Back to Journals » Drug Design, Development and Therapy » Volume 19
MLN4924 Suppresses Acute Myeloid Leukemia Progression by LINC01128-Driven Epigenetic Reactivation of TRIM58
Authors Guo Y ![]() , Jian J
, Jian J ![]() , Tang X
, Tang X ![]() , An S, Zhao L, Chen R
, An S, Zhao L, Chen R ![]() , Ma W
, Ma W ![]() , Liu B
, Liu B ![]()
Received 20 May 2025
Accepted for publication 1 October 2025
Published 22 October 2025 Volume 2025:19 Pages 9439—9456
DOI https://doi.org/10.2147/DDDT.S541720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yuancheng Guo,1– 3 Jinli Jian,3 Xiao Tang,3 Shujuan An,3,4 Long Zhao,1,3 Run Chen,3 Weiqing Ma,5 Bei Liu1– 3
1Department of Hematology, The First Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China; 2Gansu Provincial Clinical Medical Research Center for Molecular Diagnosis and Treatment of Hematological Diseases, Lanzhou, 730000, People’s Republic of China; 3The First Clinical Medical School, Lanzhou University, Lanzhou, 730000, People’s Republic of China; 4Department of Medical Laboratory, The First Hospital of Lanzhou University, Lanzhou, 730000, People’s Republic of China; 5School of Public Health, Lanzhou University, Lanzhou, 730000, People’s Republic of China
Correspondence: Bei Liu, Department of Hematology, The First Hospital of Lanzhou University, Lanzhou, People’s Republic of China, Email [email protected]
Purpose: The aim of this study was to elucidate the molecular mechanism by which MLN4924 affects the progression of acute myeloid leukemia (AML) by regulating TRIM58 DNA methylation.
Patients and Methods: Gene expression was analyzed by RT-qPCR and Western blot, while methylation changes were assessed via Methylation-sensitive restriction enzyme-quantitative PCR. Differentially expressed lncRNAs were identified through RNA sequencing. Subcellular localization was determined via nuclear-cytoplasmic fractionation-PCR. Protein-DNA/RNA interactions were analyzed by chromatin immunoprecipitation and RNA immunoprecipitation, respectively. In vivo experiments were conducted using a xenograft model, with tumor protein expression evaluated by immunohistochemistry.
Results: TRIM58 downregulation in AML correlated with promoter hypermethylation, reversible by MLN4924 treatment. Functional studies demonstrated TRIM58 mediated MLN4924-induced apoptosis through AKT pathway inhibition. While MLN4924 upregulated the tumor-suppressive lncRNA LINC01128, its overexpression recapitulated TRIM58-mediated anti-leukemic effects, including expansion arrest, apoptosis induction, and BAX/BCL-2 axis modulation. Mechanistically, nuclear-localized LINC011128 functionally interacted with DNMT1 to mediate TRIM58 promoter demethylation, establishing an epigenetic regulatory axis. Rescue experiments revealed TRIM58 knockdown attenuated MLN4924’s suppression of AKT phosphorylation and associated pro-apoptotic effects.
Conclusion: In this study, we show that MLN4924 can upregulate LINC01128, which binds to and segregates DNMT1, thereby inhibiting methylation modification of the TRIM58 and ultimately suppressing AML.
Keywords: DNA methylation, DNMT1, lncRNA LINC01128, proximal promoter
Introduction
Acute myeloid leukemia (AML) remains a therapeutic challenge due to limited treatment options.1 Targeting epigenetic dysregulation has emerged as a promising strategy, with DNA hypomethylating agents like azacitidine demonstrating clinical efficacy.2 MLN4924, a first-in-class NEDD8-activating enzyme (NAE) inhibitor, inactivates Cullin-RING ligases (CRLs) by blocking cullin neddylation, leading to CRL substrate accumulation and selective tumor cell death.3–5 Notably, clinical trials have validated its safety and synergistic effects when combined with azacitidine in AML,6–12 although the underlying epigenetic regulatory mechanisms remain unclear.
The therapeutic vulnerability of AML to epigenetic interventions stems from the frequent hypermethylation-mediated silencing of tumor suppressor genes.13 For instance, DNMT-driven SOCS3 hypermethylation accelerates AML pathogenesis, positioning it as a diagnostic biomarker.14 Similarly, BCL7A promoter hypermethylation suppresses AML cell differentiation while promoting lymph node infiltration, correlating with poor survival.15,16 Promoter methylation of SCIN has further been implicated in AML prognosis.17 These findings collectively establish DNA methylation alterations as a critical determinant in AML progression and treatment response.
Long non-coding RNA (lncRNA) is a key factor in the regulation of epigenetic. Functioning as molecular scaffolds, lncRNAs modulate DNA methylation through both direct and indirect mechanisms, thereby governing leukemogenesis.18–20 Our prior work revealed that MLN4924 alters DNA methylation patterns of AML cells, specifically inducing TRIM58 promoter demethylation.21 TRIM58 has been found to act as a tumor suppressor gene in a variety of solid tumors and is silenced by promoter hypermethylation.22–24 Therefore, we hypothesized that TRIM58 is also a tumor suppressor gene silenced by hypermethylation in AML, and that lncRNA is the mediator connecting MLN4924 processing with TRIM58 reactivation. By elucidating how MLN4924 reverses TRIM58 silencing through an lncRNA-dependent mechanism, this study will advance our understanding of the epigenetic regulatory network underlying the anti-leukemic activity of MLN4924.
Materials and Methods
Study Subjects
Peripheral blood samples were collected from 17 patients diagnosed with acute myeloid leukemia (AML) at the Department of Hematology, First Affiliated Hospital of Lanzhou University, between January 2018 and January 2024. Baseline characteristics are presented in Supplementary Table 1. Additionally, peripheral blood samples from 10 healthy individuals at the medical examination center were included as controls. The samples were derived from anonymized specimens archived from previous clinical consultations and were approved by the Ethics Review Committee for exemption from informed consent requirements (LDYYLL-2025-82).
Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
Centrifuge blood samples in EDTA tubes at 3000 rpm for 5 minutes at room temperature. Serum was removed, and PBMCs were isolated from the remaining blood using Ficoll-Paque density gradient centrifugation according to the manufacturer’s protocol (Solarbio, Cat. No. P8610). PBMCs were then stored at −80°C until further testing.
Cell Culture and Treatment
The human AML cell lines Kasumi-1 (purchased from the American Type Culture Collection), HL60 (purchased from Wuhan Servicebio, China), and MOLM13 (generously provided by Dr. Shuling Zhang at the First Affiliated Hospital of Lanzhou University) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, maintained at 37 °C in a humidified atmosphere containing 5% CO2. The STR identification documents for these cell lines are detailed in Supplementary Material. Mycoplasma detection was performed using a rapid mycoplasma detection kit (isothermal amplification method, Servicebio), with all results being negative.
HL60 cells were treated with 3 μM or 6 μM decitabine (DAC) (TargetMol, Shanghai, China). MOLM13 cells were treated with 5 μM SC79 (TargetMol, Shanghai, China) and 10 μM BIP-V5 (TargetMol, Shanghai, China), respectively.
Lentiviral Transfection
Kasumi-1, HL60 and MOLM13 cells were seeded in 24-well plates at 5×104 cells/well. LINC01128- and TRIM58-overexpression lentiviruses, their respective shRNAs, and corresponding negative control vectors were obtained from GeneChem (Shanghai, China), and transfected into Kasumi-1, HL60, and MOLM13 cells. To optimize transduction efficiency, we performed pre-experiments determining the multiplicity of infection (MOI) and selected MOI=50 for subsequent experiments. Following 72 hours of puromycin selection, the antibiotic concentration was reduced to a maintenance level to establish stable polyclonal cell populations. Transduction efficiency was validated by qPCR and Western blot.
RNA Isolation and Quantification
Total RNA was extracted using the M5 Universal RNA Mini Kit (Mei5bio, Beijing, China). RNA quantity and quality were evaluated using the NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized from the extracted RNA using the Hifair® III First Strand cDNA Synthesis SuperMix for qPCR (Yeasen, Shanghai, China). qPCR amplification was performed using the QuantStudio 3 system (Thermo Fisher Scientific, Waltham, MA, USA) and Hieff® qPCR SYBR Green Master Mix (Yeasen, Shanghai, China). All primers were synthesized by Servicebio Biotechnology (Wuhan, China), with sequences provided in Supplementary Table 2. mRNA expression levels were quantified using the 2−ΔΔCq method. Briefly, target gene Cq values were normalized to the endogenous control gene GAPDH (ΔCq), followed by normalization to the control group (ΔΔCq) for relative expression calculation. Three technical replicates were analyzed per sample.
RNA Sequencing (RNA-Seq)
Libraries were sequenced on the Illumina NovaSeq 6000 platform. Trimmomatic filtered out reads containing low-quality bases, and Fastp performed quality control to obtain high-quality reads. The R package DESeq2 was used to analyze differentially expressed genes, with filtering criteria of p < 0.05 and |log2(fold change)| > 0.58.
Methylation-Specific PCR (MSP)
Genomic DNA was extracted using the M5 HiPer Universal DNA Mini Kit and was bisulfite-converted with the EpiJET Bisulfite Conversion Kit. MSP was performed using the MSP Kit, followed by gel electrophoresis to analyze the PCR products. Primer details for the TRIM58 promoter are provided in Supplementary Table 2.
Western Blot Analysis
Cells or tissue samples were lysed with RadioImmunoPrecipitation Assay buffer to extract total proteins. Proteins were separated by SDS-PAGE for 1 h and then transferred to polyvinylidene difluoride membranes. The membranes were incubated with primary antibodies overnight at 4 °C, and GAPDH was used as an internal control. Next, the membranes were incubated with horseradish peroxidase (HRP)-labeled secondary antibodies. The bands were visualized by chemiluminescent imaging system (VILBER, France). The antibodies used were as follows: anti-TRIM58 (1:1000, YT6944, Immunoway, USA), anti-GAPDH (1:8000, 60004-1-Ig, Proteintech, USA), anti-AKT (1:1000, 4691, CST, USA), anti-p-AKT (Ser473) (1:2000, 4060, CST, USA), anti-BAX (1:1000, 2772, CST, USA), anti-BCL-2 (1:1000, 3498, CST, USA). ImageJ 1.54f was used to quantify the relative protein levels on immunoblots.
Subcellular Fractionation
Cytoplasmic and nuclear RNA were isolated using a nucleoplasmic isolation kit (Beibei Biotechnology, Zhengzhou, China) according to the manufacturer’s instructions.
Methylation-Sensitive Restriction Enzyme-Quantitative PCR (MSRE-qPCR)
MSRE-qPCR were performed using EpiJET DNA Methylation Analysis Kit (Thermo Fisher Scientific, Waltham, MA, USA). In brief, Genomic DNA was extracted using the M5 HiPer Universal DNA Mini Kit (Mei5bio, Beijing, China). 1 μg of DNA per group was used for restriction digestion, followed by incubation at 37 °C for 1 h and termination of the reaction at 90 °C for 10 min. The products were analyzed by qPCR (primer sequences in Supplementary Table 2). The 5-methylcytosine (5-mC) content was calculated using the formula:

Dual Luciferase Reporter Assay
The pGL3-basic vector containing wild-type and mutant TRIM58 promoter sequences, along with the pcDNA3.1 vector encoding the LINC01128 transcript (sequences provided in Supplementary Table 3), was constructed by Genecreate Biological Engineering Co (Wuhan, China). These plasmids were co-transfected into 293T cells using Lipofectamine 3000. Luciferase activity was measured 48 h post-transfection using a Dual-Luciferase Reporter Assay System (Biosino Biotechnology and Science Inc, China), with Renilla luciferase signal for normalization.
Chromatin Immunoprecipitation (ChIP)
Enrichment of DNMT1 in the TRIM58 promoter region was analyzed using the ChIP kit (Beyotime, China). Kasumi-1/MOLM13 cells were fixed with 37% formaldehyde for 10 min, and then glycine was added to terminate DNA protein cross-linking. Cell lysates were sonicated (40 W, 30 cycles, 10s on/20s off) using a sonicator (Scientz-IID, Ningbo, China) to produce 200–1000 bp chromatin fragments. Protein A+G agarose/salmon sperm DNA was pre-added to minimize non-specific binding of the mixture to target proteins or DNA. For immunoprecipitation, lysates were incubated with anti-DNMT1 antibody (ab188453, Abcam, USA) overnight at 4 °C with slow rotation, while IgG control antibody (30000-0-AP, Proteintech, USA) served as a negative control. Protein A + G agarose/salmon sperm DNA was then added and gently mixed for 60 min at 4 °C to allow the antibody-binding complex to precipitate. The precipitates were washed sequentially with low salt, high salt, LiCl immune complex wash buffer and TE buffer. Protein-DNA complexes were eluted with elution buffer (1% SDS, 0.1 M NaHCO₃), and then incubated with 5 M NaCl for 4 h at 65 °C to reverse cross-linking. Purified DNA was analyzed by qPCR, with primer sequences listed in Supplementary Table 2. Data are presented as fold enrichment over IgG controls.
RNA Immunoprecipitation (RIP)
The RIP kit (P0101, Geneseed, China) was used to detect whether LINC01128 and DNMT1 bound. The magnetic beads were pretreated and conjugated with anti-DNMT1 antibody (ab188453, Abcam, USA) or IgG control antibody (30000-0-AP, Proteintech, USA). The cell lysate and magnetic beads were then rotated overnight at 4 °C. After washing, RNA was extracted using a spin column method and analyzed by qPCR.
Cell Counting Kit-8 (CCK-8)
When cells reached the logarithmic growth phase at 80% confluency, they were counted and seeded into 96-well plates (2×104 cells/well in 100 μL medium). After 24 h of incubation, 10 μL CCK-8 reagent was added to each well, followed by 2 h of light-protected incubation at 37 °C. Absorbance was measured at 450 nm using a microplate reader (BioTek Synergy Neo2, USA).
Apoptosis and Cell Cycle Analysis
The Annexin V-APC/7-AAD Apoptosis Kit (Elabscience Biotechnology, Wuhan, China) was used to assess cell apoptosis. Briefly, cells were resuspended, stained with Annexin V-APC and 7-AAD for 15 min, and analyzed by flow cytometry at their respective excitation/emission wavelengths. For cell cycle analysis, the Cell Cycle Staining Kit (MultiSciences Biotech, Hangzhou, China) was employed. Cells were stained with propidium iodide in permeabilization solution and analyzed at a low flow rate using a flow cytometer.
Xenograft Tumor Models
Twenty female NOD/SCID mice (5 weeks, 18–22 g) were purchased from Changzhou Cavens Laboratory Animal Co. There were three or four mice in each group. For the TRIM58 overexpression group, 4×106 MOLM13 cells in 100 μL PBS were subcutaneously inoculated into the right flank of each mouse. HL60 cells (5×106 cells/mouse) were injected subcutaneously into the right inguinal region. Starting from day 7 post-inoculation, the HL60 cell group received daily intraperitoneal injections of MLN4924 (60 mg/kg) for 14 consecutive days. Tumor dimensions were measured every three days using vernier calipers and tumor volume was calculated using the formula:

The maximum tumor volume required by the Ethics Committee was 1500 mm3 and no mice exceeded the preset threshold tumor volume during the study period. All mice were euthanized by CO2 asphyxiation 3 weeks post-inoculation. Xenografts were excised and weighed for final analysis.
Immunohistochemistry
Mouse tumor tissues were deparaffinized and rehydrated after a 2 h incubation at 60 °C, and citrate buffer was used for antigen retrieval. The sections were incubated with 3% hydrogen peroxide at room temperature for 10 min to block endogenous peroxidase activity. Afterwards, the slides were incubated with 5% normal goat serum for 30 min at room temperature to reduce nonspecific binding. The slides were then incubated with primary antibodies at 4 °C overnight. Finally, after washing, the HRP-labeled secondary antibody was applied, followed by DAB chromogen development and hematoxylin counterstaining. The primary antibody dilutions were as follows: anti-PCNA (1:1000, Servicebio, Wuhan, China), anti-BCL-2 (1:500, Servicebio, Wuhan, China), anti-BAX (1:500, Servicebio, Wuhan, China), anti-AKT (1:200, CST, USA), and anti-pAKT (1:300, CST, USA).
Statistical Analysis
Statistical analyses of experimental data were performed using GraphPad Prism 10.2 and R software (version 4.0.3). For comparisons between two groups, an unpaired Student’s t-test was used to determine statistical significance. For multiple group comparisons, one-way analysis of variance was applied, followed by Dunnett’s post hoc test to assess differences among group means. All data are presented as mean ± standard deviation (SD). Statistical significance is denoted as follows: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Results
MLN4924 and DAC Demethylated the TRIM58 Proximal Promoter and Upregulated Its Expression
In a previous study using 850K methylation arrays, we found that MLN4924 altered the global methylation pattern of AML cells and decreased methylation at the cg26052730 site of the TRIM58 promoter. Meanwhile, transcriptome sequencing showed that MLN4924 upregulated the transcriptional level of TRIM58. In this study, Western blot demonstrated that MLN4924 also increased the TRIM58 protein level in AML cells (Figure 1A). Therefore, we speculated that MLN4924 may relieve the transcriptional repression of TRIM58 by reducing the methylation level of this site, thereby enhancing its transcriptional and translational activity.
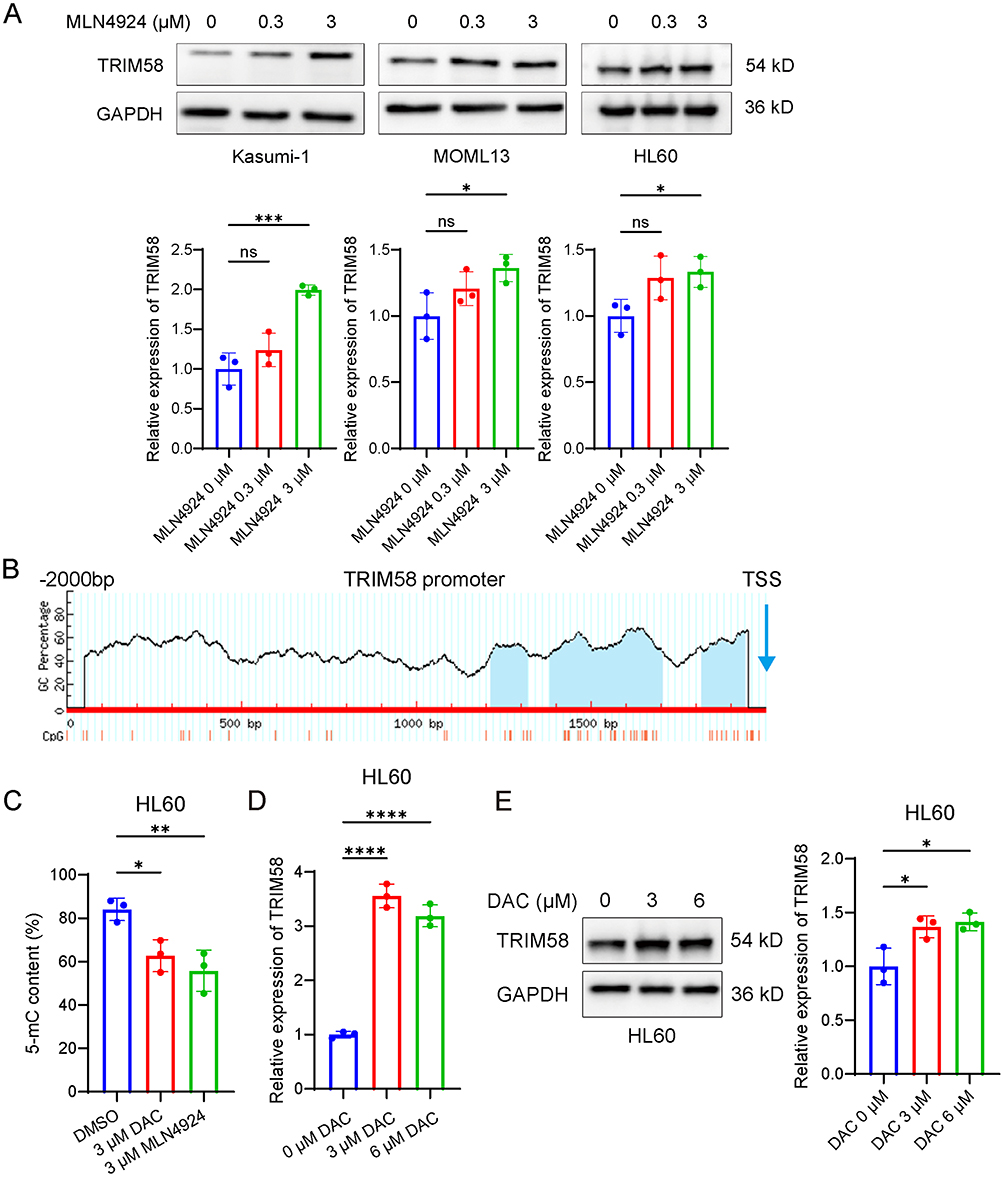 |
Figure 1 MLN4924 and DAC demethylate the proximal promoter region of TRIM58 and upregulate its expression. (A) Western blot analysis of TRIM58 protein expression in AML cell lines (Kasumi-1, MOLM13, and HL60) treated with varying concentrations of MLN4924 (0, 0.3, and 3 μM). (B) Prediction of CpG islands in the TRIM58 promoter region by MethPrimer. (C) MSRE-qPCR analysis of 5-mC content in the CpG island of the TRIM58 promoter (positions −182 to −58) in HL60 cells treated with 3 μM MLN4924 or 3 μM DAC for 24 h. (D) RT-qPCR analysis of TRIM58 expression in HL60 cells treated with varying concentrations of DAC. (E) Western blot analysis of TRIM58 protein expression in HL60 cells treated with varying concentrations of DAC (0, 3, and 6 μM). Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ****p < 0.0001. |
Using the MethPrimer database,25 we identified three CpG islands in the TRIM58 promoter region (Figure 1B), located at positions −785 to −683, −615 to −294, and −182 to −58 relative to the transcription start site (TSS). cg26052730 (−156 to −155) is located in the rightmost CpG island, which resides in the proximal promoter region closely associated with gene expression regulation. To further evaluate the relationship between methylation at this site and TRIM58 expression, we designed primers targeting this CpG island and performed MSRE-qPCR. Through digestion with HpaII (methylation-sensitive) and MspI (methylation-insensitive), we observed a decrease in 5-mC content following MLN4924 or DAC treatment, indicating significant demethylation in this region (Figure 1C). In DAC-treated HL60 cells, both mRNA and protein levels of TRIM58 were significantly increased (Figure 1D and E). Collectively, these data demonstrate that MLN4924 and DAC can demethylate the CpG island in the proximal promoter region and upregulate TRIM58 expression.
TRIM58 Inhibits Cell Expansion and Promotes Apoptosis in AML Cells in vitro
Since TRIM58 has been reported to be a tumor suppressor gene in many tumors, we investigated its biological role in AML. Analysis of the GEO database showed that TRIM58 expression was significantly lower in AML patients compared to healthy individuals (Figure 2A). We collected peripheral blood samples from 17 AML patients and 10 healthy control and confirmed the low expression of TRIM58 in AML (Figure 2B).
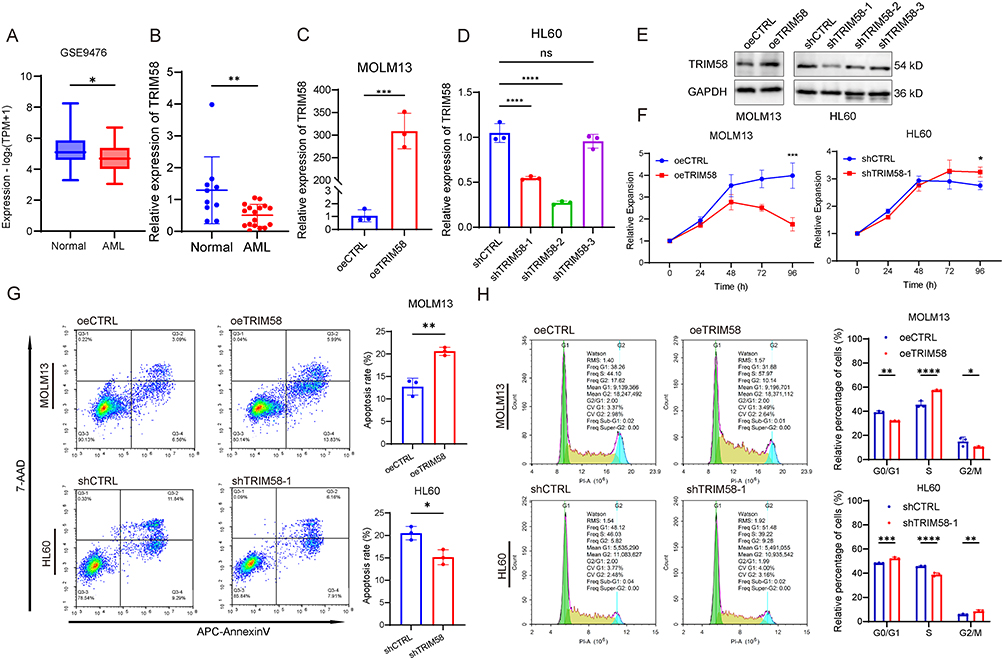 |
Figure 2 TRIM58 inhibits cell expansion and promotes apoptosis in AML cells in vitro. (A) Comparison of TRIM58 expression between AML patients (n = 26) and healthy controls (n = 38) using the GSE9476 dataset. (B) RT-qPCR quantification of TRIM58 expression levels between controls (n = 10) and AML patients (n = 17). (C and D) RT-qPCR quantification of TRIM58 expression upon its overexpression in MOLM13 cells and knockdown in HL60 cells. (E) Western blot analysis of TRIM58 expression upon its overexpression in MOLM13 cells and knockdown in HL60 cells. (F) CCK-8 assays were conducted to assess cellular expansion following TRIM58 overexpression in MOLM13 cells and knockdown in HL60 cells. (G and H) Apoptosis and cell cycle in TRIM58-overexpressing MOLM13 cells and TRIM58-knockdown HL60 cells were assessed by flow cytometry at 7 days post-transfection. Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviation: ns, not significant. |
We used lentivirus to stably overexpress TRIM58 in MOLM13 cells and knockdown TRIM58 in HL60 cells and verified the overexpression and knockdown efficiencies (Figure 2C–E). CCK-8 assay results showed that overexpression of TRIM58 inhibited cell expansion, while knockdown of TRIM58 promoted it (Figure 2F). To explore the mechanisms underlying the effects of TRIM58 on AML cell expansion, we used flow cytometry to assess apoptosis and cell cycle progression. Our findings demonstrated that overexpression of TRIM58 promoted apoptosis in AML cells, whereas its knockdown inhibited it (Figure 2G). Furthermore, analysis showed that TRIM58 overexpression increased the proportion of cells in the S phase. Conversely, TRIM58 knockdown resulted in a decrease in the proportion of cells in the S phase (Figure 2H). We repeated the above experiments in Kasumi-1 cells, and the results showed that TRIM58 overexpression and knockdown had similar effects on cell expansion and apoptosis progression, but had no significant effect on the cell cycle (Supplementary Figure 1).
MLN4924 Promotes Apoptosis Through the TRIM58/AKT/BAX Axis
Given that TRIM58 overexpression promotes apoptosis in AML cells, we investigated its effects on the apoptosis-related proteins BCL-2 and BAX. Our findings revealed that TRIM58 overexpression led to increased BAX expression, while BCL-2 levels remained unchanged (Figure 3A). Previous studies have suggested that AKT can inhibit BAX expression through the mitochondrial intrinsic pathway,26 thereby suppressing apoptosis. Therefore, we evaluated the effect of TRIM58 on the AKT pathway. The results showed decreased p-AKT levels, while total AKT expression remained unaffected (Figure 3A). These results suggest that TRIM58 overexpression inhibits the AKT pathway, potentially facilitating BAX upregulation by reducing AKT phosphorylation. It is known that MLN4924 can inhibit the phosphorylation process of AKT. To clarify whether TRIM58 was involved in the above mechanism, we performed a rescue experiment. Compared with MLN4924 alone, knocking down TRIM58 can reverse the inhibitory effect of MLN4924 on the AKT pathway, and at the same time promote cell expansion and inhibit cell apoptosis (Figure 3B–F).
 |
Figure 3 MLN4924 promotes apoptosis through the TRIM58/AKT/BAX axis. (A) Western blot analysis of AKT pathway proteins, BCL-2, and BAX in TRIM58-overexpressing MOLM13 cells. (B) RT-qPCR quantification of TRIM58 expression in each group. (C) Protein expression levels of AKT, p-AKT and TRIM58 in each group were analyzed by Western blot. (D) Cell viability was measured at 450 nm using the CCK-8 assay across experimental groups. (E and F) Apoptosis assessment by flow cytometry in each group. (G and H) Flow cytometry analysis of apoptosis in MOLM13 cells treated with DMSO, MLN4924 (0.3 μM), SC79 (5 μM), and MLN4924 + SC79. (I and J) Flow cytometry analysis of apoptosis in MOLM13 cells treated with DMSO, MLN4924 (0.3 μM), BIP-V5 (10 μM), and MLN4924 + BIP-V5. Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
To further validate the necessity of the TRIM58/AKT/BAX axis in MLN4924-induced apoptosis, we performed functional rescue experiments using the AKT activator SC79 and the BAX inhibitor BIP-V5. The results showed that activation of AKT by SC79 significantly attenuated MLN4924-induced apoptosis (Figure 3G and H), while BIP-V5 treatment similarly reduced the pro-apoptotic effects of MLN4924 (Figure 3I and J). These findings collectively demonstrate that MLN4924 promotes apoptosis in a manner partially dependent on its modulation of the TRIM58/AKT/BAX signaling axis.
MLN4924 Treatment and TRIM58 Overexpression Inhibit AML Growth in vivo by Suppressing the AKT Pathway
We further assessed the tumor-suppressive effects of MLN4924 and TRIM58 overexpression using subcutaneous AML xenograft model. Tumor volume and weight were significantly reduced in the MLN4924-treated group compared to the untreated group (Figure 4A–C). Mice injected with TRIM58-overexpressing MOLM13 cells also exhibited smaller tumors and lower tumor weight at the endpoint compared to controls (Figure 4D–F). MLN4924 treatment upregulated TRIM58 and BAX protein levels while downregulating PCNA, BCL-2, and p-AKT levels (Figure 4G and H). Similarly, TRIM58 overexpression increased BAX expression, decreased PCNA and p-AKT levels, but had minimal effect on BCL-2 and total AKT (Figure 4I). In summary, these results indicate that both MLN4924 treatment and TRIM58 overexpression inhibit the growth of AML cells in vivo by suppressing AKT.
 |
Figure 4 MLN4924 inhibits AML cell expansion by upregulating TRIM58 in vivo. (A–C) Comparison of Tumor Volume and Weight Between the Control Group and the MLN4924 Group. (D–F) Comparison of tumor volume and weight between the control group and the TRIM58 overexpression group. (G) TRIM58 protein expression levels were analyzed by Western blot in the MLN4924-treated group compared with the control group. (H and I) Representative IHC images of PCNA, BCL-2, BAX, AKT and p-AKT staining in in four groups. Scale bar: 50 μm. Data are shown as mean ± SD. **p < 0.01; ***p < 0.001; ****p < 0.0001. |
LINC01128 Mediates MLN4924-Induced TRIM58 Upregulation in AML
Substantial evidence indicates that lncRNAs regulate DNA methylation in both physiological and pathological contexts. To identify MLN4924-responsive lncRNAs potentially modulating TRIM58, we performed RNA-seq on Kasumi-1 cells treated with MLN4924 or DMSO for 24 h. This analysis identified 2553 differentially expressed lncRNAs (Figure 5A and B). We then focused on the top 500 lncRNAs ranked by |log2FC| and interrogated the Multi-Experiment Matrix (MEM) database.27 LINC01128 and CYTOR exhibited the strongest positive co-expression with TRIM58 (Figure 5C).
 |
Figure 5 RNA-seq and co-expression database analyses identify lncRNAs mediating MLN4924-induced TRIM58 upregulation. (A) Heat map of clustered differentially expressed lncRNAs comparing 3 μM MLN4924-treated and vehicle control (0.1% DMSO) groups. Red indicates upregulated transcripts, blue downregulated. (B) Volcano plot of differentially expressed lncRNAs between 3 μM MLN4924-treated and vehicle control groups. (C) Correlation analysis between differentially expressed lncRNAs and TRIM58 in the MEM database, red represents positive correlation and blue represents negative correlation. (D) Expression levels of LINC01128 and CYTOR in AML patient versus healthy controls from the GEPIA database. (E) Kaplan-Meier survival curves comparing survival probability in AML patients stratified by LINC01128 and CYTOR expression levels (median cutoff). (F) RT-qPCR quantification of LINC01128 expression levels between healthy controls (n = 10) and AML patients (n = 17). (G) Quantification of LINC01128 expression by RT-qPCR in Kasumi-1 and MOLM13 cells treated with MLN4924. (H) Spearman correlation between LINC01128 and TRIM58 transcript levels in AML patients (n = 17). Data are shown as mean ± SD. *p < 0.05; **p < 0.01. Abbreviation: ns, not significant. |
RNA-seq analysis indicated that both LINC01128 and CYTOR were upregulated following MLN4924 treatment. We then used the GEPIA and UALCAN databases to analyze the expression levels and prognostic significance of these two lncRNAs in AML. The findings revealed that both LINC01128 and CYTOR were expressed at lower levels in AML compared to healthy control (Figure 5C and D). Notably, low expression of LINC01128 was associated with poor prognosis, whereas CYTOR expression showed no significant association with overall survival (log-rank p = 0.27) (Figure 5E). RT-qPCR confirmed significantly reduced LINC01128 expression in AML patients compared to healthy controls (Figure 5F), and further demonstrated that MLN4924 treatment markedly upregulated its expression in AML cell lines (Kasumi-1 and MOLM13) (Figure 5G). Co-expression analysis revealed a strong positive correlation between LINC01128 and TRIM58 in AML cohorts (Pearson r = 0.5712, p = 0.0166) (Figure 5H).
These findings suggest that MLN4924 may enhance TRIM58 expression through upregulating LINC01128, implying a potential regulatory mechanism in AML.
LINC01128 Functions as a Tumor Suppressor in AML via AKT/BAX Axis
To explore the effect of LINC01128 on the phenotype of AML cells, we constructed stable overexpression and knockdown systems in MOLM13 and HL60 cells using lentiviral vectors, respectively, and confirmed the overexpression/knockdown efficacy (Figure 6A). CCK-8 results showed that overexpression of LINC01128 did not affect cell expansion, whereas knockdown promoted expansion (Figure 6B). Flow cytometry confirmed the pro-apoptotic effect of LINC01128: overexpression promoted apoptosis while knockdown conversely inhibited apoptosis (Figure 6C). Cell cycle analysis revealed that overexpression induced S-phase arrest, and knockdown significantly reduced the S-phase population (Figure 6D). In Kasumi-1 cells, both overexpression and knockdown of LINC01128 recapitulated the effects on apoptosis and cell cycle observed in other lines, and interestingly overexpression uniquely suppressed expansionhere (Supplementary Figure 2).
 |
Figure 6 Overexpression of LINC01128 promotes apoptosis in AML cells, and conversely knockdown inhibits apoptosis. (A) RT-qPCR quantification of LINC01128 expression upon its overexpression in MOLM13 cells and knockdown in HL60 cells. (B) CCK-8 assays were conducted to assess cellular expansion following LINC01128 overexpression in MOLM13 cells and knockdown in HL60 cells. (C and D) Apoptosis and cell cycle assessment by flow cytometry in LINC01128-overexpressing MOLM13 cells and LINC01128-knockdown HL60 cells. (E) Western blot analysis of AKT pathway proteins, BCL-2, and BAX in LINC01128-overexpressing MOLM13 cells. Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviation: ns, not significant. |
We also examined the effects of LINC01128 on AKT and apoptosis pathways. Overexpression of LINC01128 led to downregulation of p-AKT and BCL-2, increased BAX expression, and caused minimal changes in total AKT (Figure 6E). This suggests that LINC01128 can also inhibit the AKT pathway and activate BAX.
In conclusion, LINC01128 also inhibited cell expansion and promoted apoptosis by a mechanism similar to that of TRIM58.
DNMT1 Sequestration by LINC01128 Drives TRIM58 Demethylation and Expression Activation
Studies have shown that the regulation of DNA methylation by lncRNAs is one of the key mechanisms controlling gene expression during cancer development.18 To clarify the functional domain of LINC01128 in AML cells, we first predicted its subcellular localization using bioinformatics methods. Analysis based on the lncATLAS database28 showed that LINC01128 was predominantly localized to the nucleus in most cell lines, and exhibited nuclear-cytoplasmic co-distribution in the K562 cell line (Figure 7A). Further validation using the RNALOCATE database29 revealed predominant nuclear localization of LINC01128 in THP-1 and K562 cells (Supplementary Table 4). RT-qPCR after nucleoplasmic separation confirmed that (Figure 7B).
 |
Figure 7 LINC01128 binds and sequesters DNMT1, thereby inhibiting its methylation of TRIM58. (A) Prediction of subcellular localization of LINC01128 using the lncATLAS database. (B) RT-qPCR analysis of nuclear-cytoplasmic fractions revealed predominant nuclear localization of LINC01128 RNA in Kasumi-1 and MOLM-13 cells. (C) Binding regions of LINC01128 and TRIM58 promoter regions predicted by the IntaRNA database. (D) Binding of LINC01128 and TRIM58 promoter was verified using the luciferase reporter assay. (E) MSP analysis of TRIM58 promoter methylation status after LINC01128 manipulation. M: methylated; U: unmethylated. (F) RT-qPCR quantification of TRIM58 expression upon LINC01128 overexpression in MOLM13 cells and knockdown in HL60 cells. (G) Western blot analysis of TRIM58 expression upon LINC01128 overexpression in MOLM13 cells and knockdown in HL60 cells. (H) ChIP assay was performed to verify the effect of LINC01128 overexpression or knockdown on DNMT1 binding to the TRIM58 promoter. (I) RIP experiments validated the binding between LINC01128 and DNMT1. Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviation: ns, not significant. |
IntaRNA predicted two binding sites between LINC01128 and the proximal promoter region of TRIM58 (Figure 7C), and this interaction was verified by dual-luciferase reporter assays (Figure 7D). Overexpression of LINC01128 resulted in demethylation of the TRIM58 proximal promoter region (Figure 7E) and upregulation of expression (Figure 7F and G). In contrast, knockdown of LINC01128 led to hypermethylation of the TRIM58 proximal promoter (Figure 7E) and downregulation of expression (Figure 7F and G).
Hypermethylation of tumor suppressor gene in AML is mainly mediated by DNMT1, a key regulator of DNA methylation patterns during genomic DNA replication, and we therefore speculate that it may regulate the methylation of TRIM58.30 ChIP assays showed reduced DNMT1 binding to TRIM58 upon LINC01128 overexpression, whereas it was increased upon LINC01128 knockdown (Figure 7H). RIP experiments in MOLM13 and Kasumi-1 cells confirmed the physical interaction between LINC01128 and DNMT1 (Figure 7I).
Collectively, these findings suggest that LINC01128 prevents DNMT1 from targeting the TRIM58 promoter through physical isolation or competitive binding.
TRIM58 Knockdown Reverses LINC01128-Induced Expansion Arrest and Apoptosis in AML
To determine whether TRIM58 mediates the effects of LINC01128 on AML cell expansion and apoptosis, we performed rescue experiments in HL60 cells. The CCK-8 assay demonstrated that LINC01128 overexpression significantly slowed HL60 cell expansion compared to the control group. However, when TRIM58 was concurrently knocked down, the expansion rate increased (Figure 8A and B).
 |
Figure 8 Knockdown of TRIM58 reverses the tumor suppressor effect of LINC01128 on AML cells. (A) TRIM58 protein expression levels were detected in the oeCTRL group, the oeLINC01128 group, and the oeLINC01128 + shTRIM58 group, respectively. (B) Cell viability was measured at 450 nm using the CCK-8 assay in each group. (C) Apoptosis assessment by flow cytometry in each group. Data are shown as mean ± SD. ****p < 0.0001. |
Flow cytometry analysis revealed that LINC01128 overexpression significantly increased apoptosis compared to the control group, whereas concomitant TRIM58 knockdown attenuated this effect (Figure 8C). These results demonstrate that TRIM58 acts as a critical downstream mediator of LINC01128’s regulatory activity.
Discussion
Despite advances in targeted therapies, the prognosis of AML remains poor. Tumor suppressor gene promoter hypermethylation is a key driver of AML progression, which has driven the search for demethylation therapies. Our previous study found that the ubiquitination-like pathway inhibitor MLN4924 also altered the methylation pattern of AML cells, in which the TRIM58 promoter region was significantly demethylated, and hypermethylation of its promoter region was associated with poor prognosis. Here, we further investigated the biological function of TRIM58 in AML and the molecular mechanism by which MLN4924 regulates TRIM58 methylation. We found that MLN4924 could isolate DNMT1 by up-regulating LINC01128, which reactivated the expression of tumor suppressor gene TRIM58 and induced inhibition of cell expansion and apoptosis, which represents a novel mechanism for MLN4924-targeted therapy of AML.
Previous studies have shown that TRIM58 acts as a tumor suppressor gene in a variety of cancers. In colorectal cancer, TRIM58 inhibits tumor cell expansion and promotes apoptosis by promoting ubiquitination of RECQL4 to suppress the AKT pathway;31 in lung cancer, TRIM58 promotes ZEB1 degradation through the ubiquitin proteasome pathway thereby inhibiting tumor migration and survival.32 Notably, in malignant tumors such as hepatocellular carcinoma,22 lung adenocarcinoma,23 and renal cell carcinoma,24 TRIM58 expression is frequently silenced by promoter hypermethylation, a phenomenon suggesting that epigenetic inactivation of TRIM58 may be a common mechanism prevalent in different cancers. Here, we found that TRIM58 expression was significantly reduced in patients with acute myeloid leukemia compared to healthy subjects, and its downregulation was associated with hypermethylation of the proximal promoter. This is consistent with the pattern of epigenetic silencing observed in other solid tumors. We also identified TRIM58 as a novel tumor suppressor gene in AML. Mechanistically, we found that overexpression of TRIM58 inhibited AML cell expansion, promoted apoptosis, and affected BAX and PAKT expression. Rescue experiments confirmed that knockdown of TRIM58 reversed MLN4924-induced inhibition of the AKT pathway, suggesting that TRIM58 mediates the inhibitory effect of MLN4924 on the AKT pathway.
Most current reports suggest that lncRNAs regulate the methylationof target genes by recruiting DNMTs to their promoter regions. It was found that lncRNA-ZFAS1 could bind to the promoter region of Notch1 and recruit DNMT3b to induce Notch1 methylation, thus promoting myocardial ischemia/reperfusion injury in mice;33 lncRNA SNHG1 recruited DNMT1 and DNMT3B to the ZCCHC10 promoter, resulting in hypermethylation of ZCCHC10 promoter, which promoted the progression and venetoclax resistance of acute myeloid leukemia,34 leukemia progression and vinblastine resistance. Here, we found a completely opposite regulatory pattern, where LINC01128 binds and sequesters DNMT1, thereby blocking DNMT1-mediated methylation of the TRIM58 promoter and restoring TRIM58 expression. Functionally, LINC01128 is a tumor suppressor lncRNA, and overexpression of LINC01128 inhibits AML cell expansion and promotes apoptosis, which is associated with suppression of p-AKT and BCL-2 levels and activation of BAX. This pro-apoptotic effect is consistent with previous findings that nuclear-localized LINC01128 induces apoptosis in THP-1 cells through NLRP3 interaction.35
Notably, both TRIM58 and LINC01128 overexpression impeded cell cycle progression, leading to an increased proportion of S-phase cells. This S-phase arrest may trigger caspase-dependent apoptotic cell death, suggesting a potential link between cell cycle dysregulation and apoptosis in their tumor-suppressive mechanisms. However, the specific cell cycle proteins regulated by TRIM58 and LINC01128 remain unclear and require further study.
While neddylation is predominantly recognized for regulating CRLs, emerging evidence reveals that this modification also modulates the activity of non-cullin E3 ligases including VHL,36 Parkin37 and MDM2.38 Therefore, it remains to be further explored whether TRIM58, as a non-ullin E3 ligase, is similarly regulated by neddylation-mediated regulation and whether this post-translational modification contributes to its tumor suppressor function.
Conclusion
In conclusion, our study shows that MLN4924 inhibits the progression of acute myeloid leukemia by reactivating the tumor suppressor TRIM58 via LINC01128. This novel modulation provides mechanistic insights into the epigenetic dimensions of the pharmacological activity of MLN4924, and TRIM58 may be a potential therapeutic target in AML.
Abbreviations
AML, acute myeloid leukemia; CRLs, Cullin-RING ligases; lncRNA, Long non-coding RNA; RNA-seq, RNA sequencing; FC, foldchange; MSP, Methylation-Specific PCR; HRP, horseradish peroxidase; MSRE-qPCR, Methylation-sensitive restriction enzyme-quantitative PCR; 5-mC, 5-methylcytosine; ChIP, Chromatin immunoprecipitation; RIP, RNA-binding protein immunoprecipitation; CCK-8, Cell counting kit-8; SD, standard deviation; TSS, transcription start site.
Data Sharing Statement
The datasets utilized and/or analyzed in this study can be obtained from the corresponding author upon reasonable request.
Ethical Approval
This study conformed to the declaration of Helsinki. The study protocol was approved by the Ethics Committee of the First Affiliated Hospital of Lanzhou University (LDYYLL-2025-82). All animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, 2011). The MOLM13 cell line used in this study was approved by the ethics committee of the first hospital of Lanzhou University.
Acknowledgments
We are grateful to Dr. Shuling Zhang of Lanzhou University First Hospital for providing the MOLM13 cell line.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Gansu Province, China (24JRRA297).
Disclosure
The authors declare no competing interests in this work.
References
1. Forsberg M, Konopleva M. AML treatment: conventional chemotherapy and emerging novel agents. Trends Pharmacol Sci. 2024;45(5):430–448. doi:10.1016/j.tips.2024.03.005
2. Godfrey LC, Rodriguez-Meira A. Viewing AML through a new lens: technological advances in the study of epigenetic regulation. Cancers. 2022;14(23):5989. doi:10.3390/cancers14235989
3. Fu DJ, Wang T. Targeting NEDD8-activating enzyme for cancer therapy: developments, clinical trials, challenges and future research directions. J Hematol Oncol. 2023;16(1):87. doi:10.1186/s13045-023-01485-7
4. Zheng B, Qian F, Wang X, Wang Y, Zhou B, Fang L. Neddylation activated TRIM25 desensitizes triple-negative breast cancer to paclitaxel via TFEB-mediated autophagy. J Exp Clin Cancer Res. 2024;43(1):177. doi:10.1186/s13046-024-03085-w
5. Zhang Y, Du L, Wang C, et al. Neddylation is a novel therapeutic target for lupus by regulating double negative T cell homeostasis. Signal Transduct Target Ther. 2024;9(1):18. doi:10.1038/s41392-023-01709-9
6. Murthy GSG, Saliba AN, Szabo A, et al. A Phase I study of pevonedistat, azacitidine, and venetoclax in patients with relapsed/refractory acute myeloid leukemia. Haematologica. 2024;109(9):2864–2872. doi:10.3324/haematol.2024.285014
7. Short NJ, Muftuoglu M, Ong F, et al. A Phase 1/2 study of azacitidine, venetoclax and pevonedistat in newly diagnosed secondary AML and in MDS or CMML after failure of hypomethylating agents. J Hematol Oncol. 2023;16(1):73. doi:10.1186/s13045-023-01476-8
8. Adès L, Girshova L, Doronin VA, et al. Pevonedistat plus azacitidine vs azacitidine alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast-percentage AML. Blood Adv. 2022;6(17):5132–5145. doi:10.1182/bloodadvances.2022007334
9. Saliba AN, Kaufmann SH, Stein EM, et al. Pevonedistat with azacitidine in older patients with TP53-mutated AML: a Phase 2 study with laboratory correlates. Blood Adv. 2023;7(11):2360–2363. doi:10.1182/bloodadvances.2022008625
10. Fathi AT. Pevonedistat, a new partner for 5-azacitidine. Blood. 2018;131(13):1391–1392. doi:10.1182/blood-2018-02-829051
11. Visconte V, Nawrocki ST, Espitia CM, et al. Comprehensive quantitative proteomic profiling of the pharmacodynamic changes induced by MLN4924 in acute myeloid leukemia cells establishes rationale for its combination with azacitidine. Leukemia. 2016;30(5):1190–1194. doi:10.1038/leu.2015.250
12. Handa H, Cheong JW, Onishi Y, et al. Pevonedistat in East Asian patients with acute myeloid leukemia or myelodysplastic syndromes: a phase 1/1b study to evaluate safety, pharmacokinetics and activity as a single agent and in combination with azacitidine. J Hematol Oncol. 2022;15(1):56. doi:10.1186/s13045-022-01264-w
13. Schoofs T, Müller-Tidow C. DNA methylation as a pathogenic event and as a therapeutic target in AML. Cancer Treat Rev. 2011;37:S13–S18. doi:10.1016/j.ctrv.2011.04.013
14. Zhang X, Zhang K, Zhang J, Chang W, Zhao Y, Suo X. DNMTs-mediated SOCS3 methylation promotes the occurrence and development of AML. Eur J Haematol. 2024;112(3):439–449. doi:10.1111/ejh.14134
15. Li T, Gao R, Xu K, et al. BCL7A inhibits the progression and drug-resistance in acute myeloid leukemia. Drug Resist Updat. 2024;76:101120. doi:10.1016/j.drup.2024.101120
16. Patiño-Mercau JR, Baliñas-Gavira C, Andrades A, et al. BCL7A is silenced by hypermethylation to promote acute myeloid leukemia. Biomark Res. 2023;11(1):32. doi:10.1186/s40364-023-00472-x
17. Zhang ZH, Zhang W, Zhou JD, et al. Decreased SCIN expression, associated with promoter methylation, is a valuable predictor for prognosis in acute myeloid leukemia. Mol Carcinog. 2018;57(6):735–744. doi:10.1002/mc.22794
18. Huang W, Li H, Yu Q, Xiao W, Wang DO. LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond. J Exp Clin Cancer Res. 2022;41(1):100. doi:10.1186/s13046-022-02319-z
19. Hu X, Wu J, Feng Y, et al. METTL3-stabilized super enhancers-lncRNA SUCLG2-AS1 mediates the formation of a long-range chromatin loop between enhancers and promoters of SOX2 in metastasis and radiosensitivity of nasopharyngeal carcinoma. Clin Transl Med. 2023;13(9):e1361. doi:10.1002/ctm2.1361
20. Qiannan D, Qianqian J, Jiahui S, Haowei F, Qian X. LncRNA PVT1 mediates the progression of liver necroptosis via ZBP1 promoter methylation under nonylphenol exposure. Sci Total Environ. 2022;844:157185. doi:10.1016/j.scitotenv.2022.157185
21. Guo Y, Jian J, Tang X, Zhao L, Liu B. Comprehensive analysis of DNA methylation and gene expression to identify tumor suppressor genes reactivated by MLN4924 in acute myeloid leukemia. Anticancer Drugs. 2025;36(3):199–207. doi:10.1097/cad.0000000000001688
22. Qiu X, Huang Y, Zhou Y, Zheng F. Aberrant methylation of TRIM58 in hepatocellular carcinoma and its potential clinical implication. Oncol Rep. 2016;36(2):811–818. doi:10.3892/or.2016.4871
23. Kajiura K, Masuda K, Naruto T, et al. Frequent silencing of the candidate tumor suppressor TRIM58 by promoter methylation in early-stage lung adenocarcinoma. Oncotarget. 2017;8(2):2890–2905. doi:10.18632/oncotarget.13761
24. Gan Y, Cao C, Li A, et al. Silencing of the TRIM58 gene by aberrant promoter methylation is associated with a poor patient outcome and promotes cell proliferation and migration in clear cell renal cell carcinoma. Front Mol Biosci. 2021;8:655126. doi:10.3389/fmolb.2021.655126
25. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi:10.1093/bioinformatics/18.11.1427
26. Simonyan L, Renault TT, Novais MJ, et al. Regulation of Bax/mitochondria interaction by AKT. FEBS Lett. 2016;590(1):13–21. doi:10.1002/1873-3468.12030
27. Adler P, Kolde R, Kull M, et al. Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol. 2009;10(12):R139. doi:10.1186/gb-2009-10-12-r139
28. Mas-Ponte D, Carlevaro-Fita J, Palumbo E, Hermoso Pulido T, Guigo R, Johnson R. LncATLAS database for subcellular localization of long noncoding RNAs. Rna. 2017;23(7):1080–1087. doi:10.1261/rna.060814.117
29. Zhang T, Tan P, Wang L, et al. RNALocate: a resource for RNA subcellular localizations. Nucleic Acids Res. 2017;45(D1):D135–d138. doi:10.1093/nar/gkw728
30. Wong KK, Lawrie CH, Green TM. Oncogenic roles and inhibitors of DNMT1, DNMT3A, and DNMT3B in acute myeloid leukaemia. Biomark Insights. 2019;14:1177271919846454. doi:10.1177/1177271919846454
31. Sun N, Shen J, Shi Y, et al. TRIM58 functions as a tumor suppressor in colorectal cancer by promoting RECQL4 ubiquitination to inhibit the AKT signaling pathway. World J Surg Oncol. 2023;21(1):231. doi:10.1186/s12957-023-03124-4
32. Shang R, Chen J, Gao Y, Chen J, Han G. TRIM58 interacts with ZEB1 to suppress NSCLC tumor malignancy by promoting ZEB1 protein degradation via UPP. Dis Markers. 2023;2023:5899662. doi:10.1155/2023/5899662
33. Li M, Jiao L, Shao Y, et al. LncRNA-ZFAS1 promotes myocardial ischemia-reperfusion injury through DNA methylation-mediated Notch1 down-regulation in mice. JACC Basic Transl Sci. 2022;7(9):880–895. doi:10.1016/j.jacbts.2022.06.004
34. Zhou H, Zhang Q, Huang W, et al. Epigenetic silencing of ZCCHC10 by the lncRNA SNHG1 promotes progression and venetoclax resistance of acute myeloid leukemia. Int J Oncol. 2023;62(5):64. doi:10.3892/ijo.2023.5512
35. Li H, Tian X, Wang P, et al. LINC01128 resisted acute myeloid leukemia through regulating miR-4260/NR3C2. Cancer Biol Ther. 2020;21(7):615–622. doi:10.1080/15384047.2020.1740054
36. Wang K, Reichermeier KM, Liu X. Quantitative analyses for effects of neddylation on CRL2(VHL) substrate ubiquitination and degradation. Protein Sci. 2021;30(11):2338–2345. doi:10.1002/pro.4176
37. Zhou W, Xu J, Tan M, et al. UBE2M is a stress-inducible dual E2 for neddylation and ubiquitylation that promotes targeted degradation of UBE2F. Mol Cell. 2018;70(6):1008–1024.e6. doi:10.1016/j.molcel.2018.06.002
38. Xiong X, Cui D, Bi Y, Sun Y, Zhao Y. Neddylation modification of ribosomal protein RPS27L or RPS27 by MDM2 or NEDP1 regulates cancer cell survival. FASEB J. 2020;34(10):13419–13429. doi:10.1096/fj.202000530RRR
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.