):e13874. doi:10.1111/1440-1681.13874
57. Austin E, Koo E, Kabakova M, et al. Light-emitting diode red light attenuates epidermal thickening and keratinocyte proliferation in psoriasis models. Sci Rep. 2025;15:43317. doi:10.1038/s41598-025-27186-4
58. Cheng B, Peng SI, Jia YY, et al. Comprehensive secretome profiling and CRISPR screen identifies SFRP1 as a key inhibitor of epidermal progenitor proliferation. Cell Death Dis. 2025;16(1):360. doi:10.1038/s41419-025-07691-0
59. Zhang L, Wang IC, Meng S, et al. LincRNA-EPS Promotes Proliferation of Aged Dermal Fibroblast by Inducing CCND1. Int J Mol Sci. 2024;25(14):7677. doi:10.3390/ijms25147677
60. Ma Y, Liu Z, Miao L, et al. Mechanisms underlying pathological scarring by fibroblasts during wound healing. Int Wound J. 2023;20:2190–2206. doi:10.1111/iwj.14097
61. Jin C, Jin Y, Ding Z, et al. Cellular and Molecular Mechanisms of Wound Repair: from Biology to Therapeutic Innovation. Cells. 2025;14:1850. doi:10.3390/cells14231850
62. Zhou L, Hong Y, Li X, et al. Mitochondrial mGPDH Modulates Fibroblast Function in Diabetic Wound Healing via the SIRT1–c-Myc–TGF-β1 Axis. Diabetes. 2025;75(3):427–440. doi:10.2337/db25-0539
63. Li J, Zhu D, Zhang M, et al. Deciphering age-related differences in wound healing: insights from the interaction between endothelial cells and fibroblasts. Mol Med Rep. 2025;32(4):1–15. doi:10.3892/mmr.2025.13643
64. Morris NG, Woods EL, Dally J, et al. Dysfunctional pericellular hyaluronan deposition contributes to attenuated CD44/EGFR co-localization and impaired myofibroblast differentiation in chronic wound fibroblasts. Exp Cell Res. 2025;450(2):114646. doi:10.1016/j.yexcr.2025.114646
65. Gao R, Zhou P, Li YQ, et al. High glucose-induced IL-7/IL-7R upregulation of dermal fibroblasts inhibits angiogenesis in a paracrine way in delayed diabetic wound healing. J Cell Commun Signal. 2023;17(3):1023–1038. doi:10.1007/s12079-023-00754-x
66. Liu Y, Zheng B, Zheng H, et al. Resveratrol Promotes Diabetic Wound Healing by Inhibiting Notch Pathway. J Surg Res. 2024;297:63–70. doi:10.1016/j.jss.2024.02.004
67. Wang Y, Ding H, Bai R, et al. Exosomes from adipose-derived stem cells accelerate wound healing by increasing the release of IL-33 from macrophages. Stem Cell Res Ther. 2025;16:80. doi:10.1186/s13287-025-04203-x
68. Yang L, Song Y, Wang T, et al. Transcription factor c-Maf drives macrophages to promote hypertrophic scar formation. J Cosmet Dermatol. 2023;23(2):639–647. doi:10.1111/jocd.15952
69. Shen F, Cao W, Han X, et al. Microplastics impair wound healing via NAT10-mediated epigenetic dysregulation of FASN-PI3K/AKT signaling. NanoImpact. 2025;39:100580. doi:10.1016/j.impact.2025.100580
70. Lange M, Ohnesorge N, Hoffmann D, et al. Zebrafish mutants in vegfab can affect endothelial cell proliferation without altering ERK phosphorylation and are phenocopied by loss of PI3K signaling. Dev Biol. 2022;486:26–43. doi:10.1016/j.ydbio.2022.03.006
71. Matta R, Feng Y, Sansing LH, et al. Endothelial cell secreted VEGF-C enhances NSC VEGFR3 expression and promotes NSC survival. Stem Cell Res. 2021;53:102318. doi:10.1016/j.scr.2021.102318
72. Tsuji-Tamura K, Tamura M. Basic fibroblast growth factor uniquely stimulates quiescent vascular smooth muscle cells and induces proliferation and dedifferentiation. FEBS Lett. 2022;596:1686–1699. doi:10.1002/1873-3468.14345
73. Meng D, Zou J, Xu Q, et al. Endothelial cells promote the proliferation and migration of Schwann cells. Ann translat Med. 2022;10:78. doi:10.21037/atm-22-81
74. Zhang W, Xia S, Weng T, et al. Antibacterial coaxial hydro-membranes accelerate diabetic wound healing by tuning surface immunomodulatory functions. Mater Today Bio. 2022;16:100395. doi:10.1016/j.mtbio.2022.100395
75. Tefft JB, Chen CS, Eyckmans J. Reconstituting the dynamics of endothelial cells and fibroblasts in wound closure. APL Bioeng. 2021;5(1):016102. doi:10.1063/5.0028651
76. Chen L, Tredget EE, Wu PYG, et al. Correction: paracrine Factors of Mesenchymal Stem Cells Recruit Macrophages and Endothelial Lineage Cells and Enhance Wound Healing. PLoS One. 2024;19:e0302417. doi:10.1371/journal.pone.0001886
77. Radwan B, Rocchetti S, Matuszyk E, et al. EdU sensing: the Raman way of following endothelial cell proliferation in vitro and ex vivo. Biosens Bioelectron. 2022;216:114624. doi:10.1016/j.bios.2022.114624
78. Lykov AP, Surovtseva MA, Poveshchenko OV, et al. Evaluation of the Effect of Plasma from Patients with Trophic Ulcers on the Function of Dermal Fibroblasts, Mesenchymal Stem Cells, and Endothelial Cells. Bull Exp Biol Med. 2020;169(4):558–563. doi:10.1007/s10517-020-04929-z
79. Sim SL, Kumari S, Kaur S, et al. Macrophages in Skin Wounds: functions and Therapeutic Potential. Biomolecules. 2022;12(11):1659. doi:10.3390/biom12111659
80. Sousa AB, Águas AP, Barbosa MA, et al. Immunomodulatory biomaterial-based wound dressings advance the healing of chronic wounds via regulating macrophage behavior. Regenerat biomat. 2022;9:rbac065. doi:10.1093/rb/rbac065
81. Lou R, Chen J, Zhou F, et al. Exosomal miRNA-155-5p from M1-polarized macrophages suppresses angiogenesis by targeting GDF6 to interrupt diabetic wound healing. Mol Ther Nucleic Acids. 2023;34:102074. doi:10.1016/j.omtn.2023.102074
82. Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80(9):915–925. doi:10.1038/ki.2011.217
83. de Brito Sousa K, Rodrigues MFSD, de Souza Santos D, et al. Differential expression of inflammatory and anti-inflammatory mediators by M1 and M2 macrophages after photobiomodulation with red or infrared lasers. Lasers Med Sci. 2020;35(2):337–343. doi:10.1007/s10103-019-02817-1
84. Zhu X, Zhang C, Jiang W, et al. Trem2 acts as a non-classical receptor of interleukin-4 to promote diabetic wound healing. Clin transl med. 2024;14:e70026. doi:10.1002/ctm2.70026
85. Peña OA, Martin P. Cellular and molecular mechanisms of skin wound healing. Nat Rev Mol Cell Biol. 2024;25(8):599–616. doi:10.1038/s41580-024-00715-1
86. Chuhuaicura P, Rodríguez-Niklitschek C, Oporto GH, et al. Distinct Molecular Mechanisms in Oral Mucosal Wound Healing: translational Insights and Future Directions. Int J Mol Sci. 2025;26(21):10660. doi:10.3390/ijms262110660
87. Gao K, Xie Y, Xu F, et al. Silk fibroin promotes H3K9me3 expression and chromatin reorganization to regulate endothelial cell proliferation. APL Bioeng. 2024;8:026115. doi:10.1063/5.0203858
88. Li X, Zhou F, Li Y, et al. LncRNA H19-EZH2 interaction promotes liver fibrosis via reprogramming H3K27me3 profiles. Acta Pharmacol Sin. 2023;44:2479–2491. doi:10.1038/s41401-023-01145-z
89. Golebiewski C, Gastaldi C, Vieu D, et al. Identification and functional validation of SRC and RAPGEF1 as new direct targets of miR-203, involved in regulation of epidermal homeostasis. Sci Rep. 2023;13:14006. doi:10.1038/s41598-023-40441-w
90. Chang W, Xiao D, Fang X, et al. Oxidative modification of miR-30c promotes cardiac fibroblast proliferation via CDKN2C mismatch. Sci Rep. 2024;14(1):13085. doi:10.1038/s41598-024-63635-2
91. Zhang J, Yang P, Liu D, et al. Inhibiting Hyper-O-GlcNAcylation of c-Myc accelerate diabetic wound healing by alleviating keratinocyte dysfunction. Burns Trauma. 2021;9:tkab031. doi:10.1093/burnst/tkab031
92. Wang S, Ye T, Shi L, et al. Discovery of FBP1 as novel therapeutic target and asiatic acid-hydrogen sulfide donors accelerate diabetic wound healing. J Adv Res. 2025. doi:10.1016/j.jare.2025.12.003
93. Draz H, Goldberg AA, Titorenko VI, et al. Diindolylmethane and its halogenated derivatives induce protective autophagy in human prostate cancer cells via induction of the oncogenic protein AEG-1 and activation of AMP-activated protein kinase (AMPK). Cell Signalling. 2017;40:172–182. doi:10.1016/j.cellsig.2017.09.006
94. Huang Z, Huang X, Wang Q, et al. Extract of Euryale ferox Salisb exerts antidepressant effects and regulates autophagy through the adenosine monophosphate-activated protein kinase-UNC-51-like kinase 1 pathway. IUBMB Life. 2018;70:300–309. doi:10.1002/iub.1731
95. Shu F, Gao H, Wu W, et al. Amniotic epithelial cells accelerate diabetic wound healing by protecting keratinocytes and fibroblasts from high-glucose-induced senescence. Cell Biol Int. 2022;46:755–770. doi:10.1002/cbin.11771
96. Liu D, Cheng Y, Tang Z, et al. Potential Mechanisms of Methylglyoxal-Induced Human Embryonic Kidney Cells Damage: regulation of Oxidative Stress, DNA Damage, and Apoptosis. Chem Biodivers. 2021;19(2):e202100829. doi:10.1002/cbdv.202100829
97. Tavakoli S, Vaziri H, Hodjat M, et al. Expression of long non-coding RNAs DINO and ROR in bone marrow stem cells under hyperglycemic conditions. J Diabetes Metab Disord. 2026;25(1). doi:10.1007/s40200-025-01841-z
98. Wolf SJ, Audu CO, Joshi A, et al. IFN-κ is critical for normal wound repair and is decreased in diabetic wounds. JCI Insight. 2022;7:e152765. doi:10.1172/jci.insight.152765
99. Zhang L, Hu J, Meshkat BI, et al. LncRNA MALAT1 modulates TGF-β1-induced EMT in keratinocyte. Int J Mol Sci. 2021;22(21):11816. doi:10.3390/ijms222111816
100. Huang Z, Guan J, Zhang X, et al. METTL14-dependent regulation of HMGB1 attenuates inflammation in diabetic retinopathy. Acta diabetologica. 2025;63(2):245–257. doi:10.1007/s00592-025-02623-y
101. Choi W, Park DJ, Dorschner RA, et al. CDK1-loaded extracellular vesicles promote cell cycle to reverse impaired wound healing in diabetic obese mice. Mol Ther. 2025;33(3):1118–1133. doi:10.1016/j.ymthe.2025.01.039
102. Deng J, Wu X, He W, et al. Targeting DNA methylation and demethylation in diabetic foot ulcers. J Adv Res. 2023;54:119–131. doi:10.1016/j.jare.2023.01.009
103. Yang C, Lian H, Luo H, et al. The Diminution of R-Loops Generated by LncRNA DSP-AS1 Inhibits DSP Gene Transcription to Impede the Re-Epithelialization During Diabetic Wound Healing. Adv Sci. 2025;12:e2406021. doi:10.1002/advs.202406021
104. Singh K, Koroma AK, Pandey RK, et al. Restoring Histone Acetylation Accelerates Diabetic Wound Repair by Improving the Spatiotemporal Dynamics of Macrophages. Adv Sci. 2025;12:e04920. doi:10.1002/advs.202504920
105. Lin MOY, Sampath D, Bosykh DA, et al. YAP/TAZ Drive Agrin–Matrix Metalloproteinase 12–Mediated Diabetic Skin Wound Healing. J Invest Dermatol. 2024;145(1):155–170. doi:10.1016/j.jid.2024.05.005
106. Qu H, Miao T, Wang Y, et al. Dedicator of Cytokinesis 5 Regulates Keratinocyte Function and Promotes Diabetic Wound Healing. Diabetes. 2021;70(5):1170–1184. doi:10.2337/db20-1008
107. Joseph J, Chandranaath A, Louis J, et al. Comparison of Serum Matrix Metalloproteinase-9 Levels in Diabetic and Non-diabetic Chronic Wounds. Cureus. 2025;17(10):e93672. doi:10.7759/cureus.93672
108. Aghayants S, Zhou S, Zhu K, et al. Integrative analysis reveals a key role for cdkn1a in impaired wound healing in diabetic patients. Clin Cosmet Invest Dermatol. 2025;Volume 18:2149–2166. doi:10.2147/CCID.S534876
109. Bogadi S, Uddin ME, Karri VV, et al. Hypoxia Inducible Factor HIF-1α Stabilization as a Transformative Approach in Overcoming Diabetic Wound Healing Challenges. J Drug Delivery Sci Technol. 2025;113:107296. doi:10.1016/j.jddst.2025.107296
110. Yang B, Alimperti S, Gonzalez MV, et al. Reepithelialization of Diabetic Skin and Mucosal Wounds Is Rescued by Treatment With Epigenetic Inhibitors. Diabetes. 2024;73:120–134. doi:10.2337/db23-0258
111. Yue H, Umehara Y, Nguyen HL, et al. 617 AMP-IBP5 improves diabetic wound healing via activation of EGFR/STAT/MAPK pathways. J Invest Dermatol. 2021;141:S107. doi:10.1016/j.jid.2021.02.646
112. Bauer TM, Moon JY, Shadiow J, et al. Mechanisms of Impaired Wound Healing in Type 2 Diabetes: the Role of Epigenetic Factors. Arteriosclerosis Thrombosis Vasc Biol. 2025;45(5):632–642. doi:10.1161/ATVBAHA.124.321446
113. Bauer T, Barrett E, Mangum KD, et al. Inflammatory Macrophages Dictate Fibroblast Function Via Epigenetic Reprogramming in Diabetic Wounds. JVS Vascul Sci. 2023;4:100143. doi:10.1016/j.jvssci.2023.100143
114. Wolf SJ, Melvin WJ, Gallagher K. Macrophage-mediated inflammation in diabetic wound repair. Semin Cell Dev Biol. 2021;119:111–118. doi:10.1016/j.semcdb.2021.06.013
115. Zhou X, Yu D, Sun X, et al. Hypoxia-inducible Factor-1α in Diabetic Foot Ulcers: plain but Not Simple. Gene Expr. 2023;22(4):306–320. doi:10.14218/ge.2023.00051
116. Pastar I, Balukoff NC, Sawaya AP, et al. Physiology and Pathophysiology of Wound Healing in Diabetes. Contemporary Diabetes. Springer; 2024:109–134. doi:10.1007/978-3-031-55715-6_7
117. You BR, Park WH. The Effects of Mitogen-Activated Protein Kinase Inhibitors or Small Interfering RNAs on Gallic Acid-Induced HeLa Cell Death in Relation to Reactive Oxygen Species and Glutathione. J Agric Food Chem. 2010;59(2):763–771. doi:10.1021/jf103379d
118. Song M, Schnettler E, Venkatachalam A, et al. Increased expression of collagen prolyl hydroxylases in ovarian cancer is associated with cancer growth and metastasis. Am J Cancer Res. 2023;13(12):6051.
119. Ke, T, Cao, T, Tong, H. Mechanisms and intervention strategies for angiogenesis disorders in diabetic foot ulcers. 2025;41(2):120–126. doi:10.3760/cma.j.cn501225-20241204-00474
120. Zhang Z, Yang D, Shen F, et al. Epidermal keratinocytes-specific PD-L1 knockout causes delayed healing of diabetic wounds. Int Immunopharmacol. 2024;143(Pt 3):113540. doi:10.1016/j.intimp.2024.113540
121. Lebtig M, Scheurer J, Muenkel M, et al. Keratinocytes use FPR2 to detect Staphylococcus aureus and initiate antimicrobial skin defense. Front Immunol. 2023;14:1188555. doi:10.3389/fimmu.2023.1188555
122. Skov L, Baadsgaard O. Ultraviolet B-exposed major histocompatibility complex class ii positive keratinocytes and antigen-presenting cells demonstrate a differential capacity to activate T cells in the presence of staphylococcal superantigens. British j dermatol. 1996;134:824–830. doi:10.1046/j.1365-2133.1996.112847.x
123. Chen J, Ye P, Gu R, et al. Neuropeptide substance P: a promising regulator of wound healing in diabetic foot ulcers. Biochem Pharmacol. 2023;215:115736. doi:10.1016/j.bcp.2023.115736
124. Albrecht PJ, Houk G, Ruggiero E, et al. Keratinocyte Biomarkers Distinguish Painful Diabetic Peripheral Neuropathy Patients and Correlate With Topical Lidocaine Responsiveness. Front Pain Res. 2021;2:790524. doi:10.3389/fpain.2021.790524
125. Xing L, Chen B, Qin Y, et al. The role of neuropeptides in cutaneous wound healing: a focus on mechanisms and neuropeptide-derived treatments. Front Bioeng Biotechnol. 2024;12:1494865. doi:10.3389/fbioe.2024.1494865
126. Luo J, Wu T, Zhang J, et al. D-mannose promotes diabetic wound healing through inhibiting advanced glycation end products formation in keratinocytes. Mol Med. 2025;31:15. doi:10.1186/s10020-025-01070-3
127. Hu B, Li X, Li Y, et al. A comprehensive characterization of metabolic signatures-hypoxia, glycolysis, and lactylation-in non-healing diabetic foot ulcers. Front Mol Biosci. 2025;12:1593390. doi:10.3389/fmolb.2025.1593390
128. Dulcic D, Dumancic L, Gregorek AC, et al. Trimetazidine modulates angiogenesis, inflammation, and metabolism-related gene expression to promote diabetic foot ulcer healing: a transcriptomic analysis. Pharmacol Rep. 2026. doi:10.1007/s43440-026-00841-x
129. Waters JA, Robinson M, Lujano-Olazaba O, et al. Omental Preadipocytes Stimulate Matrix Remodeling and IGF Signaling to Support Ovarian Cancer Metastasis. Cancer Res. 2024;84(13):2073–2089. doi:10.1158/0008-5472.can-23-2613
130. Liu P, Liu S, Gao T, et al. Potential Role of Odanacatib in the Treatment of Postmenopausal Intervertebral Disc Degeneration. J Biomed Nanotechnol. 2024;20(4):786–792. doi:10.1166/jbn.2024.3820
131. Trojanek J. The specific role of extracellular matrix metalloproteinases in the pathology and therapy of hard-to-heal wounds. Acta Biochim Pol. 2023;70(4):745–750. doi:10.18388/abp.2020_6934
132. Abdelwahab MM, Abohashema DM, Abdelwahab MS, et al. The Role of the Extracellular Matrix (ECM) in Wound Healing (IE, Matrix Metalloproteinases (MMPs) and Growth Factors). Nanotechnol Wound Heal. 2025:124–148. doi:10.1201/9781003605966-6
133. Seo G, Hyun C, Choi S, et al. The wound healing effect of four types of beta-glucan. Appl Biol Chem. 2019;62(1). doi:10.1186/s13765-019-0428-2
134. Wang X, Guo W, Qiu W, et al. Fibroblast-like cells Promote Wound Healing via PD-L1-mediated Inflammation Resolution. Int J Bio Sci. 2022;18(11):4388–4399. doi:10.7150/ijbs.69890
135. Sami Shafiek M. The role of Cytokines in Influencing the Healing of Wounds: mechanisms, Therapeutic Targets, and Advancements. Transl Health Sci. 2025;1(1):25–39. doi:10.21608/ths.2025.344997.1002
136. Chen L, Cheng L, Chen T, et al. Macrophage Polarization in Skin Wound Healing: progress in Biology and Therapeutics. J Shanghai Jiaotong Univ. 2021;27(2):264–280. doi:10.1007/s12204-021-2276-6
137.. Mazurek Ł, Rybka M, Zajdel M, et al. Keratin-acetate dressing accelerates diabetic wound healing, promotes m2 macrophage polarization and increases cytokeratins 16 and 17 expression-in vitro and in vivo studies. Macromol Biosci. 2025;2025:e351. doi:10.1002/mabi.202500351
138. Gupta S, Mujawdiya P, Maheshwari G, et al. Dynamic Role of Oxygen in Wound Healing: a Microbial, Immunological, and Biochemical Perspective. Arch Razi Inst. 2022;77(2):513–523. doi:10.22092/ari.2022.357230.2003
139. Lin K, Xu H, Liu H, et al. Bioprinted dressing with symbiotic microbes for oxygen supply and antibacterial therapy for enhanced diabetic wound healing. J Control Release. 2026;392:114653. doi:10.1016/j.jconrel.2026.114653
140. White EK, Uberoi A, Pan JTC, et al. Alcaligenes faecalis corrects aberrant matrix metalloproteinase expression to promote reepithelialization of diabetic wounds. Sci Adv. 2024;10(26):eadj2020. doi:10.1126/sciadv.adj2020
141. Lou J, Xiang Z, Zhu X, et al. Skin microbiota and diabetic foot ulcers. Front Microbiol. 2025;16:1575081. doi:10.3389/fmicb.2025.1575081
142. Lutsenko AV, Samotrueva MA, Poroyskiy SV. Wound microbiome: mechanisms of pathogenicity and intermicrobial interactions of Pseudomonas aeruginosa and Staphylococcus aureus. SSMJ. 2026;45(6):84–96. doi:10.18699/ssmj20250608
143. Feng J, Lai S, Tang D. Mechanisms and Interventions of Diabetic Wound Healing. Current Diabetes Rev. 2025;22:E15733998356905. doi:10.2174/0115733998356905250706203510
144. Ahmad-Mansour N, Plumet L, Pouget C, et al. The ROSA-Like Prophage Colonizing Staphylococcus aureus Promotes Intracellular Survival, Biofilm Formation, and Virulence in a Chronic Wound Environment. J Infect Dis. 2023;228:1800–1804. doi:10.1093/infdis/jiad218
145. Hammad AS, Zahedy SH, Elqasass SS, et al. In Vitro Analysis of the Dynamic Role of the Bacterial Virulence Factors in Skin Wound Healing. Int J Mol Sci. 2025;26:10472. doi:10.3390/ijms262110472
146. Al-Taweel R, Hammad AS, Tajammul A, et al. Wounds and the Microbiota: the Healing Interplay Between Host and Microbial Communities. Int J Mol Sci. 2025;26:11365. doi:10.3390/ijms262311365
147. Fu Y, Leng C, Fan Y, et al. In Vitro and In Vivo Activity of 14-O-[(4,6-Diamino-pyrimidine-2-yl) thioacetyl] Mutilin against Methicillin-Resistant Staphylococcus aureus. Molecules. 2021;26(11):3277. doi:10.3390/molecules26113277
148. Konca M, Korkmaz M. Comparison of Effects of Administration of Oral or Topical Boron on Wound Healing and Oxidative Stress in Rats. Kocatepe Veter J. 2020;1. doi:10.30607/kvj.646939
149. Chang M, Nguyen TT. Strategy for Treatment of Infected Diabetic Foot Ulcers. Acc Chem Res. 2021;54:1080–1093. doi:10.1021/acs.accounts.0c00864
150. Li J, Wei C, Yang Y, et al. Apoptotic bodies extracted from adipose mesenchymal stem cells carry microRNA-21–5p to induce M2 polarization of macrophages and augment skin wound healing by targeting KLF6. Burns. 2022;48(8):1893–1908. doi:10.1016/j.burns.2021.12.010
151. Liechty C, Hu J, Zhang L, et al. Role of microRNA-21 and Its Underlying Mechanisms in Inflammatory Responses in Diabetic Wounds. Int J Mol Sci. 2020;21(9):3328. doi:10.3390/ijms21093328
152. Xuan A, Liu M, Zhang L, et al. SZC-6 Promotes Diabetic Wound Healing in Mice by Modulating the M1/M2 Macrophage Ratio and Inhibiting the MyD88/NF-χB Pathway. Pharmaceuticals. 2025;18:1143. doi:10.3390/ph18081143
153. Versey Z. Unveiling Macrophage-Biofilm Interactions: Implications for Cellular Metabolism and Wound Healing. Carleton University; 2023.
154. Zhang S, Zhao X, Zhang W, et al. Zn-DHM nanozymes regulate metabolic and immune homeostasis for early diabetic wound therapy. Bioact Mater. 2025;49:63–84. doi:10.1016/j.bioactmat.2025.02.041
155. Pouget C, Dunyach-Remy C, Magnan C, et al. Polymicrobial Biofilm Organization of Staphylococcus aureus and Pseudomonas aeruginosa in a Chronic Wound Environment. Int J Mol Sci. 2022;23(18):10761. doi:10.3390/ijms231810761
156. Yamberla I, Pupiales C, Chiliquinga AJ, et al. Pseudomonas aeruginosa Pathogenicity and Its Interaction with Other Microorganisms During the Skin Wound Healing Process. Int J Mol Sci. 2025;26:9677. doi:10.3390/ijms26199677
157. Short B, Bakri A, Baz A, et al. There is more to wounds than bacteria: fungal biofilms in chronic wounds. Current Clin Microbiol Rep. 2023;10(1):9–16. doi:10.1007/s40588-022-00187-x
158. Van Genechten W, Wijnants S, Vreys J, et al. Differential sensing by the C. albicans Gpr1 receptor results in morphogenesis, β-glucan masking and survival in macrophages. BioRxiv. 2022;2022:518566. doi:10.1101/2022.11.30.518566
159. Versey Z, Da Cruz Nizer WS, Russell E, et al. Biofilm-Innate Immune Interface: contribution to Chronic Wound Formation. Front Immunol. 2021;12:648554. doi:10.3389/fimmu.2021.648554
160. Khalid A, Beeton M, Maddocks S. Assessing the Interspecies Relationship Between Pseudomonas Aeruginosa and Staphylococcus Aureus in Mixed Biofilms Grown in Alginate Beads and Collagen Scaffolds. Microbiology Society; 2022.
161. Wardell SJT, Yung DBY, Gupta A, et al. DJK-5, an anti-biofilm peptide, increases Staphylococcus aureus sensitivity to colistin killing in co-biofilms with Pseudomonas aeruginosa. NPJ biofilmsmicrobio. 2025;11:8. doi:10.1038/s41522-024-00637-y
162. Coelho MDMF, Avelino BMA, de Oliveira BA, et al. Prevalence of Biofilm in Chronic Wounds: systematic Review With Meta-Analysis. Wounds. 2025;37(8):283–291. doi:10.25270/wnds/24124
163. Ohmann A, Guschin D, Spies P, et al. A recombinant fusion protein that mimics the inflammatory biomarker calprotectin as a tool to harmonize Mrp-8/Mrp-14 immunoassays. Clin Chim Acta. 2024;558:119801. doi:10.1016/j.cca.2024.119801
164. Namuganga AR, Nsereko M, Bagaya BS, et al. Differential expression of host protein biomarkers among symptomatic clinic attendees finally diagnosed with tuberculosis and other respiratory diseases with or without latent Mycobacterium tuberculosis infection. Immunol Lett. 2022;253:8–18. doi:10.1016/j.imlet.2022.11.006
165. Wu Q, Zhao M, Li D, et al. Cholinergic drugs reduce metabolic inflammation and diabetic myocardial injury by regulating the gut bacterial component lipopolysaccharide-induced ERK /Egr-1 pathway. FASEB J. 2023;37:e22917. doi:10.1096/fj.202202108
166. Sharma V, Singh J, Kumar Y, et al. Integrated insights into gene expression dynamics and transcription factor roles in diabetic and diabetic-infectious wound healing using rat model. Life Sci. 2025;368:123508. doi:10.1016/j.lfs.2025.123508
167. Lukiw WJ, Pelaez RP, Martinez J, Bazan NG. Budesonide epimer R or dexamethasone selectively inhibit platelet-activating factor-induced or interleukin 1beta-induced DNA binding activity of cis-acting transcription factors and cyclooxygenase-2 gene expression in human epidermal keratinocytes. Proc Natl Acad Sci U S A. 1998;95(7):3914–3919. doi:10.1073/pnas.95.7.3914
168. Fiskus W, Masarova L, Mill CP, et al. Preclinical efficacy of tasquinimod-based combinations in advanced myeloproliferative neoplasms in blastic phase. Blood Adv. 2025;9:5598–5609. doi:10.1182/bloodadvances.2025016898
169. Haegerstrand A, Jonzon B, Dalsgaard CJ, Nilsson J. Vasoactive intestinal polypeptide stimulates cell proliferation and adenylate cyclase activity of cultured human keratinocytes. Proc Natl Acad Sci U S A. 1989;86(15):5993–5996. doi:10.1073/pnas.86.15.5993
170. Yang SL, Zhu LY, Han R, et al. Effect of Negative Pressure Wound Therapy on Cellular Fibronectin and Transforming Growth Factor-β1 Expression in Diabetic Foot Wounds. Foot and Ankle Int. 2017;38(8):893–900. doi:10.1177/1071100717704940
171. Lai CY, Liu H, Tin KX, et al. Identification of UAP1L1 as a critical factor for protein O-GlcNAcylation and cell proliferation in human hepatoma cells. Oncogene. 2018;38(3):317–331. doi:10.1038/s41388-018-0442-6
172. Xue JJ, Sun H. The Role of O-GlcNAc Modification in the Immune System. Chin J Biochem Mol Biol. 2024;40:2.
173. Qiu S, Jia Y, Sun Y, et al. Von Hippel-Lindau (VHL) Protein Antagonist VH298 Improves Wound Healing in Streptozotocin-Induced Hyperglycaemic Rats by Activating Hypoxia-Inducible Factor- (HIF-) 1 Signalling. J Diabetes Res. 2019;2019:1897174. doi:10.1155/2019/1897174
174. Zhang LN. O-GlcNAc Modification Regulates NLRP3 Inflammasome and Its Effect on Hypoxia-reoxygenation Injury in H9c2 Rat Cardiomyocytes. Chinese J Pathophysiol. 2021;37:2139–2146.
175. Wang J, Lv X, Aniwan A, et al. O-GlcNAcylation regulates HIF-1α and induces mesothelial-mesenchymal transition and fibrosis of human peritoneal mesothelial cells. Heliyon. 2023;9(12):e22916. doi:10.1016/j.heliyon.2023.e22916
176. Wang J, Lv X, Lin Y, et al. Genistein inhibits HIF-1α and attenuates high glucose-induced peritoneal mesothelial-mesenchymal transition and fibrosis via the mTOR/OGT pathway. Sci Rep. 2024;14(1):24369. doi:10.1038/s41598-024-74879-3
177. Jiang A, Li J, Wang L, et al. HIF1A regulates follicular atresia through O-GlcNAcylation-mediated VEZF1/ET-1/FOXO1/BAX signaling in porcine granulosa cells. J Anim Sci Biotechnol. 2025;16(1):127. doi:10.1186/s40104-025-01263-0
178. Tang D, Lin Q, Li P, et al. Fg-4592 combined with prp significantly accelerates the healing of refractory diabetic wounds by upregulating hif-1α. Sci Rep. 2025;15:14292. doi:10.1038/s41598-025-99356-3
179. Li G, Ko C, Li D, et al. A small molecule hif-1α stabilizer that accelerates diabetic wound healing. Nat Commun. 2021;12:3363. doi:10.1038/s41467-021-23448-7
180. Xu C, Liu X, Zha H, et al. A pathogen-derived effector modulates host glucose metabolism by arginine GlcNAcylation of HIF-1α protein. PLoS Pathog. 2018;14(8):e1007259. doi:10.1371/journal.ppat.1007259
181. Giogha C, Scott NE, Wong Fok Lung T, et al. NleB2 from enteropathogenic Escherichia coli is a novel arginine-glucose transferase effector. PLoS Pathogens. 2021;17(6):e1009658. doi:10.1371/journal.ppat.1009658
182. Geng K, Ma X, Jiang Z, et al. High glucose-induced sting activation inhibits diabetic wound healing through promoting m1 polarization of macrophages. Cell Death Discov. 2023;9:136. doi:10.1038/s41420-023-01425-x
183. Feng Z, Zang C, Zhang L, et al. Sting activation promotes inflammatory response and delays skin wound healing in diabetic mice. Biochem Biophys Res Commun. 2022;611:126–131. doi:10.1016/j.bbrc.2022.04.085
184. Geng K, Ma X, Jiang Z, et al. Innate immunity in diabetic wound healing: focus on the mastermind hidden in chronic inflammatory. Front Pharmacol. 2021;12:653940. doi:10.3389/fphar.2021.653940
185. Ren X, Wang R, Yu X, et al. Regulation of histone h3 lysine 9 methylation in inflammation. All Life. 2021;14(1):492–508. doi:10.1080/26895293.2021.1931477
186. Audu CO, Melvin WJ, Joshi AD, et al. Macrophage-specific inhibition of the histone demethylase JMJD3 decreases STING and pathologic inflammation in diabetic wound repair. Cell Mol Immunol. 2022;19(11):1251–1262. doi:10.1038/s41423-022-00919-5
187. Zhang X, Guo Z, Li T, et al. Dietary capsaicin normalizes cgrp peptidergic drg neurons in experimental diabetic peripheral neuropathy. Sci Rep. 2021;11(1):1704. doi:10.1038/s41598-021-81427-w
188. Li X, Yuan D, Zhang P, et al. A neuron-mast cell axis regulates skin microcirculation in diabetes. Diabetes. 2024;73:1728–1741. doi:10.2337/db23-0862
189. Xie RM, Li X, Li QQ, et al. Hmgb1: effects on inflammation and mechanism analysis in rat diabetic foot ulcers. Prog Mod Biomed. 2020;20:2430–2434.
190. Li T, Guo Z, Liu C, et al. Association of down-regulation of calcitonin gene-related peptide and substance p with increase of myocardial vulnerability in diabetic neuropathic rats. Peptides. 2017;96:1–7. doi:10.1016/j.peptides.2017.08.007
191. Gylfadottir SS, Itani M, Kristensen AG, et al. Analysis of macrophages and peptidergic fibers in the skin of patients with painful diabetic polyneuropathy. Neurology. 2022;9:e1111. doi:10.1212/NXI.0000000000001111
192. Li X, Yi M, Song Z, et al. A calcitonin gene-related peptide co-crosslinked hydrogel promotes diabetic wound healing by regulating M2 macrophage polarization and angiogenesis. Acta Biomater. 2025;196:109–122. doi:10.1016/j.actbio.2025.02.046
193. Ullah S, Iqbal K, Rizwan M. Gait and Postural Control Deficits in Diabetic Patients with Peripheral Neuropathy Compared to Healthy Controls. Bioengineering. 2025;12(10):1034. doi:10.3390/bioengineering12101034
194. Li F, Chen W, Luo Z, et al. Bone marrow mesenchymal stem cell-derived exosomal micrornas target pi3k/akt signaling pathway to promote the activation of fibroblasts. World J Stem Cells. 2023;15:248. doi:10.4252/wjsc.v15.i4.248
195. Mahheidari N, Alizadeh M, Rashidi M, et al. Potential of mesenchymal stem cells and exosomes in tissue engineering: emerging strategies for skin regeneration and advanced wound healing. Tissue Cell. 2025;98:103201. doi:10.1016/j.tice.2025.103201
196. Grigoryeva ES, Tashireva LA, Savelieva OE, et al. The association of integrins β3, β4, and αvβ5 on exosomes, ctcs and tumor cells with localization of distant metastasis in breast cancer patients. Int J Mol Sci. 2023;24(3):2929. doi:10.3390/ijms24032929
197. Tang W, Du X, Wu Z, et al. Circ-erbb2ip from adipose-derived mesenchymal stem cell-derived exosomes promotes wound healing in diabetic mice by inducing the mir-670-5p/nrf1 axis. Cell Signal. 2024;121:111245. doi:10.1016/j.cellsig.2024.111245
198. Li B, Qian L, Pi L, et al. A therapeutic role of exosomal lncrna h19 from adipose mesenchymal stem cells in cutaneous wound healing by triggering macrophage m2 polarization. Cytokine. 2023;165:156175. doi:10.1016/j.cyto.2023.156175
199. Kasowanjete P, Abrahamse H, Houreld NN. Photobiomodulation at 660 nm stimulates in vitro diabetic wound healing via the ras/mapk pathway. Cells. 2023;12(7):1080. doi:10.3390/cells12071080
200. Rajendran NK, Houreld NN. Photobiomodulation hastens diabetic wound healing via modulation of the pi3k/akt/foxo1 pathway in an adipose derived stem cell-fibroblast co-culture. J Photochem Photobiol. 2022;12:100157. doi:10.1016/j.jpap.2022.100157
201. Chen G, Chen G, Lu J, Hu S. Exosomal non-coding RNAs: a new avenue for treating diabetic foot ulcers. Front Mol Biosci. 2025;12:1701879. doi:10.3389/fmolb.2025.1701879
202. Morabbi A, Karimian M. Therapeutic potential of exosomal lncRNAs derived from stem cells in wound healing: focusing on mesenchymal stem cells. Stem Cell Res Ther. 2025;16(1):62. doi:10.1186/s13287-025-04200-0
203. Gumede DB, Abrahamse H, Houreld NN. Targeting wnt/β-catenin signaling and its interplay with tgf-β and notch signaling pathways for the treatment of chronic wounds. Cell Commun Signaling. 2024;22:244. doi:10.1186/s12964-024-01623-9
204. Priyadarshi A, Keshri GK, Gupta A. Effect of combination of photobiomodulation 904 nm superpulsed laser therapy and Hippophae rhamnoides L. On third-degree burn wound healing. J Cosmet Dermatol. 2023;22:2492–2501. doi:10.1111/jocd.15806
205. Priyadarshi A, Keshri GK, Yadav D, et al. Dual near-infrared wavelength photobiomodulation accelerates transdermal burn wound repair via anti-inflammatory, pain relief and redox-regulating mechanisms. J Photochem Photobiol B Biol. 2025;272:113267. doi:10.1016/j.jphotobiol.2025.113267
206. Liu S, Bamodu OA, Kuo K, et al. Adipose-derived stem cell induced-tissue repair or wound healing is mediated by the concomitant upregulation of mir-21 and mir-29b expression and activation of the akt signaling pathway. Arch Biochem Biophys. 2021;705:108895. doi:10.1016/j.abb.2021.108895
207. Wang Y, Zhu J, Chen J, et al. The signaling pathways induced by exosomes in promoting diabetic wound healing: a mini-review. Curr Issues Mol Biol. 2022;44:4960–4976. doi:10.3390/cimb44100337
208. Hsu H, Wang AYL, Loh CYY, et al. Therapeutic potential of exosomes derived from diabetic adipose stem cells in cutaneous wound healing of db/db mice. Pharmaceutics. 2022;14:1206. doi:10.3390/pharmaceutics14061206
209. Liu N, Xie Y, Zhen Y, et al. Free-cell therapeutics and mechanism of exosomes from adipose-derived stem cells in promoting wound healing: current understanding and future applications. Chin Med J. 2022;135:1803–1805. doi:10.1097/CM9.0000000000001857
210. Jinyi L, Xianguang D, Yuhan D, et al. Metabolic reprogramming in diabetic foot ulcers: mechanisms, therapeutic implications and future perspectives. Metabolism. 2026;175:156455. doi:10.1016/j.metabol.2025.156455
211. Zhang P, Yang J, Liu X, et al. Fbp1 orchestrates keratinocyte proliferation/differentiation and suppresses psoriasis through metabolic control of histone acetylation. Cell Death Dis. 2024;15:392. doi:10.1038/s41419-024-06706-6
212. Luanpitpong S, Rodboon N, Samart P, et al. A novel trpm7/o-glcnac axis mediates tumour cell motility and metastasis by stabilising c-myc and caveolin-1 in lung carcinoma. Br J Cancer. 2020;123:1289–1301. doi:10.1038/s41416-020-0991-7
213. Wang H, Sun J, Sun H, et al. The ogt–c-myc–pdk2 axis rewires the tca cycle and promotes colorectal tumor growth. Cell Death Differ. 2024;31:1157–1169. doi:10.1038/s41418-024-01315-4
214. Xie D, Yang K, Xu Y, et al. N6-methyladenosine demethylase fat mass and obesity-associated protein suppresses hyperglycemia-induced endothelial cell injury by inhibiting reactive oxygen species formation via autophagy promotion. J Diabetes Complications. 2024;38:108801. doi:10.1016/j.jdiacomp.2024.108801
215. Zhang Y, Gao L, Wang W, et al. M 6 A demethylase fat mass and obesity-associated protein regulates cisplatin resistance of gastric cancer by modulating autophagy activation through ULK1. Cancer Sci. 2022;113:3085–3096. doi:10.1111/cas.15469
216. Farooq M, Hwang M, Khan AW, et al. Identification of a novel fibroblast growth factor receptor-agonistic peptide and its effect on diabetic wound healing. Life Sci. 2025;364:123432. doi:10.1016/j.lfs.2025.123432
217. Li C, Fu Z, Jin T, et al. A frog peptide provides new strategies for the intervention against skin wound healing. Cell Mol Biol Lett. 2023;28(1):61. doi:10.1186/s11658-023-00468-3
218. Yue H, Song P, Sutthammikorn N, et al. Antimicrobial peptide derived from insulin-like growth factor-binding protein 5 improves diabetic wound healing. Wound Repair Regener. 2022;30:232–244. doi:10.1111/wrr.12997
219. Zhang Y, Chen Y, Li K, et al. Ghrelin promotes chronic diabetic wound healing by regulating keratinocyte proliferation and migration through the erk1/2 pathway. Peptides. 2025;184:171350. doi:10.1016/j.peptides.2025.171350
220. Wang T, Li Y, Hao L, et al. Coriander-Derived Exosome-Like Nanovesicles Laden Hydrogel with Antioxidant Property Accelerates Wound Healing. Macromol Biosci. 2025;25:e2400640. doi:10.1002/mabi.202400640
221. Zhao Q, Hu QX, Li JP, et al. Morinda Officinalis-Derived Extracellular Vesicle-like Particles Promote Wound Healing via Angiogenesis. ACS Appl Mater Interfaces. 2025;17:30454–30464. doi:10.1021/acsami.5c01640
222. Zanotti C, Arena S, De Pascale S, et al. Anion exchange chromatography-based purification of plant-derived nanovesicles from Brassica oleracea L.: molecular profiling and bioactivity in human cells. Front Bioeng Biotechnol. 2025;13:1617478. doi:10.3389/fbioe.2025.1617478
223. Suresh AP, Kalarikkal SP, Pullareddy B, et al. Low pH-Based Method to Increase the Yield of Plant-Derived Nanoparticles from Fresh Ginger Rhizomes. ACS Omega. 2021;6(27):17635–17641. doi:10.1021/acsomega.1c02162
224. Guo J, Hu B, Wei Y, et al. Hydrogel delivering self-assembled herbal nanoparticles accelerates diabetic wound healing through mitochondrial regulation. Mater Today Bio. 2025;35:102417. doi:10.1016/j.mtbio.2025.102417
225. Kant V, Jangir BL, Sharma M, et al. Topical application of quercetin improves wound repair and regeneration in diabetic rats. Immunopharmacol Immunotoxicol. 2021;43(5):536–553. doi:10.1080/08923973.2021.1950758
226. Suresh A, Ravilla J, Narayanan J, et al. Mango ginger-derived exosome-like nanovesicles promotes diabetic wound healing via inducing the promigratory protein, follistatin-like 1. Int J Biol Macromol. 2025;322:146991. doi:10.1016/j.ijbiomac.2025.146991
227. Liu W, Jia L, Zhao S, et al. ROS-scavenging and antimicrobial polysaccharide hydrogel for methicillin-resistant staphylococcus aureus-infected diabetic wound healing. Nano Select. 2022;3:1537–1547. doi:10.1002/nano.202200117
228. Baviskar KD, Lodhi S. Preparation, Characterization and Evaluation Of Gellan Gum/Glycol Chitosan-Based Baicalein Hydrogel For Wound Healing. Int J Appl Pharm. 2024;16:299–305. doi:10.22159/ijap.2024v16i2.49661
229. Yang W, Xing Z, Wang X, et al. Microenvironment-responsive collagen hydrogel with houttuynia cordata thunb vesicles for diabetic wound repair. Int J Biol Macromol. 2025;320:145840. doi:10.1016/j.ijbiomac.2025.145840
230. Liang X, Xian C, Du J, et al. Sprayable self-assembled curcumin-metal-polyphenol nanomedicine with anti-inflammatory, ROS-scavenging and pro-angiogenesis effects for promoting diabetic wound healing. Nano Res. 2025;18:94908001. doi:10.26599/nr.2025.94908001
231. Zhao H, Li L, Miao C, et al. Effects of Tanshinone IIA on the expression of HIF-1α and VEGF in brain tissue of rats with cerebral hemorrhage. J Apoplexy Nervous Dis. 2017;2017:34–36. doi:10.19845/j.cnki.zfysjjbzz.2017.02.009
232. Gupta J, Kumar D, Gupta R, et al. Therapeutic Potential of Traditional Chinese Medicine loaded Nanocarriers in Wound Management: current Status and their Future Perspective. Pharmacol Res Mod Chin Med. 2025;15:100622. doi:10.1016/j.prmcm.2025.100622
233. Miya MB, Dey D, Pathak V, et al. Accelerated diabetic wound healing using a chitosan-based nanomembrane incorporating nanovesicles from Aloe barbadensis, Azadirachta indica, and Zingiber officinale. Int J Biol Macromol. 2025;310:143169. doi:10.1016/j.ijbiomac.2025.143169
234. Jin E, Yang Y, Cong S, et al. Lemon-derived nanoparticle-functionalized hydrogels regulate macrophage reprogramming to promote diabetic wound healing. J Nanobiotechnol. 2025;23:68. doi:10.1186/s12951-025-03138-y
235. Kumar M, Keshwania P, Chopra S, et al. Therapeutic Potential of Nanocarrier-Mediated Delivery of Phytoconstituents for Wound Healing: their Current Status and Future Perspective. AAPS Pharm Sci Tech. 2023;24(6):155. doi:10.1208/s12249-023-02616-6
236. Song Y, Wang F, Xia J, et al. Bioactivity and multi-omics profiling of purslane-derived nanovesicles with therapeutic implications in diabetic wounds. J Adv Res. 2025. doi:10.1016/j.jare.2025.10.042
237. Liu D, Gao J, Wu X, et al. Conductive Microneedles Loaded With Polyphenol-Engineered Exosomes Reshape Diabetic Neurovascular Niches for Chronic Wound Healing. Adv Sci. 2025;12(43):e07974. doi:10.1002/advs.202507974
238. Lu Y, Zhang W, Zeng P, et al. Light modulates plant-derived extracellular vesicle properties: a photosensitive-responsive nanodelivery system. Discover Nano. 2025;20:86. doi:10.1186/s11671-025-04266-y
239. Moon K, Hur J, Kim KP, et al. Surface-Functionalizable Plant-Derived Extracellular Vesicles for Targeted Drug Delivery Carrier Using Grapefruit. Adv Mater Interfaces. 2023;10(22):2300220. doi:10.1002/admi.202300220
240. Chen J, Ma H, Meng Y, et al. Analysis of the mechanism underlying diabetic wound healing acceleration by Calycosin-7-glycoside using network pharmacology and molecular docking. Phytomedicine. 2023;114:154773. doi:10.1016/j.phymed.2023.154773
241. Fan W, Qu Y, Yuan X, et al. Loureirin B Accelerates Diabetic Wound Healing by Promoting TGFβ /Smad-Dependent Macrophage M2 Polarization: a Concerted Analytical Approach Through Single-Cell RNA Sequencing and Experimental Verification. Phytother Res. 2025;39:5450–5463. doi:10.1002/ptr.8373
242. Zhang M, Wu SX, Dong M, et al. Novel nanodelivery system: engineered small extracellular vesicles. Chin J Tissue Eng Res. 2022;26:4417.
243. Şahin F, Koçak P, Güneş MY, et al. In vitro wound healing activity of wheat-derived nanovesicles. Appl Biochem Biotechnol. 2019;188(2):381–394. doi:10.1007/s12010-018-2913-1
244. Zhang Y, Wang R, Wang H, et al. Herbal hydrogel incorporating scutellaria baicalensis georgi carbon dots and baicalein for antibacterial, anti-inflammatory, and wound healing applications. Mater Des. 2025;257:114468. (in Chinese). doi:10.1016/j.matdes.2025.114468
245. Mi Y, Zhong L, Lu S, et al. Quercetin promotes cutaneous wound healing in mice through wnt/β-catenin signaling pathway. J Ethnopharmacol. 2022;290:115066. doi:10.1016/j.jep.2022.115066
246. Rybka M, Mazurek Ł, Jurak J, et al. Keratin-tmao dressing accelerates full-thickness skin wound healing in diabetic rats via m2-macrophage polarization and the activation of pi3k/akt/mtor signaling pathway. Int J Biol Macromol. 2025;310:143313. doi:10.1016/j.ijbiomac.2025.143313
247. Zhu H, Xing C, Dou X, et al. Chiral hydrogel accelerates re-epithelization in chronic wounds via mechanoregulation. Adv Healthc Mater. 2022;11(21):e2201032. doi:10.1002/adhm.202201032
248. Tang F, Li J, Xie W, et al. Bioactive glass promotes the barrier functional behaviors of keratinocytes and improves the re-epithelialization in wound healing in diabetic rats. Bioact Mater. 2021;6:3496–3506. doi:10.1016/j.bioactmat.2021.02.041
249. Qiu M, He Y, Zhang H, et al. Platelet-rich plasma (prp) based on simple and efficient integrated preparation precises quantitatively for skin wound repair. Int J Mol Sci. 2024;25(17):9340. doi:10.3390/ijms25179340
250. Yi J, Tang Q, Sun S, et al. Exosomes in diabetic wound healing: mechanisms, applications, and perspectives. Diab Metabol Syndr Obes. 2025;Volume 18:2955–2976. doi:10.2147/DMSO.S532885
251. Ning Y, Yuan Z, Wang Q, et al. Epigallocatechin-3-gallate promotes wound healing response in diabetic mice by activating keratinocytes and promoting re-epithelialization. Phytother Res. 2024;38:1013–1027. doi:10.1002/ptr.8099
252. Januszyk M, Chen K, Henn D, et al. Characterization of Diabetic and Non-Diabetic Foot Ulcers Using Single-Cell RNA-Sequencing. Micromachines. 2020;11(9):815. doi:10.3390/mi11090815
253. Parveen K, Hussain MA, Anwar S, Elagib HM, Kausar MA. Comprehensive review on diabetic foot ulcers and neuropathy: treatment, prevention and management. World J Diab. 2025;16(3):100329. doi:10.4239/wjd.v16.i3.100329
254. Rai V, Moellmer R, Agrawal DK. Clinically relevant experimental rodent models of diabetic foot ulcer. Mol Cell Biochem. 2022;477(4):1239–1247. doi:10.1007/s11010-022-04372-w
255. Gong HP, Ren Y, Zha PP, et al. Sichuan Da Xue Xue Bao Yi Xue Ban. Sichuan da Xue Xue Bao. Yi Xue Ban = Journal of Sichuan University. Medical Science Edition. 2022;53(6):969–975. doi:10.12182/20220860105
256. Yadav JP, Singh AK, Grishina M, et al. Insights into the mechanisms of diabetic wounds: pathophysiology, molecular targets, and treatment strategies through conventional and alternative therapies. Inflammopharmacology. 2024;32(1):149–228. doi:10.1007/s10787-023-01407-6
257. Sun R, Xu Y, Ji Z, et al. Update on the impact of lipid and glucose control on diabetic wound healing. Metab Open. 2025;28:100408. doi:10.1016/j.metop.2025.100408
258. Montaser E, Abad SE, Shah VN. Changes in A1C Versus GMI Across Glycemic Categories in Clinical Trials of Type 1 Diabetes. J Clin Endocrinol Metab. 2025;110(12):e4182–e4187. doi:10.1210/clinem/dgaf211
259. Sousa P, Lopes B, Sousa AC, et al. Advancements and Insights in Exosome-Based Therapies for Wound Healing: a Comprehensive Systematic Review (2018–June 2023). Biomedicines. 2023;11(8):2099. doi:10.3390/biomedicines11082099
260. Garima, Sharma D, Kumar A, Mostafavi E. Extracellular vesicle-based biovectors in chronic wound healing: biogenesis and delivery approaches. Mol Ther Nucleic Acids. 2023;32:822–840. doi:10.1016/j.omtn.2023.05.002
261. Sun T, Li M, Liu Q, et al. Insights into optimizing exosome therapies for acute skin wound healing and other tissue repair. Front Med. 2024;18(2):258–284. doi:10.1007/s11684-023-1031-9
262. Wang F, Yao J, Zuo H, et al. Diverse-Origin Exosomes Therapeutic Strategies for Diabetic Wound Healing. Int J Nanomed. 2025;20:7375–7402. doi:10.2147/ijn.s519379
263. Munap DHFA, Bakhtiar H, Bidin N, et al. Wavelength and dose-dependent effects of photobiomodulation therapy on wound healing in rat model. Laser Physics. 2018;28(11):115602. doi:10.1088/1555-6611/aad84b
264. Eber J, Schohn A, Carinato H, et al. A Pilot Study Comparing Intraoral and Transcutaneous Photobiomodulation for Oral Mucositis in Head and Neck Cancer Patients Undergoing Radiotherapy or Chemoradiotherapy. J Clin Med. 2025;14(7):2430. doi:10.3390/jcm14072430
265. Samak MM. Comment on “Post-transcriptional regulation of diabetic wound healing by junctional adhesion molecule A/miR-106b axis”. Burns. 2025;51(9):107733. doi:10.1016/j.burns.2025.107733
266. Amjadian S, Fatemi MJ, Moradi S, et al. miR-182-5p regulates all three phases of inflammation, proliferation, and remodeling during cutaneous wound healing. Archiv Dermatological Res. 2024;316(6):274. doi:10.1007/s00403-024-03079-w
267. Ni D, Liu N, Peng Y, et al. MiR-181d-5p affects skin wound healing processes via the Ikbkg/NF-κB axis. Int J Biol Macromol. 2025;322(Pt 4):147007. doi:10.1016/j.ijbiomac.2025.147007
268. Jeyaraman N, Roy M, Muthu S, et al. Preclinical Insights into Wound Healing: models, Mechanisms, and Translational Strategies. Stem Cell Biol Regener Med. 2026:287–309. doi:10.1007/978-3-032-12442-5_12
269. Zheng D, Zhu M, Liu X, et al. Stimuli-responsive hydrogels: an intelligent tool for wound management. Interdiscip Med. 2026;4. doi:10.1002/inmd.70104
270. Tang Y, Xu H, He S, et al. Ultrasound-Stimulated BMSCs Promote Regenerative Healing in Refractory Foot Ulcer by Paracrine Effect. Ultrasound Med Biol. 2025;51(12):2292–2302. doi:10.1016/j.ultrasmedbio.2025.08.005
271. Sun T, Jiang C. Stimuli-responsive drug delivery systems triggered by intracellular or subcellular microenvironments. Adv Drug Delivery Rev. 2023;196:114773. doi:10.1016/j.addr.2023.114773
272. Kumar R, Kumar V, Mohan A, et al. Translational research in the generation of therapeutic medicine for wound healing: a review. Discover Med. 2024;1(1). doi:10.1007/s44337-024-00142-3
273. Rembe JD, Garabet W, Augustin M, et al. Immunomarker profiling in human chronic wound swabs reveals IL-1 beta/IL-1RA and CXCL8/CXCL10 ratios as potential biomarkers for wound healing, infection status and regenerative stage. J Transl Med. 2025;23(1):407. doi:10.1186/s12967-025-06417-2
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.