Back to Journals » Drug Design, Development and Therapy » Volume 19
Mucoadhesive Mini-Containers with Unidirectional Drug Release Capacity for Macromolecular Therapeutics
Authors Han CS ![]() , Choi YR, Jung WY, Kang JH
, Choi YR, Jung WY, Kang JH ![]() , Kim DW, Park CW
, Kim DW, Park CW ![]()
Received 10 February 2025
Accepted for publication 16 July 2025
Published 1 August 2025 Volume 2025:19 Pages 6619—6635
DOI https://doi.org/10.2147/DDDT.S521900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Chang-Soo Han,1,* Ye-Rin Choi,1,* Woong-Young Jung,1 Ji-Hyun Kang,2 Dong-Wook Kim,3 Chun-Woong Park1
1Department of Pharmacy, Chungbuk National University, Cheongju, 28644, Republic of Korea; 2Department of Pharmacy, Jeonbuk National University, Jeonju, 54896, Republic of Korea; 3Department of Pharmacy, Wonkwang University, Iksan, 54670, Republic of Korea
*These authors contributed equally to this work
Correspondence: Chun-Woong Park, Department of Pharmacy, Chungbuk National University, Cheongju, 28644, Republic of Korea, Email [email protected]
Purpose: Peptide-based therapeutics have gained widespread attention for their high specificity and efficacy. However, their oral delivery remains challenging owing to their poor stability and bioavailability in the gastrointestinal environment and limited membrane permeability. To address these barriers, we have designed a novel mini-container system with unidirectional drug release and enhanced mucoadhesion capacities.
Methods: Mini-containers composed of ethyl cellulose shells of varying degrees of viscosity were fabricated using a simple molding process and integrated with catechol-conjugated chitosan (CC) to improve their mucosal adhesion capacity and structural stability.
Results: The catechol substitution levels were optimized (CC-A, CC-B, and CC-C), with the CC-C formulation exhibiting the highest degree of substitution (20.93%) and superior adhesion capacity, maintaining 80% attachment on porcine small intestinal mucosa after 72 h. Insulin, a model peptide drug, was successfully loaded into the CC-C mini-containers, and circular dichroism spectroscopy analysis confirmed that its secondary structure remained intact. The insulin content in the mini-containers, as determined by HPLC-UV analysis, demonstrated consistency across formulations: 101.1 ± 2.4% for 1% CC-C, 95.4 ± 3.8% for 2% CC-C, and 100.0 ± 1.8% for 3% CC-C, while in-vitro dissolution and Franz diffusion cell studies demonstrated its sustained and unidirectional release. After 12 hours of dissolution, the 3% CC-C formulation showed a release rate of 26.22 ± 2.23%, while the 1% CC-C formulation exhibited a release rate of 53.11 ± 0.25%. Catechol-mediated crosslinking significantly slowed the release rate relative to that of controls. The robust structure of the mini-containers fabricated with high-viscosity ethyl cellulose exhibited a mechanical strength of 13.21 ± 0.50 N, comparable to that of commercial enteric capsules (10 N), ensuring durability under gastrointestinal conditions.
Conclusion: This study shows the potential of mini-container technology for the stable and prolonged oral delivery of macromolecular therapeutics. However, further investigation is required to confirm its effectiveness in-vivo.
Keywords: mini-container shell, mold, catechol-conjugated chitosan, unidirectional, mucoadhesion
Introduction
Peptide-based therapeutics have gained significant attention for their high specificity and efficacy, addressing disease targets that small-molecule drugs cannot.1,2 In particular, the specificity of peptides enables them to act on diverse enzymes and receptors, expanding therapeutic possibilities across a range of diseases.2,3 Moreover, their metabolic degradation into amino acids ensures biocompatibility and minimal toxicity.4 Despite these advantages, the oral delivery of peptides still faces significant barriers, and most peptide drugs are currently formulated as injectable products.2 This reliance on invasive administration methods that could meet resistance from patients afraid of needles underscores the pressing need for effective oral delivery solutions to improve patient compliance.5 The major challenges associated with the oral delivery of peptides are their poor stability in the gastrointestinal (GI) environment, large molecular size, and hydrophilic nature, all of which limit their membrane permeability.6 To address these limitations, researchers have developed various approaches, including lipid-based nanocarriers such as liposomes and cell-penetrating and microneedle technologies.7–9 Although these strategies show promise in protecting peptides from degradation and improving their absorption, they are often hindered by complex fabrication processes and stability concerns, limiting their commercial viability. Recent innovations have included spatially targeted drug delivery systems to enhance peptide absorption. For example, in the oral semaglutide product “Rybelsus”, approved in 2019, sodium N-[8-(2-hydroxybenzoyl) amino] caprylate is used to enhance adsorption to the gastric mucosa, thereby facilitating local uptake of the drug.10,11 However, this approach requires a large amount of excipients to protect the drug in areas not adhered to the mucosa, increasing the complexity of the formulations.10 To address these challenges, research has shifted toward micro-container technologies that offer unidirectional drug release while shielding the drug from external environmental degradation.10,12,13
Micro-containers, which are typically a few micrometers to several hundred micrometers in size, have emerged as promising vehicles for the oral delivery of macromolecules.10,14,15 These unidirectional drug carriers protect the active pharmaceutical ingredient from external degradation while providing a large surface area for efficient absorption capability. Micro-containers improve the bioavailability of drug molecules by focusing their release at the target absorption site, as demonstrated in studies involving insulin and other macromolecular drugs.10,16 However, the fabrication of micro-containers presents challenges, as advanced techniques (eg, 3D printing) are often required to achieve precise control over the container size and shape, which are parameters that significantly affect performance.17 Moreover, micro-containers have inherent limitations, including a low drug-loading capacity due to their small size and susceptibility to damage in the harsh GI environment.18
To overcome these limitations, a mini-container system inspired by micro-container technologies was explored in this study. Mini-containers, with dimensions in the millimeter range, offer increased drug-loading capacity and enhanced structural stability compared with micro-containers. Fabricated by means of a simple molding process, the mini-container shells of this study were designed to withstand the mechanical stress of the GI tract while ensuring unidirectional drug release. Additionally, inspired by mussel adhesive proteins, we incorporated catechol-conjugated chitosan (CC) into the mini-containers to enhance their adhesion to the mucosal surface. Catechol groups form strong covalent and non-covalent bonds with mucosal tissue, providing superior adhesion even under challenging conditions, and their inherent cross-linking properties also enable controlled drug release. In addition, catechol-conjugated chitosan has been approved by the FDA as a medical device component in the InnoSEAL Hemostatic Pad, demonstrating high safety and biocompatibility.19 The development of mini-containers using such pre-approved excipients may facilitate accelerated regulatory approval of pharmaceutical products.
Insulin, a 5.8 kDa peptide hormone widely used in diabetes management, was selected as the model drug for this study to evaluate peptide stability in this delivery system.20 Diabetes is a chronic medical condition that requires continuous management, which typically involves multiple daily subcutaneous injections of insulin. However, the complications (eg, tissue necrosis) that can occur as a result of repeated injections underscore the urgent need for effective oral insulin delivery systems.21
The aim of this study was to fabricate durable mini-container shells using a straightforward molding technique, with CC integrated to enhance both adhesion and stability. By demonstrating the feasibility of this approach, we seek to establish mini-container technology as a promising platform for the oral delivery of macromolecular therapeutics.
Materials and Methods
Materials
Polylactic acid (PLA) filament (Raise3D Premium 3D Printing filament) was purchased from Raise3D Technologies (Irvine, CA, USA). Ethyl cellulose (EC) standards with viscosity values ranging from 4 to 100 cP (Ethocel Standard 4, 10, 20, 45, and 100) were obtained from Dow Chemical Company (Midland, MI, USA). 3,4-Dihydroxycinnamic acid (HCA) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were supplied by Tokyo Chemical Industry (Tokyo, Japan). Insulin, chitosan (deacetylation degree of 80% and 50–190 kDa molecular weight), and UV curable resin (CPS 1020 UV) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Acetone (extrapure grade) and ethanol (high-performance liquid chromatography (HPLC) grade) were purchased from SAMCHUN Chemicals (Seoul, Republic of Korea). Acetonitrile (HPLC grade) was purchased from Honeywell Burdick & Jackson Ltd. (Muskegon, MI, USA). All other chemicals were of analytical grade and used as received. All experiments were carried out using Milli-Q distilled water.
Fabrication of the Polylactic Acid Template and Silicone Mold
A PLA template was fabricated using 3D printing technology and used to prepare the silicone mold, as illustrated in Figure 1. The PLA template was designed using Rhino 6 software (Robert McNeel & Associates, Seattle, WA, USA), with the container size set to 5×2 mm. Each mold was designed to produce 25 containers, arranged in a 5×5 grids. The designed template was then processed using IdeaMaker software (Raise3D Technologies) and printed with a Raise3D Pro 2 printer under the following conditions: layer height, 0.25 mm; extrusion width and nozzle size, 0.4 mm; heated bed temperature, 60°C; extruder temperature, 205°C; and printing speed, 20 mm/s. The printed PLA template was cleaned with isopropanol, and UV curable resin was applied with a brush to remove stack lines. Then, the templates were cured for 10 min under a UV lamp at a wavelength of 365 nm and power of 48W.
 |
Figure 1 Mini-container shell fabrication schematic diagram and photos PLA template, silicon mold, and mini-container. |
Silicone base with a hardness value of 5 and viscosity of 3000 cP (Xinus Silicone SH2115; XinusLab, Seoul, Korea) was used to prepare the silicone mold. The silicone base was mixed with a curing agent (1:1 ratio) for 5 min, following which the mixture was degassed under vacuum to remove air bubbles. The mixture was then poured into the PLA template and degassed again under vacuum. After 24 h of curing at ambient temperature, the mold was detached from the template.
Unidirectional Mini-Container Shells
Mini-container shells were prepared using ECs with different viscosity values (4, 10, 20, 45, and 100 cP). Each type of EC was first dissolved in acetone (5%, w/v) overnight and then sonicated (Branson Bransonic CPX; Emerson, St. Louis, MO, USA) prior to degassing. Subsequently, 40 μL of each solution was dispensed into the fabricated silicone molds. These were then placed in Petri dishes, covered, and dried at 25°C. These conditions were established through preliminary experiments.
Evaluation of the Mini-Container Shells
Scanning Electron Microscopy
Scanning electron microscopy (SEM) images of the EC-based container shells were obtained using a GEMINI LEO 1530 microscope (Zeiss Ltd., Jena, Germany). In brief, samples were mounted on an aluminum plate using carbon tape, then placed inside a Hummer VI sputtering device (Anatech, Sparks, NV, USA), and subsequently coated with platinum to discharge the particles with 200 Å coating thickness. The SEM imaging parameters were set at a Signal A of SE2 and an accelerating voltage of 3 kV.
Texture Analysis
The hardness of the EC-based container shells, which was measured using a texture analyzer (TA.XTPlus; Texture Technologies Corp., South Hamilton, MA, USA), was determined from the absolute positive force measured during the compression and return phases.22 The test mode was set to “return to start”, and the pre-test speed was set to 1 mm/s. In brief, a cylindrical probe of 32 mm diameter was lowered until it contacted the container, reaching a trigger force of 0.049 N. From this point, the probe was further lowered at a test speed of 0.1 mm/s until it reached 50% of the container height, corresponding to a 1 mm distance. Upon reaching this point, the probe was retracted to the starting position at a post-test speed of 10 mm/s. The absolute positive force during this process was used to evaluate the shell hardness. All tests were repeated three times.
Preparation of the Mucoadhesive Core
Synthesis of Catechol-Conjugated Chitosan
CC was synthesized by reacting chitosan with HCA in the presence of the catalyst EDC. This method applies carbodiimide coupling chemistry to graft catechol groups onto chitosan by forming amide bonds between the carboxylic acid groups of HCA and the primary amine groups of chitosan.23–25 The molar ratios of chitosan to HCA and EDC were screened at 1:1, 1:2, and 1:3 (Table 1). Initially, 3.25 mmol of chitosan was dissolved in 45.5 mL of distilled water and the pH was adjusted to 1.6 using hydrochloric acid (HCl). Once the solution turned a clear pale yellow, the pH was gradually increased to 5.5 using 1 N sodium hydroxide, resulting in a cloudy appearance. Subsequently, HCA was added to the chitosan solution at molar ratios of 1:1, 1:2, and 1:3 (3.25, 6.5, and 9.75 mmol, respectively). The pH of the reaction mixtures initially dropped to approximately 3, and the solutions were stirred until they returned to a clear pale yellow. Next, EDC at the same molar ratios as those of HCA (3.25, 6.5, and 9.75 mmol) was dissolved in 50 mL of 50% ethanol and slowly added to the respective reaction mixtures with vigorous stirring. The pH of the mixtures was maintained in the range of 5.0–5.5. The reaction mixtures were stirred vigorously under these conditions for 24 h, after which they were transferred to dialysis tubes (Spectra/Por 3 RC MWCO 3.5 kD; Spectrum Labs, Piraeus, Greece) and dialyzed in 100 mM sodium chloride solution at pH 3.0 for 48 h. Subsequently, additional dialysis was performed in distilled water for 4 h. Following dialysis, the CC solutions were lyophilized to obtain the pure solid products. The lyophilization process involved preliminary freezing at –40°C for 24 h, followed by drying at –40°C and 80 mTorr until all moisture was removed.
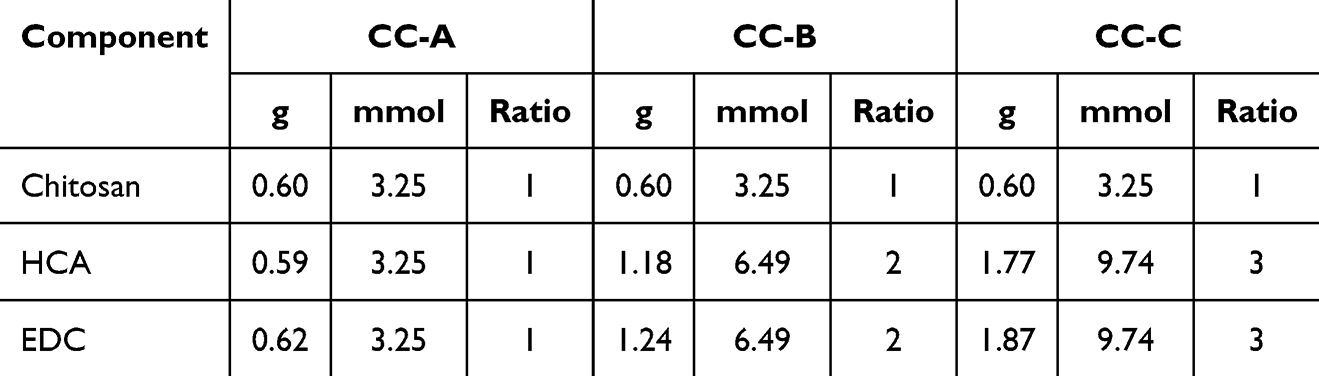 |
Table 1 Synthesis of Catechol-Conjugated Chitosan |
Proton Nuclear Magnetic Resonance Spectroscopy of Catechol-Conjugated Chitosan
To verify the synthesis of CC and the degree of catechol conjugation (DOC), proton nuclear magnetic resonance (H-NMR) spectroscopy analysis was performed using an NMR spectrometer (AVANCE III 400MHz; Bruker, Manning Park Billerica, MA, USA). The samples were dissolved in D2O for analysis. The scan number was set to 16, the spectral width to 20 ppm, and the probe temperature to 27°C. The acquisition time was set to 4s and the recycle delay (D1) time to 1s. The spectra were processed using TopSpin software (Bruker), with the chemical shifts referenced to the residual solvent peak of D2O (4.79 ppm). The DOC was calculated by dividing the peak area of the catechol group (δ 6.87, 3H, aromatic ring proton) by the peak area of the acetyl group on the chitosan backbone (δ 1.95, 3H, COCH3) and multiplying by 5, which accounts for the 80% degree of deacetylation of the chitosan used:26
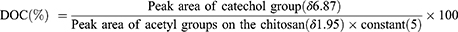
Shear Test of Mucoadhesive Strength
The shear test of the mucoadhesive strength of the CC samples was performed using chitosan as a control and the synthesized CC-A, CC-B, and CC-C at solid mass concentrations of 1%, 2%, and 3%. Porcine small intestinal mucosa was used for ex-vivo testing. Porcine intestinal mucosa was sourced from a local slaughterhouse after routine food processing, and no animals were sacrificed specifically for this study. The use of the tissue complied with institutional and ethical guidelines. Mucosal tissue samples were washed with phosphate-buffered saline (PBS; pH 7.4) and stored at –70°C. The tissues were thawed in PBS (pH 7.4) before use. The prepared porcine small intestine was cut into pieces measuring 5×2 cm, and the opposite side of the villi was attached to a slide glass (25 × 75×1 mm) using cyanoacrylate adhesive. Ten containers from each fabricated sample were attached to the mucosal tissue. To ensure uniform attachment, the mini-containers were dropped vertically from a height of 1 cm above the mucosa without applying any additional force. Then, the tissue with the attached containers was fixed in 100 mL of PBS (pH 7.4) in a 100 mL beaker. The temperature was maintained at 37°C using a magnetic stirrer with heating function, with stirring performed at 1000 rpm using a 2 cm magnetic bar. During the experiment, the evaporated PBS was replenished to maintain the buffer volume. Observations were made every hour for 12 h and additionally at 24, 48, and 72 h, and the number of remaining containers was recorded by visually inspecting the mucosal surface.
Preparation of Insulin-Loaded Mini-Containers
Insulin was selected as the peptide model drug, with a target dose of 10 IU per container. Chitosan and CC (synthesized at molar ratios of 1:1, 1:2, and 1:3) were prepared at solid mass concentrations of 1%, 2%, and 3%. Chitosan was dissolved in 1% acetic acid, whereas CC was dissolved in an HCl solution (pH 2.0). These solutions were stirred overnight using a magnetic stirrer, after which they were respectively mixed with the appropriate amount of insulin to achieve a concentration of 10 IU and further stirred for an additional 6 h. The homogeneous insulin-containing chitosan and CC solutions were then sonicated for 1 h to remove any air bubbles, and 40 µL of the solution was dispensed into the prepared EC-based containers. The drug-loaded containers were pre-frozen at –40°C for 24 h and then freeze-dried at –40°C and 80 mTorr for 24 h (LP03; ilShinBioBase, Dongducheon-si, Korea). The resulting drug-loaded mini-containers were stored at –20°C until analysis.
Circular Dichroism and Content Analyses of the Insulin-Loaded Mini-Containers
The stability of the insulin molecules within the chitosan and CC matrices, particularly at the manufacturing concentration, was evaluated using circular dichroism spectroscopy (Chirascan VX; Applied Photophysics, Surrey, UK). Native insulin, chitosan, and CC-C were analyzed in this experiment, with HCl (pH 2.0) used to dilute all three samples to an insulin concentration of 0.1 mg/mL. All samples were filtered through a 0.45 µm polyvinylidene difluoride (PVDF) filter. The wavelength range was set as 185–260 nm, with a bandwidth of 1.0 nm. The time-per-point was set to 0.5s, resulting in an approximate scan time of 69s. The analysis was performed at a controlled temperature of 25°C, using a 0.5 mm path length cell. Each sample was measured three times, and the average value was calculated. The mean residue ellipticity (MRE) was calculated according to the following equation (eq).
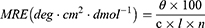
Where, θ is observed ellipticity in millidegrees measured by the CD spectropolarimeter, c is concentration in mol/L, l is path length of cuvette in centimeters (cm), n is the number of amino acid residues in the protein or peptide.
The contents of the prepared insulin-loaded mini-containers were measured using a validated HPLC-UV method. The analysis was performed using an UltiMate 3000 system (Thermo Fisher Scientific, Waltham, MA, USA) fitted with a 5 μm C18 column (100Å, 250×4.6 mm; Phenomenex Ltd., Torrance, CA, USA). The mobile phases consisted of 0.1% (v/v) trifluoroacetic acid (TFA) in water as phase A and 0.1% TFA (v/v) in acetonitrile as phase B. The gradient conditions started with a ratio of 75:25 (A) for the first 0–3 min, then a change to 20:80 from 3 to 3.5 min, and finally a reversion to 75:25 from 3.5 to 4.5 min. The flow rate was set at 0.5 mL/min, and the column oven was maintained at 25°C. The detection wavelength was 214 nm, and the injection volume was 20 μL. Mobile phase A was used as the sample diluent. The calibration curve was linear in the range of 2–100 μg/mL (r2 = 0.999). The mean percentage of recovery was found to be 99.2% and RSD was 0.5%. The proposed analytical method exhibited good reproducibility, intermediated precision and repeatability. RSD values were 0.87%. The limit of quantification (LOQ) and limit of detection (LOD) were calculated to be 1.977 μg/mL and 0.653 μg/mL, respectively.
Dissolution Test
The drug dissolution tests of the insulin-loaded chitosan containers and CC-C containers, each prepared at solid mass concentrations of 1, 2, 3% (w/v), were performed at 37°C in a water bath set to rotate at 100 rpm. In brief, 10 containers from each sample were attached to the wall of a 50 mL conical tube using double-sided tape to prevent them from detaching from the walls during dissolution, thereby ensuring accurate measurement of core dissolution. Each conical tube was filled with 50 mL of pre-warmed PBS (pH 7.4) at 37°C. The tubes were then placed in the water bath, and samples were collected at 0.5, 1, 2, 4, 6, 8, 10, and 12 h. At each time point, 1 mL of the dissolution medium was sampled using a syringe and replaced with 1 mL of fresh dissolution medium to maintain the volume. The collected samples were filtered using a 0.45 µm PVDF filter, with half of the filtrate discarded and the other half retained for analysis. The retained samples were analyzed without further dilution using the previously established HPLC-UV method used to determine the drug content. All tests were performed in triplicate.
To analyze the in-vitro release data various kinetics models were used to describe the release kinetics.27,28
Zero-Order Model
Zero-order model describes the system where the release rate of drug is independent of its concentration.
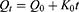
Where, Q is Amount of drug release or dissolved, Q0 is Initial amount of drug in solution, K0 is Zero order rate constant, t is the time.
First-Order Model
This model describes the absorption and removal of some drugs which depends on the concentration of the drug.
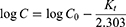
Where, C0 is Initial concentration of drug, K1 is First order constant, t is the time.
Higuchi Model
Higuchi described mathematical equation to describe the release of drugs from insoluble matrix as a square root of time dependent process based on Fickian diffusion equation.29
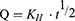
Korsmeyer-Peppas Model
Korsmeyer derived a simple relationship which describes the release of drug from a polymeric system equation.30 To explain the mechanism of drug release, first 60% of drug release data was fitted in Korsmeyer-Peppas model.
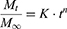
Where, Mt/M∞ is the fraction of drug release at time, K is the rate constant, n is the release exponent. The n value is used to characterize different release mechanism.
Unidirectional Release Test Using the Franz Diffusion Cell System
Drug release from the containers through the membrane was evaluated using the Franz diffusion cell system (Phoenix DB-6 Diffusion Testing System). Samples, including Insulin solution, 3% (w/v) chitosan solution, chitosan mini-containers (normal and reverse direction), CC-C solution, and CC-C mini-containers (normal and reverse direction), were prepared with insulin and subjected to testing. Additionally, to verify that the release was unidirectional, the containers were tested in two configurations: one with the drug-release side facing the membrane and the other with the bottom side facing the membrane. A 0.45 µm regenerated cellulose membrane (GVS, Bologna, Italy) was used. During the experiment, the receptor chamber was filled with PBS (pH 7.4), the containers were loaded into the donor chamber, and the system was set to rotate at 200 rpm. Samples (400 μL) were collected at intervals of 0.5, 1, 2, 4, 8, and 12 h and replaced with 400 µL of fresh PBS (pH 7.4). The collected samples were filtered through a 0.45 µm PVDF filter and analyzed directly without further dilution using the established HPLC-UV method described above. All tests were performed in triplicate.
Statistical Analysis
All statistical analyses were conducted using a one-way ANOVA, followed by Tukey’s post-hoc test with GraphPad Prism 8 (release 8.4.2; San Diego, CA, USA). Statistical significance was considered at p < 0.05 and p < 0.005.
Results and Discussion
Fabrication and Characterization of the Mini-Container Shells
The PLA template was fabricated using the fused deposition modeling 3D printing technique. As shown in Figure 1, the PLA template had visible stack lines that were created by the layer-by-layer deposition process, which persisted even after production of the silicone mold. These imperfections were anticipated to affect the hardness and unidirectional properties of the mini-container shell. To improve this, UV resin was applied to the PLA template to fill the stack lines, which resulted in silicone molds with improved surface smoothness and no stack lines.
EC-based mini-container shells with varying degrees of viscosity were prepared using the silicone molds. The bottom and side surfaces of the prepared shells were observed using SEM as shown in Figure 2. The shells made using molds with unmodified stack lines (EC viscosity values of 10, 25, and 45) showed unfilled areas at the bottom. Although most of the sides were filled, unfilled areas were observed at the stripe boundaries in the EC 10 and 25 shells. Conversely, shells produced using molds with UV resin-modified stack lines exhibited fully filled bottoms and sides, presenting a denser surface structure. Even though faint stripe patterns were visible in some low-viscosity shells (eg, EC 4, 10, and 25), the boundary areas were completely filled. These findings suggest that silicone molds with improved stack lines facilitate the production of shells with densely packed surfaces, indicating their potential for unidirectional drug release. Additionally, with increase in the EC viscosity, the impact of surface imperfections diminished. This phenomenon can be attributed to the lower flowability of highly viscous EC solutions into uneven surfaces.
 |
Figure 2 SEM images of the bottom and side surfaces of mini-container shells prepared with EC of different viscosities using stripe and non-stripe molds: (A) Mini-container shells manufactured using a stripe mold, (B) Mini-container shells manufactured using a non-stripe mold. |
The effects of the EC viscosity and presence of stack lines on shell strength were evaluated using a texture analyzer as shown in Figure 3. The properties of these shells are summarized in Table 2. All prepared shells using EC exhibited a diameter of approximately 5.5 mm and a height of about 2.0 mm. In terms of mass, a statistically significant increase was observed in EC 100 shells (3.42 ± 0.06 mg) compared to EC 4 (3.01 ± 0.25 mg) and EC 10 shells (3.05 ± 0.05 mg). This difference is attributed to the density variation caused by viscosity differences, as the solutions were dispensed based on equal volume using a pipette. The strength of the shells with stack lines was weaker than that of those without the lines. Shells fabricated with EC 100 exhibited the highest mechanical strength (5.81 ± 0.12 N), regardless of the presence or absence of stack lines, which was statistically significant. At other viscosities (eg, 10, 25, and 45 cP), both types of shells exhibited comparable strengths of 2.41 ± 0.29, 2.27 ± 0.27, and 2.58 ± 0.47 N, respectively. EC 4, EC 10, EC 20, and EC 45 shells without stack lines exhibited mechanical strengths of 4.96 ± 0.78, 4.68 ± 0.75, 4.73 ± 0.52, and 5.47 ± 1.54 N, respectively. EC 100 shells without stack lines were the strongest, with a measured strength of approximately 13.21 ± 0.50 N, showing statistically significant differences compared to shells made with other EC viscosities. Commercial soft gel capsules are typically designed to withstand mechanical stress of approximately 10 N, ensuring their durability in GI environments.31 Accordingly, the EC 100 shell without stack lines is expected to withstand the physical conditions of the GI tract. This trend indicates that EC of higher viscosity contributes to stronger intermolecular bonds, enhancing the structural integrity of the containers. Overall, the shells fabricated using high-viscosity ECs (eg, EC 100) exhibited comparable or superior mechanical strength to that of shells made with low-viscosity EC, making them well suited to better endure mechanical stress conditions in the GI tract and ensuring stable unidirectional drug release in the small intestine while providing protection against the harsh gastric environment. In addition, the EC100 shells fabricated via the molding method exhibited a relative standard deviation (RSD) of less than 5% in size, weight, and hardness. This indicates that the molding process enables the production of uniform shells, and it is expected that consistent shell quality can be maintained even during scale-up using larger molds.
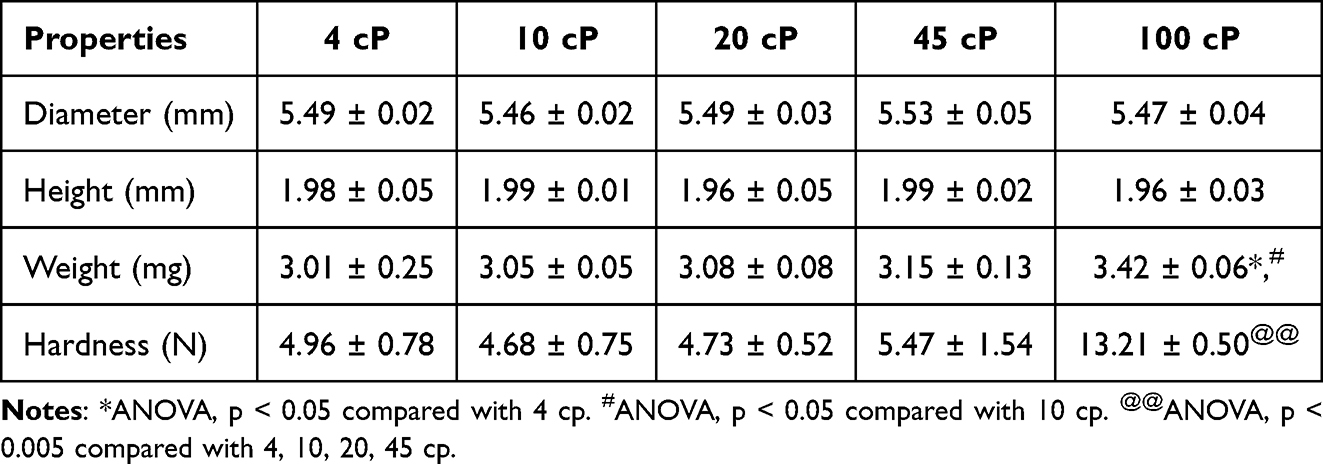 |
Table 2 Comparison of Mini-Container Shell Properties Fabricated Using Different Viscosity Grades of Ethyl Cellulose Without Stack Lines (n=3) |
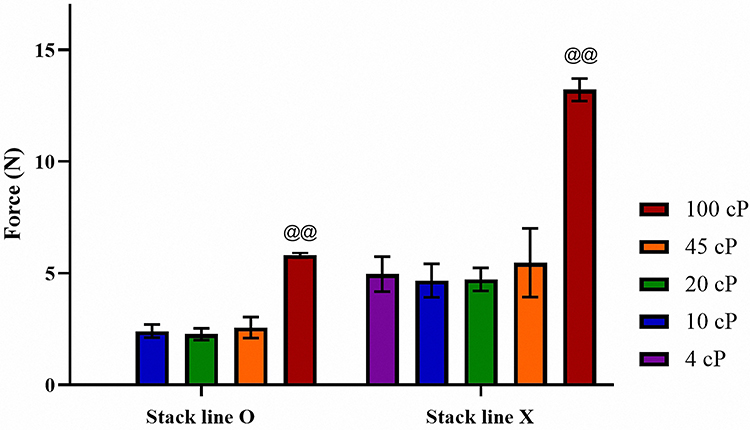 |
Figure 3 Hardness of mini-container shells with striped and non-striped surfaces prepared using EC of different viscosities (n=3). Notes: @@ANOVA, p < 0.005 compared with 4, 10, 20, 45 cp. |
Catechol-Conjugated Chitosan and Mucoadhesive Strength
CC was synthesized by reacting chitosan with HCA at molar ratios of 1:1, 1:2, and 1:3, yielding CC-A, CC-B, and CC-C, respectively. The degree of catechol substitution on the chitosan amine groups was analyzed using H-NMR spectroscopy. Distinct peaks corresponding to catechol groups were observed at δ 6.87, confirming the successful conjugation of catechol to the chitosan backbone as shown in Figure 4. The degrees of substitution, calculated by comparing the peak areas of the catechol groups to those of the acetyl groups at δ 1.95, were determined to be as follows: CC-A, 8.84%; CC-B, 14.48%; and CC-C, 20.93%. These results indicate that with increase in the molar ratio of HCA to chitosan, the degree of catechol substitution also increases. A higher degree of catechol substitution is expected to enhance the mucoadhesive property of the container owing to the greater availability of catechol groups, which are known to form strong interactions with mucosal surfaces.32,33 This suggests that CC-C, with the highest degree of substitution, would possess the best mucoadhesive strength among the synthesized formulations.
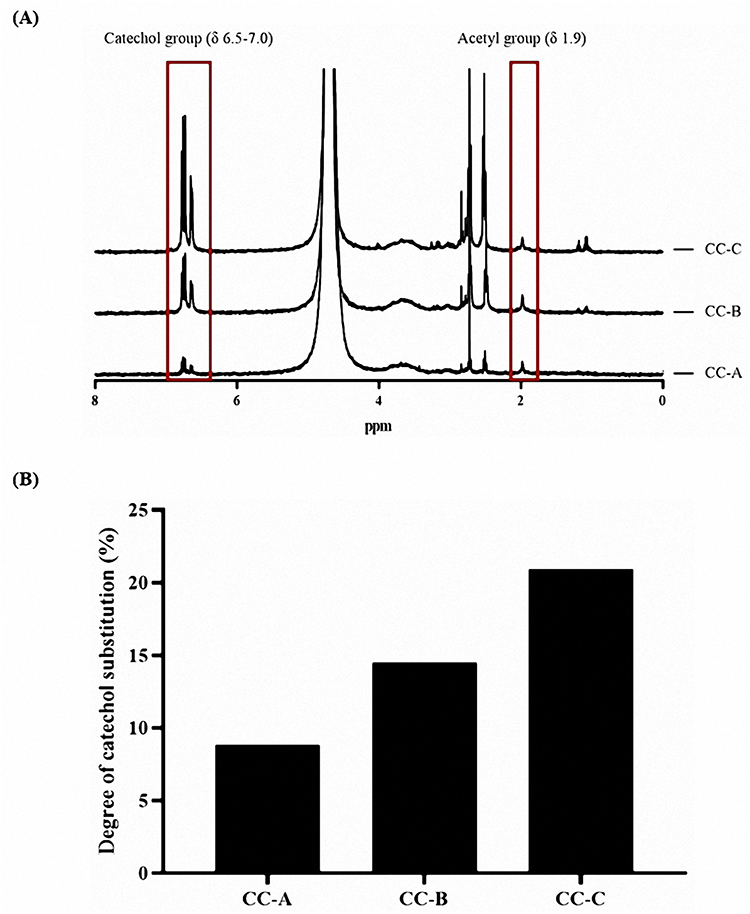 |
Figure 4 H-NMR results of CC-A, CC-B, and CC-C with different degrees of substitution: (A) H-NMR spectrum and (B) Degree of catechol substitution. |
The mucoadhesive performance of CC-A, CC-B, and CC-C was evaluated by loading each formulation into mini-containers and testing their adhesion on porcine small intestinal mucosa, as shown in Figure 5. Adhesion was assessed by counting the number of mini-containers remaining attached over 72 h, as summarized in Table 3. Mini-containers made with chitosan detached completely within 72 hours at all concentrations. Formulations made with CC-A exhibited 10%, 20%, and 40% residual mini-containers at 1%, 2%, and 3% concentrations, respectively. CC-B showed 20%, 30%, and 60%. CC-C retained 50%, 80%, and 80% under the same concentration conditions. The number of remaining mini-containers increased with the degree of catechol conjugation. This trend was shown in Figure 6A. CC-C, with the highest degree of catechol substitution, showed the strongest mucoadhesive performance. At 3%, only one mini-container detached after 6 hours. At 2%, detachment occurred after 3 hours. Adhesion improved with increasing CC concentrations (3% > 2% > 1%), as shown in Figure 6B. Across all concentrations, the duration of adhesion increased with increase in the degree of catechol substitution, following the order CC-C > CC-B > CC-A > chitosan. These findings confirm that the duration of mucoadhesion is directly proportional to the number of catechol groups, likely owing to strong catechol–mucosa interactions. According to the results, CC-C exhibited the most extended mucoadhesion among the formulations tested. Although further in-vivo evaluation using imaging techniques such as CT is required to assess the gastrointestinal residence time, these findings support the potential application of CC-C mini-containers for stable and prolonged mucosal drug delivery.
 |
Table 3 Properties of Synthesized Catechol-Conjugated Chitosan |
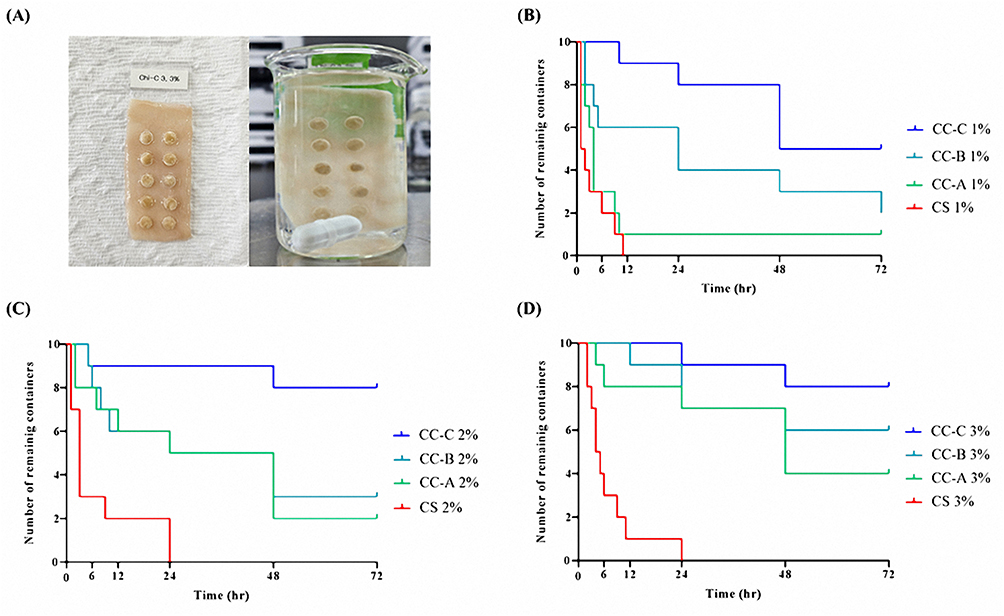 |
Figure 5 Ex-vivo attachment duration of mini-containers on porcine small intestine depending on catechol conjugation and polymer concentration: (A) Representative images from the mucoadhesive shear test; (B) Mini-containers with 1% concentration of chitosan, CC-A, CC-B, and CC-C; (C) with 2% concentration; and (D) with 3% concentration. |
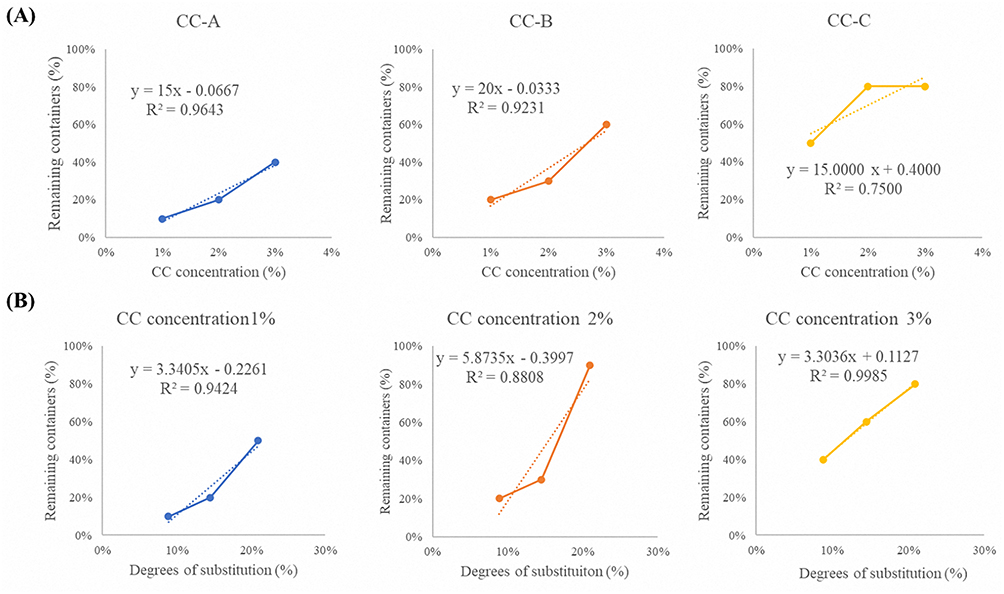 |
Figure 6 Correlation of CC concentration and catechol substitution degree with the number of remaining containers: (A) Effect of CC concentration on remaining containers for CC-A, CC-B, and CC-C formulations and (B) Effect of catechol substitution degree on remaining containers at CC concentrations (1%, 2%, and 3%). |
Insulin-Loaded Mini-Containers
Prior to the fabrication of insulin-loaded mini-containers, the stability of the drug’s secondary structure in the catechol-conjugated chitosan (CC) formulation was evaluated using circular dichroism (CD) spectroscopy. The mean residue ellipticity (MRE) was subsequently calculated to quantitatively assess structural retention. The spectrum showed a strong positive peak at 195 nm and negative peaks at 208 and 222 nm, characteristic of the alpha-helix structure of insulin, as shown in Figure 7. All tested samples displayed MRE consistent with that of native insulin, with comparable ellipticity values at the characteristic peaks. These results confirm that the alpha-helix structure of insulin had remained unchanged, suggesting that the drug retains its activity when loaded onto the CC formulations.
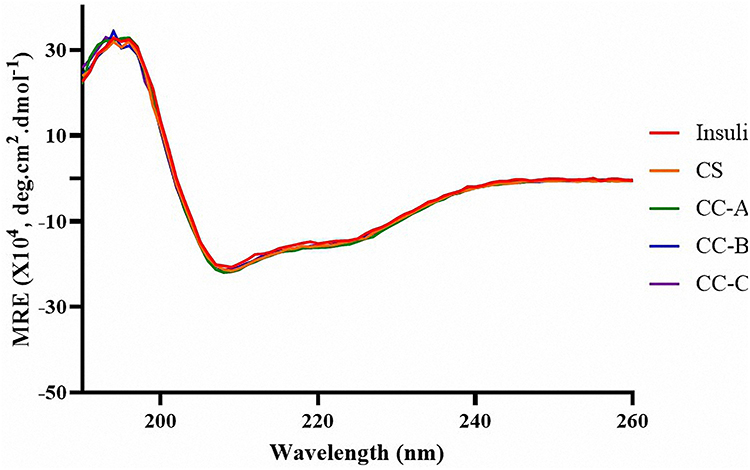 |
Figure 7 The mean residue ellipticity (MRE) spectra calculated from circular dichroism (CD) measurements of insulin in HCl solution (pH 2.0) and in catechol-conjugated chitosan (CC) solutions. The spectra demonstrate the preservation of insulin’s secondary structure upon interaction with CC formulations, as indicated by comparable MRE profiles. |
To prepare the insulin-loaded mini-containers, CC-C solutions at concentrations of 1%, 2%, and 3% were prepared, and insulin was dissolved within these solutions. The resulting formulations were dispensed into EC 100 shells at 10 IU insulin per shell and the mixtures were freeze-dried to obtain the insulin-loaded mini-containers. The insulin content in the mini-containers was evaluated using a validated HPLC-UV method. The results confirmed successful drug loading, with the following insulin contents: 1% CC-C (mini-container), 101.1 ± 2.4%; 2% CC-C (mini-container), 95.4 ± 3.8%; and 3% CC-C (mini-container), 100.0 ± 1.8%. Further investigation is required to evaluate the long-term stability of the CC-C formulation. However, it is expected to exhibit high stability due to the inherent biocompatibility and film-forming properties of chitosan, which can provide a protective barrier against external environmental factors such as moisture, oxygen, and pH fluctuations.34
Dissolution of the Mini-Containers
The dissolution test results demonstrated that insulin release from the CC containers was delayed compared to that from the chitosan containers, as shown in Figure 8. Additionally, with increase in the solid mass, the release rate further decreased. Examining the dissolution profiles in detail, the 1% chitosan sample released 16.67% of the drug at 0.5 h and 90.79% after 12 h. By contrast, the 1% CC-C sample released 8.89% of the drug at 0.5 h and 53.11% after 12 h, indicating that drug release from the catechol-containing samples was significantly delayed. A similar trend was observed with increasing solid mass. The 3% chitosan sample released 77.03% of the drug after 12 h, whereas the 3% CC-C sample only released 26.22% after 12 h. This suggests that a higher solid mass leads to stronger crosslinking between catechol groups, further slowing the drug release rate. These results can be explained by the effect of the catechol groups, which form crosslinked matrices through the hydrogen bonding of phenolic groups, thereby delaying dissolution. These crosslinked matrices regulate the drug release rate, allowing for more sustained and stable drug delivery. With increase in the solid mass, the molecular interactions within the CC matrix strengthen, resulting in slower drug release.
 |
Figure 8 Dissolution profiles of mini-container formulations composed of chitosan (CS) and catechol-conjugated chitosan C (CC-C) at polymer concentrations of 1%, 2%, and 3%, conducted in a water bath at 37 °C with agitation at 100 rpm (n = 3). |
To further investigate the drug release behavior, mathematical modeling was performed, and the calculated parameters were summarized in Table 4. Drug release profiles of CS 1%, 2%, and 3% formulations showed the highest fitting with the first-order model, excluding the Korsmeyer–Peppas equation. Analysis of the n value from the Korsmeyer–Peppas model indicated that CS 1% and CS 2% exhibited values of 0.694 and 0.931, respectively, suggesting that drug release was governed by a combination of diffusion and polymer swelling. In contrast, the CS 3% formulation exhibited a release exponent of 1.132, which may be attributed to weak intermolecular interactions between chitosan chains. This limited cohesion likely led to matrix disintegration during the swelling process, resulting in a steeper initial drug release profile. The strong correlation with both the first-order and Korsmeyer–Peppas models in CS formulations indicates that drug release was driven by a swelling induced drug concentration gradient and concentration dependent diffusion within the polymer matrix.
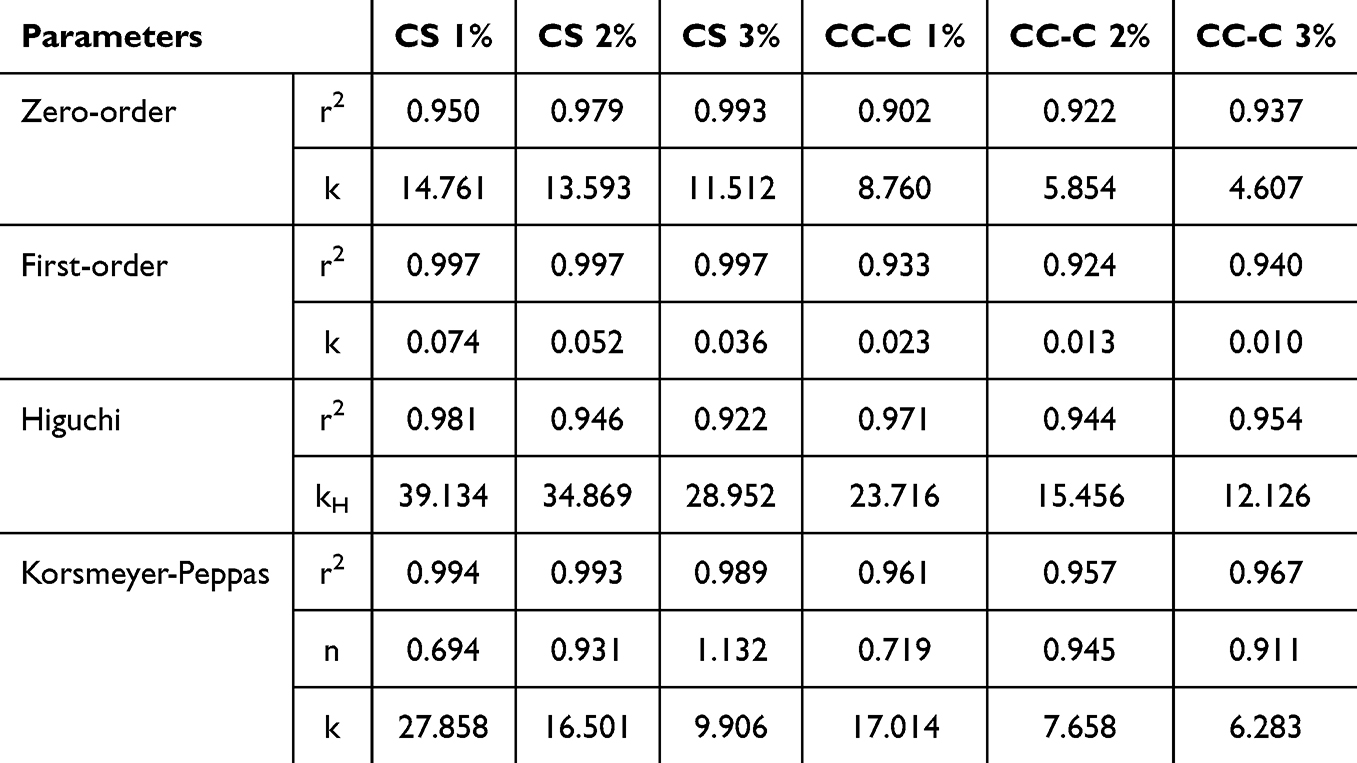 |
Table 4 Release Kinetics Parameters of Mini-Container Formulations Prepared with Chitosan and CC-C at 1%, 2%, and 3% Concentrations |
In the case of CC-C 1%, 2%, and 3% formulations, the release profiles were best fitted to the Higuchi model. This implies that catechol-mediated crosslinking reinforced the polymer network, resulting in a more pronounced diffusion mechanism compared to the CS formulations. The n values for CC-C 1%, 2%, and 3% were 0.719, 0.945, and 0.911, respectively. Notably, none of the CC-C formulations exhibited a value greater than 1.0, in contrast to CS 3%. All CC-C formulations exhibited release exponent values between 0.45 and 1.0, indicating a non-Fickian transport mechanism involving both diffusion and matrix swelling. The high correlation with both Higuchi and Korsmeyer–Peppas models further suggests that drug release from CC-C formulations was governed by diffusion through a swelling polymer matrix. These results suggest that catechol modification of chitosan enhances matrix integrity via crosslinking, contributing to sustained release behavior predominantly governed by diffusion.
Unidirectional Release
Unidirectional release behavior was assessed using a Franz diffusion cell with samples comprising Insulin solution, 3% (w/v) chitosan solution, chitosan mini-containers (normal and reverse direction), CC-C solution, and CC-C mini-containers (normal and reverse direction), were prepared with insulin, as shown in Figure 9. The insulin solution exhibited the fastest release rate, with almost all the drug molecules released within 0.5 h and over 100% released after 12 h. Both the 3% chitosan and 3% CC-C solutions showed more sustained release patterns compared with the insulin solution. The 3% chitosan solution exhibited an initial insulin release of approximately 39% at 0.5 h and reached 104% at 12 h, while the 3% CC-C solution showed a higher initial release of approximately 49% at 0.5 h and reached 102% at 12 h. The chitosan mini-container showed no drug release after 0.5 h, and released approximately 70% after 12 h. By contrast, the CC-C mini-container also showed no drug release after 0.5 h but only released approximately 26% after 12 h. While the CS and CC-C formulations exhibited similar release behavior in solution form, the mini-container form of CC-C showed a slower release rate compared to CS. Although CS and CC-C solutions exhibited similar release behavior, the CC-C mini-containers showed a slower release rate compared to those made with CS. This suggests that the difference in drug diffusion is not due to the polymer type itself, but rather to the slower diffusion of the drug through the swollen CC-C core compared to the CS core. Consistent with previous dissolution results, this slower release is likely attributed to the formation of crosslinked matrices induced by the catechol moieties in CC-C. Notably, both the chitosan container (reverse) and CC-C container (reverse) did not release any drug during the experimental period, confirming that the containers possess unidirectional release properties and ensuring that the drug is released only in the intended direction. The mini-container fabricated via the molding method demonstrated unidirectional drug release. When combined with a mucoadhesive core, this unidirectionality is expected to reduce exposure to intestinal fluids containing enzymes by adhering to the mucosal surface, thereby enhancing the stability and bioavailability of peptides and proteins such as insulin. Further studies, including confirmation of mucosal adhesion and in-vivo evaluation, are warranted.
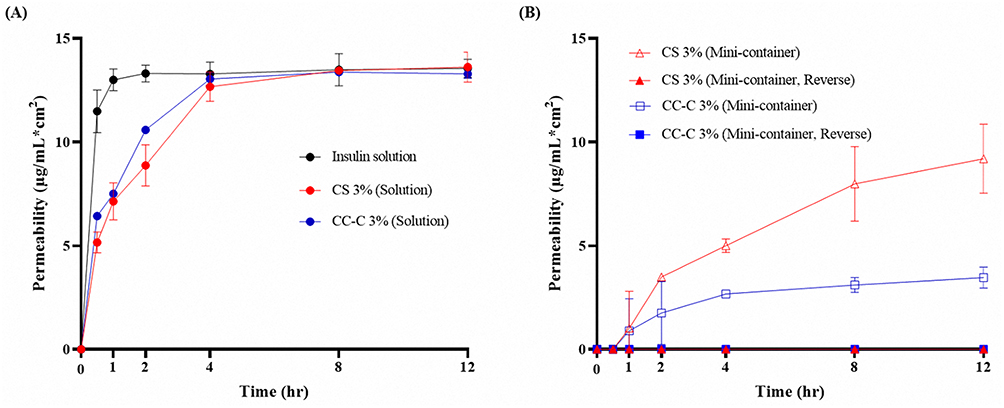 |
Figure 9 Evaluation of unidirectional release of mini-container using the Franz diffusion cell: (A) Insulin-containing solutions of distilled water (DW), 3% chitosan (CS), and 3% catechol-conjugated chitosan C (CC-C) and (B) Mini-containers prepared with 3% CS (normal and reverse direction) and 3% CC-C (normal and reverse direction) (n = 3). |
Conclusion
A mini-container shell capable of unidirectional drug release was successfully fabricated using a molding technique. CC was screened at varying ratios, and its adhesion performance was evaluated using a porcine small intestinal mucosa model. The CC-C formulation maintained an 80% adhesion rate over 72 h. Insulin was successfully loaded onto the CC-C formulation, and subsequent analyses confirmed no degradation or alteration of the content or secondary structure of the drug. Additionally, sustained drug release was demonstrated through dissolution testing, and unidirectional release was validated using the Franz diffusion cell system. The mucoadhesive mini-containers exhibited unidirectional drug release and mucoadhesive potential, highlighting their high applicability as carriers for macromolecular drug delivery by protecting the drug from external environments. However, further investigation is needed to confirm in-vivo whether the formulation adheres to the mucosal surface and contributes to improved absorption. In addition, storage stability should be evaluated to ensure that the formulation maintains its structural integrity and drug content over time.
Acknowledgments
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2021R1A2C4002746), and this work was supported by the National Research Foundation of Korea(NRF) grant funded by the Korea government.(MSIT) (No. RS-2025-02273102).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rossino G, Marchese E, Galli G, et al. Peptides as therapeutic agents: challenges and opportunities in the green transition era. Molecules. 2023;28(20):7165. doi:10.3390/molecules28207165
2. Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Trans Targeted Ther. 2022;7(1):48 doi:10.1038/s41392-022-00904-4.
3. Petta I, Lievens S, Libert C, Tavernier J, De Bosscher KJ. Modulation of protein–protein interactions for the development of novel therapeutics. Mol Ther. 2016;24(4):707–718. doi:10.1038/mt.2015.214
4. Cao S-J, Lv Z-Q, Guo S, Jiang G-P, Liu H-L. An update-prolonging the action of protein and peptide drugs. J Drug Delivery Sci Technol. 2021;61:102124 doi:10.1016/j.jddst.2020.102124.
5. Shaji J, Patole VJ. Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci. 2008;70(3):269. doi:10.4103/0250-474X.42967
6. Aungst BJ, Saitoh H, Burcham DL, Huang S-M, Mousa SA, Hussain M. Enhancement of the intestinal absorption of peptides and nonpeptides. J Controlled Release. 1996;41(1–2):19–31 doi:10.1016/0168-3659(96)01353-3.
7. Viegas C, Seck F, Fonte PJ. An insight on lipid nanoparticles for therapeutic proteins delivery. J Drug Delivery Sci Technol. 2022;77:103839.
8. Sun Z, Huang J, Fishelson Z, Wang C, Zhang S. Cell-penetrating peptide-based delivery of macromolecular drugs: development, strategies, and progress. Biomedicines. 2023;11(7):1971. doi:10.3390/biomedicines11071971
9. Haddadzadegan S, Dorkoosh F, Bernkop-Schnürch A. Oral delivery of therapeutic peptides and proteins: technology landscape of lipid-based nanocarriers. Adv Drug Delivery Rev. 2022;182:114097. doi:10.1016/j.addr.2021.114097
10. Jørgensen JR, Jepsen ML, Nielsen LH, et al. Microcontainers for oral insulin delivery–in vitro studies of permeation enhancement. European J Pharmaceutics Biopharm. 2019;143:98–105. doi:10.1016/j.ejpb.2019.08.011
11. Buckley ST, Bækdal TA, Vegge A, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. 2018;10(467):eaar7047. doi:10.1126/scitranslmed.aar7047
12. Mazzoni C, Tentor F, Strindberg SA, et al. From concept to in vivo testing: microcontainers for oral drug delivery. J Controlled Release. 2017;268:343–351. doi:10.1016/j.jconrel.2017.10.013
13. Jørgensen JR, Yu F, Venkatasubramanian R, et al. In vitro, ex vivo and in vivo evaluation of microcontainers for oral delivery of insulin. Pharmaceutics. 2020;12(1):48. doi:10.3390/pharmaceutics12010048
14. Nielsen LH, Keller SS, Boisen A, Müllertz A, Rades T. A slow cooling rate of indomethacin melt spatially confined in microcontainers increases the physical stability of the amorphous drug without influencing its biorelevant dissolution behaviour. Drug Delivery Transl Res. 2014;4(3):268–274. doi:10.1007/s13346-013-0166-7
15. Nielsen LH, Keller SS, Boisen A. Microfabricated devices for oral drug delivery. Lab Chip. 2018;18(16):2348–2358. doi:10.1039/c8lc00408k
16. von Halling Laier C, Gibson B, Moreno JAS, et al. Microcontainers for protection of oral vaccines, in vitro and in vivo evaluation. J Controlled Release. 2019;294:91–101. doi:10.1016/j.jconrel.2018.11.030
17. Madou MJ. Fundamentals of Microfabrication: The Science of Miniaturization. CRC press; 2018.
18. Chen G, Kawazoe N, Ito Y. Photo-crosslinkable hydrogels for tissue engineering applications. Photochemistr Biomed Appl. 2018:277–300 doi:10.1007/978-981-13-0152-0_10.
19. Choi G-S, Kim SH, Seo HI, et al. A multicenter, prospective, randomized clinical trial of marine mussel-inspired adhesive hemostatic materials, InnoSEAL Plus. Ann Surg Treatment Res. 2021;101(5):299–305. doi:10.4174/astr.2021.101.5.299
20. Sriram N, Parasakthi N. Overcoming challenges in oral insulin delivery: innovations in nanocarrier systems for enhanced bioavailability. Scientific Hub Applied Res Emerg Med Sci Technol. 2024;3(4):1–7.
21. Xiao Y, Tang Z, Wang J, et al. Oral insulin delivery platforms: strategies to address the biological barriers. Angewandte Chemie. 2020;59(45):19787–19795. doi:10.1002/anie.202008879
22. Bogdan C, Hales D, Cornilă A, et al. Texture analysis–a versatile tool for pharmaceutical evaluation of solid oral dosage forms. Int J Pharm. 2023;638:122916. doi:10.1016/j.ijpharm.2023.122916
23. Costa PM, Learmonth DA, Gomes DB, et al. Mussel-inspired catechol functionalisation as a strategy to enhance biomaterial adhesion: a systematic review. Polymers. 2021;13(19):3317. doi:10.3390/polym13193317
24. Li J, Zhang C, He W, et al. Coordination-driven assembly of catechol-modified chitosan for the kidney-specific delivery of salvianolic acid B to treat renal fibrosis. Biomat Sci. 2018;6(1):179–188 doi:10.1039/c7bm00811b.
25. Guyot C, Cerruti M, Lerouge S. Injectable, strong and bioadhesive catechol-chitosan hydrogels physically crosslinked using sodium bicarbonate. Mat Sci Engineer. 2021;118:111529. doi:10.1016/j.msec.2020.111529
26. Kim K, Ryu JH, Lee DY, Lee HJ. Bio-inspired catechol conjugation converts water-insoluble chitosan into a highly water-soluble, adhesive chitosan derivative for hydrogels and LbL assembly. Biomat Sci. 2013;1(7):783–790. doi:10.1039/c3bm00004d
27. Gangane PS, Pachpute T, Mahapatra DK, Mahajan NM. HPMC polymers and xanthan gum assisted development and characterization of stavudine extended release floating tablets. Indian J Pharm Educ Res. 2021;55:S681–S692 doi:10.5530/ijper.55.3s.175.
28. Dash S, Murthy PN, Nath L, Chowdhury PJ. Kinetic modeling on drug release from controlled drug delivery systems. Acta Poloniae Pharmaceutica. 2010;67(3):217–223.
29. Higuchi T. Mechanism of sustained‐action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharmaceut Sci. 1963;52(12):1145–1149. doi:10.1002/jps.2600521210
30. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
31. Zheng B-D, Yu Y-Z, Yuan X-L, et al. Sodium alginate/carboxymethyl starch/κ-carrageenan enteric soft capsule: processing, characterization, and rupture time evaluation. Int J Biol Macromolecules. 2023;244:125427. doi:10.1016/j.ijbiomac.2023.125427
32. Pornpitchanarong C, Rojanarata T, Opanasopit P, Ngawhirunpat T, Patrojanasophon P. Catechol-modified chitosan/hyaluronic acid nanoparticles as a new avenue for local delivery of doxorubicin to oral cancer cells. Colloids Surfaces B. 2020;196:111279. doi:10.1016/j.colsurfb.2020.111279
33. Kim K, Kim K, Ryu JH, Lee HJB. Chitosan-catechol: a polymer with long-lasting mucoadhesive properties. Biomaterials. 2015;52:161–170. doi:10.1016/j.biomaterials.2015.02.010
34. Barbosa FC, Silva MC, Silva HN, et al. Progress in the development of chitosan based insulin delivery systems: a systematic literature review. Polymers. 2020;12(11):2499. doi:10.3390/polym12112499
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.