Back to Journals » International Journal of Nanomedicine » Volume 11
Multicomponent, peptide-targeted glycol chitosan nanoparticles containing ferrimagnetic iron oxide nanocubes for bladder cancer multimodal imaging
Authors Key J ![]() , Dhawan D, Cooper CL, Knapp DW, Kim K, Kwon IC, Choi K, Park K, Decuzzi P, Leary JF
, Dhawan D, Cooper CL, Knapp DW, Kim K, Kwon IC, Choi K, Park K, Decuzzi P, Leary JF
Received 30 March 2016
Accepted for publication 22 May 2016
Published 29 August 2016 Volume 2016:11 Pages 4141—4155
DOI https://doi.org/10.2147/IJN.S109494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Jaehong Key,1,2 Deepika Dhawan,3 Christy L Cooper,3,4 Deborah W Knapp,3 Kwangmeyung Kim,5 Ick Chan Kwon,5 Kuiwon Choi,5 Kinam Park,1,6 Paolo Decuzzi,7–9 James F Leary1,3,4
1Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN, USA; 2Department of Biomedical Engineering, Yonsei University, Wonju, Republic of Korea; 3School of Veterinary Medicine-Department of Basic Medical Sciences, Purdue University, West Lafayette, 4Birck Nanotechnology Center at Discovery Park, Purdue University, West Lafayette, IN, USA; 5Biomedical Research Center, Korea Institute of Science and Technology, Sungbook-Gu, Seoul, Republic of Korea; 6Department of Pharmaceutics, Purdue University, West Lafayette, IN, 7Department of Translational Imaging, 8Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX USA; 9Laboratory of Nanotechnology for Precision Medicine, Fondazione Istituto Italiano di Tecnologia (IIT), Genova, Italy
Abstract: While current imaging modalities, such as magnetic resonance imaging (MRI), computed tomography, and positron emission tomography, play an important role in detecting tumors in the body, no single-modality imaging possesses all the functions needed for a complete diagnostic imaging, such as spatial resolution, signal sensitivity, and tissue penetration depth. For this reason, multimodal imaging strategies have become promising tools for advanced biomedical research and cancer diagnostics and therapeutics. In designing multimodal nanoparticles, the physicochemical properties of the nanoparticles should be engineered so that they successfully accumulate at the tumor site and minimize nonspecific uptake by other organs. Finely altering the nano-scale properties can dramatically change the biodistribution and tumor accumulation of nanoparticles in the body. In this study, we engineered multimodal nanoparticles for both MRI, by using ferrimagnetic nanocubes (NCs), and near infrared fluorescence imaging, by using cyanine 5.5 fluorescence molecules. We changed the physicochemical properties of glycol chitosan nanoparticles by conjugating bladder cancer-targeting peptides and loading many ferrimagnetic iron oxide NCs per glycol chitosan nanoparticle to improve MRI contrast. The 22 nm ferrimagnetic NCs were stabilized in physiological conditions by encapsulating them within modified chitosan nanoparticles. The multimodal nanoparticles were compared with in vivo MRI and near infrared fluorescent systems. We demonstrated significant and important changes in the biodistribution and tumor accumulation of nanoparticles with different physicochemical properties. Finally, we demonstrated that multimodal nanoparticles specifically visualize small tumors and show minimal accumulation in other organs. This work reveals the importance of finely modulating physicochemical properties in designing multimodal nanoparticles for bladder cancer imaging.
Keywords: MRI, NIRF, multimodal imaging, chitosan, iron oxide, bladder cancer
Introduction
Over the last 2 decades, “nanomedicine” has grown rapidly, representing a promising new paradigm for cancer diagnosis and therapy. Nanoparticles ranging from tens to a few hundred nanometers have improved the preferential delivery of imaging agents and drugs to tumors by enhanced permeability and retention (EPR) effect.1,2 Moreover, specific binding ligands on nanoparticles have a high affinity for tumor microenvironments and vasculature.3–5 Several studies have shown that the physicochemical properties of nanoparticles, including size, stiffness, and zeta-potential, can play an important role in the efficacy of their cell binding and in vivo biodistribution.6–8 However, the following limitations still restrict the clinical use of nanoparticles: 1) high toxicity, 2) limited visualization effect, and 3) nonspecific accumulation to organs and tissues.
Iron oxide nanoparticles have received great attention in nanomedicine due to their unique magnetic properties and low toxicity.9 While untargeted superparamagnetic iron oxide nanoparticles were commonly demonstrated for liver cancer imaging in magnetic resonance imaging (MRI), mainly due to their uptake in nontumor areas by phagocytic Kupffer cells, their clinical applications for other cancers, such as bladder cancer, are still limited due to their low contrast to background and nonspecificity to normal cells. Ferrimagnetic iron oxide nanoparticles show higher contrast and hyperthermia, but they are not widely used as MRI and therapeutic agents due to their magnetic dipole interactions, which result in poor stability in aqueous solution. Interestingly, 22 nm ferrimagnetic iron oxide nanocubes (NCs) formed closely at superparamagnetic range could be utilized as MRI or effective hyperthermia agents by encapsulating them in polymers that minimize the aggregation of ferrimagnetic iron oxides.10–12 Unlike superparamagnetic nanoparticles, these ferrimagnetic NCs are difficult to maintain in a dispersed state by themselves. However, as we describe in this paper, multiple 22 nm ferrimagnetic NCs can be contained within a larger chitosan-coated nanoparticle that can be maintained in a dispersed state. NCs 22 nm in size are ferrimagnetic rather than superparamagnetic, have weak coercivity, and readily aggregate. We can further minimize the coercivity by encapsulating them in polymers. The ferrimagnetic iron oxide NCs show high MRI contrast and hyperthermia.10,13 However, ferrimagnetic iron oxide NCs are not easy to use in biological applications due to magnetic memory after exposure to an external magnetic field. Magnetic memory leads the iron oxide particles to cluster quickly, which could boost precipitation of the particles in biological conditions. Here, we encapsulated multiple ferrimagnetic iron oxide NCs in our modified glycol chitosan polymer, which has excellent results for in vivo cancer imaging. The encapsulation and conjugation to bladder cancer-targeting peptide (CSNRDARRC) increased both the circulation time in the blood and tumor specificity. While we did not optimize relaxivities in this paper, Hyeon’s group demonstrated that NCs can potentially provide transversal relaxivities r2, as high as 761 mM−1s−1.10 This result is about fivefold better than commercialized iron oxide nanospheres, Feridex,10,13 indicating that NCs have potential advantages over nanospheres.
Chitosan is a linear cationic polysaccharide with abundant amino groups that has been identified as a potential transporter in nanomedicine. Chitosan has extensive biomedical applications in drug and gene delivery and tissue engineering due to its biocompatibility and biodegradability.14,15 Chitosan can be also used as a responsive polymer to increase solubility at acidic pH, which can improve drug release in cancer environments.16–19 In particular, glycol chitosan and its derivatives have been exploited to deliver therapeutic agents, such as paclitaxel and doxorubicin, owing to its enhanced solubility in various physiological conditions. Glycol chitosan modified with hydrophobic moieties forms spherical nanoparticles in aqueous solutions where self-assembled nanoparticles effectively hold hydrophobic drugs (~20 wt%) and release them in a controlled manner.20 In addition, chitosan copolymer and its derivatives can transport hydrophobic iron oxide nanoparticles for dark contrast in T2-weighted magnetic resonance (MR) images.21–24 Chitosan nanoparticles (CNPs) have many advantages as a potential transporter for human cancer imaging and therapy. They also serve as an effective “stealth” factor to improve circulation time in vivo. Chitosan and its derivatives have several advantages due to their excellent biocompatibility, biodegradability, and low immunogenicity. Hydrophobically-modified chitosan polymers form nano-sized particles that can deliver hydrophobic drugs and inorganic compounds. Further, chitosan has primary amino groups that can interact with negatively charged molecules, such as peptides and genes.14,25
Single-modality imaging has limitations in detecting small tumors in the body. Moreover, while delineating the tumor is important to prevent recurrence and perform minimally invasive surgery, human eyes are inadequate to find the edge of tumors. Therefore, dual-modality imaging can be a promising solution to provide more visual information by simultaneously improving image resolution, sensitivity, and tissue penetration depth. Development of nanoparticles to enhance dual-modality imaging can provide even more detail on tumor locations.26 Recently, we reported on dual-modality CNPs with nanospheres, expanding their function as MRI agents as well as optical imaging agents (with near infrared fluorescence [NIRF] probes, such as cyanine 5.5 [Cy5.5]) and positron emission tomography imaging agents. We performed in vivo dual-modality imaging, probing specific tumor areas by using the nanoparticles.27–29 However, optimizing the design of dual-modality nanoparticles to satisfy both the imaging function and a good biodistribution remains a challenge. While dual-modality nanoparticles can expand the imaging capabilities, they can also increase nonspecific accumulation to organs and tissues due to the altered physicochemical characteristics of the nanoparticles. Also, visualizing small tumors (<5 mm in diameter) in both imaging modalities is even more challenging, requiring more specific accumulation at tumor sites. Moreover, dual-modality nanoparticles, including metals such as iron oxide or gold, can easily accumulate in the liver or spleen. Thus, maintaining a successful in vivo biodistribution is critical as the imaging functions of the nanoparticles expand.
In this study, we varied the physicochemical properties and functions of glycol CNP by conjugating them to a CSNRDARRC and loading ferrimagnetic iron oxide NCs into them.11,30 The biodistribution and tumor accumulation of the modified nanoparticles in each step were compared in parallel through in vivo systems. We demonstrated that adding new components, such as new MR contrast agents or targeting ligands, clearly changed its in vivo biodistribution by altering their physicochemical properties of the particles. Finally, we suggested a design of NC-loaded CNPs (peptide-conjugated chitosan nanoparticles, pMCNP) that showed excellent MRI and optical dual-modality in vivo imaging of bladder cancer. For therapeutic applications, the nanoparticles represent the high loading efficacy of a clinically available bladder cancer drug, vinblastine, which had sustained release for 3 days. All animal experiments in this study were approved by the Purdue Animal Care and Use Committee (PACUC) of Purdue University. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act.
Materials and methods
Chemicals
Glycol chitosan (molecular weight =250 kDa, degree of deacetylation =82.7%), 5β-cholanic acid, 1-ethyl-3-(3-dimethylamniopropyl)-carbodiimide (EDC) hydrochloride, and N-hydroxysuccinimide (NHS) were ordered from Sigma-Aldrich Co. (St Louis, MO, USA) as reagent grade and used without further purification. Mono-reactive NHS ester of Cy5.5 (Cy5.5-NHS) was purchased from GE Healthcare Corp (Piscataway, NJ, USA). Methanol and dimethyl sulfoxide were anhydrous grade. Iron (III) acetylacetonate, 4-biphenylcarboxylic acid, and benzyl ether were ordered from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Oleic acid was purchased from Sigma-Aldrich Co. CSNRDARRC peptide sequence with 90% purity was manufactured and validated by EZBiolab Inc (Carmel, IN, USA).
Self-assembled CNP
Glycol chitosan was conjugated with hydrophobic 5β-cholanic acid, following previous studies.28,31,32 Glycol chitosan (250 mg) and EDC (31 mg) were dissolved in water (65 mL). 5β-Cholanic acid (39 mg) and NHS (17 mg) were suspended in methanol (65 mL). The EDC/NHS reaction was performed for 20 hours at room temperature. The solution was dialyzed (molecular weight cut-off [MWCO] =10 kDa) for 3 days and lyophilized. NHS ester of Cy5.5 was also conjugated to the glycol chitosan complex (4:1 mole ratio Cy5.5-NHS:glycol chitosan complex) in dimethyl sulfoxide in the dark. The solution was dialyzed (MWCO =10 kDa) and lyophilized.
Peptide-conjugated CNP
CNP (20 mg) was dissolved in water (5 mL). CSNRDARRC (2.8 mg) was mixed with the CNP solution overnight in the dark. EDC (2 mg/mL) and NHS (7.8 mg/mL) were reacted together. Each mole of peptide had ~4% mole of free amines on CNP. The final pCNP solution was lyophilized for 3 days in the dark. Dried pCNPs were stored at −20°C freezer.
Monodispersed ferrimagnetic iron oxide NC
NCs of 22 nm were synthesized by following a previously developed protocol.33 Iron (III) acetylacetonate (0.71 g), oleic acid (1.27 g), benzyl ether (10.40 g), and 4-biphenylcarboxylic acid (0.41 g) were mixed and degassed for 1 hour with nitrogen gas in a three-necked flask. The mixture was heated at 290°C for 30 minutes. The mixture was then cooled down and mixed with ethanol and then purified with a magnetic separator of Dexter Magnetic Technologies Inc. (Elk Grove Village, IL, USA). The NCs were stored in chloroform at room temperature. The final concentration of NCs was 433 μg/μL. The NCs were physically loaded into pCNPs by using a probe-sonicator at 10–12 W for 1–2 minutes in ice water. The solution was centrifuged at 13,000 rpm for 3 minutes. The pellet was resuspended in water and stored at −2°C. The NC-loaded pCNP is called pMCNP here. The nanoparticles were stable and well dispersed in aqueous solution.
Transmission electron microscopy
pMCNPs were prepared in distilled water. The suspension was sonicated for 10 minutes in a Branson 2510 bath-type sonicator (Danbury, CT, USA) and for 1 minute with a Sonics® Vibra-Cell™ probe-type sonicator (Newtown, CT, USA) in ice water. A drop of pMCNP was deposited onto holey carbon transmission electron microscopy (TEM) grids. After drying, TEM images were acquired with a FEI/Philips CM-100 (FEI Company, Hillsboro, OR, USA).
Dynamic light scattering
The hydrodynamic diameter of pMCNPs was determined at 25°C by dynamic light scattering (DynaPro99, Wyatt Technology Corp., Santa Barbara, CA, USA). The data were analyzed with Dynamics v5.26.60 software (Wyatt Technology Corp.).
Cell lines and culture conditions
Targeting of pMCNPs to K9TCC canine bladder cancer cells in vitro was observed with an inverted confocal microscopy from Nikon (A1R, Melville, NY, USA). The K9TCC cell line was provided by Dr Deborah Knapp. K9TCC cells were cultured in DMEM/F12 (Cellgro, Manassas, VA, USA) supplemented with 10% (v/v) fetal bovine serum in a humidified incubator at 37°C and 5% CO2. The bladder cancer cells (~1×105) were grown in cover glass-bottom dishes (SPL Life Sciences, Pochen, South Korea). When they exceeded above 60% confluency, cells were washed three times with phosphate-buffered saline (PBS), including calcium and magnesium. pMCNPs were dissolved in Opti-MEM I with Glutamax (Thermo Fisher Scientific. Waltham, MA, USA) (1 mg/mL), and mixed well by sonication. This suspension (100 μL) was mixed with Opti-MEM (900 μL) and added to each cell culture dish. The cells were incubated with pMCNPs for 3 hours at 37°C. The cells were washed with PBS three times and fixed in 4% paraformaldehyde (Sigma-Aldrich Co.) for 30 minutes at room temperature in the dark. Confocal microscopy was performed with a 40× magnification lens, a 640 nm laser, and a band-pass emission filter for Cy5.5 (660–740 nm).
NIRF and MRI of nanoparticles
Nanoparticles were prepared in water and added to a 96-well plate. Contrast enhancement of NIRF imaging in the water phantom was confirmed by a Cary Eclipse spectrophotometer (Agilent Technologies, Santa Clara, CA, USA) and IVIS® II Lumina (Caliper Life Sciences). IVIS images were acquired under the following conditions: excitation (675 nm), emission (695 nm), exposure time (1 second), binning (small), F/stop (8), lamp power (high), Field of View (FOV) (D), and sample height (1.98 cm). MR images of the phantom were obtained with a Signa® EXCITE 3.0T HD system using 8HRBRAIN coil (GE Healthcare, Milwaukee, WI, USA). The MR sequences were spin echo, repetition time (TR)=1,500 ms, echo time (TE)=100 ms, frequency/phase=256/256, and FOV=100 mm.
In vivo and ex vivo NIRF imaging
Nanoparticle accumulation in K9TCC bladder tumors was evaluated by using the IVIS® II Lumina, which measured the amount of Cy5.5 NIRF in different organs and tissues of live anesthetized animals. Athymic mice (n=4, 4–5 weeks old, average 20 g, male) were purchased from NCI-Frederick (Frederick, MD, USA). K9TCC tumor bearing mice were prepared by subcutaneously injecting K9TCC cells (1.0×106 cells suspended in 100 μL of medium) in the flank. When the tumor reached ~5 to 7 mm in diameter on the skin, the mice were administered with nanoparticles (100 μL) via the tail-vein. Mice were anesthetized and whole body in vivo NIRF imaging was performed 24 hours after the injection. The mice were necropsied and ex vivo NIRF imaging of the major organs (liver, lung, heart, spleen, kidneys, and tumors) was performed. IVIS NIRF imaging was performed with the following parameters: excitation (640 nm), emission filter (Cy5.5), exposure time (10 seconds), binning factor (4), F number (2), lamp power (high), FOV (12.5 cm), and sample height (1.98 cm). All NIRF images were normalized before the comparison and quantified with imaging software, NIS Elements (Nikon). However, due to nonlinear aspects of the measuring process, NIRF imaging is only semiquantitative. Nonetheless, it can provide simple and fast measurements in both living and anesthetized animals, and also necropsied organs.
In vivo MRI
3T MR images were obtained at 24 hours postinjection of pMCNPs under the following conditions: T2-weighted image, axial plane, 3D acquisition, TR (2,000 ms), TE (82 ms), Knee coil, and frequency/phase (512/512). Knee coils helped improve the S/N for the mice in this 3T system designed for humans. For in vivo MRI, mice were anesthetized by injecting with a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg) in saline. DICOM files of MR images were exported with Sante DICOM viewer software (Athens, Greece). The contrast intensity at the tumor sites was quantified with ImageJ software developed by the National Institutes of Health (Bethesda, MD, USA).
Results and discussion
Self-assembled glycol CNPs were synthesized by partially conjugating hydrophobic 5β-cholanic acid to glycol chitosan polymers. For NIRF imaging, Cy5.5 was conjugated to free amine groups on the CNPs. The CSNRDARRC peptide sequences were used to specifically target bladder cancer cells. Twenty-two nm sized iron oxide NCs were synthesized for high MR contrast and physically loaded into the CNPs by using a probe-type sonicator. For drug loading, hydrophobic vinblastine was prepared and loaded into the CNPs by using solvent evaporation (Figure 1).
 | Figure 1 Chemical structures of glycol chitosan conjugated to hydrophobic 5β-cholanic acid, peptide (CSNRDARRC), and iron oxide NCs (A) and schematic diagram of pMCNPs containing iron oxide NCs and vinblastine (B). |
Physicochemical characterization of self-assembled glycol CNPs
Iron oxide NCs were prepared by thermal decomposition and stored in chloroform for further reactions.33 The NCs were dried at room temperature and observed by TEM. TEM images showed that the NCs had keen edges on each face. The length of each face was 22 nm (Figure 2). The NCs were physically loaded into CSNRDARRC peptide-conjugated CNPs (pCNPs), which were stable in aqueous solution (Figure 3A). The loading condition and distribution of the NCs in the pCNPs were measured by TEM. Numerous NCs were encapsulated and concentrated in each pCNP (Figure 2C and D). The hydrodynamic diameter of the bare CNPs without peptide and NCs was 289 nm and had a polydispersity index of 0.2 (meaning the nanoparticles were highly monodispersed). After peptide conjugation, the hydrodynamic diameter was 360 nm, an increase of ~70 nm from the bare CNPs, and the polydispersity index was similar (0.2). After loading NCs into pCNP (pMCNP), the average hydrodynamic diameter was 482 nm as measured by dynamic light scattering on a Malvern Zetasizer (Malvern Instruments, Malvern, UK), while the polydispersity index did not change (0.2) (Figure 3A and B). The physicochemical properties of the particles were changed continuously after loading NCs; in particular, the size increased. The dramatic change in physicochemical properties can affect both in vitro cell binding and in vivo biodistribution. It would be a big limitation for in vivo application if the large pMCNPs make lots of sequestration in organs or vessels. In terms of EPR effect, this size might not be optimal considering that many tumor fenestrations are around 100–200 nm, but this does not mean all tumor fenestrations are in this range. Previous studies demonstrated that a size was not the only critical factor for better biodistribution and tumor-targeting. Mechanical properties, zeta-potential, and immune responses were also important, which changed the biodistribution and tumor accumulation.34 In this study, we demonstrated that smaller-sized CNPs without NCs had a higher accumulation than pMCNPs in the liver and kidneys (Figures 4 and 5).
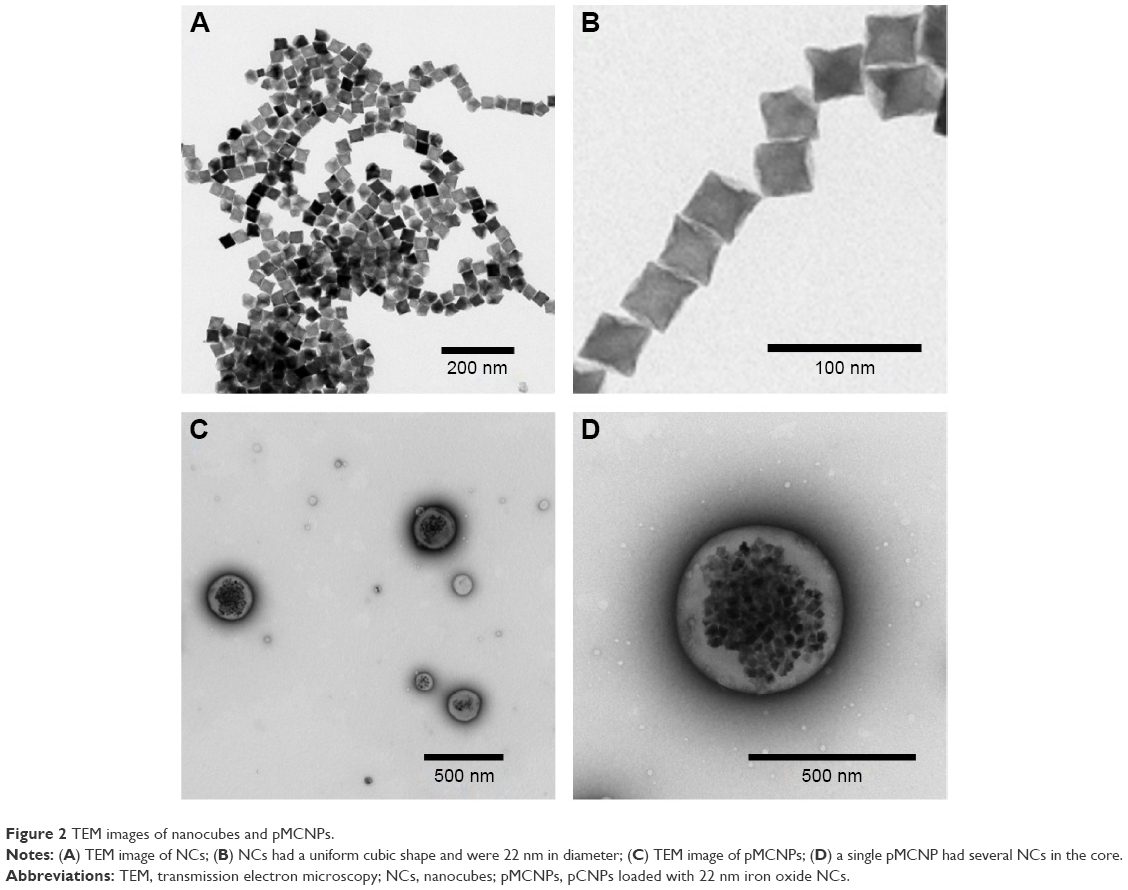 | Figure 2 TEM images of nanocubes and pMCNPs. |
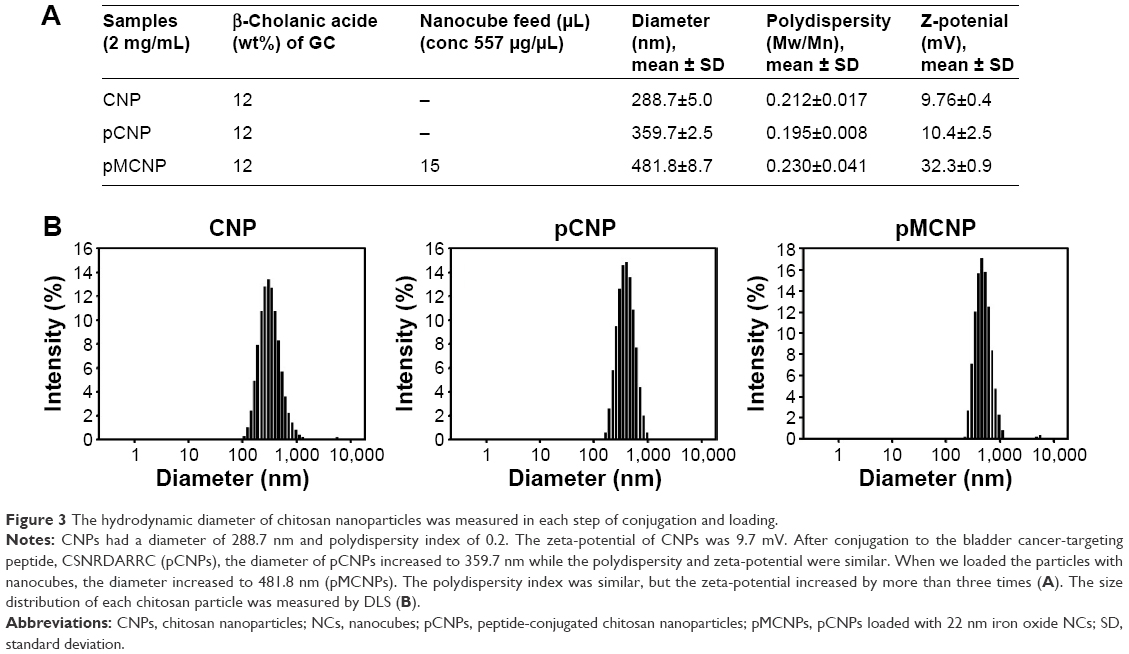 | Figure 3 The hydrodynamic diameter of chitosan nanoparticles was measured in each step of conjugation and loading. |
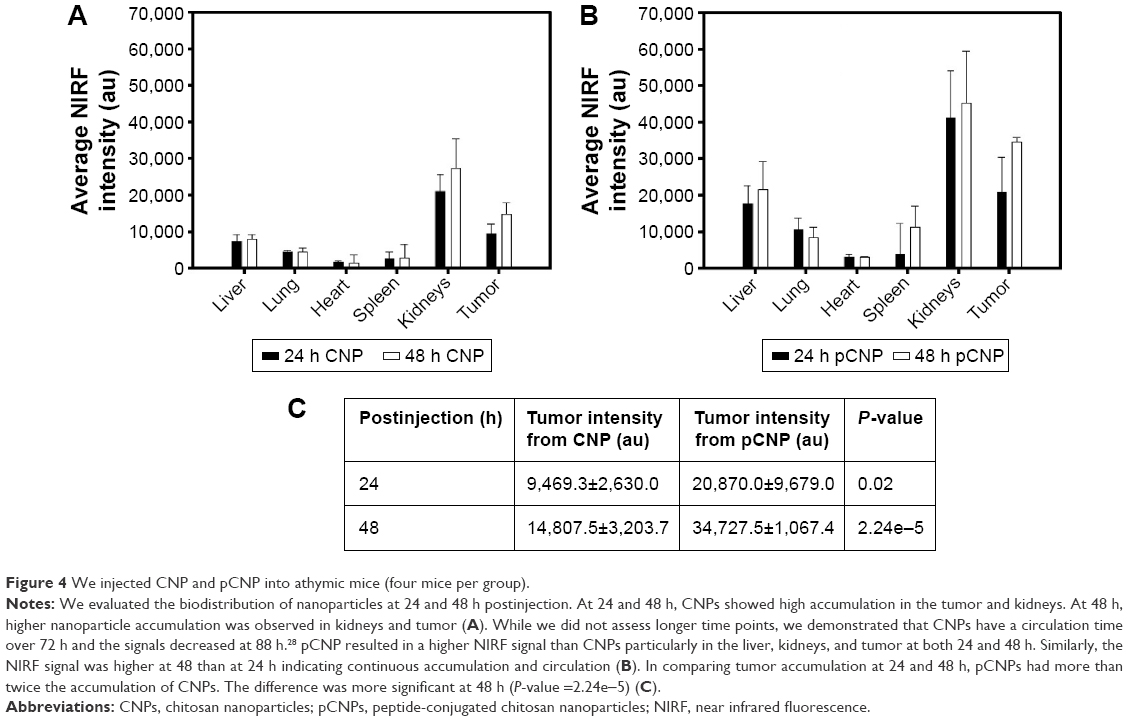 | Figure 4 We injected CNP and pCNP into athymic mice (four mice per group). |
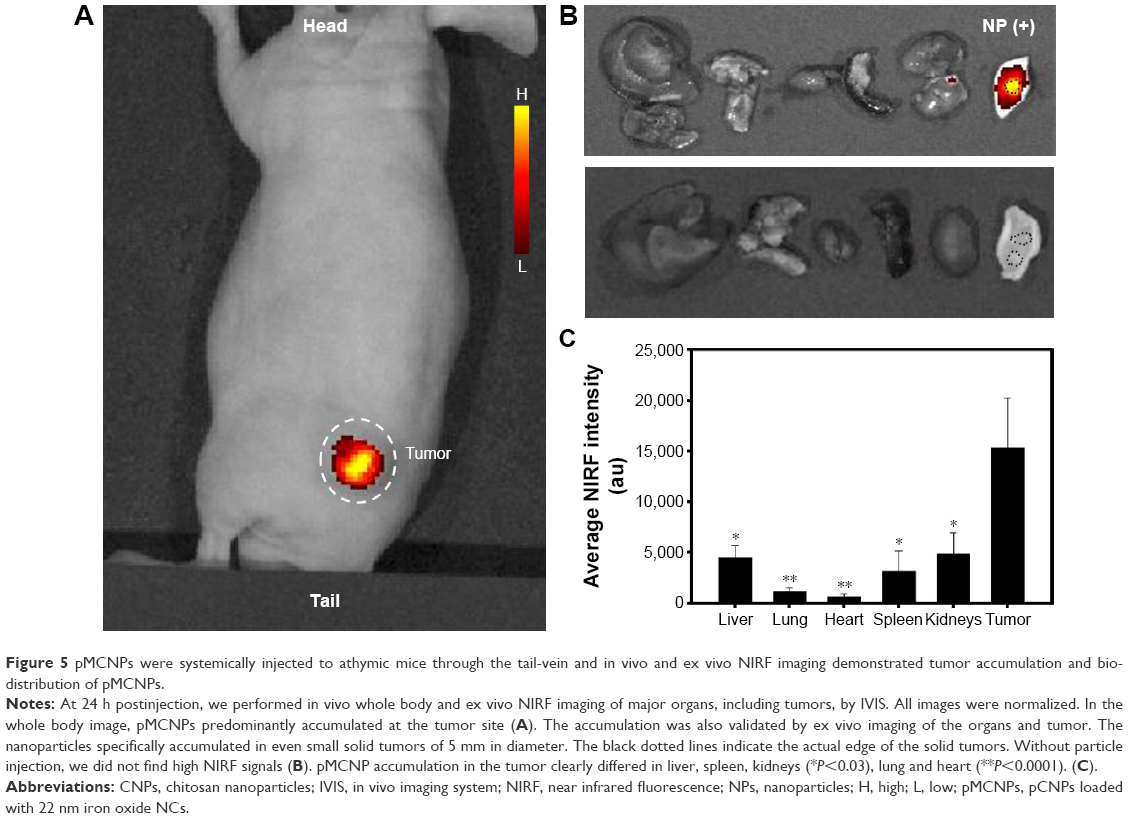 | Figure 5 pMCNPs were systemically injected to athymic mice through the tail-vein and in vivo and ex vivo NIRF imaging demonstrated tumor accumulation and bio-distribution of pMCNPs. |
Magneto-optical properties of pMCNPs
Ferrimagnetic iron oxide NCs ranging from 20 to 160 nm demonstrated much higher magnetization than Superparamagnetic Iron Oxide Nanoparticles (SPIOs) below 20 nm in diameter, so using the ferrimagnetic iron oxide NCs, very high MR contrast effects and hyperthermia effects can be achieved together.13,33 The only limitation is that ferrimagnetic iron oxide NCs have size-dependent coercivity as the particle size increases while SPIOs are zero-coercive.13,33 Therefore, when we engineer the iron oxide NCs with proper stabilizers, such as amphipathic polymers, it is possible to satisfy both high relaxivity and specific targeting. In this study, we demonstrated a good biodistribution of the nanoparticles (NPs) for bladder cancer imaging using 22 nm iron oxide NCs.
The iron oxide NCs were well dispersed in chloroform but not in water, so pCNPs were used to encapsulate those NCs that were stable in PBS 7.0 with 10% serum for 24 hours without aggregation. Our previous studies also demonstrated the stable condition of iron oxide NCs in modified chitosan polymers in a PBS solution for 7 days (Figure 6A).13 pMCNPs were evaluated for use in both NIRF imaging and MRI. pMCNPs (200 μL) in a 1.5 mL tube emitted NIRF signals at excitation (675 nm) and emission (695 nm) due to Cy5.5 labeling (Figure 6B). pMCNPs (1 mg/mL) were prepared in water and added to tissue culture wells in serial twofold dilutions. With increases in dilution, the pMCNPs emitted weaker NIRF signals, indicating that more pMCNPs emitted a higher NIRF signal (Figure 6C). The NIRF signals were quantified by in vivo imaging system (IVIS; IVIS®II Lumina, Caliper Life Sciences, Hopkinton, MA, USA) based on the total amount of Fe ions in each well. A decreased number of pMCNPs represents the decreased NIRF intensity (Figure 6D).
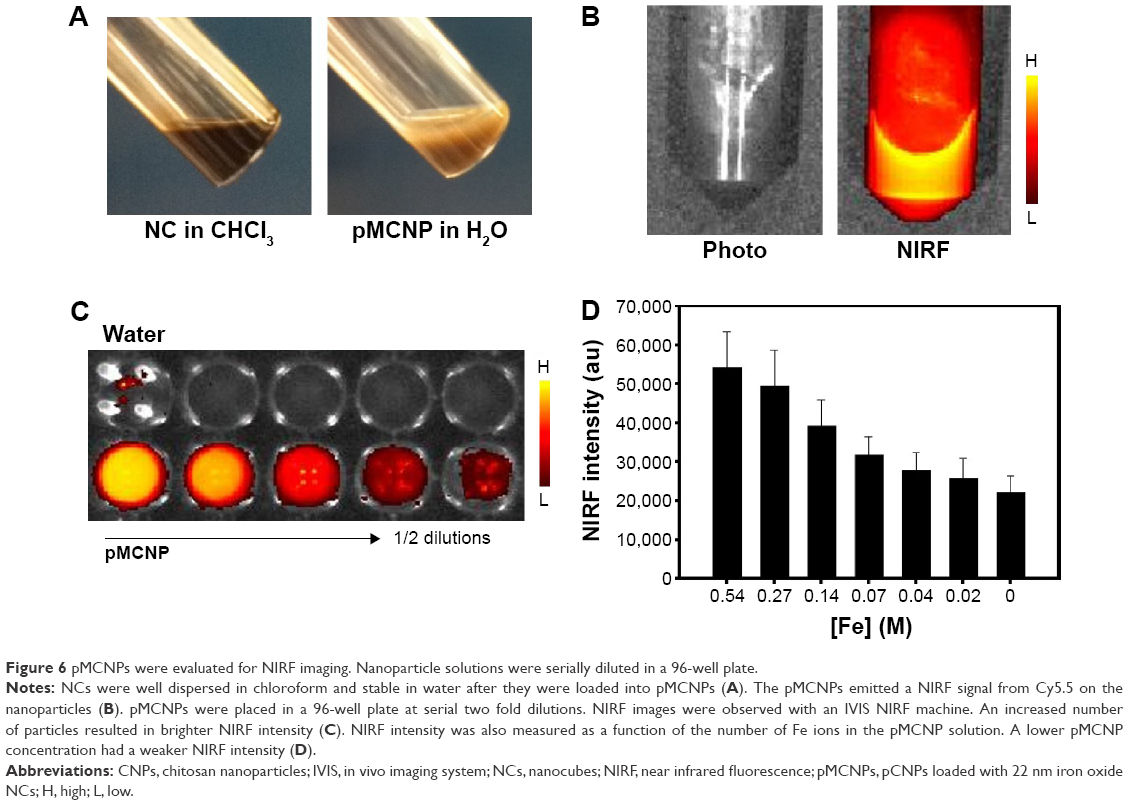 | Figure 6 pMCNPs were evaluated for NIRF imaging. Nanoparticle solutions were serially diluted in a 96-well plate. |
The MRI contrast effects of pMCNPs were observed by MRI on a Signa EXCITE 3.0T HD (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). With increases in dilution, the MR signal decreased due to fewer NCs. The result was clearly different from water alone (Figure 7A). Average MR signals were calculated based on the total amount of Fe ions in each well. The Fe concentration was almost linearly related to MR intensity (Figure 7B). A pMCNP (0.04 M of Fe) solution was compared with water at different echo times. As the echo time increased, only the pMCNP solution continuously decreased MR intensity, representing a T2 shortening effect (Figure 7). The r2 relaxivity of pMCNPs was 180 mM−1s−1. pMCNPs did not have the best r2 relaxivity when compared with many previous reports, but the r2 value was still 1.5 times higher than Feridex.9,13 More importantly, pMCNPs showed focused bladder cancer accumulation with low nonspecific accumulation in other organs, particularly liver and kidneys. The high specificity of pMCNPs could be useful for cancer therapy and diagnosis because the performance of theranostic NPs is not only determined by the r2 value but also by factors, such as targeting specificity, biocompatibility, drug loading, and drug release profile.
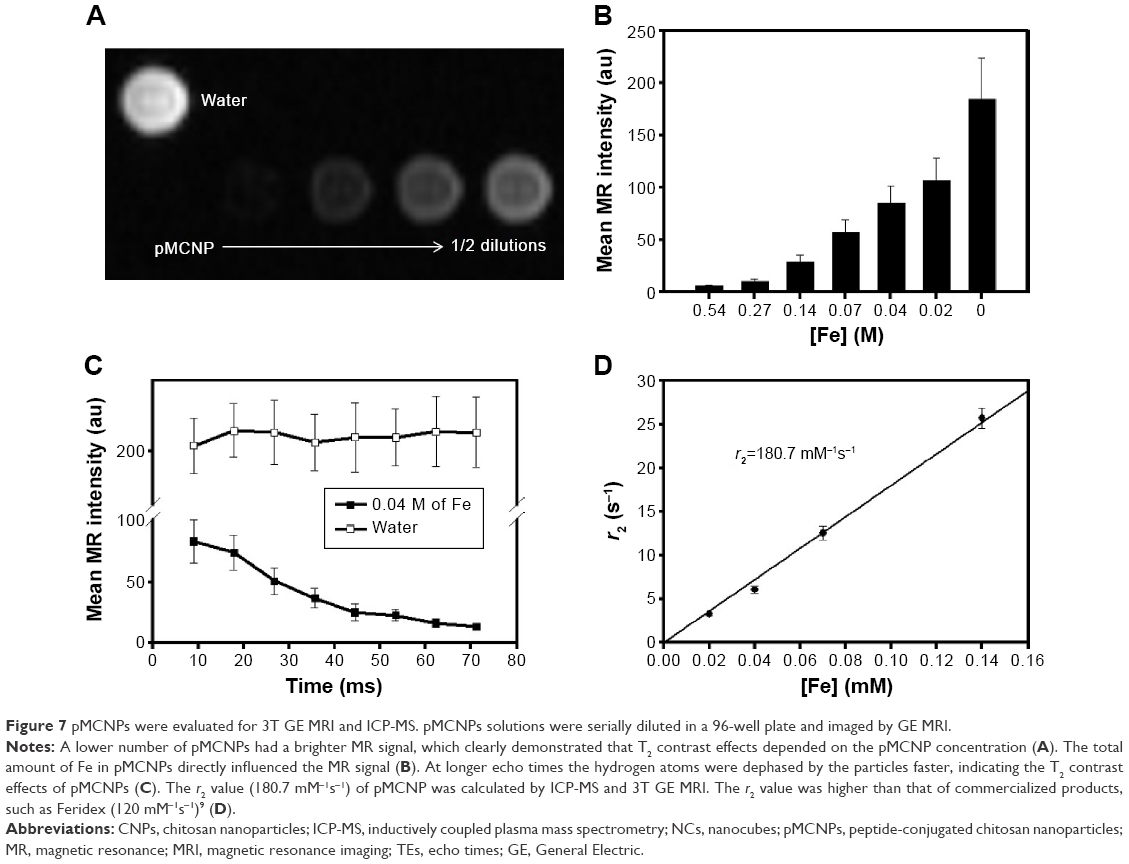 | Figure 7 pMCNPs were evaluated for 3T GE MRI and ICP-MS. pMCNPs solutions were serially diluted in a 96-well plate and imaged by GE MRI. |
Intracellular uptake of pMCNPs by K9TCC bladder cancer cells
Labeling pMCNPs with Cy5.5 allowed the nanoparticles to be visualized by NIRF fluorescent imaging (Figure 8). The overlapped bright-field and NIRF fluorescent images represent cell binding and internalization of pMCNPs by the K9TCC cells. This result was demonstrated in previous free peptide studies, showing that CSNRDARRC has high affinity for human bladder tumors.30,35 We previously reported the high affinity of this peptide to various bladder cancer cells, including human, murine, and canine cells36 (Figure 8A and B). Here, we incubated the pMCNPs with canine bladder cancer cells (K9TCC). After 0–4 hours incubation, the cell culture medium was washed three times. Many pMCNPs remained in the K9TCC cells, implying that pMCNPs were successfully delivered to the K9TCC tumors and internalized or bound to these cells (Figure 8C).
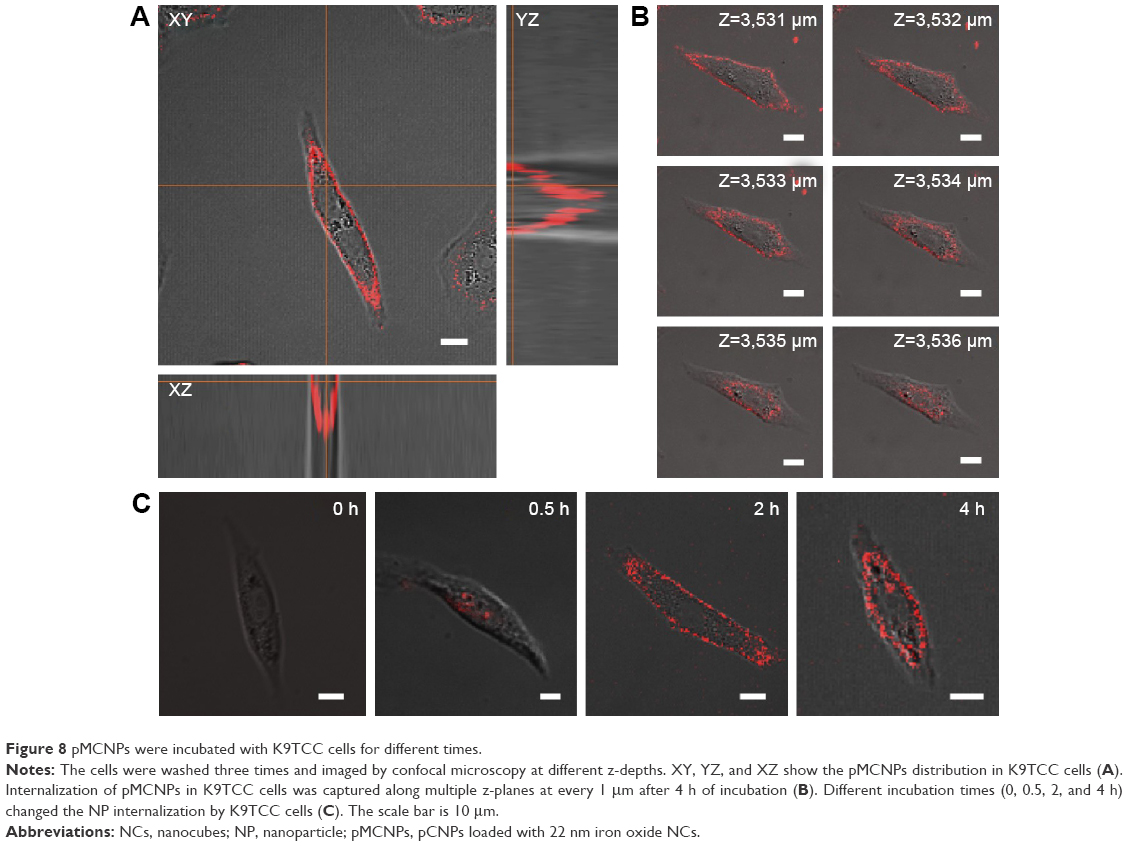 | Figure 8 pMCNPs were incubated with K9TCC cells for different times. |
Biodistribution and tumor uptake of various CNPs
NIRF intensities of bare CNPs and pCNPs were compared, and both nanoparticles had similar NIRF intensities (P-value =0.99) (Figure S1). We compared the in vivo biodistribution of CNPs and pCNPs at 24 and 48 hours postinjection (n=4 for each group). Both nanoparticles had a long circulation time and nanoparticles continuously accumulated in the tumor for 48 hours, indicating that many nanoparticles were still circulating at 2 days. The pCNPs had more accumulation in the tumor than did CNPs at both 24 and 48 hours. pCNPs also had higher accumulation in the kidneys and liver than CNPs (Figure 4A and B). These results might be explained by either bigger size of pCNPs or nonspecific binding by the peptide. However, the higher tumor accumulation by pCNPs was still clear at 48 hours (P-value <0.0002) (Figure 4C). As shown in the result, without peptide, many CNPs accumulated in the tumor that should be related to the EPR effect. We previously showed the EPR effect with 4 nm SPIO-loaded glycol CNPs.28 Thus, this result demonstrated that the peptide conjugation increased tumor accumulation by both EPR effect and peptide-targeting as shown in Figure 4. However, the CNPs with peptide conjugation also had a higher accumulation in the liver and kidneys. The nonspecific accumulation of pMCNPs could be related to the side effect of the peptide itself. Conjugation of the peptide changed the hydrodynamic size and zeta-potential of the CNPs as shown in Figure 3, and this slight change could affect the macrophage response and biodistribution.
Next, we tested pMCNPs in K9TCC bearing mice (n=4). At 24 hours postinjection, in vivo whole body NIRF images and ex vivo NIRF images of major organs, including tumors (~5 mm in diameter), were collected (Figure 5). In vivo whole body images showed specific probing effects at the tumor site by pMCNPs (Figure 5A). Ex vivo images also showed the probing effect by pMCNPs at the small tumor and also intensities at other organs were very low (Figure 5B). The result clearly showed the major accumulation of pMCNPs in the tumor site, emphasizing the highest tumor accumulation. To better understand the zeta-potential under more realistic conditions, we measured the zeta-potential of pMCNPs in canine blood serum and plasma, rather than water. The results clearly show opposite zeta-potentials: −5.17 mV in serum and −2.5 mV in plasma (Table S1). In the blood, counter ions and proteins cover the pMCNP surface resulting in a slightly negative zeta-potential, which is desirable.34 Therefore, specific pMCNPs accumulation might be explained by the change in zeta-potential. The bigger sizes reduced rapid renal excretion and the larger pMCNPs were not susceptible to the reticuloendothelial system in the liver or spleen (Figure 5).
pMCNPs for MRI contrast
Before in vivo NIRF imaging, the pMCNPs were injected into athymic mice and imaged by 3T MRI (Figure 9). The red dotted lines indicate the tumor area, which was formed by a subcutaneous injection of K9TCC bladder cancer cells. The yellow arrows indicate the negative contrast effects of pMCNPs in T2-weighted images. The contrast intensity at the tumor sites (described by the red dotted lines) was quantified (Figure 9). At the tumor sites, pMCNPs reduced the signal by an average of 67% compared with control tumors. The minimum value was also 37% darker. The normalized intensity was 57% darker per pixel. Thus, the pMCNPs improved negative MR contrast effects at the tumor site.
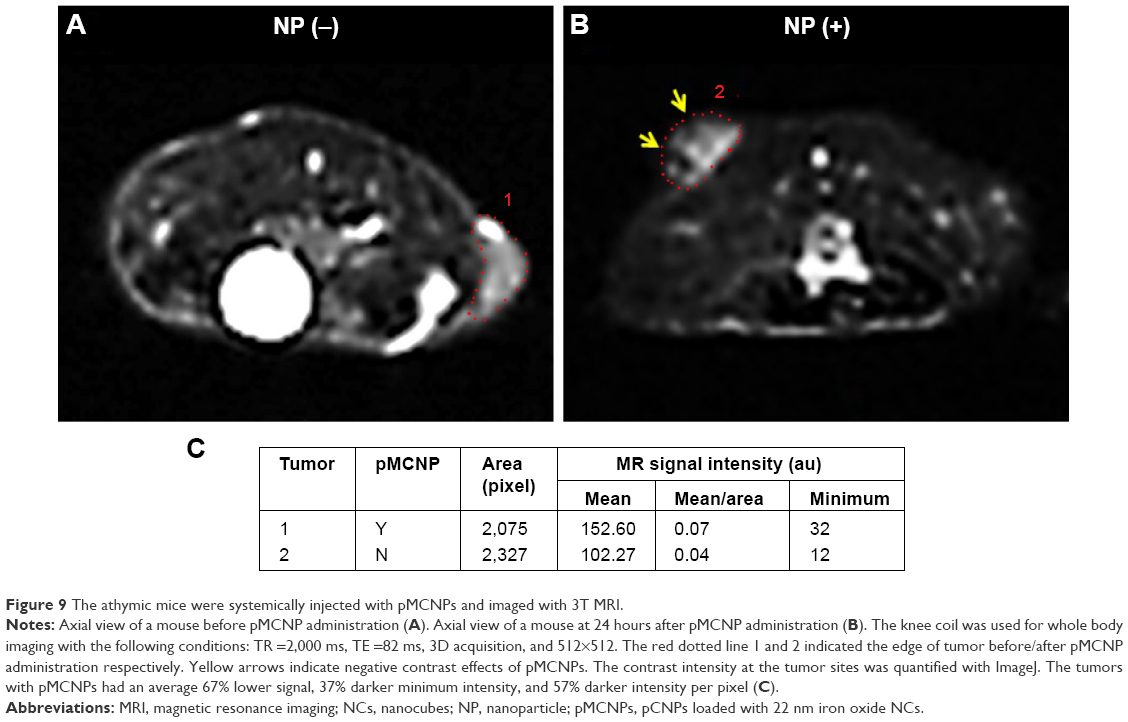 | Figure 9 The athymic mice were systemically injected with pMCNPs and imaged with 3T MRI. |
Previously, CNPs have been shown to have a long circulation time beyond 1–2 days in the blood stream of mice. Many CNPs passively accumulate through tumor fenestrations over time to deliver both imaging agents and therapeutic drugs. However, untargeted CNPs stayed at the tumor sites for a limited time and were washed out through blood or lymph drainage.28 Given that CNPs are needed for fluorescence-guided surgery of bladder tumors beyond normal optical penetration, a longer duration at cancer cells or tumor microenvironments will be highlighted. Although both targeted and untargeted nanoparticles have EPR effects, conjugating specific targeting peptides to pCNPs further increased the affinity for cancer cells or tumor vasculature, improving the retention time compared with bare CNPs. The pCNPs had twice the tumor accumulation of CNPs at 48 hours postinjection (Figure 4A and B). For MRI and NIRF dual-modality imaging, pCNPs loaded with 22 nm iron oxide NCs (pMCNPs) were tested in vitro and in vivo. The NC loading significantly changed the physicochemical properties of the nanoparticles and resulted in specific visualization of the tumor areas and margins. The pMCNPs have shown to be specifically targeted to the K9TCC bladder tumors (~5 mm diameter) and had low accumulation in other organs (Figure 5). In treating bladder cancer, this targeting could lead to an imaging approach to more accurately detect early lesions in the bladder and in metastases. We also tested the use of the nanoparticles as a drug delivery carrier. CNPs were loaded with ~20 wt% commercial bladder cancer drug, vinblastine (Table S2). The vinblastine in CNPs showed controlled release for up to 50 hours (Figure S2), indicating the potential of CNPs as an improved drug transporter for therapeutics.
Conclusion
In summary, we developed new MRI and NIRF dual-modality nanoparticles with a CSNRDARRC, iron oxide NCs, and glycol chitosan derivatives. We demonstrated improved tumor accumulation and biodistribution of these glycol CNPs. Our improved design of targeted pMCNPs had excellent accumulation in small tumors, minimal nonspecific binding to other organs, and longer retention making them better 3T MRI contrast agents. The nanoparticles will be further developed as a targeted theranostic drug (eg, vinblastine) transporter.
Acknowledgments
This work was supported by the Intramural Research Program (Theragnosis) of KIST, the Christopher Columbus Foundation support to JFL, and a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No 2015R1C1A1A01052592). We would like to especially thank Patty I Bonney, Lindsey M Fourez, Jane C Stewart, and Carol Ann Dowell at Purdue University for their assistance with this work. We also express our appreciation to Dr Aaron Taylor of the Bindley Bioscience Imaging Facility, Dr Tom Talavage and Greg Tamer of the Purdue MRI facility at Purdue Research Park, Dr Debby Sherman and Mr Chia-Ping Huang of the Life Sciences Microscopy Facility at Purdue University for the TEM images, and Lisa Reece of the Bionano Facility in the Birck Nanotechnology Center who was supported in part by the Indiana Clinical and Translational Sciences Institute from the National Institutes of Health, National Center for Research Resources, Clinical and Translational Sciences Award.
Disclosure
The authors report no conflicts of interest in this work.
References
Maeda H, Matsumura Y. EPR effects. Adv Drug Deliv Rev. 2011;63(3):161–169. | ||
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | ||
Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. | ||
Zhang C, Jugold M, Woenne EC, et al. Specific targeting of tumor angiogenesis by RGD-conjugated ultrasmall superparamagnetic iron oxide particles using a clinical 1.5-T magnetic resonance scanner. Cancer Res. 2007;67(4):1555–1562. | ||
Zhang H, Aina OH, Lam KS, et al. Identification of a bladder cancer-specific ligand using a combinatorial chemistry approach. Urol Oncol. 2012;30(5):635–645. | ||
Ferrari M. Nanovector therapeutics. Curr Opin Chem Biol. 2005;9(4):343–346. | ||
Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–327. | ||
Thorek DLJ, Tsourkas A. Size, charge and concentration dependent uptake of iron oxide particles by non-phagocytic cells. Biomaterials. 2008;29(26):3583–3590. | ||
Laurent S, Forge D, Port M, et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108(6):2064–2110. | ||
Lee N, Choi Y, Lee Y, et al. Water-dispersible ferrimagnetic iron oxide nanocubes with extremely high r2 relaxivity for highly sensitive in vivo MRI of tumors. Nano Lett. 2012;12(6):3127–3131. | ||
Lee N, Kim H, Choi SH, et al. Magnetosome-like ferrimagnetic iron oxide nanocubes for highly sensitive MRI of single cells and transplanted pancreatic islets. Proc Natl Acad Sci U S A. 2011;108(7):2662–2667. | ||
Bae KH, Park M, Do MJ, et al. Chitosan oligosaccharide-stabilized ferrimagnetic iron oxide nanocubes for magnetically modulated cancer hyperthermia. ACS Nano. 2012;6(6):5266–5273. | ||
Cervadoro A, Cho M, Key J, et al. Synthesis of multifunctional magnetic nanoflakes for magnetic resonance imaging, hyperthermia, and targeting. ACS Appl Mater Interfaces. 2014;6(15):12939–12946. | ||
Park JH, Kwon Sl, Lee M, et al. Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin: in vivo biodistribution and anti-tumor activity. Biomaterials. 2006;27(1):119–126. | ||
Lee SJ, Huh MS, Lee SY, et al. Tumor-homing poly-siRNA/glycol chitosan self-cross-linked nanoparticles for systemic siRNA delivery in cancer treatment. Angew Chem Int Ed Engl. 2012;51(29):7203–7207. | ||
Crayton SH, Tsourkas A. pH-titratable superparamagnetic iron oxide for improved nanoparticle accumulation in acidic tumor microenvironments. ACS Nano. 2011;58(12):9592–9601. | ||
Bao H, Pan Y, Ping Y, et al. Chitosan-functionalized graphene oxide as a nanocarrier for drug and gene delivery. Small. 2011;7(11): 1569–1578. | ||
Costa RR, Custodio CA, Arias FJ, Rodriguez-Cabello JC, Mano JF. Layer-by-layer assembly of chitosan and recombinant biopolymers into biomimetic coatings with multiple stimuli-responsive properties. Small. 2011;7(18):2640–2649. | ||
Saboktakin MR, Tabatabaie RM, Maharramov A, Ramazanov MA. Synthesis and characterization of pH-dependent glycol chitosan and dextran sulfate nanoparticles for effective brain cancer treatment. Int J Biol Macromol. 2011;49(4):747–751. | ||
Park J, Saravanakumar G, Kim K, Kwon I. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv Drug Deliv Rev. 2010;62(1):28–41. | ||
Szpak A, Kania G, Skorka T, Tokarz W, Zapotoczny S, Nowakowska M. Stable aqueous dispersion of superparamagnetic iron oxide nanoparticles protected by charged chitosan derivatives. J Nanopart Res. 2013;15(1):1372. | ||
Javid A, Ahmadian S, Saboury AA, Kalantar SM, Rezaei-Zarchi S. Chitosan coated superparamagnetic iron oxide nanoparticles for doxorubicin delivery: synthesis and anticancer effect against human ovarian cancer cells. Chem Biol Drug Des. 2013;82(3):296–306. | ||
Kievit FM, Veiseh O, Bhattarai N, et al. PEI-PEG-chitosan copolymer coated iron oxide nanoparticles for safe gene delivery: synthesis, complexation, and transfection. Adv Funct Mater. 2009;19(14): 2244–2251. | ||
Lee C-M, Jang D, Kim J, et al. Oleyl-chitosan nanoparticles based on a dual probe for optical/MR imaging in vivo. Bioconjug Chem. 2011;22(2):186–192. | ||
Huh M, Lee S, Park S, et al. Tumor-homing glycol chitosan/polyethylenimine nanoparticles for the systemic delivery of siRNA in tumor-bearing mice. J Control Release. 2010;144(2):134–143. | ||
Cheon J, Lee JH. Synergistically integrated nanoparticles as multimodal probes for nanobiotechnology. Acc Chem Res. 2008;41(12):1630–1640. | ||
Lee DE, Na JH, Lee S, et al. Facile method to radiolabel glycol chitosan nanoparticles with (64)Cu via copper-free click chemistry for microPET imaging. Mol Pharm. 2013;10(6):2190–2198. | ||
Key J, Cooper C, Kim A, et al. In vivo NIRF and MR dual-modality imaging using glycol chitosan nanoparticles. J Control Release. 2012;163(2):249–255. | ||
Nam T, Park S, Lee SY, et al. Tumor targeting chitosan nanoparticles for dual-modality optical/MR cancer imaging. Bioconjug Chem. 2010;21(4):578–582. | ||
Lee SM, Lee EJ, Hong HY, et al. Targeting bladder tumor cells in vivo and in the urine with a peptide identified by phage display. Mol Cancer Res. 2007;5(1):11–19. | ||
Park K, Kim JH, Nam YS, et al. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. J Control Release. 2007;122(3):305–314. | ||
Kwon S, Park J, Chung H, Kwon I, Jeong S. Physicochemical characteristics of self-assembled nanoparticles based on glycol chitosan bearing 5-cholanic acid. Langmuir. 2003;19(24):10188–10193. | ||
Kim D, Lee N, Park M, Kim BH, An K, Hyeon T. Synthesis of uniform ferrimagnetic magnetite nanocubes. J Am Chem Soc. 2009;131(2):454–455. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Jia XY, Yu Q, Zhang ZH, Yang XF. Targeting bladder tumor cells in voided urine of Chinese patients with FITC-CSNRDARRC peptide ligand. Onco Targets Ther. 2012;5:85–90. | ||
Key J, Dhawan D, Knapp DW, et al. Design of peptide-conjugated glycol chitosan nanoparticles for near infrared fluorescent (NIRF) in vivo imaging of bladder tumors. SPIE BIOS. Paper presented at SPIE2012. |
Supplementary materials
We observed the in vivo biodistribution of peptide-conjugated chitosan nanoparticles (pCNP) and chitosan nanoparticle (CNP). Before injecting the nanoparticles into the athymic mice, the near infrared fluorescent (NIRF) intensity was measured by in vivo imaging system. We prepared 1 mL each of pCNP and CNP in water and observed the NIRF intensities. Figure S1 showed that the average NIRF intensities of the two nanoparticles were not significantly different (P-value =0.968).
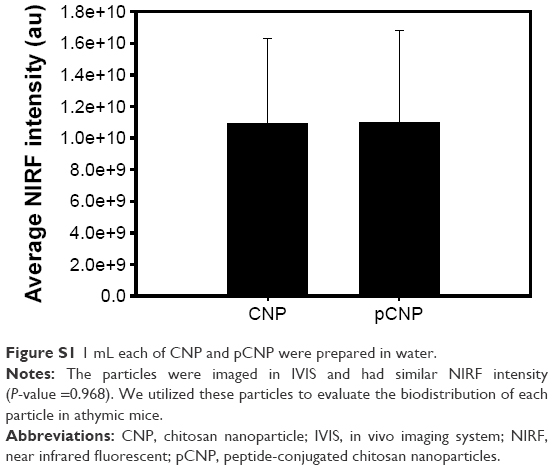 | Figure S1 1 mL each of CNP and pCNP were prepared in water. |
The high positive-charged nanoparticles might cause higher toxic effects to cells by nonspecific binding. It is possible that the positive nanoparticles are accumulated at organs.1 We also observed a high accumulation of CNP and pCNP in kidneys at 24 and 48 hours (Figure 7). However, interestingly, peptide-conjugated chitosan nanoparticles (pMCNP) showed a very low accumulation in kidneys as well as the other organs only except for tumor. Therefore, we measured the zeta-potential of pMCNP in water, serum, and plasma, respectively. The serum and plasma were obtained from the blood of a dog. Table S1 showed the change of zeta-potential of pMCNP. The zeta-potential of the particles in water was highly positive (26.7 mV). However, when we suspended the particles in serum and plasma, the zeta-potential was altered to negative charges (−5.37 and −2.5 mV, respectively). The results might indicate that numerous counter ions or proteins in the blood can rapidly cover the surface of the pMCNP changing the overall zeta-potential. The change might explain the highly specific accumulation of pMCNPs in tumor minimizing the nonspecific accumulation by the positive surface at major organs. This shows why the zeta-potential should be measured in the actual fluid environment (eg, in serum) rather than in a totally different microenvironment (eg, water).
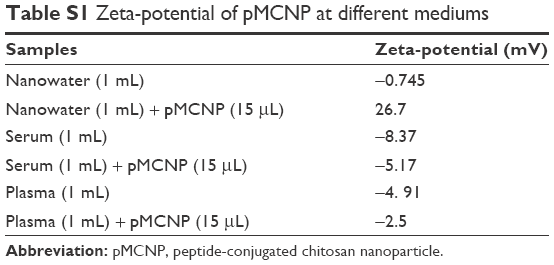 | Table S1 Zeta-potential of pMCNP at different mediums |
Table S2 and Figure S2 show the vinblastine (Vin) loading efficiency and release profile of pCNP. Vin is a commercially available bladder cancer drug. We loaded Vin of 20 mg into the pCNP. Briefly, we added 1M NaOH in Vin solution and sonicated them for 10 minutes at 4°C. The mixture was centrifuged at 13,000 rpm for 5 minutes and the supernatant was removed. We repeated this process three times. The pellet was resuspended in distilled water and centrifuged again at 13,000 rpm for 5 minutes two times. We removed the supernatant and lyophilized the pellet. We got 12 mg of Vin without sulfate. pCNP of 10 mg was dissolved in 5 mL water and 5 mL Methanol was added. Two milligram of Vin was dissolved in water and Methanol (1:1). The CNP and Vin were stirred overnight. Methanol was evaporated at 50°C for 2–3 minutes. All the remaining solution was centrifuged again at 3,000 rpm for 10 minutes and supernatants were collected, which were lyophilized. Vin loading condition and drug release profile of the particles were evaluated by high-performance liquid chromatography analysis. We used Column (C18, 5 μm, 3.9×150 mm, Symmetry®, Waters Corporation, Milford, MA, USA). Samples were dissolved in 0.8 M sodium salicylate solution for better flow of Vin without sulfate. Gradient combination of solvents was applied by four steps: Methanol (5%) and water (95%) for 0–5 minutes, Methanol (95%) and water (5%) for 5–35 minutes, Methanol (95%) and water (5%) for 35–40 minutes, and Methanol (5%) and water (95%) for 40–45 minutes. The signals of Vin were taken at 38.5–39 minutes at 215 nm. For release test, Vin-loaded pCNP (4 mg/mL) was prepared and 100 kDa dialysis bags were utilized.
 | Table S2 Loading efficiency of vinblastine by pCNP |
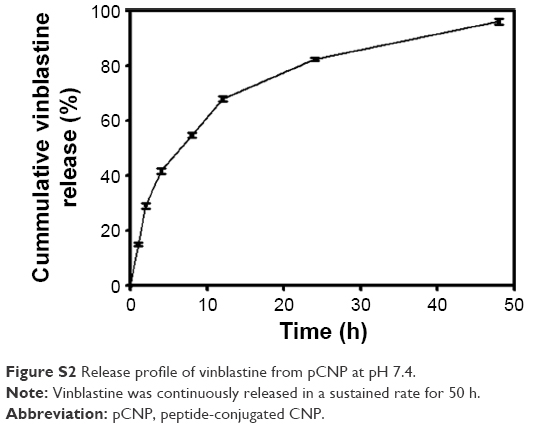 | Figure S2 Release profile of vinblastine from pCNP at pH 7.4. |
The loading content of Vin was ~19 wt% and loading efficiency was 94%. After loading Vin, the size was measured by 421 nm. The high loading content and efficiency indicate the potential of CNP to deliver Vin. We also demonstrated superiority of CNPs to deliver anticancer drugs.2–4 Figure S2 shows the sustained release of Vin from the pCNPs.
References
Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem. 2010;53(19):66–76. | ||
Park J, Saravanakumar G, Kim K, Kwon I. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv Drug Deliv Rev. 2010;62(1):28–41. | ||
Saravanakumar G, Min KH, Min DS, et al. Hydrotropic oligomer-conjugated glycol chitosan as a carrier of paclitaxel: synthesis, characterization, and in vivo biodistribution. J Control Release. 2009;140(3):210–217. | ||
Min KH, Park K, Kim YS, et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J Control Release. 2008;127(3):208–218. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.