Back to Archived Journals » Research and Reports in Biology » Volume 6
Mitochondrial dysfunction in breast cancer
Authors Riar AK, Kenny T, Takabatake Y, Papa L, Germain D
Received 22 April 2015
Accepted for publication 9 July 2015
Published 11 September 2015 Volume 2015:6 Pages 137—145
DOI https://doi.org/10.2147/RRB.S62507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Zvi Kelman
Amanjot Kaur Riar,* Timothy C Kenny,* Yukie Takabatake,* Luena Papa, Doris Germain
Division of Hematology/Oncology, Icahn School of Medicine at Mount Sinai, Tisch Cancer Institute, New York, NY, USA
*These authors have contributed equally to this work
Abstract: The reprogramming of the mitochondria of cancer cells is essential for their growth. However, such reprogramming leads to the increased formation of reactive oxygen species, which can lead to damage to the organelle. This review summarizes the mechanisms that protect the mitochondrial network against oxidative stress. Further, since several transcription factors have now been linked to the mitochondrial unfolded protein response (UPRmt) and regulate the expression of one or more genes involved in these "mito-protective" mechanisms, the UPRmt is a prime candidate to coordinate the maintenance of the integrity of the mitochondria under oxidative stress conditions.
Keywords: mitochondrial unfolded protein response (UPRmt), mitochondrial DNA, SOD2, reactive oxygen species, breast cancer, mitomiRs, mitochondrial protein quality controls
Introduction
The mitochondria are complex and fascinating organelles with unique features including the fact that they are the only organelle with two subcompartments, are dependent on both the nuclear and mitochondrial genomes and are highly dynamic. Mitochondrial dynamics results from the constant balance between fusion and fission of individual mitochondrion, creating a mitochondrial network.
Originally, the mitochondria were considered mainly for their role in the generation of ATP. Otto Warburg was the first to describe the elevated levels of lactate in cancer cells compared to normal cells, despite the presence of oxygen. This shift from oxidative phosphorylation to aerobic glycolysis for the production of ATP has led to the generalized misconception that the mitochondria of cancer cells are inactive. In marked contrast, it is now evident that the mitochondrial network of cancer cells is essential for their viability. For instance, deletion of mitochondrial DNA (mtDNA) severely reduces their growth. Therefore, far from being inactive, the mitochondrial dysfunction of cancer cells allows their reprogramming to generate metabolites that are critical for the production of amino acids and lipids necessary for the rapid growth of cancer cells. While the mitochondrial dysfunction discussed in this review applies to breast cancer, several aspects are not restricted to breast cancer and apply to all solid tumors.
Mitochondrial mutations in breast cancer
The metabolic reprogramming of the mitochondrial network of cancer cells is achieved through several mechanisms, including mutations in both nuclear genes encoding mitochondrial proteins and mtDNA.
The mtDNA encodes seven subunits of complex I of the electron transport chain, one subunit of complex III, three subunits of complex IV, and two subunits of complex V. In addition to these polypeptides, mtDNA encodes 22tRNA and 2rRNA. Since mtDNA is not protected by histones, it is more prone to acquiring mutations. Another feature of mtDNA is that approximately 2–10 copies are present per mitochondrion. Further, mutation of each copy may vary from one another. Homoplasmy refers to a state where all copies of mtDNA in a cell share identical sequences. However, once a somatic mutation is introduced, it can propagate either randomly or by positive selection. The latter case results in a heteroplasmic state, which after several rounds of replication can lead to the selection of a dominant clone in the population. As discussed later in this review, such selection can have important consequences on the response of cancer cells to treatment. Because mtDNA encodes for subunits of the electron transport chain, on one hand, mutations in these subunits affect the ability of electrons to be transported efficiently and result in the formation of reactive oxygen species (ROS). ROS, on the other hand, can promote further mutations in mtDNA. Such a vicious cycle supports the notion that mutations in mtDNA may be an early event in tumorigenesis. This has led to an interest in monitoring the number of mtDNA mutations as a prognostic marker of breast cancer.
While the sequencing of mtDNA in cancer has been of keen interest, the role of somatic mutation in mtDNA remains of debate. A recent study by McMahon and LaFramboise1 has used next-generation sequencing for the analysis of 99 breast cancer samples and their adjacent normal tissue. They found that somatic mutations were found in 73.9% of patients, with a total of 141 different mutations being identified.1 In general, mutations were found in individual patients with a few exceptions where the same mutation was found in more than one patient. Interestingly, all somatic mutations were found in a heteroplasmic state, with a heteroplasmy ranging from 5.7% to 97.4% (median was 44.1%).1 This finding raises the possibility that heteroplasmy may be used as a marker of clonal heterogeneity of individual tumors.
In term of genomic location of the somatic mutations, most (55.3%) were found in the protein-coding regions.1 NADH dehydrogenase 5 of complex I was the most frequently mutated gene. This observation supports the finding from another study.2 As sequencing was performed in several samples from both tumor and adjacent normal tissues, the comparison of these samples allowed the determination of whether mtDNA content increases or decreases in breast cancer. The results revealed a general decrease in mtDNA content.
Most importantly, this study also revealed an intriguing correlation between the number of mtDNA somatic mutations and overall survival. They found that patients with no or less than two mutations were doing significantly worse than patients carrying more than three mutations.1 One interpretation of the correlation between the number of mutations and survival is that on one hand, primary breast cancers carrying multiple mutations may have major defects in oxidative phosphorylation, elevated ROS, and as a result may be more sensitive to chemotherapy leading to good clinical outcome (Figure 1). On the other hand, primary breast cancers carrying few mutations in their mtDNA may have only minor defects in oxidative phosphorylation, moderate levels of ROS, leading to increased resistance to chemotherapy and poorer clinical outcomes (Figure 1). Further, mutations in the mtDNA affecting specifically NADH dehydrogenase 6 subunit have been shown to promote metastasis3–5 in breast cancer cell lines, although the mechanism remains unclear. However, since McMahon and LaFramboise’s1 study has revealed an array of somatic mutations rather than hot spots, whether specific mutations in NADH dehydrogenase 6 holds true in human breast cancers remains to be determined in the future.
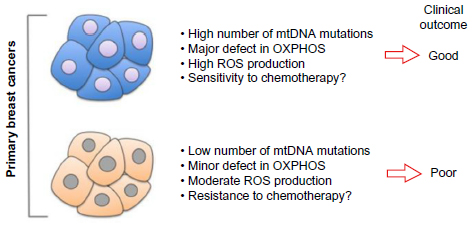 | Figure 1 Number of mutations in mtDNA correlates with clinical outcome. |
In addition to mutations in mtDNA, several mutations in nuclear DNA that encoded mitochondrial enzymes have been reported. These have been extensively reviewed in detail elsewhere.6 For example, mutations in succinate dehydrogenase, isocitrate dehydrogenase 1 and 2, and fumarate hydratase result in elevation in metabolites that contribute to cellular proliferation.6
Therefore, mutations in mitochondrial proteins encoded by both nuclear and mitochondrial genomes contribute to elevation of ROS levels in cancer cells. While increase in ROS promotes proliferation, ROS also leads to oxidation of proteins and their misfolding. Since mitochondria are the main source of ROS and also the main target of ROS, this organelle is prone to proteotoxic stress. As such, mechanisms must exist to limit ROS and the accumulation of misfolded proteins in the mitochondria. We will now describe some of these mechanisms.
Protein quality controls
The mitochondrial matrix contains proteases (LonP, ClpXP, AAA-proteases) and chaperones (hsp10, hsp60) that act to either refold or degrade unassembled or damaged proteins.7–10 The importance of these proteases is best illustrated by the fact that mutations in these proteases are associated with several neurological defects.10
Cancer cells overexpress the LonP protease indicating that it may be an important player against proteotoxic stress.11,12 One of the best characterized substrate of the LonP protease is the oxidized formed of aconitase 2, an enzyme involved in the citric acid cycle. The severity of the oxidation is critical, however, since excessive oxidation of aconitase 2 causes its aggregation and inhibits its degradation by LonP. Further, oxidative stress was reported to inactivate LonP itself.8,13 In addition to its role in proteolysis, the LonP protease associates with mtDNA and influences both accessibility to and replication of mtDNA.14–16 Therefore, these latter roles of the LonP protease may be critical in regulating the integrity of mtDNA as it is not protected by histones.
The m-AAA protease regulates the degradation of damaged or unassembled respiratory chain complexes as well as the regulation of mitochondrial dynamics through degradation of the GTPase OPA1.17–19
In addition to the proteases of the matrix, hsp90 is also overexpressed in cancer cells and localizes specifically to the matrix.9,20 The finding of the remarkable selectivity of mitochondrial hsp90 to cancer cells opens the possibility that inhibitors of hsp90 may represent a novel therapeutic option while sparing normal tissues.20 In support of the fact that disruption of mitochondrial hsp90 promotes cell death, hsp90 may be a viable target for cancer therapy.20
Hsp70 may also play a protective role as it localizes to the mitochondria where it has been reported to regulate mitochondrial protein import21 and prevent mitochondrial protein aggregation.22
In contrast to the matrix, the intermembrane space (IMS) does not contain chaperones. Folding activity toward selected proteins has been associated with Mia40.23 Further, the proteases that localize to the IMS are involved either in the processing of mitochondrial targeting signals in an ATP-dependent manner or cleavage of peptides generated by the processing proteases.24 The i-AAA-protease, which faces the IMS, acts mainly on protein assembly of the respiratory chain complex in the inner membrane.24 The protease Omi/HtrA2 appears to be the only protease in the IMS that has the ability to cleave misfolded proteins. If so, this observation suggests that compared to the matrix, the IMS has limited proteolytic activity against misfolded proteins.25 Since the outer membrane of the mitochondria contains several ubiquitin ligases,26 it is tempting to postulate that similar to the endoplasmic reticulum (ER), these ubiquitin ligases are involved in the elimination of misfolded proteins. ER-associated degradation promotes the ubiquitination and retrotranslocation of misfolded proteins from the lumen of the ER into the cytoplasm for their degradation by the proteasome. While evidence supporting the possibility of mitochondria-associated degradation has been reported,27 data obtained in our group favor the view that the effect of the proteasome on the protein quality control of the IMS proteins takes place prior to their import.25 Since we also found that Omi promotes the degradation of misfolded proteins in the IMS, we proposed a two-step checkpoint where the proteasome plays a major primary role in limiting the import of misfolded proteins in the IMS. But, if proteins escape this first step and enter the IMS, then as a secondary step, Omi promotes their degradation.25 It remains entirely possible, however, that the proteasome plays a role in both pre- and postimport mechanisms such as mitochondria-associated degradation.
Collectively these findings indicate that the overexpression of the proteasome, chaperones, and proteases may play a critical role in protecting the mitochondria of cancer cells against proteotoxic stress.28 The importance of these proteases and chaperones is further supported by the fact that, as will be described in “The antioxidant machinery” section, the antioxidant machinery of the matrix is downregulated in breast cancer.
The antioxidant machinery
The matrix manganese dismutase SOD2 promotes the conversion of superoxide to hydrogen peroxide, and its activity is regulated by acetylation.29 The matrix deacetylase SIRT3 deacetylates SOD2 and promotes its activation.29 Therefore, SIRT3 and SOD2 represent an important mechanism to limit the accumulation of superoxide in the matrix, and consequently, the oxidation of proteins.30
The expression of SIRT3 is reduced in the vast majority of breast cancers, and the decrease in SIRT3 was proposed to be essential for the metabolic reprogramming of cancer cells toward glycolysis.31 We reported that SOD2 is also reduced in breast cancers.32,33 Therefore, the low expression of both SIRT3 and SOD2 contribute to elevation in ROS levels in the mitochondrial matrix of cancer cells.
While superoxide produced by complex I localizes to the matrix, complex III produced superoxide in both the matrix and the IMS. The observation that the IMS is more oxidized than either the cytoplasm or the matrix suggests that as for protein quality control, the antioxidant capacity of the IMS is limited and relies on SOD1. Factors contributing to this limitation include the reduced diffusion of reduced glutathione over the outer membrane and the contribution of oxidative folding by the Erv1-Mia40 disulfide relay machinery to the generation of additional ROS.24
One additional factor that may also contribute to the oxidized milieu of the IMS is the proposed peroxidase activity of cytochrome c. Hydrogen peroxide produced by SOD1 can act as a substrate for the peroxidase activity of cytochrome c and be converted to the highly toxic peroxidase compound 1.34
Further, oxidative folding of proteins in the IMS promotes the formation of hydrogen peroxide,35,36 and misfolding of proteins stimulates the peroxidase activity of cytochrome c.34,37 Strikingly, numerous proteins that localize to the IMS contain multiple cysteine residues and therefore are prone to the formation of abnormal disulfide bonds.24
We previously reported that SOD1 is overexpressed in breast cancer and that the mitochondrial fraction is increased.33 We proposed that the increase in SOD1 may counterbalance the decrease in SOD2 in the matrix and thereby limit the increase in ROS to the matrix.
Mitophagy and breast cancer
The role of autophagy in general in cancer biology has been intensively studied and debated over the last decade. Based on the data from various cancer types and stages of progression, the picture that emerges is that the role of autophagy may be different at different times during tumor progression. Initially, autophagy may be suppressive of tumor formation. For instance, deletion of the autophagy gene Beclin in mice accelerates Wnt-1 driven mammary tumor formation.38 However, in established tumors, efficient autophagy may be essential to support survival of cancer cells in an adverse microenvironment. Adding to this complexity is the fact that the effect of autophagy may be tissue specific and therefore affect different tumor types differently. This topic has been extensively reviewed recently.39
Mitophagy, or selective autophagy of mitochondria, may also affect the growth of cancer differently depending on the tissue of origin. For instance, defect in Atg7 led to the conversion of aggressive adenocarcinoma of the lung to benign oncocytomas, characterized by excessive mitochondrial mass.40 Oncocytic tumors of the thyroid, kidney, salivary glands, adrenal cortex, and parathyroid glands are most frequently benign.41 In contrast, oncocytic and mitochondria-rich breast cancers were found to be associated with worst prognosis.42 This finding raises the possibility that mitochondrial mass alone may not be an accurate measure of mitochondrial health. Increased content of damaged mitochondria is likely to lead to benign tumors, while increased content of healthy mitochondria is likely to increase resistance to therapy.
Mechanistically, the work from the Youle laboratory has led to major advances in our understanding of the role of PINK and Parkin in the elimination of damaged mitochondria from the network.43–48 More recently, the phosphorylation of ubiquitin chain at the mitochondrial surface was found to act as a receptor of Parkin to damaged mitochondria.49
While a detailed review of mitophagy is beyond the focus of this review, it is nevertheless essential to emphasis the importance of mitophagy in the context of its role in the quality control of the mitochondrial network. At the molecular level, the selective elimination of irreversibly damaged mitochondrion from the mitochondrial network can be viewed as a complementary mechanism to protein quality control and the antioxidant machinery.
microRNAs in mitochondria and breast cancer
One of the latest and most exciting developments in terms of regulation of the activity of the mitochondria is the finding that microRNAs (miRNAs) can not only regulate the translation of mitochondrial proteins that are encoded by the nuclear genome, but that miRNAs are found in the organelle itself.
miRNAs are transcribed by RNA polymerase II from the nuclear genome as a primary miRNA transcript (pri-miRNA). The 1,000 nucleotides (nt) long pri-miRNA are then processed into an approximately 70 nt precursor miRNA (pre-miRNA) by the Dorsha/Pasha microprocessor complex in the nucleus. Export of the pre-miRNA is achieved by the recognition of the 2 nt overhang by exportin-5. Once in the cytoplasm, the RNA-induced silencing complex ribonucleoprotein processes the pre-miRNA into mature miRNA (for review, refer Ambros50). Typically, miRNAs decrease translation of their target mRNAs by either promoting the degradation of the mRNA or inhibiting the translation. Each miRNA can affect multiple target genes, and the repression of translation is tightly regulated to fine-tune the level of mRNA/protein in a tissue- and condition-specific manner. Therefore, the complexity of miRNAs offers a possible explanation for the pathogenesis of multifactorial diseases such as cancer.
In terms of the mitochondria, miRNA can affect the mitochondria in three distinct ways; first, miRNA encoded by the nuclear genome targeting nuclear-genome-encoded mitochondrial proteins through the repression of the translation of their mRNA in the cytoplasm. These miRNAs therefore affect the level of import of specific mitochondrial proteins into the mitochondria. Second, miRNAs encoded by the nuclear genome but imported in the mitochondria to regulate the translation of mitochondria-genome-encoded proteins. Third, miRNA encoded by mitochondria-genome affecting translation of mitochondria-genome-encoded mitochondria proteins. miRNAs that localize to the mitochondria are termed mitomiRs.51–53 The mechanism by which mitomiRs are imported in the mitochondria remains unclear, but it has been postulated that the import may involve the same translocases involved in the import of mitochondrial proteins.
Since an extensive review of the functions of each miRNA affecting the mitochondria is beyond the scope of this review, only a few miRNA will be discussed here as to illustrate the extent by which they can affect the mitochondria. miRNA-149 affects mitochondria biogenesis by inhibiting poly(ADP-ribose) polymerase-2 (PARP-2). As PARP-2 activity is dependent on NAD+, the decrease in PARP-2 results in an increase in NAD+ leading to the activation of sirtuin-1 (SIRT1), which is also dependent on NAD+ for its activity. SIRT1 is required for the activity of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1 α), which in turn is responsible for the transcription of several genes involved in mitochondrial biogenesis. Oxidative phosphorylation, ROS production, antioxidant machinery, as well as fusion/fission of the mitochondria are also regulated by miRNA.53 Interestingly, miRNAs were reported to increase rather than decrease mitochondrial protein translation.54
The characterization of miRNA in the regulation of the mitochondria of cancer cells specifically is an emerging field. miR23b and miR-210 affect ROS production and were identified in renal, breast and colorectal cancers, respectively (for review, refer Duarte et al53). In term of breast cancer, the only miRNA that has been characterized is miR-200a.
The miR200 family of miRNAs is transcribed by two chromosomal locations miR-200a and miR-200b. miR-200a was found to bind directly to the mRNA of the mitochondrial transcription factor TFAM and reduce its levels. miR-200a is reduced in breast cancer compared with normal tissue, while TFAM is increased.55 Since TFAM has a dual role in mtDNA replication and transcription, it is considered a key player in the maintenance of the integrity of the mitochondrial genome in cancer cells.
The functions of the mitomiRs in general remain largely unknown, and even less is known about their role in cancer, including breast cancer. Barrey et al56 have identified 243 miRNA in the mitochondrial RNA fraction. Intriguingly, three miRNAs, miR-146a, miR-103, and miR-16, were found in the IMS. Considering the small size of the mitochondrial genome and the limited number of mitochondrial-encoded mRNA, the task of identifying the targets of mitomiRs appears to be within reach. The discovery of their targets represents a novel and exciting new avenue of mitochondrial research. In addition, it is important to note that the expression pattern of mitomiRs may be tissue specific. In their study, Barrey et al56 identified mitomiRs that are enriched in myoblasts, while another study reported cardiac-specific mitomiRs.57 Therefore, it is possible that some mitomiRs may be expressed specifically in the breast. However, as our current knowledge of the mitomiRs is still in its infancy, more research is needed to determine their role in breast cancer.
Summary of the “mito-protective” mechanisms against oxidative stress
The collective array of mutations affecting mitochondrial proteins encoded by both the nuclear and mitochondrial genomes is critical in promoting the reprogramming of the mitochondrial network of cancer cells. The resulting increase in oxidative stress caused by this reprogramming, however, puts the mitochondria at risk of irreversible damage. The mechanisms that allow the mitochondria to be protected from such damage include: 1) the protein quality control, chaperones, proteases, and the proteasome, 2) the antioxidant machinery, 3) mitochondrial biogenesis and oxidative phosphorylation, 4) mitophagy, and possibly 5) miRNA-mediated alteration in mitochondrial functions (Figure 2).
 | Figure 2 Summary of the protective mechanisms that protect the mitochondria from the damage of oxidative stress. |
The question that remains is what is the mechanism by which several components of these “mito-protective” outcomes are simultaneously upregulated in cancer cells. The transcriptional program activated by mitochondrial unfolded protein response (UPRmt) is a prime candidate toward achieving this goal.
The UPRmt orchestrates mitochondrial recovery
Identification of the UPRmt
The effect of the accumulation of misfolded proteins in the mitochondrial matrix was first described by Martinus et al.58 Their pioneer study of UPRmt used the expression of OTC delta, which misfolded and formed aggregates in the mitochondrial matrix. They reported that the resulting proteotoxic stress leads to the activation of the transcription factor CHOP and the subsequent expression of matrix LonP protease and the hsp10 and 60 chaperones.59 They further described that CHOP binding to the promoter of these genes required the presence of AP-1.60–62 Since CHOP is implicated in ER-stress, but none of the UPRER genes are activated by matrix proteotoxic stress, the binding to AP-1 offers a potential explanation for the dual function of CHOP in both mitochondrial and ER-stress. Martinus et al58 is also the first to have identified the endonuclease G (EndoG), which resides in the IMS of the mitochondria as a gene upregulated by UPRmt. While the identification of EndoG remained enigmatic at that time, EndoG has subsequently been identified as a transcriptional target of ERRα.63 As ERRα/PGC-1α is a major transcriptional complex involved in the regulation of a large number of mitochondrial genes involved in biogenesis and oxidative phosphorylation, their findings also suggested that ERRα/PGC-1α is also activated by mitochondrial proteotoxic stress.
Using the same OTC delta model, we subsequently reported that mitochondrial proteotoxic stress activates the expression of the matrix deacetylase SIRT3.64 We found that downstream of SIRT3, the transcription factor Foxo3A and its transcriptional target SOD2 and catalase are activated.64 Our results indicate that induction of SOD2 and catalase is dependent on SIRT3, but not affected by the inhibition of CHOP. Conversely, inhibition of SIRT3 had no effect on the level of LonP, hsp10, or hsp60. We therefore concluded that SIRT3 and CHOP represent distinct branches of UPRmt upon accumulation of mutant OTC in the matrix.
This work also led to the finding, using electron microscopy of cells undergoing matrix proteotoxic stress, that a fraction of the mitochondrion was targeted for elimination by mitophagy.64 In agreement with this finding, we found that several markers of mitophagy were induced under these conditions. Since SIRT3, but not CHOP, was found to be essential for the induction of these markers, we conclude that the SIRT3 branch of UPRmt has the dual role of activating the antioxidant machinery and mitophagy. This finding raises the possibility that SIRT3 may play an important role in a mitochondrial checkpoint where a moderately stressed mitochondrion may be repaired through an upregulation of the antioxidant machinery while irreversibly damaged organelles are selectively eliminated from the network by mitophagy. If so, the question that emerges is what distinguishes moderately from irreversibly damaged mitochondrion. Work from the Jin and Youle study44 indicates that accumulation of misfolded OTC in the matrix of the mitochondria leads to the accumulation of PINK and translocation of the ubiquitin ligase PARK2 to the mitochondria, therefore offering a potential mechanism of selection.
In addition to CHOP, Foxo3α, and PGC-1α, we also reported that the estrogen receptor alpha (ERα) plays a role in UPRmt by regulating the activity of the proteasome and the IMS protease Omi.65
Further, the work from Benedetti et al66 and Haynes et al67 has led to the identification of several players of UPRmt in Caenorhabditis elegans. Using chromatin immunoprecipitation (ChIP)-sequencing, they reported that the transcription factor ATFS-1 bonds directly to the promoters of oxidative phosphorylation genes in both the nuclear and mitochondrial genomes and therefore promotes oxidative phosphorylation.68 They found that ATFS-1 also upregulates chaperones, antioxidant machinery, and glycolysis.68 Mechanistically, ATFS-1 is normally found in the mitochondria and degraded. However, upon accumulation of misfolded proteins in the mitochondria, ATFS-1 gets imported into the nucleus to promote transcription of several mitochondrial genes.
These findings indicate that mitochondrial proteotoxic stress activates an array of transcription factors that collectively result in the upregulation of all “mito-protective” outcomes (Figure 3).
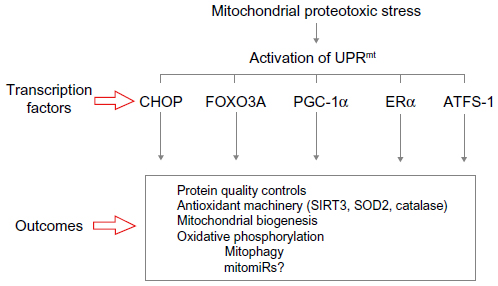 | Figure 3 Diagram of how mitochondrial stress leads to the activation of UPRmt. |
Since the expression of SIRT3 and SOD2 were reported to be decreased in breast cancer, how can these observations be reconciled with the activation of the UPRmt? We propose a dual role of SIRT3 and SOD2 in breast cancer. On the one hand, a decrease in their expression is necessary to increase the levels of ROS to moderate levels and assist the metabolic reprogramming during malignant transformation. On the other hand, when levels of ROS reach elevated levels, UPRmt is activated and induces the expression of SIRT3 and SOD2. We propose that by activating the antioxidant machinery and mitophagy, SIRT3 acts to lower ROS levels under a critical threshold so that ROS levels are kept in the moderate range so that cellular viability is maintained.
Overexpression of SOD1 has an additional mitochondria-protective mechanism
While we found that SOD1 is overexpressed in breast cancer, we also found that the transcription of SOD1 is not increased by UPRmt. Therefore, the elevation in SOD1 may result from a posttranscriptional effect. However, since SOD1 accumulates in the IMS, it may act to activate the UPRmt. We therefore proposed that SOD1 may have two protective functions: first, a dismutase-dependent function to reduce ROS, and second, a dismutase-independent function to activate UPRmt.32
Mitochondrial biomarkers for breast cancer progression
Sequencing of mtDNA to either predict outcome or monitor progression has been the focus of several studies and has been extensively reviewed recently.69 Mutations in mtDNA including point mutation, deletion, insertion changes in copy number as well as homoplasmy and heteroplasmy have been analyzed.69 More specific focus on the prognostic value of mutations in genes encoding subunits of the electron transport chain complexes have also been analyzed.69 Polymorphism in NADH dehydrogenase 3 has been linked to increased risk of breast cancer.69 But other studies did not find an association.69
More recently, high throughput deep sequencing has facilitated the analysis of mtDNA to trace clonal heterogeneity in tumors including breast cancer. The advance of this new technology allows to uncover the fact that different metastatic sites are populated by different clones. However, how selection of mitochondrial clones affects the behavior of the tumors remains to be determined. Most likely, based on the work of LaFramboise’ s group, clones with elevated levels of mutations are likely to respond better to treatment, while clones with few mutations and a trend toward low level of heteroplasmy are likely to be more resistant to treatment.
Conclusion
The term mitochondrial dysfunction is widely used in the literature. Dysfunction however has led to the general misconception that mitochondria of cancer cells are dispensable. Metabolic reprogramming of the mitochondria is now well established as being critical for the survival of cancer cell. Therefore, mitochondrial dysfunction should be used with the understanding that such dysfunction is necessary for metabolic reprogramming.
More recent advances now suggest that the reprogramming may in fact be reversible and may vary depending on the stage of the disease, and therefore also play a role in the adaptation of cancer cells to the changing microenvironment. In support of this possibility, several studies indicated that oxidative phosphorylation and mitochondrial biogenesis are upregulated in different models of recurrent tumors70,71 or circulating cancer cells.72 Therefore, whether mitochondrial metabolism and morphology changes with progressive disease conditions represent an important aspect of mitochondrial biology that remains to be determined. These findings indicate that we have yet much more to learn about the complex role of mitochondria in cancer biology.
Targeting the mitochondria for cancer therapy has gained a lot of interest in the last few years.73–78 In view of several exciting new developments regarding the role of mitochondria in breast cancer, this interest is likely to continue to grow.
Disclosure
The authors report no conflicts of interest in this work.
References
McMahon S, LaFramboise T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis. 2014;35:1046–1054. doi:10.1093/carcin/bgu012. | |
Shen L, Wei J, Chen T, et al. Evaluating mitochondrial DNA in patients with breast cancer and benign breast disease. J Cancer Res Clin Oncol. 2011;137:669–675. doi:10.1007/s00432-010-0912-x. | |
He X, Zhou A, Lu H, et al. Suppression of mitochondrial complex I influences cell metastatic properties. PLoS One. 2013;8:e61677. doi:10.1371/journal.pone.0061677. | |
Ishikawa K, Takenaga K, Akimoto M, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi:10.1126/science.1156906. | |
Santidrian AF, Matsuno-Yagi A, Ritland M, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068–1081. doi:10.1172/JCI64264. | |
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi:10.1038/nrc3365. | |
Arnold I, Langer T. Membrane protein degradation by AAA proteases in mitochondria. Biochim Biophys Acta. 2002;1592:89–96. | |
Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–680. | |
Altieri DC. Mitochondrial HSP90s and tumor cell metabolism. Autophagy. 2013;9:244–245. doi:10.4161/auto.22527. | |
Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi:10.1038/sj.emboj.7601972. | |
Zhu Y, Wang M, Lin H, et al. Epidermal growth factor up-regulates the transcription of mouse lon homology ATP-dependent protease through extracellular signal-regulated protein kinase- and phosphatidylinositol-3-kinase-dependent pathways. Exp Cell Res. 2002;280:97–106. | |
Bernstein SH, Venkatesh S, Li M, et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119:3321–3329. doi:10.1182/blood-2011-02-340075. | |
Stanyer L, Jorgensen W, Hori O, Clark JB, Heales SJ. Inactivation of brain mitochondrial Lon protease by peroxynitrite precedes electron transport chain dysfunction. Neurochem Int. 2008;53:95–101. doi:10.1016/j.neuint.2008.06.004. | |
Lu B, Yadav S, Shah PG, et al. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J Biol Chem. 2007;282:17363–17374. doi:10.1074/jbc.M611540200. | |
Liu T, Lu B, Lee I, et al. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J Biol Chem. 2004;279:13902–13910. doi:10.1074/jbc.M309642200. | |
Lu B, Lee J, Nie X, et al. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol Cell. 2013;49:121–132. doi:10.1016/j.molcel.2012.10.023. | |
Ehses S, Raschke I, Mancuso G, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi:10.1083/jcb.200906084. | |
Duvezin-Caubet S, Koppen M, Wagener J, et al. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell. 2007;18:3582–3590. doi:10.1091/mbc.E07-02-0164. | |
Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi:10.1038/sj.emboj.7601184. | |
Siegelin MD, Dohi T, Raskett CM, et al. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest. 2011;121:1349–1360. doi:10.1172/JCI44855. | |
Kang PJ, Ostermann J, Shilling J, et al. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990;348:137–143. doi:10.1038/348137a0. | |
Liu Q, Krzewska J, Liberek K, Craig EA. Mitochondrial Hsp70 Ssc1: role in protein folding. J Biol Chem. 2001;276:6112–6118. doi:10.1074/jbc.M009519200. | |
Banci L, Bertini I, Cefaro C, et al. MIA40 is an oxidoreductase that catalyzes oxidative protein folding in mitochondria. Nat Struct Mol Biol. 2009;16:198–206. doi:10.1038/nsmb.1553. | |
Herrmann JM, Riemer J. The intermembrane space of mitochondria. Antioxid Redox Signal. 2010;13:1341–1358. doi:10.1089/ars.2009. 3063. | |
Radke S, Chander H, Schäfer P, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. | |
Germain D. Ubiquitin-dependent and -independent mitochondrial protein quality controls: implications in ageing and neurodegenerative diseases. Mol Microbiol. 2008;70:1334–1341. doi:10.1111/j. 1365–2958.2008.06502.x. | |
Bragoszewski P, Gornicka A, Sztolsztener ME, Chacinska A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol Cell Biol. 2013;33:2136–2148. doi:10.1128/MCB.01579-12. | |
Goard CA, Schimmer AD. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene. 2014;33:2690–2699. doi:10.1038/onc.2013.228. | |
Tao R, Coleman MC, Pennington JD, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi:S1097-2765(10)00969-X [pii] 10.1016/j.molcel.2010.12.013. | |
Lombard DB, Tishkoff DX, Bao J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb Exp Pharmacol. 2011;206:163–188. doi:10.1007/978-3-642-21631-2_8. | |
Finley LW, Carracedo A, Lee J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19:416–428. doi:10.1016/j.ccr.2011.02.014. | |
Papa L, Manfredi G, Germain D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer. 2014;5:15–21. | |
Papa L, Hahn M, Marsh EL, Evans BS, Germain D. SOD2 to SOD1 switch in breast cancer. J Biol Chem. 2014;289:5412–5416. doi:10.1074/jbc.C113.526475. | |
Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, et al. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J Biol Chem. 2008;283:8446–8452. doi:M706111200 [pii] 10.1074/jbc.M706111200. | |
Sideris DP, Tokatlidis K. Trapping oxidative folding intermediates during translocation to the intermembrane space of mitochondria: in vivo and in vitro studies. Methods Mol Biol. 2010;619:411–423. doi:10.1007/978-1-60327-412-8_25. | |
Deponte M, Hell K. Disulphide bond formation in the intermembrane space of mitochondria. J Biochem. 2009;146:599–608. doi:mvp133 [pii] 10.1093/jb/mvp133. | |
Diederix RE, Ubbink M, Canters GW. Peroxidase activity as a tool for studying the folding of c-type cytochromes. Biochemistry. 2002;41: 13067–13077. doi:bi0260841 [pii]. | |
Cicchini M, Chakrabarti R, Kongara S, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10:2036–2052. doi:10.4161/auto.34398. | |
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi:10.15252/embj.201490784. | |
Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461. doi:10.1101/gad.219642.113. | |
Maximo V, Rios E, Sobrinho-Simoes M. Oncocytic lesions of the thyroid, kidney, salivary glands, adrenal cortex, and parathyroid glands. Intl J Surg Pathol. 2014;22:33–36. doi:10.1177/1066896913517938. | |
Ragazzi M, de Biase D, Betts CM, et al. Oncocytic carcinoma of the breast: frequency, morphology and follow-up. Hum Pathol. 2011;42:166–175. doi:10.1016/j.humpath.2010.07.014. | |
Hasson SA, Kane LA, Yamano K, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi:10.1038/nature12748. | |
Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi:10.4161/auto.26122. | |
Lazarou M, Narendra DP, Jin SM, et al. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013;200:163–172. doi:10.1083/jcb.201210111. | |
Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. doi:10.1242/jcs.093849. | |
Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi:10.1016/j.devcel.2011.12.014. | |
Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012;4(11):a011338. doi:10.1101/cshperspect.a011338. | |
Okatsu K, Koyano F, Kimura M, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209(1):111–128. doi:10.1083/jcb.201410050. | |
Ambros V. The functions of animal microRNAs. Nature. 2004;431: 350–355. doi:10.1038/nature02871. | |
Bandiera S, Mategot R, Girard M, Demongeot J, Henrion-Caude A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic Biol Med. 2013;64:12–19. doi:10.1016/j.freeradbiomed.2013.06.013. | |
Sripada L, Tomar D, Singh R. Mitochondria: one of the destinations of miRNAs. Mitochondrion. 2012;12:593–599. doi:10.1016/j.mito.2012.10.009. | |
Duarte FV, Palmeira CM, Rolo AP. The role of microRNAs in mitochondria: small players acting wide. Genes. 2014;5:865–886. doi:10.3390/genes5040865. | |
Zhang X, Zuo X, Yang B, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi:10.1016/j.cell.2014.05.047. | |
Yao J, Zhou E, Wang Y, et al. microRNA-200a inhibits cell proliferation by targeting mitochondrial transcription factor A in breast cancer. DNA Cell Biol. 2014;33:291–300. doi:10.1089/dna.2013.2132. | |
Barrey E, Saint-Auret G, Bonnamy B, et al. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6:e20220. doi:10.1371/journal.pone.0020220. | |
Das S, Ferlito M, Kent OA, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. 2012;110:1596–1603. doi:10.1161/CIRCRESAHA.112.267732. | |
Martinus RD, Garth GP, Webster TL, et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240:98–103. | |
Zhao Q, Wang J, Levichkin IV, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. | |
Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PloS One. 2007;2:e874. doi:10.1371/journal.pone.0000874. | |
Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PloS One. 2007;2:e835. doi:10.1371/journal.pone.0000835. | |
Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–722. doi:10.1146/annurev.biochem. 76.052305.091720. | |
Ranhotra HS. Estrogen-related receptor alpha and mitochondria: tale of the titans. J Recept Signal Transduct Res. Epub 2014 Sep 15. doi:10.3109/10799893.2014.959592. | |
Papa L, Germain D. SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. 2014;34:699–710. doi:10.1128/MCB.01337-13. | |
Papa L, Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124:1396–1402. doi:10.1242/jcs.078220. | |
Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. | |
Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. | |
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58:123–133. doi:10.1016/j.molcel.2015.02.008. | |
Yadav N, Chandra D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim Biophys Acta. 2013;1836:336–344. doi:10. 1016/j.bbcan.2013.10.002. | |
Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi:10.1038/nature13611. | |
Hu J, Hwang SS, Liesa M, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148: 651–663. doi:10.1016/j.cell.2011.12.028. | |
LeBleu VS, O’Connell JT, Gonzalez Herrera KN, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992–1003, 1001–1015. doi:10.1038/ncb3039. | |
Altieri DC. Hsp90 regulation of mitochondrial protein folding: from organelle integrity to cellular homeostasis. Cell Mol Life Sci. 2013;70:2463–2472. doi:10.1007/s00018-012-1177-0. | |
Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2012;1823:767–773. doi:10.1016/j.bbamcr.2011.08.007. | |
Valtieri M, Venturelli D, Carè A, et al. Antisense myb inhibition of purified erythroid progenitors in development and differentiation is linked to cycling activity and expression of DNA polymerase alpha. Blood. 1991;77:1181–1190. | |
Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–464. doi:10.1038/nrd3137. | |
Lamb R, Harrison H, Hulit J, et al. Mitochondria as new therapeutic targets for eradicating cancer stem cells: quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014;5: 11029–11037. | |
Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6: 4569–4584. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.