Back to Journals » Clinical Optometry » Volume 11
Monitoring and Management of the Patient with Stargardt Disease
Authors Cicinelli MV ![]() , Battista M
, Battista M ![]() , Starace V
, Starace V ![]() , Battaglia Parodi M, Bandello F
, Battaglia Parodi M, Bandello F ![]()
Received 24 September 2019
Accepted for publication 16 November 2019
Published 28 November 2019 Volume 2019:11 Pages 151—165
DOI https://doi.org/10.2147/OPTO.S226595
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Mr Simon Berry
Maria Vittoria Cicinelli, Marco Battista, Vincenzo Starace, Maurizio Battaglia Parodi, Francesco Bandello
Department of Ophthalmology, University Vita-Salute, IRCCS Ospedale San Raffaele, Milan, Italy
Correspondence: Maria Vittoria Cicinelli
Department of Ophthalmology, San Raffaele Vita-Salute University, Via Olgettina, 60, Milano 20132, Italy
Tel +39 02 26432648
Fax +39 02 26483643
Email [email protected]
Abstract: Stargardt disease (STGD1) represents one of the major common causes of inherited irreversible visual loss. Due to its high phenotypic and genotypic heterogeneity, STGD1 is a complex disease to understand. Non-invasive imaging, biochemical, and genetic advances have led to substantial improvements in unveiling the disease processes and novel promising therapeutic landscapes have been proposed. This review recapitulates the modalities for monitoring patients with STGD1 and the therapeutic options currently under investigation for the different stages of the disease.
Keywords: Stargardt disease, inherited retinal dystrophy, multimodal imaging, optical coherence tomography, gene therapy
Introduction
Stargardt disease (STGD1) or Stargardt macular dystrophy is a recessive inherited retinal disease with an incidence of 8–10 per 100.000 persons.1 First reported by Stargardt in 1909, it is caused by an autosomal mutation in the adenosine triphosphate binding cassette transporter 4 (ABCA4) gene.2 Autosomal dominant pattern of inheritance of STGD1 has also been described, in association with mutations in PRPH2, ELOVL4, and PROM1 genes.3
Yellow or white fish-shaped flecks and photoreceptors, retinal pigment epithelium (RPE) and choriocapillaris (CC) atrophy are the pathognomonic clinical features of STGD1.4 Flecks result from the accumulation of lipofuscin at the level of the RPE within or outside the main vascular arcades; flecks are thought to be the epiphenomenon of the RPE cell metabolic impairment in the clearance of photoreceptors debris.
STGD1 symptoms typically develop within the first two decades of life and include progressive, irreversible loss of central and color vision and delayed dark adaptation. The peripheral visual field is normal and there is no complaint of night blindness. Nevertheless, the natural course of the disease is characterized by marked clinical variability with regards to the age of onset, the pattern of fundus lesions, and the rate of progression.5 Unknown mechanisms as genotype-phenotype interaction or environment factors could modify the anatomical fate and the functional prognosis. To shed light on these differences, STGD1 patients have been extensively monitored by means of non-invasive imaging techniques.
Fundus autofluorescence (FAF) classically shows hyperautofluorescence corresponding to the flecks and hypoautofluorescence at the level of the RPE atrophy.6 Angiographic examinations, as fundus fluorescein angiography (FFA) and indocyanine green angiography (ICGA) show specific features but are poorly applicable in the first diagnosis of the disease. Optical coherence tomography (OCT) enlightens on the changes in the outer nuclear layer, as photoreceptor loss, RPE abnormalities, or the rare occurrence of choroidal neovascularization (CNV).7 Little is still known about the degree to which retinal and choroidal vascular networks are involved in STGD1, but new exciting information is coming from OCT angiography (OCTA) studies.8–10
To date, no treatment is currently approved for STGD1 patients; however, stem cell therapy, gene replacement, and pharmacological strategies are the latest therapeutic promises intended to restore the RPE damage or slow down the advancement of the disease.11 Recent trials are aiming to correlate clinical and functional factors with different rates of RPE atrophy enlargement, helping in stratifying clusters of patients and fixing clearer endpoints.5,12 The aim of this review is to recapitulate the modalities for monitoring patients with STGD1 and the therapeutic options presently under investigation for the different stages of the disease.
Molecular Basis
ABCA4 encodes for a retinal ATP-binding cassette protein located on the membrane of the outer segment discs of the cones and the rods.13,14 ABCA4 works as a transporter that utilizes the energy of ATP hydrolysis to unidirectionally translocate retinoids (N-retinylidene-PE and all-trans-retinal) generated after photobleaching-induced isomerization of 11-cis-retinal, from the luminal to the cytoplasmic side of the disk membrane.13
Following isomerization and release from the cell, all-trans retinal travels to the RPE to be first esterified by lecithin-retinol acyltransferase (LRAT) and then converted to 11-cis-retinol by the isomerohydrolase RPE65. Finally, it is oxidized 11-cis-retinal before being transported back to the photoreceptor outer segment, where it is again conjugated to rhodopsin or cone opsin to form new, functional visual pigment.13 Failure of this transport results in the accumulation of lipofuscin, the main by-product of the photoreceptor visual cycle, into the RPE during the process of disk shedding. Lipofuscin and its components, especially N-retinylidene-N-retinylethanolamine (A2E), turn out to be toxic to epithelial and neuronal cells, with consequent RPE and photoreceptor degeneration.15 ABCA4 knockout mice supported the hypothesis: accelerated deposition of lipofuscin in RPE and increased levels of N-ret-PE have been observed after light exposure in abca4−/− mice.16
ABCA4 is a large complex gene in chromosome 1 consisting in 50 exons and has a causative role in numerous retinal diseases; mutations have been found in STGD1, cone-rod dystrophy, retinitis pigmentosa, and age-related macular degeneration (AMD).17,18 The extreme complexity of the gene makes it difficult to establish a thorough analysis of the genotype-phenotype interactions. Its high heterogenicity is reflected in the almost 6000 variant types reported in the literature, the majority located in the coding region of ABCA4;19 discriminating between normal and pathological variants is not always possible.19 To add complexity, heterogeneity was observed to vary among different populations.20–24 Therefore, genetic test represents a crucial inclusion criterion for access to clinical trials.
The most common genetic variants are missense and null mutations: the former are generally associated with a milder presentation, while the latter have severe visual prognosis and an early vision loss onset, with some exceptions. Some missense variants could also lead to fast-progressive disease, as p.Leu541Pro and p.Ala1038Val.25 On the other hand, homozygous p.Gly1961Glu missense mutation phenotypically corresponds to adult-onset disease, with atrophy confined to limited areas of the macula, mild retinal dysfunction and variable visual acuity.26
This large number of pathogenic variants in ABCA4 only explain 60–75% of STGD1 phenotypes; other types of variants in non-coding regions, hardly identified through the most commonly-used strategies of genetic screening, may account for missing hereditability in ABCA4-related diseases (that is, the gap between heritability estimates from genotype data and heritability estimates from twin studies).27,28 Some of these mutations include hypomorphic29,30 and deep intronic variants, which have a presumed effect on splicing.31–34 Non-coding deep-intronic splice variants represent potential interesting therapeutic targets for gene therapy approaches (i.e., CRISPR/Cas9 and antisense oligonucleotide-based splicing correction).31
So far, the use of a variety of mutation detection techniques for STGD have been reported, such as single-strand conformation polymorphism (SSCP)/heteroduplex analysis,35 high-resolution melting,36 microarray,37 and direct Sanger sequencing.38 These approaches are labor- and cost-intensive or low throughput. Next-generation sequencing (NGS) and whole exome sequencing (WES) strategies, on the contrary, allows for a comprehensive molecular diagnosis of STGD1 in a more rapid, cost-effective, and high throughput way.39,40 Nevertheless, a fair amount of patients with clinical diagnosis of STGD1 remains molecularly “unsolved”,29,41 and this could be explained by the presence of yet unknown variants, unidentified regulatory systems, or inaccurate diagnosis (phenocopies).
Clinical Presentation
STGD1 clinical spectrum varies widely.4,42 Progressive bilateral loss can occur at any age; generally, two phenotypes can be distinguished on the basis of symptoms onset: a first peak around 20 years and a second peak around 40 years. Age of onset could be indicative of disease severity, with earlier onset being associated with a more aggressive disease course; however, this parameter should be interpreted cautiously, as it is mainly dependent on the degree of involvement of the fovea. Adult-onset STGD1 is generally associated with missense mutations and better prognosis, while childhood-onset is largely linked to null variants with the poorest prognosis. Cases of very late-onset disease (i.e., after the sixth decade) are extremely rare.
Many attempts have been published to categorize the clinical spectrum of the disease; the most widely used classification belongs to Fishman, who described 4 stages based on clinical findings, and electrooculogram (EOG), electroretinogram (ERG), and psychophysical testing (Table 1).43
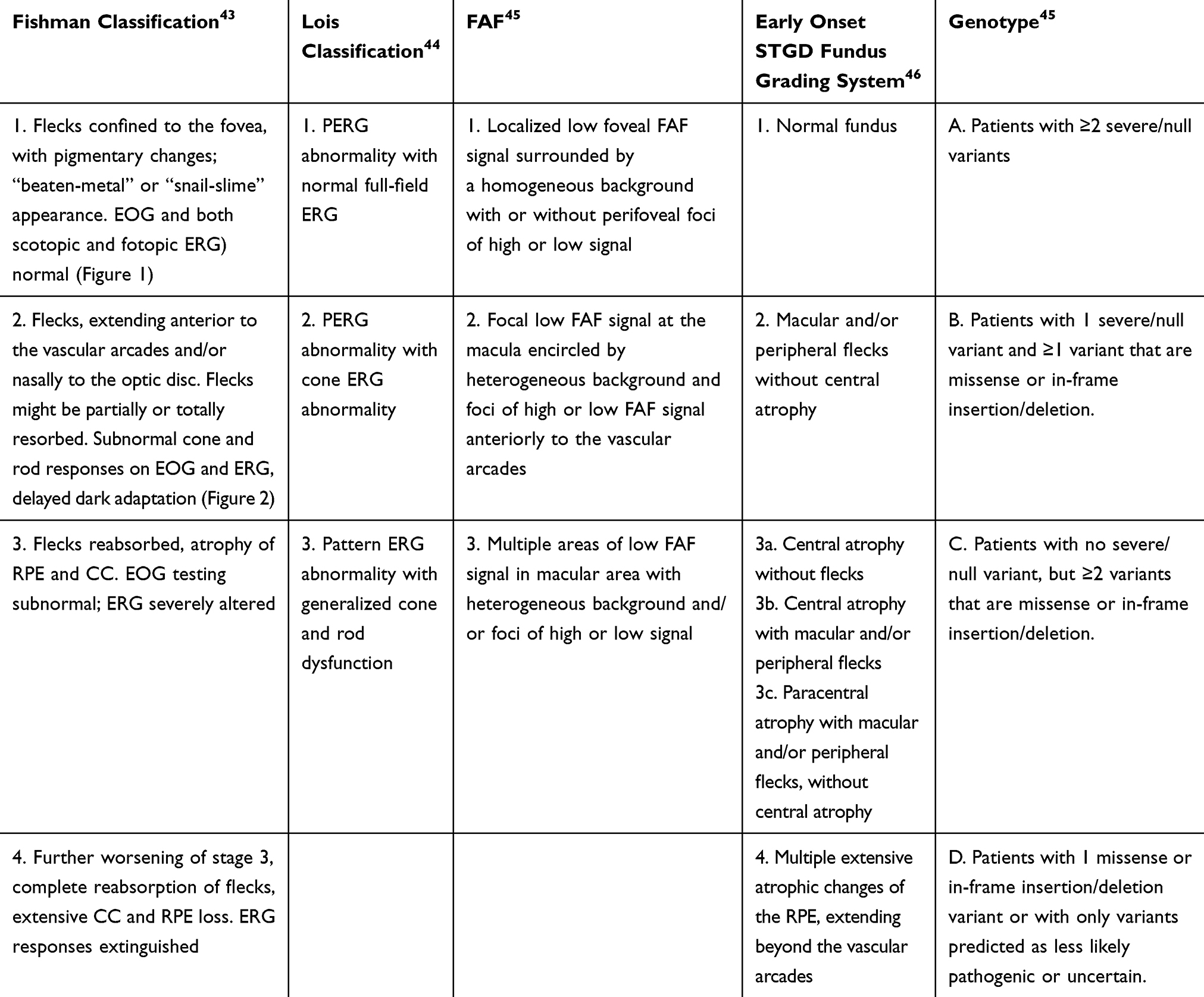 |
Table 1 Classifications of Stargardt Disease. The Groups Proposed Do Not Always Correspond to Different Progressive Stages of the Disease but Have Important Implications in the Clinical and Functional Assessment of the Patients |
 |
Figure 1 Multimodal imaging of a patient with STGD1 and macular involvement. Color fundus photograph (A), near-infrared-autofluorescence (B) and short wavelength fundus autofluorescence (C). Absence of RPE is visible as an area of irregular hyopoautofluorescence on fundus autofluorescence. Several hyperautofluorescence lesions at the macula correspond to the flecks. The OCT scan (D) centered on the fovea shows atrophy of RPE and ellipsoid zone and backscattering. |
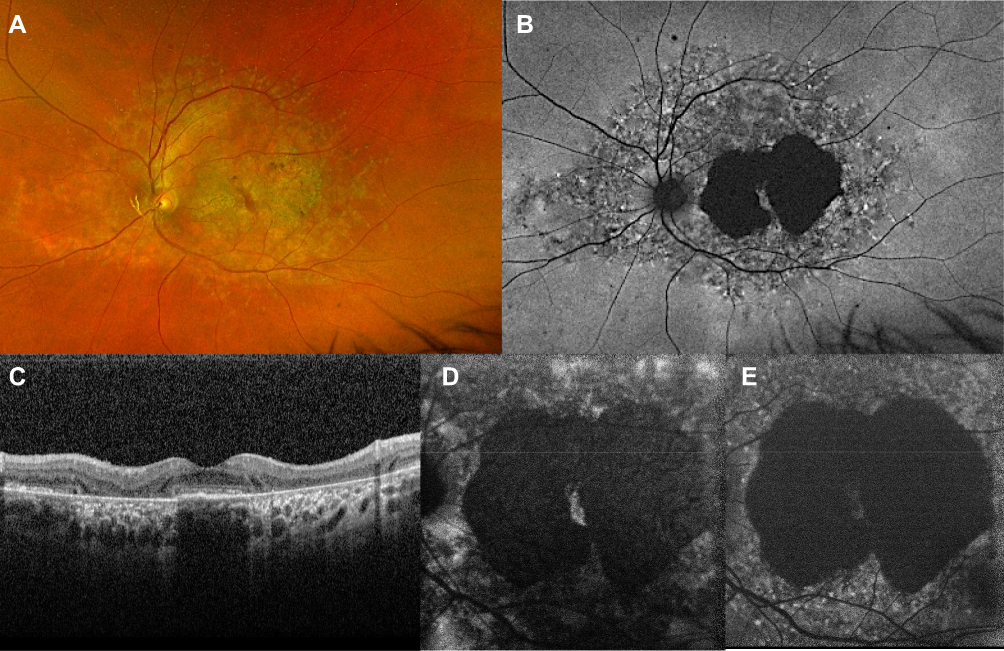 |
Figure 2 Ultra-widefield fundus photograph and autofluorescence of an advanced form of STGD1. The area of RPE mottling is comprised between the vascular arcades and broadens nasally to the optic disc (A, B). The macular area shows a marked hypofluorescence, with a little amount of foveal sparing (C). The OCT scan (C) demonstrates irregularity in the subfoveal RPE-photoreceptors complex and extensive atrophy in RPE and external retinal layers in the peri- and para-foveal areas. Foveal sparing is better seen on near-infrared autofluorescence (D) rather than short wavelength autofluorescence (E). |
Presenting best-corrected visual acuity (BCVA) is also heterogeneous. Moderate to high correlations between BCVA and both subjective onset (r = 20.64, P = 0.001) and duration of symptoms (r = +0.71, P = 0.001) have been recently reported.47 Patients with disease onset later than 20 years of age are more likely to maintain lifetime visual acuities better than 20/200 compared to patients with STGD1 presenting before 20 years of age. As predictable, the presence of foveal sparing ophthalmoscopically is associated with better visual acuity in diverse cohorts of patients.48
The Progression of Atrophy Secondary to Stargardt Disease (ProgStar) study, a landmark project involving a large international cohort of STGD1 patients based on both prospective and retrospective analyses, reported no significant change in BCVA during a 12-month follow-up49 and a clinically small BCVA loss over 24 months.50 The change rate varied depending on baseline BCVA and visual involvement; eyes with no or modest acuity impairment but with a foveal lesion at baseline had the fastest loss rate. Nevertheless, due to its slow rate of change over time, BCVA might not be looked at as a reliable endpoint for putative STGD1 treatments.
Monitoring Progression of STGD1
Ophthalmoscopic signs and the visual function might not be helpful in monitoring STGD1, either in the first phase or in the later stage of the disease. The classic yellowish-white fundus flecks, the alteration of the foveal reflex, and the RPE and choroidal irregularities generally appear late. On the other hand, high BCVA might be maintained if a small island of foveal photoreceptors is preserved.
Conversely, novel non-invasive imaging techniques, as FAF and OCT, might attain valuable information on the metabolic state of the photoreceptor/RPE complex since the very early stages.
FFA reveals blockage of the choroidal fluorescence, referred to as the dark choroid sign, due to the lipofuscin accumulation into the RPE cells; however, the dark choroid is a late-onset feature and is not detectable in all cases. Moreover, in cases of advanced disease, it might be difficult to identify due to the presence of extensive window defects in regions of confluent RPE atrophy.51 ICGA provides useful information about the alterations of the choroidal vasculature, revealing hypofluorescence within the area of macular atrophy (dark atrophy).52 This sign has been interpreted as extended damage of the CC, and (when present) is useful to distinguish late-onset STGD1 from geographic atrophy (GA) secondary to age-related macular degeneration.
SW-FAF and NIR-FAF
Short-wavelength FAF (SW-FAF) imaging, based on the signal derived from lipofuscin excited with a 488-nm wavelength beam, has been used as a fast and reproducible technique to calculate the progression of RPE atrophy in both monocentric and multicentric studies.12 Reported rates of progression are slightly different across different investigations, as mainly dependent on the age of the patients, the advancement of the disease, and the method of hypo-FAF quantification.
In a large report on 68 patients, the rate of atrophy enlargement was calculated stratifying patients into 3 classes, according to the SW-FAF phenotype (Table 1).45 The rate of progression was significantly faster in type 3 lesions (4.37 mm2/year) compared to type 1 (0.06 mm2/year) and type 2 (0.67 mm2/year) ones (Figure 3).45
 |
Figure 3 Progression of STGD1. First row: Short wavelength autofluorescence, Multicolor imaging, and OCT at baseline. Second row: Short wavelength autofluorescence, Multicolor imaging, and OCT after 12 months, showing enlargement of the area of RPE and photoreceptor loss on autofluorescence and central retinal thinning the foveal area. |
Indeed, two different types of decreased autofluorescence have been proposed based on the measurement of relative of darkness with respect to the optic nerve head (or, if not present in the image, the retinal vessels), considered as the reference for 100% blackness on the gray scale: definitely decreased autofluorescence (DDAF), defined as a signal more than 90% black in reference to the optic nerve head, and questionably decreased autofluorescence (QDAF), defined as 50% to 90% black, as compared to the reference.53 Areas of QDAF tend to be unstable on consecutive examinations; they have been interpreted as a transition state between healthy retina and regions of advanced disease (ie, DDAF). Therefore, lesions of QDAF may be still amenable to rescue, and may be of interest for future therapeutic interventions. DDAF and QDAF can be summed up to calculate the total decreased autofluorescence (TDAF). Mean progression of TDAF has been estimated as 0.35 mm2/year (95% CI, 0.42–0.61) and 0.64 mm2/year (95% CI, 0.54–0.97) in the retrospective54 and in the prospective12 branch of the ProgStar study, respectively. The growth rates were dependent on initial lesion size, with larger lesions at baseline expanding faster than smaller ones. Unifocal lesions tended to display a milder course with respect to multifocal atrophy, regardless of lesion size at baseline.54
Alongside the observation of hypo-FAF consequent to the RPE loss, FAF allows for the identification of other lesions pathognomonic of STGD1, as the hyperautofluorescent border surrounding the RPE atrophy in certain phenotypes,55 which is believed to represent accelerated bisretinoid production in metabolically-deranged RPE/photoreceptor complex. This feature has been identified as a predictor of selective atrophy enlargement in STGD1, even though no association between the rate of progression and the hyper-FAF ring surrounding RPE atrophy has been proven.12 Another distinctive feature of STGD1 is the peripapillary sparing, which often helps to distinguish this disease from other phenocopies (e.g. PRPH2-related retinopathies).56,57
Near-infrared FAF (NIR-FAF, 787 nm excitation), originating from melanin in the choroid and RPE, is another convenient method to visualize the alterations secondary to STGD1; NIR-FAF has been found to correlate consistently with SW-FAF and the loss of the photoreceptor band on OCT.58,59
Theoretically, NIR-FAF has potential advantages over SW-FAF in the evaluation of STGD 1 progression. Because degenerating photoreceptor cells produce an abnormally higher amount of lipofuscin, SW-FAF signal might be falsely normal in correspondence of atrophic RPE lesions; on the other hand, NIR-FAF provides a better delineation of RPE cell loss and photoreceptor damage compared to shorter wavelength imaging techniques.60 Also, fovea involvement may be better assessed with NIR-FAF (Figure 4). Nevertheless, NIR-FAF has not met the same enthusiasm in the international scientific community in comparison to SW-FAF. One of the drawbacks of NIR-FAF includes the technical difficulty of obtaining gradable images in the absence of adequate pupil dilatation and media transparency. Moreover, interpretation of NIR-FAF is challenging in cases of evident RPE atrophy due to the presence of melanin both within the RPE cells and the underlying choroid.
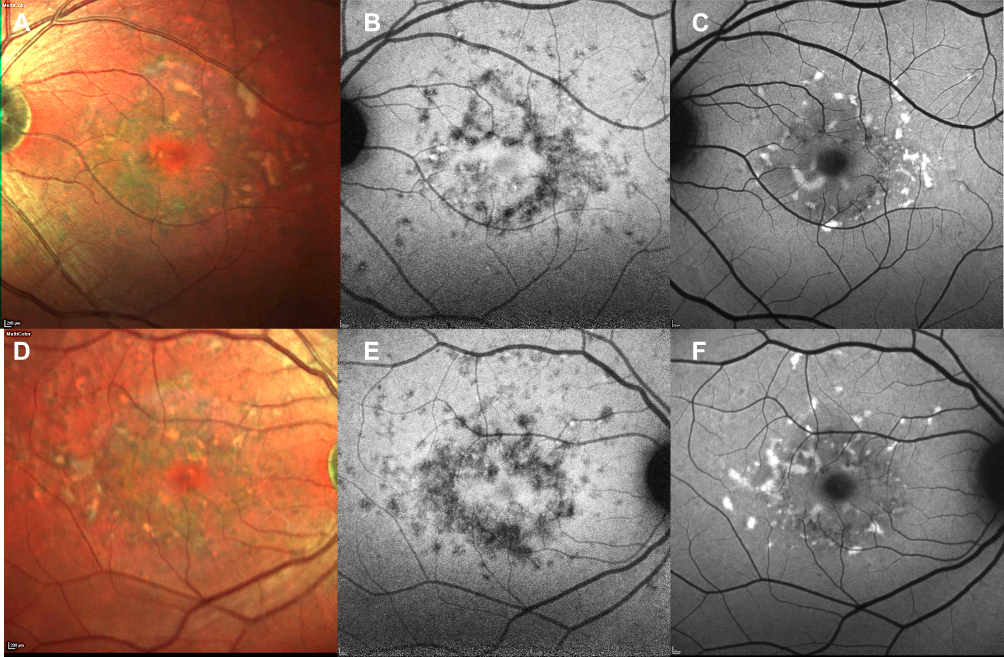 |
Figure 4 Comparison between short wavelength autofluorescence and near-infrared autofluorescence. Preservation of RPE in the foveal region is clearer on multicolor (A, B) and near-infrared autofluorescence (C, D) than on short wavelength autofluorescence (E–F), as the dark signal in the fovea in the latter due to blockage of normal choroidal fluorescence might be misinterpreted as RPE atrophy in the macular region. |
Relatively new automated or semi-automated tools to quantify FAF signal,61 as quantitative autofluorescence (qAF),55,62 and new techniques, as fluorescence lifetime imaging ophthalmoscopy (FLIO),63 have allowed more precise, objective and reliable methods of assessing the course of the disease. In particular, FLIO is a novel non-invasive imaging method detecting anatomic and metabolic changes of the human retina in vivo.64 The analysis of retinal fluorescence lifetimes has been used to characterize and differentiate hyperfluorescent lesions and hypofluorescent atrophic areas in STGD1 and is potentially able to predict disease progression and assess therapeutic effects in upcoming clinical trials.64
Optical Coherence Tomography and Optical Coherence Tomography Angiography
Spectral-domain OCT (SD-OCT) and swept-source OCT (SS-OCT) have led to consistent improvements in spatial and depth resolution in comparison to previous generation time-domain instruments. Both devices allow visualization of the individual layers of the retina and the choroid, en face reconstruction, or simultaneous imaging with infrared, color, or FAF acquisition.
A generalized thinning of the neuroepithelium and diffuse atrophic changes involving the outermost retinal layers (namely, the external limiting membrane (ELM), the ellipsoid zone (EZ) and the EPR) are the hallmarks of the disease. The EZ and the ELM are two distinct hyperreflective layers in the outer retina strongly associated with photoreceptor integrity. The degree of EZ and ELM disruption correlates well with the BCVA65 and the retinal function as measured on multifocal ERG (mfERG) or microperimetry (MP),60,66 consistent with the findings in other macular conditions. Using quantification of the EZ defect on serial SD-OCT imaging as the main predictor of disease progression, a rate of RPE atrophy expansion between 0.2865 and 0.31 mm2/year67 has been calculated. The vectors of atrophy enlargement were not uniform, and EZ changes were detected also in areas of apparently normal FAF signal. In fact, the extent of photoreceptor damage on SD-OCT appeared to exceed area of RPE loss according to different studies.59,68,69
In a limited subgroup of patients, a peculiar macular phenotype has been recognized by means of SD-OCT, referred to as foveal cavitation (FC) (Figure 5).7 FC consists in the focal absence of the outer segments and EZ of the foveal photoreceptors in patients with general preservation of the ELM; this feature is generally stable versus time, but is associated with a punctual profound loss of retinal sensitivity on MP and mfERG.
 |
Figure 5 Foveal cavitation in STGD1. Short wavelength autofluorescence (A) and near-infrared autofluorescence (B) show macular mottling of RPE. In OCT (C), foveal cavitation appears as a hollow subfoveal space due to focal loss of RPE and photoreceptors with backscattering effect. External limiting membrane is preserved. |
Finally, SD-OCT has shown reliability in the diagnosis and follow-up of patients developing macular CNV (Figure 6).70
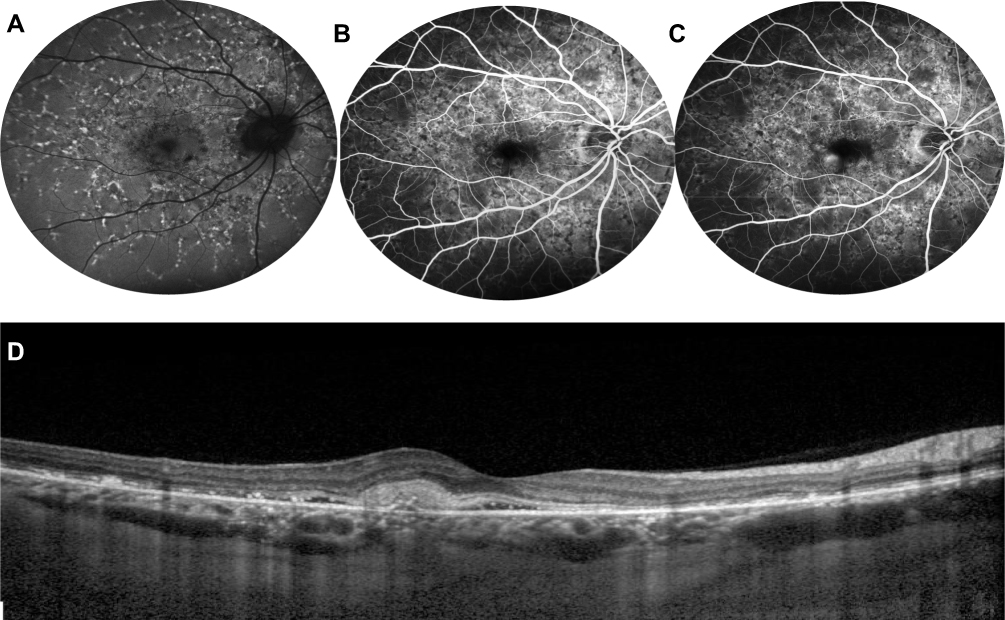 |
Figure 6 Choroidal neovascularization in STGD1. Fundus autofluorescence (A) shows hyperautofluorescent flecks s and irregular macular hypoautofluorescence. Early (B) and late (C) fluorescein angiography frames demonstrate a parafoveal focal dye leakage, corresponding to a type 1 choroidal neovascularization (CNV). OCT scan (D) reveals growth of the CNV above the RPE with mild exudation, along with focal irregularity/atrophy of RPE/ellipsoid zone layers. |
Enhanced depth imaging (EDI) OCT and SS-OCT allowed for deep range imaging of the CC and the choroid in STGD1 patients. Previous studies have found contradictive results with regards to subfoveal choroidal thickness (SFCT) in patients with STGD1 compared with matched normal controls.71–73 The analysis relying on SS-OCT revealed a generalized thinning of the choroid affecting mainly the mall choroidal vessels.74 Similarly, the choroidal vascularity index, calculated using the ratio of luminal area/total choroidal area, was significantly reduced in STGD1 patients as compared with normal eyes, with a significant negative correlation with BCVA.75 Recently, four choroidal patterns have been described, with the following distribution: pattern 1 (normal appearing choroid) (15%), pattern 2 (reduced Sattler or Haller layer) (29%), pattern 3 (reduced Sattler and Haller layers) (26%), pattern 4 (Pattern 3 + choroidal caverns) (30%). These patterns positively correlated with loss of retinal structural integrity on OCT. Furthermore, only advanced patterns (ie, pattern 3 and pattern 4) showed remarkable signs of progression after one-year follow-up.76
OCTA allows to study retinal and choroidal microvasculature without dye injection and has been used to assess the superficial and the deep retinal vascular plexa as well as the CC in several retinal vascular diseases, including STGD1. There is overall agreement that patients with STGD1 show a reduction of the superficial capillary plexus (SCP), the deep capillary plexus (DCP), and the CC compared to healthy eyes (Figure 7).8,9,77 Comparing paired FAF and OCTA examinations, it appeared that RPE damage on FAF was significantly larger than CC loss on OCTA, which might suggest RPE damage precedes CC loss in the natural history of the disease.10 Moreover, OCTA permits an overall differentiation between STGD1 cases and GA secondary to AMD patients on the basis of CC impairment: while STGD1 eyes display a severe loss of CC with a clear demarcation between diseased CC and normal areas, GA revealed rarefied but still present CC within the area of RPE atrophy, with no detectable transition zone.78 Longitudinal studies in GA secondary to AMD have demonstrated a significant correlation between CC impairment and disease progression;79,80 as longitudinal studies involving OCTA in patients with STGD1 disease do not exist yet, the role of this technique in assessing the rate of progression of STGD1 is still vague.
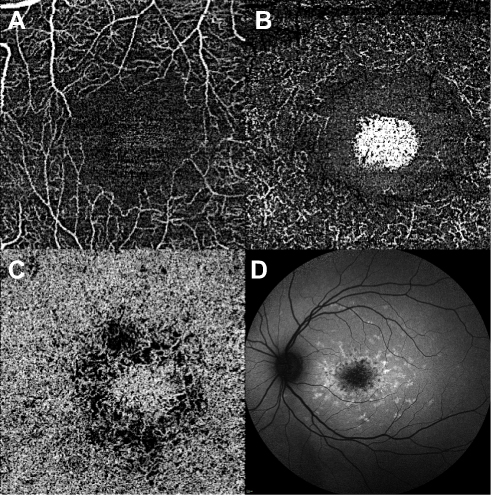 |
Figure 7 Optical coherence tomography angiography of STGD1. OCT angiography (left) shows foveal avascular zone enlargement and reduced vessel density on superficial capillary plexus (A) and deep capillary plexus (B), as well as damaged choriocapillaris (C). (D) Shows the corresponding autofluorescence. |
Microperimetry
MP is a suitable tool to measure the residual function of the macula, the location, and the stability of fixation in STGD1 patients, thanks to the fundus tracking and fixation monitoring features embedded in most of the commercially-available instruments.81,82 These parameters have been used to quantify the decline of visual function in longitudinal studies of patients with STGD. As RPE atrophy progresses into the fovea, STGD1 patients begin to use an eccentric location of the retina to fixate, corresponding to a healthier retinal locus. This area is often characterized by reduced retinal sensitivity and poor fixation stability.
MP has also been used to examine the visual function changes related to the presence of retinal flecks; a significant difference in the mean retinal sensitivity has been found between flecked and “unflecked” areas, with most flecked areas having a decrease in sensitivity.83
Although significant heterogeneity in retinal sensitivity and preferred retinal locus (PRL) of fixation have been recorded depending on the instrument used for the examination,84 retinal sensitivity tends to be inversely correlated to disease duration and directly correlated with BCVA, even with some exceptions.81 Moreover, a linear relationship has been found between fixation location and BCVA (1° farther PRL eccentricity is associated with a 2.3-letter loss of BCVA) and between fixation stability and BCVA. In accordance with previous studies,65 Schönbach et al demonstrated an association between earlier onset of disease symptoms and a longer duration of STGD1 with a more eccentric location of the PRL in a cross-sectional analysis of the ProgStar patients.85
In conclusion, MP equipped with eye-tracking can provide a quantifiable evaluation of the remaining visual function, developing a better understanding of vision changes in STGD1 patients, and may be used as a secondary outcome measure for therapeutic trials in STGD1. These results may be important also in the rehabilitation of patients with central vision loss; biofeedback techniques coupled with MP have shown significant improvements in fixation stability, mean BCVA, mean reading speed, and contrast sensitivity for STGD1 subjects with unstable eccentric fixation.86
Management and Treatment of STGD1
There is lack of consensus regarding the most appropriate management of patients with STGD1, and the actual therapeutic opportunities are scarce. Latest studies have focused on achieving a better understanding of the underlying genetic defects and the pathogenetic mechanisms leading to visual damage in STGD1, rather than proposing guidelines for follow-up and treatment.
Even if there is no approved treatment for STGD1, different therapies are under investigation (Table 2); the most promising are visual cycle modulators, complement inhibitors, gene therapy, and stem-cell therapy.
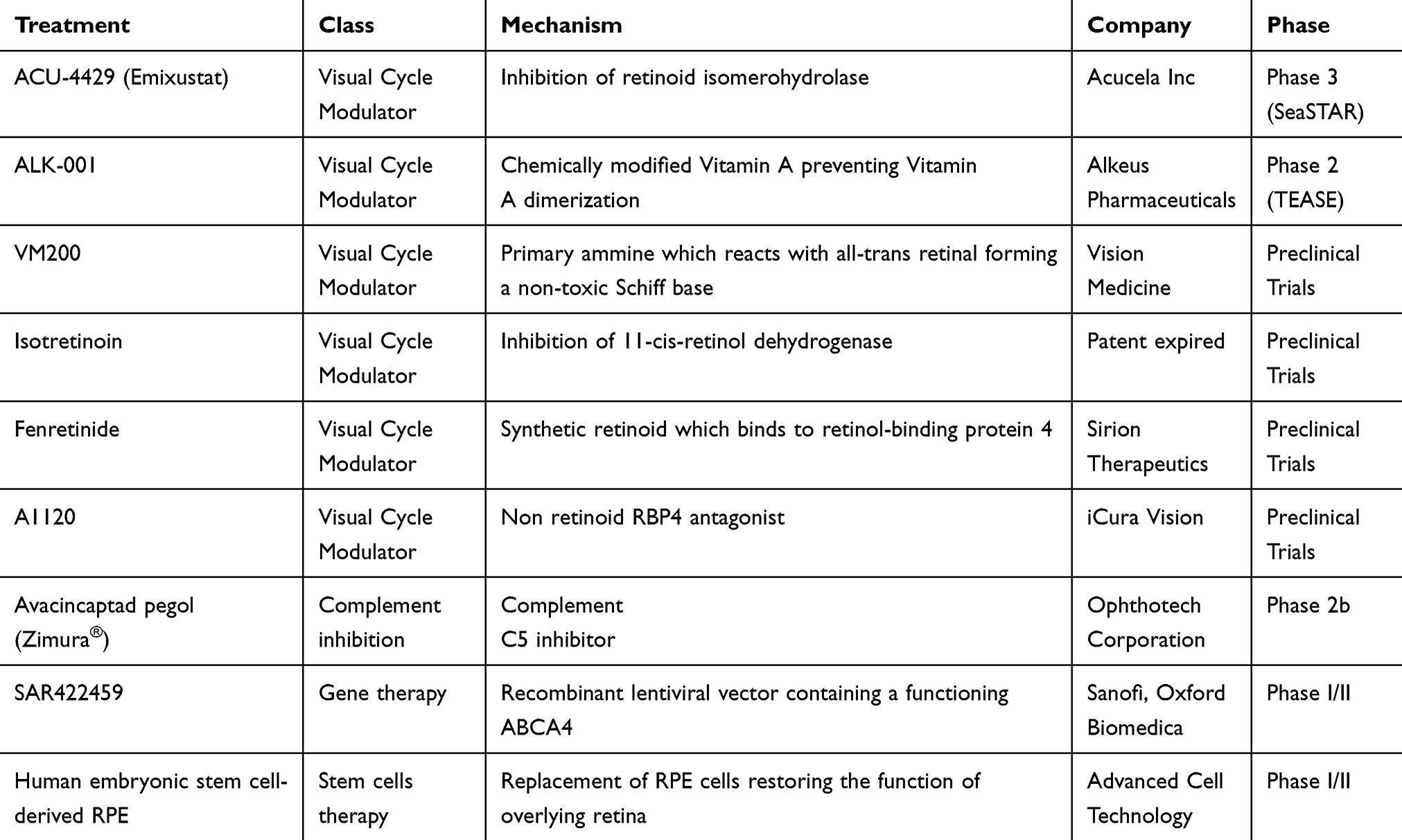 |
Table 2 Treatment Currently Under Investigation for Stargardt Disease |
Visual Cycle Modulators
The aim of Visual Cycle Modulators (VCM) is to prevent the accumulation of toxic Vitamin A dimers at the level of the RPE.87 Emixustat inhibits retinoid isomerohydrolase and decreases the conversion of all-trans-retinyl ester to 11-cis-retinol, with the aim to slow down the formation A2E. The drug has been recently tested in patients with GA secondary to AMD; the drug was overall well-tolerated,88 but failed to exert a therapeutic effect on the rate of progression of GA in the treated arm.89 A multicenter, randomized, double-masked, placebo-controlled study to evaluate the efficacy and safety of emixustat compared to placebo in subjects who have macular atrophy secondary to STGD1 (SeaSTAR) is ongoing.90
ALK-001 is a chemically modified form of vitamin A, in which three hydrogen atoms have been replaced with deuterium atoms (heavy hydrogen). In preclinical studies, ALK-001 reduced the buildup of lipofuscin and the decline in ERG amplitudes.91 It is currently being evaluated in a double-masked, placebo-controlled, clinical trial in Stargardt disease.92
VM200 is a primary ammine which reacts with all-trans retinal and forms a non-toxic Schiff base, preventing the formation of A2E and cellular damage. VM200 has been tested in preclinical trials and showed positive results in murine models. Isotretinoin,93 fenretinide, and other non-retinoids compounds were shown to inhibit the accumulation of lipofuscin in mouse models of recessive STGD1 by inhibiting different enzymatic steps of the visual cycle.94
Complement Inhibition
Inflammation is thought to have a role in the progression of STGD1. A2E and other bisretinoids could activate the complement system in RPE cell, inducing inflammation and promoting RPE and photoreceptors degeneration.95,96 Complement inhibition has demonstrated a protective role in cultured RPE cells against apoptosis and a reduction in the accumulation of lipofuscin in a mouse model of STGD1.97
Avacincaptad pegol (Zimura, Ophthotech Corporation) is an inhibitor of complement component 5 (C5), administered by intravitreal injection. C5 plays a pivotal role in the assemblage of membrane attack complex (MAC). Its inhibition is thought to prevent the formation of the MAC and reduce the rate of photoreceptor and RPE loss in STGD1. A phase 2b trial is ongoing.
Gene Therapy
In the majority of cases, STGD1 is caused by loss-of-function mutations of the gene ABCA4. Gene therapy aims to introduce a functional gene in the retina, supplementing the role of the impaired one in order to restore the normal cycling of 11-cis-retinal and preventing the accumulation of lipofuscin. There are two main ways to administer retinal gene therapy: intravitreal injection or subretinal delivery.98 In the intravitreal injection, the vector is injected in the vitreous cavity and then diffuses to the retina. Subretinal injections are more invasive, requiring pars plana vitrectomy and a small neurosensory retinal detachment, but deliver the vectors directly between RPE cells and photoreceptors. In inherited retinal diseases, adeno-associated virus (AAV) and lentivirus have been used as viral vectors to deliver the intact target genes.99 As the length of the ABCA4 gene (6.8 kb) exceeds the transportation capacity of AAV (4.5–5 kb), lentiviruses have been considered the vector of choice for STGD1 trials. SAR422459 (Oxford Biomedica, Sanofi), formerly known as Stargen, is a recombinant lentiviral vector based on Equine Infectious Anemia Virus containing a functioning ABCA4 gene. It was preclinically tested in mice, macaques, and rabbits, with encouraging results.100,101 A phase I/II clinical trial has started in 2011, but up to date, no preliminary results have been published.102
Stem Cells Transplantation
Stem cells are a promising therapeutic option in STGD1, whose purpose is to replace lost RPE cell in degenerated tissue. Preclinical studies of RPE cells derived from human embryonic stem cell (hESC) showed encouraging results: the subretinal transplantation of these cells in mouse models improved visual function and photoreceptor survival in a dose-dependent way.103 A phase I/IIa clinical trial was conducted to test the safety profile of transplanted RPE cells originated from hESC in patients with STGD1, enrolling 13 patients. The trial met its primary endpoint, demonstrating the tolerability of stem cell therapy. A longer follow-up (5-years) of this study is still uncompleted.
Treatment of Complications
The development of CNV in patients with STGD1, although rare, can lead to rapid and severe vision loss. Laser photocoagulation104 and photodynamic therapy105 have been attempted for the treatment of choroidal neovascular membranes, without encouraging results.
The efficacy of vascular endothelial growth factor (VEGF) inhibition has been demonstrated for neovascularization of other rarer etiologies rather than AMD, including STGD1.70,106 Ranibizumab has shown slowing down CNV progression, apparently without significant visual improvement. However, due to the infrequent rate of CNV development in STGD1, it is difficult to achieve robust evidence for the long-term management of neovascular membranes in these patients.
Conclusion
STGD1 still represents one of the major common causes of inherited childhood and adulthood irreversible visual impairment. Due to its high phenotypic and genotypic heterogeneity, STGD1 is a complex disease to understand. Increasingly improved non-invasive imaging techniques, biochemical and genetic advances have led to substantial forward steps in the monitoring and management of patients with STGD1. Optimization in study design and implementation of interventional trials will be pivotal in reaching the ambitious goal to shed complete light on this disease.
Disclosure
FB is a consultant for: Alcon (Fort Worth,Texas,USA), Alimera Sciences (Alpharetta, Georgia, USA), Allergan Inc (Irvine, California,USA), Farmila-Thea (Clermont-Ferrand, France), Bayer Shering-Pharma (Berlin, Germany), Bausch and Lomb (Rochester, New York, USA), Genentech (San Francisco, California, USA), Hoffmann-La-Roche (Basel, Switzerland), NovagaliPharma (Évry, France), Novartis (Basel, Switzerland), Sanofi-Aventis (Paris, France), Thrombogenics (Heverlee,Belgium), Zeiss (Dublin, USA). He also reports personal fees from Allergan Inc, Bayer, Boehringer Ingelheim, Fidia Sooft, Hofmann La Roche, Novartis, NTC Pharma, SIFI, Thrombogenics, and Zeiss, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101(1):25–30. doi:10.1136/bjophthalmol-2016-308823
2. Lewis RA, Shroyer NF, Singh N, et al. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64(2):422–434. doi:10.1086/302251
3. Yi J, Li S, Jia X, et al. Evaluation of the ELOVL4, PRPH2 and ABCA4 genes in patients with Stargardt macular degeneration. Mol Med Rep. 2012;6(5):1045–1049. doi:10.3892/mmr.2012.1063
4. Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999;117(4):504–510. doi:10.1001/archopht.117.4.504
5. Strauss RW, Ho A, Munoz B, et al. The natural history of the progression of atrophy secondary to stargardt disease (ProgStar) studies: design and baseline characteristics: progStar report no. 1. Ophthalmology. 2016;123(4):817–828. doi:10.1016/j.ophtha.2015.12.009
6. Parodi MB, Iacono P, Triolo G, et al. Morpho-functional correlation of fundus autofluorescence in Stargardt disease. Br J Ophthalmol. 2015;99(10):1354–1359. doi:10.1136/bjophthalmol-2014-306237
7. Parodi MB, Cicinelli MV, Iacono P, Bolognesi G, Bandello F. Multimodal imaging of foveal cavitation in retinal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2017;255(2):271–279. doi:10.1007/s00417-016-3450-7
8. Battaglia Parodi M, Cicinelli MV, Rabiolo A, Pierro L, Bolognesi G, Bandello F. Vascular abnormalities in patients with Stargardt disease assessed with optical coherence tomography angiography. Br J Ophthalmol. 2017;101(6):780–785. doi:10.1136/bjophthalmol-2016-308869
9. Mastropasqua R, Toto L, Borrelli E, et al. Optical coherence tomography angiography findings in Stargardt disease. PLoS One. 2017;12(2):e0170343. doi:10.1371/journal.pone.0170343
10. Guduru A, Lupidi M, Gupta A, Jalali S, Chhablani J. Comparative analysis of autofluorescence and OCT angiography in Stargardt disease. Br J Ophthalmol. 2018;102(9):1204–1207. doi:10.1136/bjophthalmol-2017-311000
11. Hussain RM, Ciulla TA, Berrocal AM, Gregori NZ, Flynn HW
12. Strauss RW, Kong X, Ho A, et al. Progression of stargardt disease as determined by fundus autofluorescence over a 12-month period: progStar report no. 11. JAMA OPHTHALMOL. 2019;137:1134. doi:10.1001/jamaophthalmol.2019.2885
13. Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv Exp Med Biol. 2010;703:105–125.
14. Lenis TL, Hu J, Ng SY, et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci U S A. 2018;115(47):E11120–E11127. doi:10.1073/pnas.1802519115
15. Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ, Sparrow JR. The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci U S A. 2007;104(49):19273–19278. doi:10.1073/pnas.0708714104
16. Radu RA, Yuan Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49(9):3821–3829. doi:10.1167/iovs.07-1470
17. Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243(2):90–100. doi:10.1007/s00417-004-1079-4
18. Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci. 2002;43(6):1980–1985.
19. Cornelis SS, Bax NM, Zernant J, et al. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum Mutat. 2017;38(4):400–408. doi:10.1002/humu.2017.38.issue-4
20. Salles MV, Motta FL, Martin R, et al. Variants in the ABCA4 gene in a Brazilian population with Stargardt disease. Mol Vis. 2018;24:546–559.
21. Riveiro-Alvarez R, Lopez-Martinez MA, Zernant J, et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology. 2013;120(11):2332–2337. doi:10.1016/j.ophtha.2013.04.002
22. Nassisi M, Mohand-Said S, Dhaenens CM, et al. Expanding the mutation spectrum in ABCA4: sixty novel disease causing variants and their associated phenotype in a large French Stargardt cohort. Int J Mol Sci. 2018;19:8. doi:10.3390/ijms19082196
23. Schulz HL, Grassmann F, Kellner U, et al. Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter german cohort-impact of selected deep intronic variants and common SNPs. Invest Ophthalmol Vis Sci. 2017;58(1):394–403. doi:10.1167/iovs.16-19936
24. Di Iorio V, Orrico A, Esposito G, et al. Association between genotype and disease progression in italian stargardt patients: a retrospective natural history study. Retina. 2019;39(7):1399–1409. doi:10.1097/IAE.0000000000002151
25. Garces F, Jiang K, Molday LL, et al. Correlating the expression and functional activity of ABCA4 disease variants with the phenotype of patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2018;59(6):2305–2315. doi:10.1167/iovs.17-23364
26. Burke TR, Fishman GA, Zernant J, et al. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2012;53(8):4458–4467. doi:10.1167/iovs.11-9166
27. Young AI. Solving the missing heritability problem. PLoS Genet. 2019;15(6):e1008222. doi:10.1371/journal.pgen.1008222
28. Bauwens M, Garanto A, Sangermano R, et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet Med. 2019;21(8):1761–1771. doi:10.1038/s41436-018-0420-y
29. Bauwens M, De Zaeytijd J, Weisschuh N, et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum Mutat. 2015;36(1):39–42. doi:10.1002/humu.2015.36.issue-1
30. Zernant J, Lee W, Collison FT, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 2017;54(6):404–412. doi:10.1136/jmedgenet-2017-104540
31. Sangermano R, Garanto A, Khan M, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019;21(8):1751–1760. doi:10.1038/s41436-018-0414-9
32. Zernant J, Lee W, Nagasaki T, et al. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud. 2018;4:4. doi:10.1101/mcs.a002733
33. Braun TA, Mullins RF, Wagner AH, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22(25):5136–5145. doi:10.1093/hmg/ddt367
34. Fadaie Z, Khan M, Del Pozo-Valero M, et al. Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum Mutat. 2019. doi:10.1002/humu.23890
35. September AV, Vorster AA, Ramesar RS, Greenberg LJ. Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2004;45(6):1705–1711. doi:10.1167/iovs.03-1167
36. Aguirre-Lamban J, Riveiro-Alvarez R, Garcia-Hoyos M, et al. Comparison of high-resolution melting analysis with denaturing high-performance liquid chromatography for mutation scanning in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2010;51(5):2615–2619. doi:10.1167/iovs.09-4518
37. Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22(5):395–403. doi:10.1002/humu.v22:5
38. Xiang Q, Cao Y, Xu H, et al. Identification of novel pathogenic ABCA4 variants in a Han Chinese family with Stargardt disease. Biosci Rep. 2019;39:1. doi:10.1042/BSR20180872
39. Zhang X, Ge X, Shi W, et al. Molecular diagnosis of putative Stargardt disease by capture next generation sequencing. PLoS One. 2014;9(4):e95528. doi:10.1371/journal.pone.0095528
40. Xin W, Xiao X, Li S, Jia X, Guo X, Zhang Q. Identification of genetic defects in 33 probands with Stargardt disease by WES-based bioinformatics gene panel analysis. PLoS One. 2015;10(7):e0132635. doi:10.1371/journal.pone.0132635
41. Zernant J, Xie YA, Ayuso C, et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum Mol Genet. 2014;23(25):6797–6806. doi:10.1093/hmg/ddu396
42. Burke TR, Tsang SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 2011;32(3):165–174. doi:10.3109/13816810.2011.565397
43. Fishman GA. Fundus flavimaculatus. A clinical classification. Arch Ophthalmol. 1976;94(12):2061–2067. doi:10.1001/archopht.1976.03910040721003
44. Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359–369. doi:10.1001/archopht.119.3.359
45. Fujinami K, Lois N, Mukherjee R, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. 2013;54(13):8181–8190. doi:10.1167/iovs.13-12104
46. Fujinami K, Zernant J, Chana RK, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122(2):326–334. doi:10.1016/j.ophtha.2014.08.012
47. Collison FT, Fishman GA. Visual acuity in patients with Stargardt disease after age 40. Retina. 2018;38(12):2387–2394. doi:10.1097/IAE.0000000000001903
48. Rotenstreich Y, Fishman GA, Anderson RJ. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology. 2003;110(6):1151–1158. doi:10.1016/S0161-6420(03)00333-6
49. Kong X, Strauss RW, Cideciyan AV, et al. Visual acuity change over 12 months in the prospective progression of atrophy secondary to stargardt disease (ProgStar) study: progStar report number 6. Ophthalmology. 2017;124(11):1640–1651. doi:10.1016/j.ophtha.2017.04.026
50. Kong X, Fujinami K, Strauss RW, et al. Visual acuity change over 24 months and its association with foveal phenotype and genotype in individuals with Stargardt disease: progStar study report no. 10. JAMA Ophthalmol. 2018;136(8):920–928. doi:10.1001/jamaophthalmol.2018.2198
51. Jayasundera T, Rhoades W, Branham K, Niziol LM, Musch DC, Heckenlively JR. Peripapillary dark choroid ring as a helpful diagnostic sign in advanced stargardt disease. Am J Ophthalmol. 2010;149(4):656–660 e652. doi:10.1016/j.ajo.2009.11.005
52. Giani A, Pellegrini M, Carini E, Peroglio Deiro A, Bottoni F, Staurenghi G. The dark atrophy with indocyanine green angiography in Stargardt disease. Invest Ophthalmol Vis Sci. 2012;53(7):3999–4004. doi:10.1167/iovs.11-9258
53. Ervin AM, Strauss RW, Ahmed MI, et al. A workshop on measuring the progression of atrophy secondary to Stargardt disease in the ProgStar studies: findings and lessons learned. Transl Vis Sci Technol. 2019;8(2):16. doi:10.1167/tvst.8.2.16
54. Strauss RW, Munoz B, Ho A, et al. Progression of Stargardt disease as determined by fundus autofluorescence in the retrospective progression of Stargardt disease study (ProgStar report no. 9). JAMA Ophthalmol. 2017;135(11):1232–1241. doi:10.1001/jamaophthalmol.2017.4152
55. Burke TR, Duncker T, Woods RL, et al. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2014;55(5):2841–2852. doi:10.1167/iovs.13-13624
56. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. 2005;46(12):4739–4746. doi:10.1167/iovs.05-0805
57. Burke TR, Rhee DW, Smith RT, et al. Quantification of peripapillary sparing and macular involvement in Stargardt disease (STGD1). Invest Ophthalmol Vis Sci. 2011;52(11):8006–8015. doi:10.1167/iovs.11-7693
58. Greenstein VC, Schuman AD, Lee W, et al. Near-infrared autofluorescence: its relationship to short-wavelength autofluorescence and optical coherence tomography in recessive stargardt disease. Invest Ophthalmol Vis Sci. 2015;56(5):3226–3234. doi:10.1167/iovs.14-16050
59. Duncker T, Marsiglia M, Lee W, et al. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Invest Ophthalmol Vis Sci. 2014;55(12):8134–8143. doi:10.1167/iovs.14-14848
60. Gomes NL, Greenstein VC, Carlson JN, et al. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009;50(8):3953–3959. doi:10.1167/iovs.08-2657
61. Kuehlewein L, Hariri AH, Ho A, et al. Comparison of manual and semiautomated fundus autofluorescence analysis of macular atrophy in Stargardt disease phenotype. Retina. 2016;36(6):1216–1221. doi:10.1097/IAE.0000000000000870
62. Sparrow JR, Duncker T, Schuerch K, Paavo M, de Carvalho JRL
63. Sauer L, Andersen KM, Li B, Gensure RH, Hammer M, Bernstein PS. Fluorescence Lifetime Imaging Ophthalmoscopy (FLIO) of macular pigment. Invest Ophthalmol Vis Sci. 2018;59(7):3094–3103. doi:10.1167/iovs.18-23886
64. Dysli C, Wolf S, Hatz K, Zinkernagel MS. Fluorescence lifetime imaging in Stargardt disease: potential marker for disease progression. Invest Ophthalmol Vis Sci. 2016;57(3):832–841. doi:10.1167/iovs.15-18033
65. Testa F, Rossi S, Sodi A, et al. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: implications for gene therapy. Invest Ophthalmol Vis Sci. 2012;53(8):4409–4415. doi:10.1167/iovs.11-8201
66. Berisha F, Feke GT, Aliyeva S, Hirai K, Pfeiffer N, Hirose T. Evaluation of macular abnormalities in Stargardt’s disease using optical coherence tomography and scanning laser ophthalmoscope microperimetry. Graefes Arch Clin Exp Ophthalmol. 2009;247(3):303–309. doi:10.1007/s00417-008-0963-8
67. Cai CX, Light JG, Handa JT. Quantifying the rate of ellipsoid zone loss in Stargardt disease. Am J Ophthalmol. 2018;186:1–9. doi:10.1016/j.ajo.2017.10.032
68. Sodi A, Mucciolo DP, Cipollini F, et al. En face OCT in Stargardt disease. Graefes Arch Clin Exp Ophthalmol. 2016;254(9):1669–1679. doi:10.1007/s00417-015-3254-1
69. Tanna P, Georgiou M, Strauss RW, et al. Cross-sectional and longitudinal assessment of the ellipsoid zone in childhood-onset Stargardt disease. Transl Vis Sci Technol. 2019;8(2):1. doi:10.1167/tvst.8.2.1
70. Battaglia Parodi M, Munk MR, Iacono P, Bandello F. Ranibizumab for subfoveal choroidal neovascularisation associated with Stargardt disease. Br J Ophthalmol. 2015;99(9):1268–1270. doi:10.1136/bjophthalmol-2014-305783
71. Vural E, Acar U, Sevinc MK, et al. Choroidal thickness in patients with Stargardt disease. Retina. 2018;38(3):614–619. doi:10.1097/IAE.0000000000001557
72. Adhi M, Read SP, Ferrara D, Weber M, Duker JS, Waheed NK. Morphology and vascular layers of the choroid in Stargardt disease analyzed using spectral-domain optical coherence tomography. Am J Ophthalmol. 2015;160(6):1276–1284 e1271. doi:10.1016/j.ajo.2015.08.025
73. Nunes RP, Rosa PR, Giani A, et al. Choroidal thickness in eyes with central geographic atrophy secondary to Stargardt disease and age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina. 2015;46(8):814–822.
74. Ratra D, Jaishankar D, Sachidanandam R, Yusufali H, Ratra V. Swept-source optical coherence tomography study of choroidal morphology in Stargardt disease. Oman J Ophthalmol. 2018;11(2):150–157. doi:10.4103/ojo.OJO_21_2017
75. Ratra D, Tan R, Jaishankar D, et al. Choroidal structural changes and vascularity index in Stargardt disease on swept source optical coherence tomography. Retina. 2018;38(12):2395–2400. doi:10.1097/IAE.0000000000001879
76. Arrigo A, Grazioli A, Romano F, et al. Choroidal patterns in Stargardt disease: correlations with visual acuity and disease progression. J Clin Med. 2019;8(9). doi:10.3390/jcm8091388.
77. Muller PL, Pfau M, Moller PT, et al. Choroidal flow signal in late-onset stargardt disease and age-related macular degeneration: an OCT-angiography study. Invest Ophthalmol Vis Sci. 2018;59(4):AMD122–AMD131. doi:10.1167/iovs.18-23819
78. Pellegrini M, Acquistapace A, Oldani M, et al. Dark atrophy: an optical coherence tomography angiography study. Ophthalmology. 2016;123(9):1879–1886. doi:10.1016/j.ophtha.2016.05.041
79. Nassisi M, Baghdasaryan E, Borrelli E, Ip M, Sadda SR. Choriocapillaris flow impairment surrounding geographic atrophy correlates with disease progression. PLoS One. 2019;14(2):e0212563. doi:10.1371/journal.pone.0212563
80. Thulliez M, Zhang Q, Shi Y, et al. Correlations between choriocapillaris flow deficits around geographic atrophy and enlargement rates based on swept-source OCT imaging. Ophthalmol Retina. 2019;3(6):478–488. doi:10.1016/j.oret.2019.01.024
81. Schonbach EM, Wolfson Y, Strauss RW, et al. macular sensitivity measured with microperimetry in Stargardt disease in the progression of atrophy secondary to Stargardt disease (ProgStar) study: report no. 7. JAMA Ophthalmol. 2017;135(7):696–703. doi:10.1001/jamaophthalmol.2017.1162
82. Cideciyan AV, Swider M, Aleman TS, et al. Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest Ophthalmol Vis Sci. 2012;53(2):841–852. doi:10.1167/iovs.11-8415
83. Verdina T, Tsang SH, Greenstein VC, et al. Functional analysis of retinal flecks in Stargardt disease. J Clin Exp Ophthalmol. 2012;3.
84. Parodi MB, Triolo G, Morales M, et al. Mp1 and maia fundus perimetry in healthy subjects and patients affected by retinal dystrophies. Retina. 2015;35(8):1662–1669. doi:10.1097/IAE.0000000000000504
85. Schonbach EM, Ibrahim MA, Strauss RW, et al. Fixation location and stability using the MP-1 microperimeter in Stargardt disease: progStar report no. 3. Ophthalmol Retina. 2017;1(1):68–76. doi:10.1016/j.oret.2016.08.009
86. Verdina T, Giacomelli G, Sodi A, et al. Biofeedback rehabilitation of eccentric fixation in patients with Stargardt disease. Eur J Ophthalmol. 2013;23(5):723–731. doi:10.5301/ejo.5000291
87. Hussain RM, Gregori NZ, Ciulla TA, Lam BL. Pharmacotherapy of retinal disease with visual cycle modulators. Expert Opin Pharmacother. 2018;19(5):471–481. doi:10.1080/14656566.2018.1448060
88. Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina. 2014;34(3):603–609. doi:10.1097/01.iae.0000434565.80060.f8
89. Rosenfeld PJ, Dugel PU, Holz FG, et al. Emixustat hydrochloride for geographic atrophy secondary to age-related macular degeneration: a randomized clinical trial. Ophthalmology. 2018;125(10):1556–1567. doi:10.1016/j.ophtha.2018.03.059
90. Safety and efficacy of emixustat in Stargardt disease (SeaSTAR); 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT03772665.
91. Ma L, Kaufman Y, Zhang J, Washington I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem. 2011;286(10):7966–7974. doi:10.1074/jbc.M110.178657
92. Phase 2 tolerability and effects of ALK-001 on Stargardt disease (TEASE). Available from: https://clinicaltrials.gov/ct2/show/NCT02402660.
93. Radu RA, Mata NL, Nusinowitz S, Liu X, PA S, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A. 2003;100(8):4742–4747. doi:10.1073/pnas.0737855100
94. Dobri N, Qin Q, Kong J, et al. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci. 2013;54(1):85–95. doi:10.1167/iovs.12-10050
95. Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50(3):1392–1399. doi:10.1167/iovs.08-2868
96. Radu RA, Hu J, Yuan Q, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286(21):18593–18601. doi:10.1074/jbc.M110.191866
97. Lenis TL, Sarfare S, Jiang Z, Lloyd MB, Bok D, Radu RA. Complement modulation in the retinal pigment epithelium rescues photoreceptor degeneration in a mouse model of Stargardt disease. Proc Natl Acad Sci U S A. 2017;114(15):3987–3992. doi:10.1073/pnas.1620299114
98. Han Z, Conley SM, Naash MI. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv Exp Med Biol. 2014;801:719–724.
99. Auricchio A, Trapani I, Allikmets R. Gene therapy of ABCA4-associated diseases. Cold Spring Harb Perspect Med. 2015;5(5):a017301. doi:10.1101/cshperspect.a017301
100. Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311–1320. doi:10.1038/gt.2008.78
101. Binley K, Widdowson P, Loader J, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci. 2013;54(6):4061–4071. doi:10.1167/iovs.13-11871
102. Phase I/II Study of SAR422459 in patients with Stargardt’s macular degeneration. Available from: https://clinicaltrials.gov/ct2/show/NCT01367444.
103. Lund RD, Wang S, Klimanskaya I, et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 2006;8(3):189–199. doi:10.1089/clo.2006.8.189
104. Klein R, Lewis RA, Meyers SM, Myers FL. Subretinal neovascularization associated with fundus flavimaculatus. Arch Ophthalmol. 1978;96(11):2054–2057. doi:10.1001/archopht.1978.03910060442009
105. Valmaggia C, Niederberger H, Helbig H. Photodynamic therapy for choroidal neovascularization in fundus flavimaculatus. Retina. 2002;22(1):111–113. doi:10.1097/00006982-200202000-00022
106. Staurenghi G, Lai TYY, Mitchell P, et al. Efficacy and safety of ranibizumab 0.5 mg for the treatment of macular edema resulting from uncommon causes: twelve-month findings from PROMETHEUS. Ophthalmology. 2018;125(6):850–862. doi:10.1016/j.ophtha.2017.12.002
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.