Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Molecular variants and mutations in medulloblastoma
Authors Schroeder K, Gururangan S
Received 4 October 2013
Accepted for publication 14 November 2013
Published 4 February 2014 Volume 2014:7 Pages 43—51
DOI https://doi.org/10.2147/PGPM.S38698
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Kristin Schroeder, Sri Gururangan
Pediatric Clinical Services, Preston Robert Tisch Brain Tumor Center at Duke, Duke University Medical Center, Durham, NC, USA
Abstract: Medulloblastoma is the commonest malignant brain tumor in children. Treatment with surgery, irradiation, and chemotherapy has improved outcomes in recent years, but patients are frequently left with devastating neurocognitive and other sequelae following such therapy. While the prognosis has traditionally been based on conventional histopathology and clinical staging (based on age, extent of resection, and presence or absence of metastasis), it has become apparent in recent years that the inherent biology of the tumor plays a significant part in predicting survival and sometimes supersedes clinical or pathologic risk factors. The advent of deep sequencing gene technology has provided invaluable clues to the molecular makeup of this tumor and allowed neuro-oncologists to understand that medulloblastoma is an amalgamation of several distinct disease entities with unique clinical associations and behavior. This review is a concise summary of the pathology, genetic syndromes, recent advances in molecular subgrouping, and the associated gene mutations and copy number variations in medulloblastoma. The association of molecular alterations with patient prognosis is also discussed, but it should be remembered that further validation is required in prospective clinical trials utilizing uniform treatment approaches.
Keywords: medulloblastoma, children, adults, molecular subgroups, mutations
Introduction
Brain tumors are the most frequent solid tumors in children, and the leading cause of childhood cancer death.1,2 Medulloblastoma is the most common malignant brain tumor, comprising over 20% of all new central nervous system tumors.1 The last 30 years have seen significant improvements in overall survival, at around 80% for patients with average risk (children >3 years of age, less than 1.5 cm2 of residual disease in the primary site, and absence of metastatic disease) and 60%–70% for high-risk patients (those with bulky residual disease, brain stem invasion, and neuraxis metastasis) with the use of standard chemotherapy and radiotherapy (craniospinal irradiation and focal boost).3 Infants (≤3 years of age) with medulloblastoma and those with recurrent disease following standard cytotoxic therapy continue to fare poorly.4,5 In general, it has been difficult to accurately predict outcome using standard clinical risk stratification guidelines.3,6 In recent years, there has been a steady improvement in our understanding of the pathologic, genetic, and molecular heterogeneity of this tumor, that has led not only to better classification of prognostic risk but also to specific leads towards improved targeted therapies for children with this malignancy and perhaps better treatment selection in the future.7,8
Epidemiology
The incidence of medulloblastoma is 0.71/100,000 children years, with an estimated 400–500 cases annually in the US.9 Medulloblastoma can occur in infancy through adulthood, with a peak incidence at 3–4 years and 5–9 years.9 This disease is rare past 50 years of age. No specific environmental risk factors have been reported to predispose to this tumor.10 While certain genetic syndromes can predispose the individual to the development of medulloblastoma,11 less than 7% of patients with this tumor have an identified germ line genetic mutation.12 Familial occurrence of medulloblastoma has been rarely reported.10,11
Pathology
The recent World Health Organization classification of central nervous system tumors includes the following histologic types of medulloblastoma; classic medulloblastoma, desmoplastic/nodular medulloblastoma, medulloblastoma with excessive nodularity, anaplastic medulloblastoma, and large cell medulloblastoma.13 Table 1 lists the distribution of the main histologic subtypes of medulloblastoma, with a brief histologic description of each type and prognosis.14 Nearly 90% of cases are either classic or desmoplastic. The distribution of histologic subtypes is dependent on the patient’s age, with classic histology predominating in children between 3 and 16 years of age (71%) followed by large cell/anaplastic in 17%, and nodular/desmoplastic tumors in 5%.14 A paucinodular variant without desmoplasia is also observed in this age group.14 However, in striking contrast, 57% of infants with medulloblastoma have nodular/desmoplastic histology (including desmoplastic medulloblastoma with excessive nodularity [DMEN] in 3%). Patients with large cell (2%–4%) and anaplastic (10%–22%) variants are often grouped together as large cell/anaplastic in studies, as they are a histologic continuum with all large cell variants having some anaplasia.14 These are associated with a poor prognosis. In comparison, tumors of the nodular/desmoplastic and DMEN types in infants are associated with an excellent outcome.5,15,16 However, the same histologic type in older children and adults does not have a better prognosis as compared to those with classic medulloblastoma.3,17
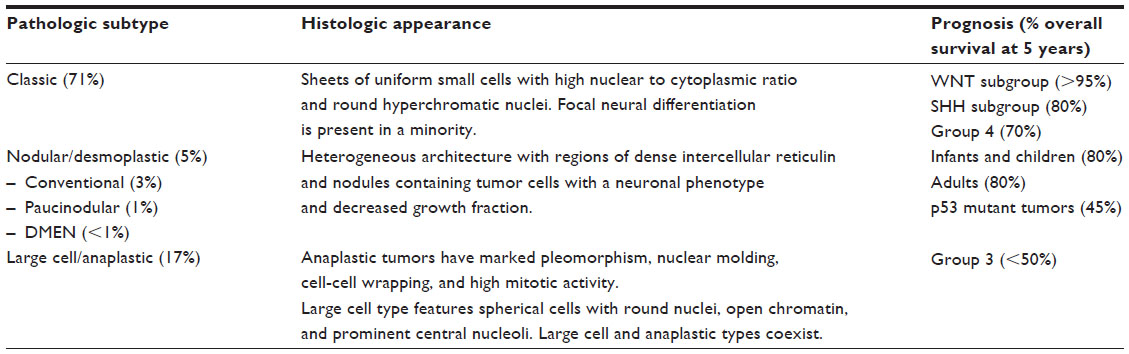 | Table 1 Pathologic subtypes of medulloblastoma |
Genetic syndromes associated with medulloblastoma
Genetic syndromes typically associated with medulloblastoma are distinctly rare (less than 7% of cases) but the specific mutations and aberrant signaling pathways that lead to the phenotypic features of these syndromes have in turn provided clues to the possible origins of this aggressive tumor.
Gorlin syndrome
This autosomal dominant genetic disorder is characterized by odontogenic keratocysts, hyperkeratosis of the palms and soles, skeletal abnormalities, intracranial ectopic calcifications, facial dysmorphism, intellectual disability, and a predilection for tumor formation, including multiple basal cell carcinomas, medulloblastomas, rhabdomyosarcomas, and ovarian fibromas.18 Medulloblastoma occurs in 2%–5% of patients with this syndrome, and usually within the first 2 years of life.18 Gorlin syndrome is caused by a germ line mutation in the PTCH1 gene that is present on chromosome 9q22.32.19 Medulloblastoma that occurs in Gorlin syndrome is typically of the nodular/desmoplastic type, hemispheric in location, and arises as a direct consequence of the PTCH mutation and activation of the Sonic Hedgehog (SHH) pathway in all cases. In contrast, SHH pathway aberrations (PTCH1, SUFU, SMO) are found in only about 15% of patients with sporadic medulloblastoma.20
Turcot syndrome
Two major inherited syndromes have been recognized in the context of occurrence of colorectal neoplasia and primary brain tumors, ie, familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer.11,21 In familial adenomatous polyposis, an autosomal dominant disorder, patients develop several thousand adenomatous colorectal polyps along with several extracolonic manifestations, including hypertrophic retinal epithelium, desmoid tumors, epidermal cysts, osteomas, hepatoblastomas, and papillary cancer of the thyroid.21 Medulloblastoma is the central nervous system tumor most associated with familial adenomatous polyposis, although astrocytoma and ependymomas have been described in kindred populations.21 On the other hand, glioblastoma is the classic central nervous system tumor associated with hereditary nonpolyposis colorectal carcinoma, and is due to mutations in the mismatch repair genes (MSH2, MSH6, PMS-1, and PMS-2).22
Li–Fraumeni syndrome
Li–Fraumeni syndrome is an autosomal dominant disorder characterized by the familial occurrence of a spectrum of tumors, including sarcomas, brain tumors, breast cancer, adrenocortical carcinoma, osteosarcoma, and leukemia.11 About 70% of patients carry a germ line p53 mutation.23 While medulloblastoma rarely occurs in p53–/– mice, homozygous loss of p53 accelerates the rate and frequency of occurrence of this tumor in PTCH+/– or Myc expressing mouse models.24 The role of germ line p53 mutation in the induction or prognosis of human medulloblastoma has been controversial over the years,23 but two recent studies have attempted to address this issue.25,26 Rausch et al performed whole genome sequencing initially on patients with Li–Fraumeni syndrome and SHH medulloblastoma and demonstrated complex intrachromosomal and interchromosomal rearrangements that were representative of a phenomenon called chromothripsis, resulting from a single catastrophic chromosomal event and subsequent defective DNA repair, all due to the inherent germ line p53 mutation.25 Furthermore, amplifications of selective SHH genes (eg, MycN, GLI2, BOC, and IGFR1) were found in the same tumor, accounting for deregulation of the SHH pathway and tumor formation. In a subsequent international study, Zhukova et al reported p53 mutations to be enriched in patients aged 8–14 years and with SHH medulloblastoma (over 50% of these patients also harbored germ line mutations) and to have a significantly worse prognosis.26 The germ line p53 mutation also puts these patients at risk for excessive toxicity from DNA-damaging agents, including chemotherapy and radiotherapy.
Molecular subgroups, mutations, and somatic copy number alterations
With the advent of technology to study the whole genome using gene arrays and whole exome sequencing, our understanding of the molecular underpinnings of medulloblastoma has been revolutionized but is yet to replace traditional clinical and pathologic markers of prognosis in this disease. It has also revealed the incredible genetic complexity of this tumor at the time of clinical presentation.27 Gene expression profiling has demonstrated that medulloblastoma is not a single entity but possibly a group of diseases that was originally categorized by Kool et al into five subtypes28,29 that has been subsequently resolved by international consensus into four nonoverlapping molecular groups;7,30,31 wingless (WNT), SHH, and two additional groups called group 3 and 4 because the specific signaling pathways leading to these two variants are yet to be conclusively determined (Table 2). Medulloblastoma is also characterized by a distinct set of novel recurrent mutations, averaging about 10–12 single nucleotide variants per medulloblastoma genome (as compared with >100 in adult solid tumors).32,33 However, the rate of occurrence of single nucleotide variants in medulloblastoma increases with age.27,33 The isodicentric chromosome 17q [idic(17)(p11.2)] is the most frequent chromosomal abnormality found in medulloblastoma (20%–50% of tumors). This mutational event might be responsible for initiation of a tumor and has been shown in some studies to be an independent adverse prognostic indicator.34–36
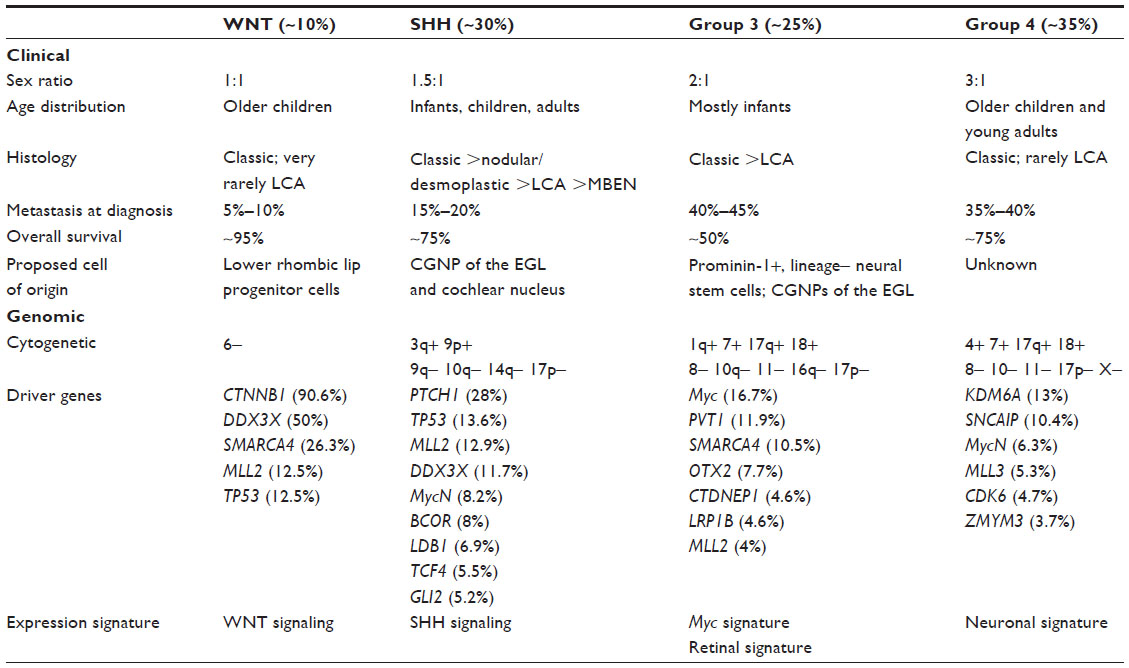 | Table 2 Clinical and molecular characteristics of subgroups of medulloblastoma |
Refinements in DNA technology, including next-generation sequencing, have revealed that each medulloblastoma subgroup is associated with specific somatic copy number aberrations that result in characteristic focal gene amplifications (oncogenic drivers) or deletions (resulting in possible loss of tumor suppressor genes) that cause deregulation of key signaling receptor tyrosine kinase pathways, histone modifiers (histone methyl transferases, methylases, acetylases, and deacetylases), and chromatin-associated genes.27 These new discoveries have dramatically shifted the understanding of this disease at the molecular level and simultaneously revealed a level of complexity that is baffling for clinicians who routinely treat patients with this malignant brain tumor.
WNT medulloblastoma
The WNT subgroup is found in 15% of cases and is associated with excellent survival (>90%) as compared with the other three groups.3,7,8 WNT pathway tumors are more commonly seen in children aged older than 3 years, with an equal sex distribution (as compared with an overall male predominance in medulloblastoma), and being rarely metastatic at diagnosis.7,27 While the tumors in this subgroup are usually of the classic histology, anaplasia is occasionally observed but does not seem to affect prognosis.8,37 WNT tumors can be reliably identified by immunohistochemical expression of DKK1, filamin-A, YAP-1, and beta-catenin (nuclear expression +/− cytoplasmic) in the tumor.8,38 Over 75% of the tumors in this subtype have a point mutation on exon 3 (CTNNB1) of the beta-catenin gene, allowing for overexpression and nuclear localization of β-catenin. The genome of the WNT tumors is relatively stable compared with that of the other groups. Copy number aberration and single nucleotide variants in this molecular variant include deletion of one copy of chromosome 6 (monosomy 6, almost all cases), mutations in the DEAD-box RNA helicase gene (DDX3X, 50%) which enhances cell proliferation by increasing the transactivating capacity of beta-catenin, SMARCA4 (26%), TP53 mutations (16%, not associated with a corresponding germ line mutation and does not affect the excellent prognosis associated with this subgroup), and MLL2 mutation (12%).27,39 Tetraploidy is present in about 14% of cases and might be an early event in tumorigenesis.40 Interestingly, a recent study sought to evaluate the cellular origins of WNT signature medulloblastoma and developed a mouse model expressing a conditional stabilized allele of CTNNB1 in progenitor cells of the lower rhombic lip of the developing mouse brain. In the context of TP53 mutation and a cell-specific promoter (brain lipid-binding protein (Blbp)-Cre; Ctnnb1+/lox(Ex3); Trp53flx/flx), tumors with a classic histology occurred following a relatively long latency and low penetrance (about 15%) only from the dorsal brainstem in contrast with the cerebellar origin of the SHH medulloblastoma.41 The addition of a mutant phosphoinositol-3-kinase allele (Pik3caE545K) to this transgenic model accelerated tumor occurrence with 100% penetrance.42 In addition to providing a better understanding of the cell of origin of human WNT tumors, this mouse model can now be used to test novel agents for the treatment of this subtype in humans. Further work is also necessary to understand the reasons why patients with this tumor subtype fare significantly better than other groups. Prospective clinical trials are evaluating reductions in cytotoxic therapy for patients with WNT medulloblastoma to minimize long-term organ damage while still maintaining excellent survival.
SHH medulloblastoma
The SHH subgroup of medulloblastoma is found in 25% of patients in a bimodal age distribution that includes infants (0–3 years) and young adults (>16 years), but occurs much less frequently in children aged 4–15 years.8,27 The sexes are equally affected. The pathology of this molecular subtype is typically nodular/desmoplastic, although over half of this subgroup can include tumors with classic and rarely anaplastic features.8 SHH medulloblastomas can be identified readily using tumor immunohistochemistry expression for GAB1, SFRP, and GLI1 proteins.8,38
This tumor subtype is initiated by either amplification or deletions of genes that are components of the SHH pathway (including PTCH1/2 [somatic or germ line mutation], SUFU [somatic or germ line mutation], SMO [somatic activating mutation], and GLI2 [amplification]) and other receptor tyrosine kinase signaling pathways.43–45 Germ line mutations in PATCH-1 (resulting in Gorlin’s syndrome) and SUFU have been linked to infant desmoplastic medulloblastoma.11 SHH medulloblastoma is also the subtype in which TP53 mutations are most enriched (21% of tumor samples) with more than half of the patients with SHH/p53 tumors having a germ line mutation.26 Copy number aberrations include amplifications of protein phosphatase ID (PPM1D, chromosome 17q23.2), IGF1R, IRS2, PIK3C2G, PIK3C2B, and YAP-1 along with deletion of PTEN on chromosome 10q23.31 and mutations in DDX3X (11%). 27 Tetraploidy is present in about 29% of samples and is associated with p53 mutations and chromothripsis.40
SHH medulloblastoma is a fairly heterogeneous tumor in terms of clinical and biologic features. Using unsupervised hierarchical clustering of a cohort of 33 SHH tumor samples, Northcott et al have recently shown that adult and pediatric tumors segregate into three fairly equal clusters, with the infants and children (groups 1 and 3) clearly separating from adults (group 2).46 Further analysis using clinical and molecular markers indicates that the pediatric and adult SHH tumors are clinically and molecularly distinct entities (Table 3). Much information regarding the genesis of medulloblastoma and its origin from cerebellar granular neuron precursors of the external granule layer has been derived from mouse models created using components of the SHH pathway, including patch+/– mice and transgenic mice that overexpress activated smoothened (SMO) under the control of a tissue specific promoter (neurogenic differentiation 2 [Neurod2-Smoa1]) and develop cerebellar tumors at variable latencies.47
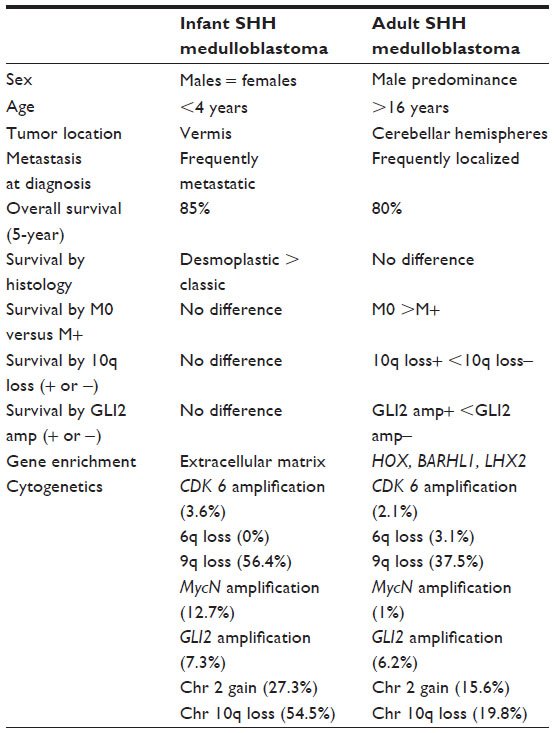 | Table 3 Clinical and biologic characteristics of infant and adult SHH medulloblastoma |
Patients with SHH medulloblastoma have an intermediate prognosis, with overall survival rates of 60%–80%.8,46 Infants and young children with SHH medulloblastoma fare better than adults with the disease.46 In addition, patients with SHH/p53 tumors have an inferior prognosis as compared with those having wild-type p53 tumors.26
Inhibitors of the SHH pathway, particularly at the level of SMO, have been developed and are being tested in the clinic. GDC-0449 (vismodegib, Genentech Corporation, San Francisco, CA, USA), an SMO inhibitor, has undergone Phase I testing in children and a Phase II study in adults with recurrent medulloblastoma, with encouraging albeit short-lived responses and acceptable toxicities.48,49 It is becoming apparent that these tumors exhibit de novo resistance to SMO antagonists due to lack of either PTCH1 or SMO mutations or rapidly develop resistance to this inhibitor due to either point mutation in the SMO gene that prevents drug binding or overactivity of the phosphoinositol-3-kinase pathway due to PTEN loss and are therefore unlikely to respond in a sustained manner to single-agent therapy.50–52 Future strategies that combine drugs targeting multiple pathways (eg, combining GDC-0449 with a phosphoinositol-3-kinase or histone deacetylase inhibitor) are more likely to provide benefit.
Group 3 medulloblastoma
Group 3 (also known as group C) medulloblastoma is found in up to 25% of cases, is more common in males (2:1), restricted to pediatric patients, and has a high incidence of metastatic disease at diagnosis.8,27 Medulloblastoma in this molecular variant is frequently of the large cell/anaplastic histology. While immunohistochemical expression of natriuretic peptide receptor 3 (NPR3) antigen has been reported to confirm this subgroup, others have questioned the validity of this marker and it should not be used for clinical purposes until further validation in prospective cohorts of newly diagnosed patients.8,34,36 There are no known germ line mutations associated with this group. However, focal high-level amplification of Myc and OTX-2 proto-oncogenes are observed in approximately 12%–16% and 7% of these tumors, respectively, and most have aberrant Myc expression.7,27 The genome in group 3 medulloblastoma is highly unstable, and frequent gains of 1q, 7, and 17q (i17q) are observed along with 10q, 11, 16q, and 17p deletions.27 Tetraploidy is seen in 54% of group 3 tumors and probably occurs as an early event in tumorigenesis.40 Tetraploid tumors also show significantly higher copy number variations compared with their diploid counterparts.40 Chromothripsis (in the absence of p53 mutations) occurs frequently in this malignancy that results in bizarre chromosomal rearrangements or fusions in an attempt at ineffective DNA repair. One such example is the Myc/PVT1 gene fusion (between exon 1 and 3 of PVT-1 and exon 2 of Myc) that is present in about 60% of cases with Myc amplification.27 There appears to be a positive feedback control between Myc and PVT-1, in that the former controls its own expression by activating the canonical E-boxes on the PVT-1 promoter.39 There also appears to be upregulation of transforming growth factor-β signaling in this group due to specific somatic copy number aberrations of the genes in this signaling pathway and interaction with downstream targets that include OTX-2.27 Recently, two laboratory groups have described mouse models that approximate the pathologic and biologic behavior of group 3 medulloblastoma.24,53 Pei et al found that expressing a stable form of Myc (MycT58A) through viral infection of CD133+ neural stem cells derived from the postnatal mouse cerebellum caused rapid cellular proliferation.24 When these cells were transplanted into the cerebellar hemisphere of immunodeficient mice, they formed large masses that resolved spontaneously due to concomitant Myc-induced apoptosis.24 However, sustained tumor formation occurred when these cells were also infected with dominant negative p53 (DNp53) to overcome Myc-induced apoptosis. These tumors had all the pathologic and molecular characteristics of group 3 medulloblastoma. Kawauchi et al have reported similar findings, but used cerebellar granule neuron progenitors (Atoh-1+) instead that had enforced expression of Myc in a p53 null background.53 Interestingly, tumors from the latter study had gene expression patterns that resembled an embryonic stem cell phenotype (Atoh-1–, Oct +/Nanog +, although the tumor cells originated from mature granule neuron precursors) suggesting dedifferentiation of these cells following Myc expression.
This subgroup is associated with the worst prognosis amongst all the four molecular subtypes of medulloblastoma, with survival rates of 50% or less.7,30 In a validation cohort reported by Kool et al, no patients in group 3 survived past 124 months.30 Novel agents are desperately needed to improve survival of the majority of patients who fail standard cytotoxic therapy. The preclinical mouse models described above could be used to screen for new agents that are likely to provide benefit in patients with this aggressive malignancy, that does not seem to respond to conventional cytotoxic therapy.
Group 4 medulloblastoma
Group 4 medulloblastoma (also called group D) constitutes 35% of all cases and is the most common of the four subgroups.7 However, much remains to be understood in terms of the cell of origin and the signaling pathways that govern these tumors. Group 4 tumors can present at any age, although it is rare in infants.8 Sex bias is most prominent in this group, with a male to female ratio of 3:1.8 No known germ line mutations exist that predispose to this subtype. Most tumors are of classic histology, but large cell/anaplastic has been observed as well. While immunohistochemical expression for potassium voltage-gated channel subfamily A member 1 (KCNA) has been reported to identify this subgroup, others have questioned the validity of this marker and this test should not be used for clinical purposes until further validation in prospective cohorts of newly diagnosed patients.8,34,36 A third of patients, especially young adults, have metastatic disease at diagnosis.27
Similar to group 3 medulloblastoma, tetraploidy occurs as an early change in group 4 tumors (40% of cases).40 Although an early study from the 1990s in a limited group of patients with medulloblastoma demonstrated a better outcome for patients with hyperdiploid tumors, the prognostic impact of tetraploidy remains unclear.54 However, it might serve as a rational target for mitotic checkpoint kinase or kinesin pathway inhibitors.40 Additionally, genes MycN and CDK6 are commonly amplified.27 Isochromosome 17q occurs in 80% of cases, as does 17p deletion. Most female group 4 medulloblastoma tumors lose one copy of the X chromosome, which raises the possibility of the existence of one or more tumor suppressor genes on the X chromosome.39 Additionally, chromatin-remodeling genes are mutated in this group and include KDM6A (a H3K27 methylase, located on chromosome Xp11.3), ZYMYM3, and CHD7. These mutations, along with overexpression of enhancer of zeste homolog 2, keep neural stem cells in an undifferentiated state and might be sustaining tumorigenesis.27 A tandem duplication on chromosome 5q23.2 has recently been identified in 25% of cases, and might serve as a signature gene (or lineage marker of a possible cell of origin) for this group.39 This involves a focal single copy gain of the gene for α-synuclein-interacting protein (SNCAIP) and is represented as a tandem duplication of a truncated portion of the SNCAIP gene adjacent to its germ line allele. SNCAIP encodes for synphilin-1 that binds to α-synuclein and is present as a major component of the Lewy bodies found in the brains of patients with Parkinson’s disease.39
There is no specific preclinical mouse model that fully recapitulates the human group 4 medulloblastoma, although one has been described that expresses MycN and luciferase under the control of a brain-specific promoter and develops either SHH or non-SHH medulloblastoma depending on the time of tumor initiation.55 It is unclear if this non-SHH medulloblastoma mouse model is an accurate representation of conventional group 4 tumors. Children with group 4 medulloblastoma have an intermediate prognosis with conventional cytotoxic therapy, although adult patients with the same tumors have a worse outcome.27
Conclusion
Since the first seminal publication describing the molecular variants of medulloblastoma using gene expression profiling and transcriptome analysis,28 there has been a veritable explosion of data in this field, adding new discoveries of novel mutations and copy number aberrations almost on a daily basis and reflecting the incredible complexity of this aggressive tumor. The emerging data suggest that medulloblastoma is possibly a mixture of different disease entities with different molecular underpinnings and underlying cell or region of origin. While these are exciting times for laboratory scientists and clinicians with the identification of several tumor targets that provides avenues for improving therapeutic efficacy, the plight of the clinical neuro-oncologist and caregivers of patients with medulloblastoma is obvious and justified when faced with this bewildering array of molecular data. One clear consensus to emerge from the recent molecular studies is the reduction of cytotoxic therapy for patients with tumors carrying the WNT signature that seem to have an excellent prognosis with standard cytotoxic therapy. Even here, there are polarizing views on what can or should be considered “safe reduction” of treatment. Faced with multiple complex signaling pathways and mutated targets, it is not clear, at any given time in the disease course, which of the identified markers within each group are currently oncogenic drivers and which ones are either just passenger mutations or those that were responsible for tumor initiation but are no longer actively contributing to disease progression. While druggable targets abound within the tumor, very few targeted therapies are currently available, much less validated in clinical practice, except for SMO antagonists for SHH tumors. Also, it is clear that no single agent is going to be effective for a sustained period of time, and combination therapy is probably the best strategy to overcome either de novo or acquired resistance to treatment. It is also unknown if potential agents would be more effective and should be used at the time of diagnosis during a period of treatment-naive minimal residual disease (in combination with standard cytotoxic therapy) rather than at the time of relapse following multiple treatment regimens and when a tumor potentially becomes more aggressive and drug-resistant. It is of some relief that emerging data with the use of paired samples from the time of diagnosis and the time of relapse seem to suggest that the molecular group at diagnosis is maintained at relapse and patterns of relapse are group-specific.56 SHH tumors usually recur locally and group D patients almost always relapse in metastatic sites.56 This suggests that improved strategies for local control of the former and better therapies for metastatic disease in the latter are potential ways to improve disease-free survival. Finally, there is justifiable concern regarding the possible toxicities, especially in growing children, with the use of therapies that are potentially targeting pathways present in both the tumor and normal tissue (eg, the effect of SHH inhibition on growing bone57 and normal neural stem cells that are sustained by this pathway58) and should be used with caution in children with appropriate tools to monitor such toxicities.
Disclosure
The authors report no conflicts of interest in this work.
References
Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol. 2011;8:540–549. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. | |
Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820. | |
Gururangan S, Krauser J, Watral MA, et al. Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol. 2008;10:745–751. | |
Rutkowski S, von Hoff K, Emser A, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol. 2010;28:4961–4968. | |
Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127–2136. | |
Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. | |
Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. | |
Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012;14 Suppl 5: v1–v49. | |
Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232–250. | |
Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47: 888–901. | |
Evans G, Burnell L, Campbell R, Gattamaneni HR, Birch J. Congenital anomalies and genetic syndromes in 173 cases of medulloblastoma. Med Pediatr Oncol. 1993;21:433–434. | |
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114: 97–109. | |
McManamy CS, Pears J, Weston CL, et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17:151–164. | |
Massimino M, Antonelli M, Gandola L, et al. Histological variants of medulloblastoma are the most powerful clinical prognostic indicators. Pediatr Blood Cancer. 2013;60:210–216. | |
Ellison DW. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010;120: 305–316. | |
Rodriguez FJ, Eberhart C, O’Neill BP, et al. Histopathologic grading of adult medulloblastomas. Cancer. 2007;109:2557–2565. | |
Evans DG, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer. 1991;64:959–961. | |
Farndon PA, Del Mastro RG, Evans DG, Kilpatrick MW. Location of gene for Gorlin syndrome. Lancet. 1992;339:581–582. | |
Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5:209–218. | |
Skomorowski M, Taxier M, Wise W Jr. Turcot syndrome type 2: medulloblastoma with multiple colorectal adenomas. Clin Gastroenterol Hepatol. 2012;10:A24. | |
Gururangan S, Frankel W, Broaddus R, et al. Multifocal anaplastic astrocytoma in a patient with hereditary colorectal cancer, transcobalamin II deficiency, agenesis of the corpus callosum, mental retardation, and inherited PMS2 mutation. Neuro Oncol. 2008;10:93–97. | |
Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28:1345–1350. | |
Pei Y, Moore CE, Wang J, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012;21:155–167. | |
Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. | |
Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927–2935. | |
Northcott PA, Jones DT, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818–834. | |
Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088. | |
Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. | |
Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. | |
Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8: 340–351. | |
Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. | |
Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. | |
Bien-Willner GA, Lopez-Terrada D, Bhattacharjee MB, et al. Early recurrence in standard-risk medulloblastoma patients with the common idic(17)(p11.2) rearrangement. Neuro Oncol. 2012;14:831–840. | |
Barbouti A, Stankiewicz P, Nusbaum C, et al. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet. 2004;74:1–10. | |
Min HS, Lee JY, Kim SK, Park SH. Genetic grouping of medulloblastomas by representative markers in pathologic diagnosis. Transl Oncol. 2013;6:265–272. | |
Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29: 1400–1407. | |
Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–396. | |
Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488: 49–56. | |
Jones DT, Jager N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. | |
Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. | |
Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. | |
Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. | |
Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–310. | |
Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. | |
Northcott PA, Hielscher T, Dubuc A, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240. | |
Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768–1776. | |
Gajjar A, Stewart CF, Ellison DW, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a Pediatric Brain Tumor Consortium Study. Clin Cancer Res. October 24, 2013. [Epub ahead of print.] | |
Gajjar A, Gururangan S, Qaddoumi I, et al. A prospective phase II study to determine the efficacy of GDC 0449 (vismodegib)in adults with recurrent medulloblastoma (MB): a Pediatric Brain Tumor Consortium Study (PBTC 25B). Proc Am Soc Clin Oncol. 2013;31:2035. | |
Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. | |
Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–444. | |
Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the Pi3K pathway in medulloblastoma. Sci Transl Med. 2010;2:1–8. | |
Kawauchi D, Robinson G, Uziel T, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. | |
Gajjar AJ, Heideman RL, Douglass EC, et al. Relation of tumor-cell ploidy to survival in children with medulloblastoma. J Clin Oncol. 1993;11:2211–2217. | |
Swartling FJ, Savov V, Persson AI, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–613. | |
Ramaswamy V, Remke M, Bouffet E, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013;14:1200–1207. | |
Kimura H, Ng JMY, Curran T. Transient inhibition of the hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249–260. | |
Sanai N, Buylla AA, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.