Back to Journals » International Journal of Nanomedicine » Volume 12
Mithramycin-loaded mPEG-PLGA nanoparticles exert potent antitumor efficacy against pancreatic carcinoma
Authors Liu XJ ![]() , Li L, Liu XJ, Li Y, Zhao CY, Wang RQ, Zhen YS
, Li L, Liu XJ, Li Y, Zhao CY, Wang RQ, Zhen YS
Received 12 April 2017
Accepted for publication 26 June 2017
Published 24 July 2017 Volume 2017:12 Pages 5255—5269
DOI https://doi.org/10.2147/IJN.S139507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Xu-Jie Liu, Liang Li, Xiu-Jun Liu, Yi Li, Chun-Yan Zhao, Rui-Qi Wang, Yong-Su Zhen
Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
Abstract: Previous studies have shown that mithramycin A (MIT) is a promising candidate for the treatment of pancreatic carcinoma through inhibiting transcription factor Sp1. However, systemic toxicities may limit its clinical application. Here, we report a rationally designed formulation of MIT-loaded nanoparticles (MIT-NPs) with a small size and sustained release for improved passive targeting and enhanced therapeutic efficacy. Nearly spherical MIT-NPs with a mean particle size of 25.0±4.6 nm were prepared by encapsulating MIT into methoxy poly(ethylene glycol)-block-poly(D,L-lactic-co-glycolic acid) (mPEG-PLGA) nanoparticles (NPs) with drug loading of 2.11%±0.51%. The in vitro release of the MIT-NPs lasted for >48 h with a sustained-release pattern. The cytotoxicity of MIT-NPs to human pancreatic cancer BxPC-3 and MIA Paca-2 cells was comparable to that of free MIT. Determined by flow cytometry and confocal microscopy, the NPs internalized into the cells quickly and efficiently, reaching the peak level at 1–2 h. In vivo fluorescence imaging showed that the prepared NPs were gradually accumulated in BxPC-3 and MIA Paca-2 xenografts and retained for 168 h. The fluorescence intensity in both BxPC-3 and MIA Paca-2 tumors was much stronger than that of various tested organs. Therapeutic efficacy was evaluated with the poorly permeable BxPC-3 pancreatic carcinoma xenograft model. At a well-tolerated dose of 2 mg/kg, MIT-NPs suppressed BxPC-3 tumor growth by 96%. Compared at an equivalent dose, MIT-NPs exerted significantly higher therapeutic effect than free MIT (86% versus 51%, P<0.01). Moreover, the treatment of MIT and MIT-NPs reduced the expression level of oncogene c-Myc regulated by Sp1, and notably, both of them decreased the protein level of CD47. In summary, the novel formulation of MIT-NPs shows highly therapeutic efficacy against pancreatic carcinoma xenograft. In addition, MIT-NPs can downregulate CD47 expression, implying that it might play a positive role in cancer immunotherapy.
Keywords: mithramycin, pancreatic cancer, mPEG-PLGA nanoparticles, Sp1, CD47
Introduction
Pancreatic cancer is a major cause of cancer-associated mortality. In 2015, an estimated 367,000 new cases of pancreatic cancer were diagnosed worldwide and 359,000 people died from pancreatic cancer in the same year.1 In spite of the decline in mortality from the most common cancers in the past few decades, the mortality from pancreatic cancer has remained high. As the incidence of pancreatic cancer is rising globally, it is likely to become the second leading cause of cancer mortality in the near future. Pancreatic cancer is curable only in 5%–10% of the cases with localized and resectable tumors, and the 5-year survival rate is only 10%–20% after surgery.2 Several reasons lead to extremely poor prognosis of malignant neoplasms of the pancreas. Owing to perineural and vascular local growth and early distant metastases, pancreatic cancer is usually unresectable in most patients diagnosed at first. Effective chemotherapy is needed for patients who undergo surgical resection or are unable to have a curative surgery. However, pancreatic cancer is generally associated with a remarkable resistance to most conventional therapies.1 Thus, novel chemotherapeutic methods are urgently needed to treat pancreatic cancer.
Mithramycin A (MIT), also called plicamycin, is a natural aureolic acid-type polyketide isolated from various strains of streptomyces.3 MIT has been used clinically as a chemotherapeutic agent to treat several cancer types, including testicular embryonal carcinoma and glioblastoma, although systemic toxicities limited its clinical use. In recent years, there has been renewed interest in the capability of MIT to bind to the minor groove of guanine and cytosine-rich DNA regions, since pharmacologically mediated modulation of DNA/protein complex formation represents a promising approach to control gene expression. The main mechanism of MIT to inhibit tumor growth may be related to its action to block the binding of transcription factor Sp1 to gene regulatory elements.
Sp1, a zinc finger transcription factor, is overexpressed in a number of cancers and plays a critical role in the regulation of numerous genes that mediate proliferation, differentiation, DNA damage response, apoptosis, angiogenesis, invasion, and metastasis of cancer cells.4 MIT can reduce the expression level of oncogenes c-Myc and c-Src with the Sp1-binding site in the promoter.5,6 By blocking the binding of Sp1 to promoter, MIT has been shown to inhibit the transcriptional activity of MDM2, an important negative regulator of p53, and then lead to the activation of the p53 pathway.7 MIT was found to reduce drug resistance through downregulation of the xenobiotic pump ABCG2 and the multidrug resistant gene MDR1.8,9 It was also shown that MIT inhibited DNA methyltransferase (DNMT), histone deacetylase (HDAC), and antiapoptotic protein XIAP.10–12 Preclinical studies demonstrated that MIT can inhibit the generation and progression of several cancers, including carcinomas of the lung, breast, pancreas, stomach, and prostate, as well as sarcoma and glioblastoma.8,13–16 Meanwhile, its prominent in vitro and in vivo activities linked to specific transcription regulation have triggered the clinical trials of MIT in lung cancer, esophagus cancer, and Ewing sarcoma, sponsored by the National Cancer Institute (NCT01610570, NCT01624090, and NCT02859415, www.ClinicalTrials.gov).
Previous studies have shown that a high level of Sp1 identifies a subset of pancreatic ductal adenocarcinoma with aggressive clinical behavior and Sp1 overexpression increased the probability of cancer metastasis.17 MIT can exhibit an inhibitory effect on pancreatic cancer growth through distinct mechanisms of Sp1 inhibition. Treatment with MIT resulted in the inhibition of Sp1 recruitment onto vascular endothelial growth factor (VEGF) and transforming growth factor (TGF)-β type II receptor (TGF-β RII) promoters, leading to downregulation of VEGF and TGF-β RII protein expression in pancreatic cancer cells.15,18,19 Meanwhile, MIT can sensitize pancreatic cancer cells to TRAIL-induced apoptosis.20 In spite of promising preclinical findings, the clinical use of MIT, especially as an anticancer drug requiring higher doses, has been hampered by its systemic toxicity. In order to enhance its safety and efficacy, one of the feasible strategies is to develop efficient formulations of drug delivery systems.
Nanotechnology has attracted growing interest in cancer therapies due to its uniquely appealing features for drug delivery. The nanoparticle (NP) platforms for the targeted delivery of therapeutic drugs to solid tumors hold great promise for improving drug bioavailability and minimizing systemic toxicity. NPs are widely used as drug carriers because large molecules can avoid renal clearance and circulate in the body for prolonged time in comparison to small molecules. NPs accumulate in the tumor through the enhanced permeability and retention (EPR) effect, which enter the tumor interstitial space by enhanced permeability of the abnormal tumor microvasculature and retain in the location because of the suppressed lymphatic drainage.21 Some therapeutic NPs (eg, Doxil, MM-398, and Abraxane) have received clinical approval for cancer treatment; in addition, a number of nanomedicines have entered into clinical trials. The design of a delivery system for MIT based on NPs could result in a series of advantages, including the ameliorative release kinetics of drug molecules, protection of drug from degradation, alteration of drug circulation time, the increase in the amount of drug delivered to tumor tissue, and potential to bypass multidrug resistance mechanisms.22
Therefore, we developed a formulation of MIT-loaded nanoparticles (MIT-NPs) to deliver MIT to tumor with decreased off target side effects. The NPs were based on the copolymer of poly(D,L-lactic-co-glycolic acid) (PLGA) and polyethylene glycol (PEG), which are biodegradable, biocompatible, and US Food and Drug Administration (FDA)-approved components. PLGA, a controlled release polymer, has been used in the preparation of a wide variety of drug delivery systems, because its safety in the clinic is well established.23,24 PLGA can protect loaded drug from degradation. Sustained release of drug from NPs based on PLGA is likely a result of both the gradual degradation of PLGA and diffusion of drug through the PLGA matrix.25 In vivo, the reticuloendothelial system (RES) recognizes hydrophobic particles and takes them up in the liver or the spleen where NPs are degraded and eliminated. Because the injected NPs may bind with opsonin proteins in the blood serum leading to recognition and phagocytosis by macrophages,21,22 the surface of NPs coated with PEG is a commonly used approach for improving the efficiency of drug and gene delivery to target cells and tissues. PEGylation on NPs shielded the surface from opsonization, aggregation, and phagocytosis and hence significantly prolonged the systemic circulation time.26,27
In this study, we designed and prepared a formulation of MIT-NPs with a small size and sustained release for improved passive targeting and enhanced therapeutic efficacy. In vitro study showed that the prepared MIT-NPs can keep a controlled released in phosphate-buffered saline (PBS). The prepared NPs were taken up rapidly in pancreatic cancer cells and displayed intensive cytotoxicity of MIT-NPs. In pancreatic cancer xenograft models, MIT-NPs showed specific accumulation at the tumor tissue site and potent therapeutic efficacy. In addition, MIT and MIT-NPs significantly reduced CD47 expression levels in pancreatic cancer cells. Taken together, our results support that the application of MIT-NPs is an effective therapeutic strategy for pancreatic cancer.
Materials and methods
Materials and agents
MIT was isolated and collected from Streptomyces olivochromogenes No 684 strain (CPCC 200397) preserved in China Pharmaceutical Culture Collection (Beijing, China) and verified by quadrupole-time of flight mass spectrometry (QTOF-MS; AB SCIEX QSTAR; AB Sciex, Navarre, FL, USA). mPEG-PLGA (PEG: molecular weight [MW] 5,000 Da, PLGA: MW 15,000 Da, lactide/glycotide was 50/50) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). Cyanine 5 (Cy5) was provided by Fanbo Biochemicals Co., Ltd (Beijing, China). Poloxamer 188 and fetal bovine serum (FBS) were products of Thermo Fisher Scientific (Waltham, MA, USA). Roswell Park Memorial Institute (RPMI)-1640 and DMEM were purchased from GE Healthcare Life Sciences (South Logan, UT, USA). Sulforhodamine B (SRB) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Anti-Sp1 antibody, anti-PARP antibody, and anti-GAPDH antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-c-Myc antibody and anti-CD47 antibody were purchased from Abcam (Cambridge, UK).
Cells lines and experimental animals
The human pancreatic carcinoma BxPC-3 and PANC-1 cell lines were obtained from the Cell Resource Center, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS/PUMC; Beijing, China), which is the center of National Cell Line Resource, National Sciences and Technology Infrastructure in China. The human pancreatic carcinoma cell lines MIA Paca-2 and AsPC-1 were purchased from the American Type Culture Collection (Manassas, VA, USA). BxPC-3 and AsPC-1 cells were maintained in the RPMI-1640 medium. MIA Paca-2 and PANC-1 cells were cultured in the DMEM. All the media contained 10% FBS, penicillin G (100 U/mL), and streptomycin (100 μg/mL; North China Pharmaceutical, Shijiazhuang, China). The cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C. All procedures were approved by the ethics committee of Institute of Medicinal Biotechnology, Peking Union Medical College.
Animal experiments were conducted using 18–20 g female BALB/c nude mice ordered from Beijing Anikeeper Biotech Co. Ltd. (Beijing, China) after a 7-day period acclimatization. All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the animal care and use committee of Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College.
Preparation of NPs
MIT-NPs were prepared using a modified single-emulsion solvent evaporation method (Figure 1A). Briefly, methoxy PEG5k-PLGA15k was dissolved in ethyl acetate at a concentration of 35 mg/mL. MIT was also dissolved in ethyl acetate at a weight ratio of 1:14 to the polymers. In all, 2 mL of the blend solution was added dropwise into the aqueous phase (10 mL double-distilled water with 3% poloxamer 188) using a homogenizer and then emulsified for 4 min using a probe sonicator in ice bath. The resulting emulsion was subjected to remove the organic solvent under vacuum at room temperature (RT). After consecutive ultracentrifugation/washing steps with distilled water, the MIT-NPs were ultimately passed through a 0.22 μm syringe filter and stored at 4°C. The blank NPs and Cy5-labeled NPs were prepared using a similar procedure.
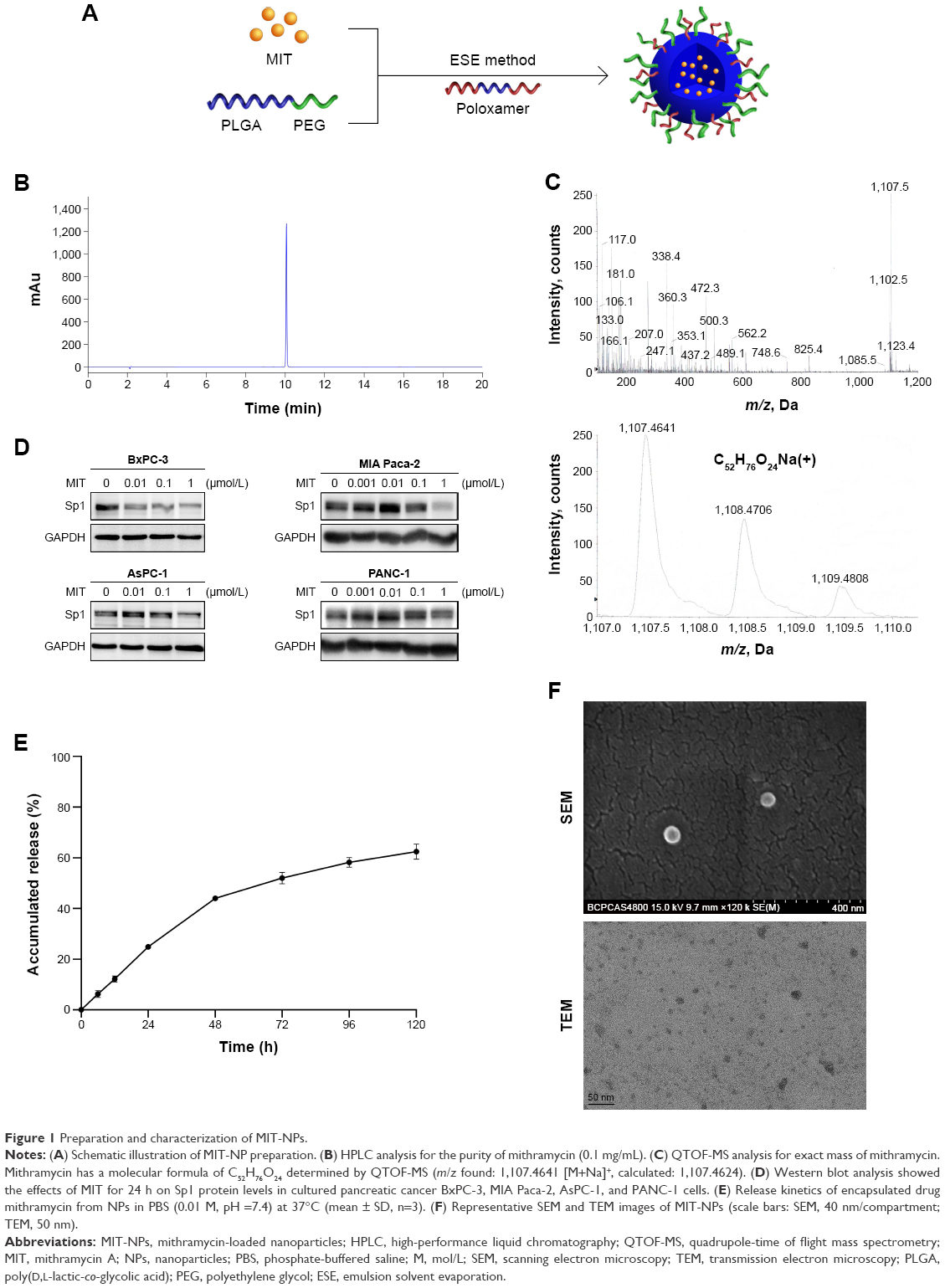 | Figure 1 Preparation and characterization of MIT-NPs. |
Particle size, zeta potential (ZP), and morphology
The mean particle size and ZP of the prepared NPs were measured in distilled water (ddH2O) at 25°C by dynamic light scattering using ZetaPlus (Brookhaven, Holtsville, NY, USA). All results were the mean of three different samples. The morphology of MIT-NPs was examined by field emission scanning electron microscopy (FESEM; S-4800; Hitachi Ltd., Tokyo, Japan) and transmission electron microscopy (TEM; Tecnai G2 F30; FEI, Hillsboro, OR, USA). Before analysis, the prepared NPs were diluted with ddH2O. The sample for TEM was placed on a copper grid with carbon film and then negatively stained with a 2% (w/v) phosphotungstic acid solution.
Drug loading (DL) and encapsulation efficiency (EE)
The DL and EE were evaluated through extraction of MIT from freeze-dried NPs. Briefly, 1 mL of prepared NPs were freeze-dried and then weighted. Dimethyl sulfoxide (DMSO; 1 mL) was added to NPs to dissolve both polymer and drug. The mixture was subjected to bath sonication for 30 min and then centrifuged for 15 min at 12,000 × g. The supernatant was separated and preserved. MIT in the supernatant was measured using a reverse-phase C18 column (250×4.6 mm; Phenomenex, Torrance, CA, USA) by high-performance liquid chromatography (HPLC; 1200 Series; Agilent Technologies, Santa Clara, CA, USA). The amount of drug was then calculated using a previously prepared standard curve for a varying amount of MIT. The DL and EE were determined from the following equation:
|
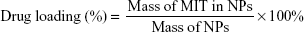
|

In vitro drug release profiles
After removing unencapsulated MIT, 1 mL of MIT-NPs was added into a dialysis tube (D-Tube Dialyzer Maxi; EMD Millipore, Billerica, MA, USA), which released MIT in 100 mL of PBS (pH 7.4). The PBS was gently stirred at 150 rpm at 37°C. At designated time points, 1 mL of the solution outside the dialysis bag was extracted and then the same volume of fresh PBS was added. The drug concentration in the solution was measured by HPLC following the methodology described before.
Growth inhibition of pancreatic cancer cells
Cell viability was examined by using the SRB assay, which determined cell number indirectly by measuring total basic amino acids.28 Briefly, the cells were incubated in 96-well microtiter plates for 24 h. Following the addition of different concentrations of free MIT or corresponding concentrations of MIT-NPs, the plates were incubated at 37°C for an additional 24, 48, or 72 h in a humidity-controlled 5% CO2 cell culture incubator. After an incubation period, the culture medium was discarded and cell monolayers were fixed with 100 μL of cold 10% (w/v) trichloroacetic acid and incubated for 60 min at 4°C. The supernatant was discarded, and the plates were washed five times with double distilled water and air-dried. Then, 100 μL of SRB solution at 0.4% (w/v) in 1% acetic acid was added and the plates were incubated for 5 min at RT. After staining, unbound dye was removed by washing five times with 1% acetic acid and the plates were air-dried. Bound stain was subsequently solubilized with 10 mmol/L Tris (pH 10.5), and the absorbance was read at 570 nm on a microplate reader (Infinite F50; Tecan Inc., Grödig, Austria).
Nanocarrier internalization by pancreatic cancer cells
The visualized process of cellular internalization of Cy5-labeled NPs was performed using fluorescence microscopy (Zeiss LSM 710; Carl Zeiss Meditec AG, Jena, Germany), while the quantitative analyses were measured using FACSCalibur flow cytometry (BD Biosciences, San Jose, CA, USA).
BxPC-3 and MIA Paca-2 cells were seeded into eight-well chambered coverglass (Nunc Lab-Tek II; Thermo Fisher Scientific) at a density of 2×104 cells/well for fluorescence microscopy. After culturing for 24 h, NP solutions were diluted in the culture medium to the determined concentrations and then added at 300 μL/well. After incubating at 37°C for several time points, the cells were fixed and stained using Dil (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine) and 4′,6-diamidino-2-phenylindole (DAPI) to label the cell membrane and nucleus, respectively. Finally, the cells were imaged under a confocal microscope. The images were focused on the focal plane where the largest nucleus was seen.
For flow cytometry, BxPC-3 and MIA Paca-2 cells were cultured in a six-well plate for 24 h to reach 70% confluence before the treatment. After incubated with fresh media containing Cy5-labeled NPs for 0.25, 0.5, 1, and 2 h, respectively, the cells were washed three times with cold PBS, trypsinized, and centrifuged at 800 rpm for 3 min. Then, the samples resuspended in PBS were analyzed by flow cytometry.
Western blot analysis
BxPC-3 and MIA Paca-2 cells were seeded in six-well plates at a density of 3×105 cells/well and allowed to adhere for 24 h before treatment. After incubation with free MIT or MIT-NPs for 24 h, total cell proteins were extracted using the radioimmunoprecipitation assay (RIPA) lysis buffer. Equal amounts of protein samples were separated on 10%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel followed by electrotransfer to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% bovine serum albumin (BSA), incubated with primary and appropriate secondary antibodies, and visualized using a chemiluminescent HRP substrate kit (Millipore).
Biodistribution study
The pancreatic carcinoma BxPC-3 and MIA Paca-2 xenograft tumor models were generated by subcutaneously injecting 1×107 cells in 100 μL of PBS into the right flank of nude mice (n=3). When the tumor size reached 300–400 mm3, the mice were injected with 200 μL at 15 mg/mL of Cy5-labeled NPs through tail veins. Mice were imaged at designated time points using IVIS (PerkinElmer Inc., Waltham, MA, USA). After 168 h, mice were sacrificed and the liver, heart, lung, stomach, small intestine, colon, kidney, pancreas, spleen, femur, and tumor were collected and imaged with the IVIS system.
In vivo therapeutic efficacy
The pancreatic carcinoma BxPC-3 xenograft tumor model was generated by subcutaneously injecting 1×107 cells in 100 μL of PBS into the right flank of mice. Tumor volume (mm3) was calculated according to the equation V = lw2/2, in which l and w indicate the length and width of the tumor. When the average tumor size reached 100 mm3, the tumor-bearing mice were randomly divided into five groups (n=6). Each group was treated by tail vein injection of free MIT at 1 mg/kg of body weight, MIT-NPs at an equivalent MIT dose of 1 or 2 mg/kg, blank NPs, or saline, once a week for 4 weeks. Tumor dimensions and mouse weights were measured every 4 days. The inhibition rate of each treatment was calculated as [1- (Vtf − Vti)/(Vcf − Vci)] ×100%, where Vtf and Vti represent the final and initial tumor volumes of the treatment group, while Vcf and Vci represent the final and initial tumor volumes of the control group.29,30
After 29 days post transplantation, mice were sacrificed and specimens were taken from the tumor and various organs for histopathological examination. The specimens were fixed in 4% (v/v) formalin and then embedded in paraffin. The paraffin-embedded specimens were cut into 6-μm-thick sections and stained with hematoxylin and eosin (H&E); some particular sections were stained with Alcian blue (AB). The images of specimens were obtained using microscopy with a digital camera (Leica Microsystems, Wetzlar, Germany). Immunohistochemical analyses were performed on paraffin-embedded sections of tumor tissues. Briefly, the sections were deparaffinized and heat induced epitope retrieval. Following endogenous peroxidase inactivated by 3% H2O2, the sections were blocked with 5% BSA and incubated with primary and appropriate secondary antibodies. Finally, the sections were visualized using 3,3′-diaminobenzidine (DAB) solution and captured using Leica microscopy.
Statistical analysis
All the experimental data were calculated using Microsoft Excel or GraphPad Prism 7 software (GraphPad Software, Inc., La Jolla, CA, USA), which were presented as mean ± SD. One-way analysis of variance (ANOVA) followed by Student’s t-test was used to determine significant differences between two groups of data. P-values <0.05 were considered as statistically significant.
Results
Characterization of MIT
MIT was successfully produced by the S. olivochromogenes No 684 strain and isolated through an established procedure developed in our laboratory. MIT was collected at >95% purity as analyzed using HPLC (Figure 1B). Mithramycin has a molecular formula of C52H76O24 determined by QTOF-MS (m/z found: 1,107.4641 [M+Na]+, calculated: 1,107.4624) as shown in Figure 1C. Western blot analysis revealed that MIT treatment inhibited the protein level of Sp1 in all the tested pancreatic cancer cell lines, including BxPC-3, MIA Paca-2, AsPC-1, and PANC-1 (Figure 1D).
In vitro NP characteristics
Table 1 presents the characteristics of the blank NPs, MIT-NPs, and Cy5-labeled NPs. As shown, MIT-NPs had an average diameter of 25.0±4.6 nm (polydispersity index [PDI] =0.267±0.015). The morphology of NPs was nearly spherical as shown in the SEM image (Figure 1F). TEM image further revealed that MIT-NPs were <40 nm in size and homogeneous in aqueous solution, which are in good accordance with the narrow particle size distribution (Figure 1F). Similar to MIT-NPs, the mean sizes of the blank NPs and Cy5-labeled NPs were 22.2±5.6 nm (PDI =0.245±0.032) and 22.7±2.9 nm (PDI =0.270±0.017), respectively. As shown in Table 1, the ZP values of the blank NPs and Cy5-labeled NPs were observed to be −17.2±4.3 mV and −24.2±5.6 mV, respectively, being similar to that of MIT-NPs (−19.6±4.6 mV). Besides, the DL and EE of MIT-NPs were calculated to be 2.11%±0.51% and 29.48%±7.2%, respectively.
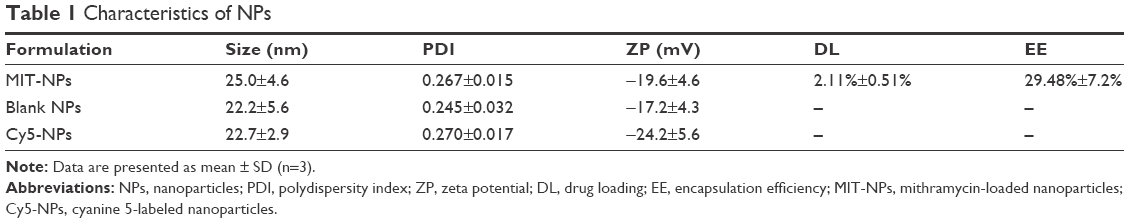 | Table 1 Characteristics of NPs |
In vitro drug release
In vitro MIT release from NPs was investigated in pH 7.4 PBS at 37°C. As shown in Figure 1E, a relative fast release of MIT from MIT-NPs was observed in the initial 48 h (44.0%±0.1%). After that, MIT-NPs exhibited sustained release with the total release in 5 days reaching 62.4%±3.0% in PBS.
Cellular uptake of Cy5-labeled NPs
The cellular uptake profile of the NPs was investigated. Flow cytometry was used for the quantitative analyses of cellular uptake (Figure 2A and B). As shown, NP internalization appeared at 15 min of incubation and then increased quickly in the following 30 min.
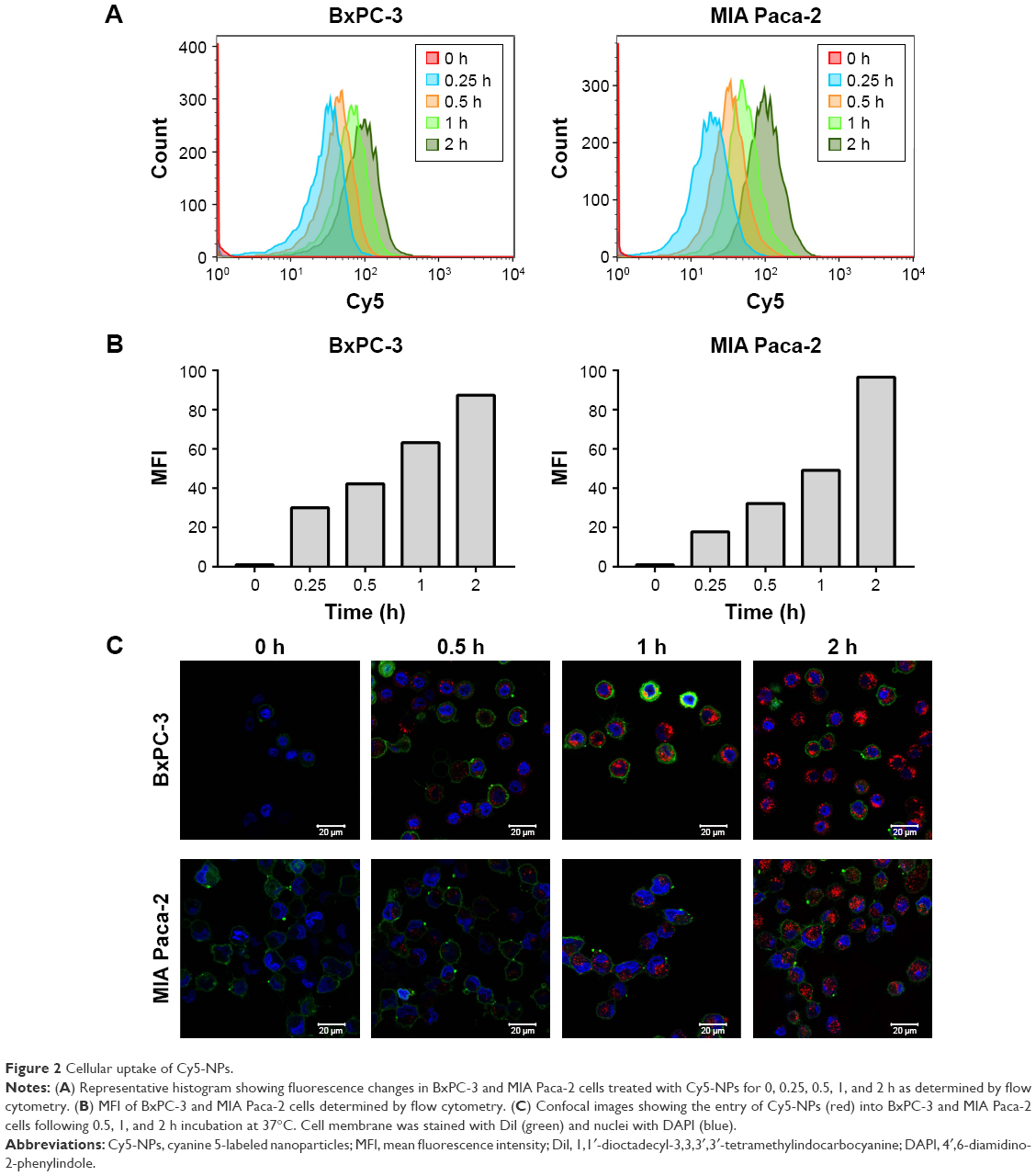 | Figure 2 Cellular uptake of Cy5-NPs. |
To further confirm the process of increased NP internalization, Figure 2C displays the confocal laser scanning microscope images of BxPC-3 and MIA Paca-2 cells after incubation (0.5, 1, and 2 h) with Cy5-labeled NPs. The fluorescence intensity of both BxPC-3 and MIA Paca-2 cells, which represents the cellular uptake efficiency, was low in the initial 0.5 h but increased rapidly by 1 h. However, the fluorescence intensity had less difference between 1 and 2 h of incubation, indicating that cellular uptake can reach the peak level in a short time. In line with these findings, BxPC-3 and MIA Paca-2 cells showed similar uptake levels for Cy5-labeled NPs. These data indicate that the prepared NPs can be taken up by BxPC-3 and MIA Paca-2 cells quickly and efficiently.
In vitro cytotoxicity of MIT-NPs
SRB assay was used to evaluate the cytotoxicity of MIT and MIT-NPs in vitro. Pancreatic cancer BxPC-3, MIA Paca-2, PANC-1, and AsPC-1 cells were treated at the concentrations of 0.0001, 0.001, 0.01, 0.1, and 1 μmol/L equivalent MIT concentrations for 24, 48, and 72 h, respectively. Figure 3A shows that both MIT and MIT-NPs exhibited the inhibitory effect on the growth of BxPC-3, MIA Paca-2, PANC-1, and AsPC-1 cells in a dose- and time-dependent manner. With the increment in the concentration of MIT or the prolongation of the exposure time, the survival percentages of BxPC-3 and AsPC-1 cells decreased significantly at the range of 0.01–1 μmol/L MIT, while the survival rates of MIA Paca-2 and PANC-1 cells decreased at the range of 0.1–1 μmol/L MIT. Meanwhile, the results indicated that the cytotoxicity of MIT-NPs was close to that of free MIT at a wide range of concentrations and exposure time intervals. The results showed that MIT inhibited the growth of BxPC-3, AsPC-1, PANC-1, and MIA Paca-2 cells and the prepared formulation of NPs did not reduce the cytotoxicity of MIT.
 | Figure 3 Cytotoxicity of MIT-NPs in pancreatic cancer cells. |
To assess the effects of MIT on Sp1-dependent expression of genes, BxPC-3 and MIA Paca-2 cells were grown in the absence or in the presence of MIT and MIT-NPs at different concentrations for 24 h. Western blot analysis revealed that both MIT and MIT-NP treatments inhibited the protein level of Sp1 in two cell lines (Figure 3B and C). In addition, MIT and MIT-NPs reduced the expression of c-Myc and, notably, decreased the level of CD47, a c-Myc-downstream gene. MIT and MIT-NPs also induced cleavage of poly(ADP-ribose) polymerase (PARP) in a concentration-dependent manner, suggesting that MIT can induce apoptosis in both BxPC-3 and MIA Paca-2 cells.
Biodistribution of Cy5-labeled NPs
BALB/c nude mice bearing pancreatic cancer BxPC-3 and MIA Paca-2 xenograft models were used for the study of in vivo biodistribution and tumor accumulation of Cy5-labeled NPs. The variations in fluorescence intensity at the tumor region were monitored for 7 days. As shown in Figure 4A, the fluorescence intensity in both tumors increased gradually with time during the initial phase after the injection of Cy5-NPs. The fluorescence signals in the MIA Paca-2 tumor site appeared after 2 h post injection and reached the peak during 10–12 h. Meanwhile, BxPC-3 tumor, a hypopermeable pancreatic xenograft model, showed the distinct fluorescence signals at 4 h, reached the peak at 12 h post injection, and kept the highest intensity until 72 h. After 7 days (168 h) post injection, tumors and various organs were dissected and imaged ex vivo using the IVIS system (Figure 4B). As shown, the fluorescence intensity of both BxPC-3 and MIA Paca-2 tumors was much stronger than all other examined organs, including the liver, heart, lung, stomach, small intestine, colon, kidney, pancreas, spleen, and femur. This provides evidence that the prepared NPs can specifically accumulate in both BxPC-3 and MIA Paca-2 tumors and retain for a prolonged period of time.
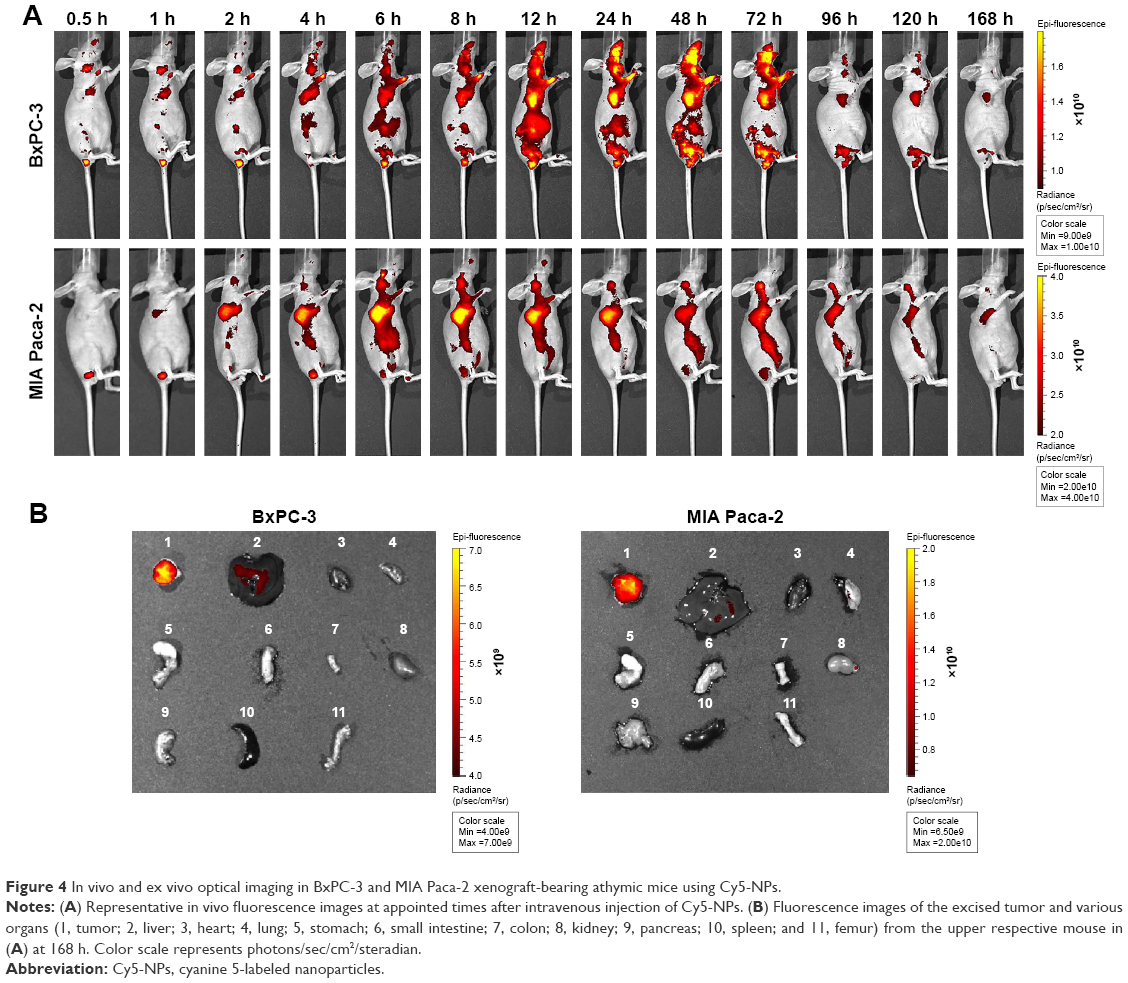 | Figure 4 In vivo and ex vivo optical imaging in BxPC-3 and MIA Paca-2 xenograft-bearing athymic mice using Cy5-NPs. |
In vivo tumor growth inhibition by MIT-NPs
The antitumor activity of MIT-NPs was evaluated in nude mice bearing a poorly permeable BxPC-3 tumor xenograft.31 As shown in Figure 5A, the mice with free MIT treatments (1 mg/kg) showed slower tumor growth than the mice in the control group; as indicated, MIT suppressed tumor growth by ~51% (P<0.001). By contrast, MIT-NPs caused 86% tumor suppression at the equivalent MIT dose of 1 mg/kg (P<0.01 versus MIT 1 mg/kg) and 96% tumor suppression at the dose of 2 mg/kg (P<0.001 versus free MIT treatment). No deaths and no significant body weight changes were found in the mice of the treatment groups (Figure 5B).
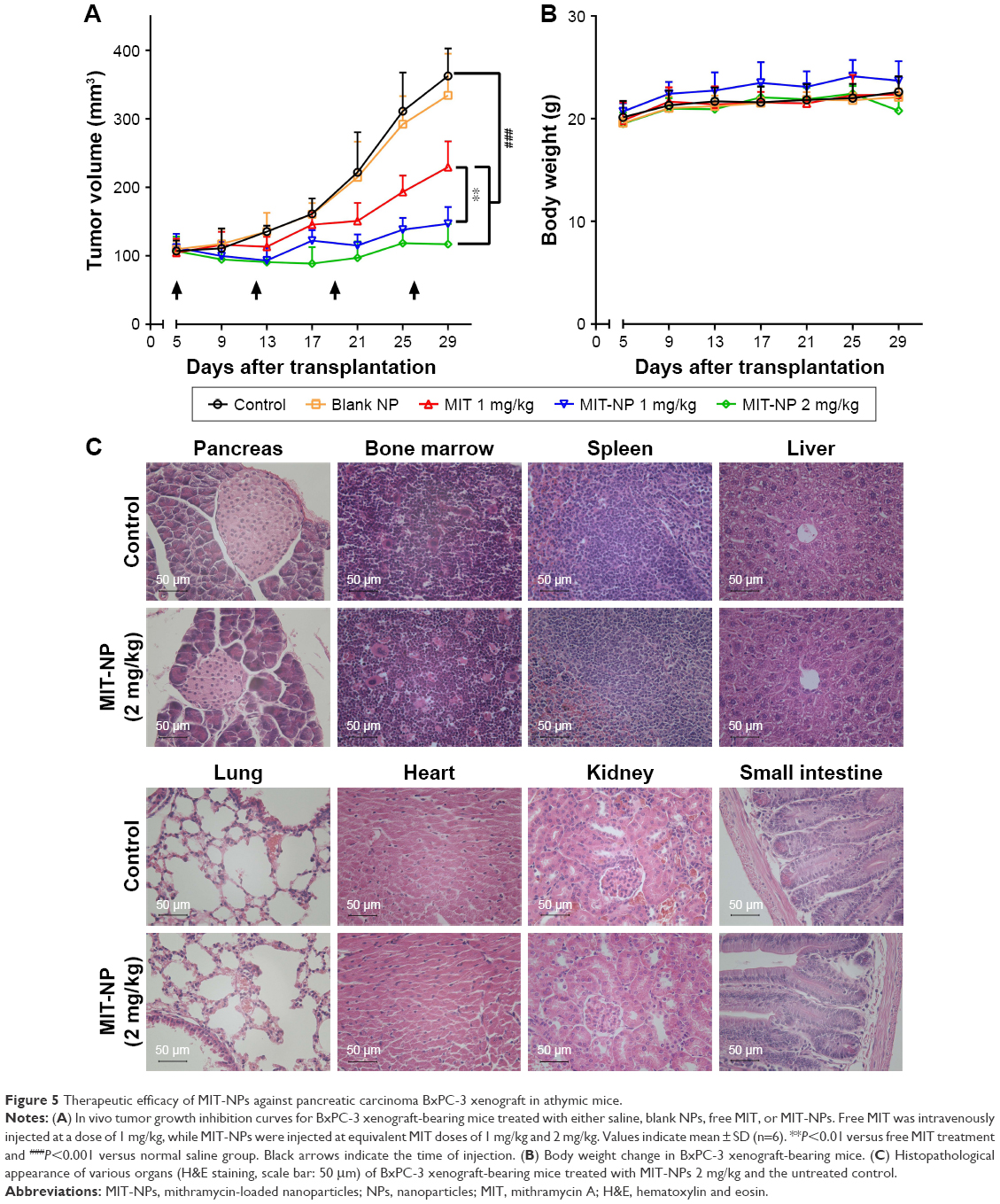 | Figure 5 Therapeutic efficacy of MIT-NPs against pancreatic carcinoma BxPC-3 xenograft in athymic mice. |
Histopathological appearance of major organs from the mice of both control and MIT-NP (2 mg/kg) groups was examined on sections with H&E staining (Figure 5C). No toxicopathological changes were found in the tested tissues and organs, including the pancreas, bone marrow, spleen, heart, lung, liver, kidney, small intestine, and colon, indicating that the highly effective dose level of 2 mg/kg MIT-NPs was well tolerated.
Immunohistochemical observation of the tumor
The results of immunohistochemistry showed that the tumor proliferation marker Ki-67 level in tumor tissue treated with MIT-NPs decreased obviously as compared with the control group (Figure 6A). This suggested that MIT-NPs could inhibit the tumor proliferation marker in association with the inhibition of tumor growth in BxPC-3 xenograft. Notably, the treatment of MIT-NPs reduced the protein level of CD47 in the BxPC-3 tumors, further confirming that MIT-NPs could induce downregulation of CD47 (Figure 6A), a membrane protein that took part in innate immune checkpoint in cancer.32 In tumor tissue of the MIT-NPs group (2 mg/kg), a more striking number of vacuoles existed as compared with that of the control group. These vacuoles appeared in blue with AB mucin staining (Figure 6B), indicating that acidic glycan occurs in the vacuole structure. Compared to the control group, a larger blue staining area could be detected in the tumor of the MIT-NPs-treated group.
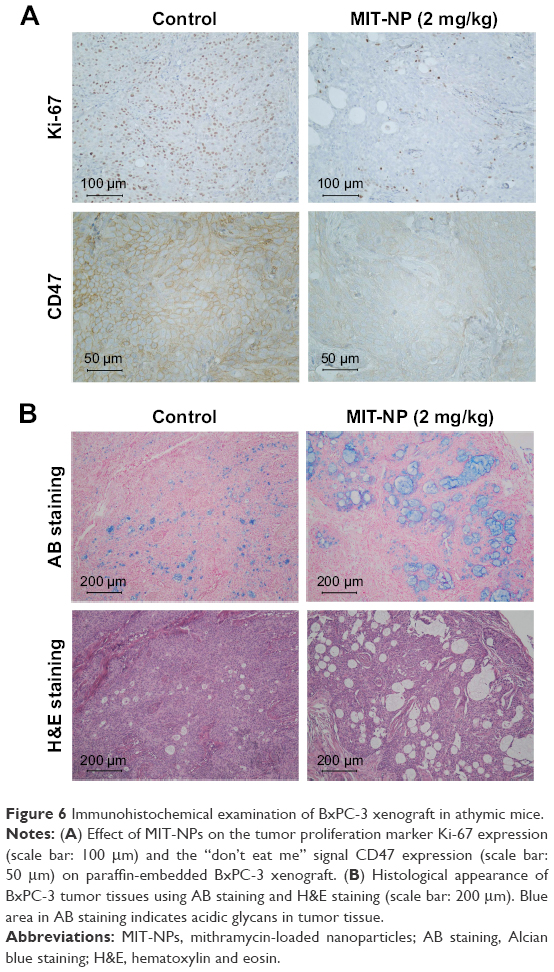 | Figure 6 Immunohistochemical examination of BxPC-3 xenograft in athymic mice. |
Discussion
In this study, we have explored for the possibility to enhance the chemotherapeutic efficacy of MIT on pancreatic cancer by improving its bioavailability and tumor accumulation using rationally designed NPs based on the copolymer of PLGA and PEG. In particular, we demonstrated that the prepared NPs were accumulated at the tumor site and the antitumor efficacy was enhanced as compared with free MIT using a pancreatic cancer BxPC-3 cell-derived xenograft model. In addition, the protein level of CD47 in pancreatic carcinoma cells was reduced by treatment with MIT and MIT-NPs both in vitro and in vivo.
MIT is a DNA-binding aureolic acid natural product approved by the FDA. Since the 1970s, MIT has been used clinically as a chemotherapeutic agent to treat various malignant tumors, including testicular embryonal carcinoma and glioblastomas, as well as hypercalcemia and Paget’s disease of bone. Recently, researchers have shown that MIT interferes with the transcription of genes that bear GC-rich motifs in their promoters. The transcription regulation of MIT depends on a non-covalent interaction with GC-rich DNA regions, especially the site of union of Sp1 transcription factor.11 MIT was shown to have antiangiogenic effect in human pancreatic cancer xenograft models due to decreased expression of VEGF regulated by Sp1.33 Meanwhile, Sp1 as a partner involved in SMAD-mediated signal transduction of TGFβ, a key regulator of epithelial-to-mesenchymal transition (EMT), in pancreatic carcinoma.17 In our study, MIT and MIT-NPs exhibited the inhibitory effect on the growth of pancreatic cancer cell lines BxPC-3 and MIA Paca-2 in a dose- and time-dependent manner. Furthermore, the protein level of Sp1 in pancreatic cancer cells decreased after free MIT or MIT-NP treatment. This result could be caused by mechanisms that Sp1 gene was autoregulated with multiple Sp1 binding sites in the proximal promoter of Sp1 and that MIT could decrease Sp1 protein by inducing proteasome-dependent degradation.34,35 A previous report showed that PARP might be a potential target of pancreatic tumors due to their unstable genomes.1 In our study, the increased level of cleaved PARP indicated that both MIT and MIT-NPs could cause apoptosis of pancreatic cancer cells. It is of interest that both MIT and MIT-NPs downregulated the protein level of c-Myc in pancreatic cells. As reported, c-Myc plays a significant role in the oncogenesis of mutant K-Ras-driven tumors in the pancreas, and c-Myc overexpression can also contribute to the progression of pancreatic ductal adenocarcinoma.36 Furthermore, our study has found that the protein level of CD47 in pancreatic cancer cells was downregulated by both MIT and MIT-NPs. At a well-tolerated dose of 2 mg/kg, MIT-NPs exerted highly therapeutic efficacy against the BxPC-3 tumor; in association, a marked decrease in the CD47 level was detected in the tumor. The study results demonstrated that MIT-NPs are active in decreasing the CD47 level both in vitro and in vivo. As previously reported, c-Myc could bind to the promoters and activate the expression of Cd47 gene.37 MIT was already found to suppress c-Myc; however, it had not been reported yet to affect the expression of CD47 in the previous studies. To our knowledge, this is the first report that showed that MIT and MIT-NPs can decrease the CD47 level in cancer cells. As known, CD47 is a protein overexpressed on various cancer cells as the “don’t-eat me” signal. It plays a significant role in the innate immune checkpoint through interaction with signal regulatory protein-α (SIRPα), a myeloid inhibitory immunoreceptor.32 A previous study had suggested that CD47 was highly expressed on primary pancreatic cancer cells and the anti-CD47 monoclonal antibodies (mAbs) could enable phagocytosis of cancer cells by macrophages as well as inhibit self-renewal of cancer stem cells.38 In comparison to the relatively stable expression of SIRPα, the variable levels of CD47 may be relevant to immune checkpoint in cancer.32 Several clinical studies have shown the expression level of CD47 in tumor tissues to be positively correlated with the resistance to both chemotherapy and antibody therapy.32,39 Furthermore, the decreased level of CD47 may change tumor microenvironment via interaction with thrombospondin-1 (TSP-1), which is overexpressed in the tumor stroma and takes part in the regulation of angiogenesis and inflammation.40 Therefore, CD47 inhibition could be an effective treatment strategy for a variety of cancers. The present study suggests that MIT-NPs might be highly effective in CD47 targeting therapy.
As an emerging treatment, NP therapeutics showed great promise in clinical therapy of various solid tumors in the recent decades. However, nanocarriers have not been successful yet in exhibiting significant therapeutic effects on intractable pancreatic cancer. The theoretical basis of the passive targeting of nanomedicine in solid tumors is the EPR effect depending on leakiness of tumor vessels to nanoscale agents, but the characteristics of pancreatic cancer, including desmoplasia and hypovascularity, could exert the impact on the EPR effect.41,42 To address these challenges, different strategies have been investigated. One approach is to change the size of nanocarriers to improve the penetration into tumors. As reported, a higher MW of dextran as a model macromolecule was found to present decreased permeability into tumors and reduced clearance rate in both plasma and tumor extravascular compartment; in addition, dextrans with MWs of 40 and 70 kDa (a diameter of 11.2–14.6 nm) provided the greatest tumor penetration and accumulation.43 A previous study has suggested that only drug-loaded polymeric micelles with a diameter of 30 nm showed sufficient accumulation to achieve an antitumor effect on hypopermeable pancreatic cancer in contrast with those with larger diameters.31 Therefore, in the present study, we optimized the preparation method to prepare MIT-NP with diameters of 25.0±4.6 nm. Then, the in vivo antitumor efficacy was evaluated with BxPC-3 xenograft in nude mice, which is a model system of pancreatic carcinoma reportedly characterized by hypovascularity and thick fibrosis in the tumor microenvironment.42 The results showed that the prepared MIT-NPs exhibited significantly enhanced therapeutic effects on BxPC-3 xenograft as compared with equivalent MIT (86% versus 51%, P<0.01). At the highly effective dose, MIT-NPs (2 mg/kg) showed no toxicopathological changes in the tested organs. Moreover, the prepared NPs can accumulate specifically in the BxPC-3 xenograft, indicating the process of passive targeting.
MIT is an amphiphilic compound composed of a tricyclic aromatic polyketide moiety with a dihydroxymethoxy-oxo-pentyl hydrophilic side chain attached at C-3 and two oligosaccharide chains attached at C-2 and C-6.3 MIT is soluble in water (26 g/L at 25°C in pH 7, data from SciFinder®) and also relatively soluble in several organic solvents, including ethyl acetate, acetone, and DMSO. The hydrophilicity of MIT limits efficient encapsulation into NPs by nanoprecipitation. MIT shows a logP of 1.290±1.454, suggesting a slight lipophilic property and the potential to be loaded in NPs using the emulsion solvent evaporation method.44 In the present study, we prepared the MIT-NPs with the copolymer of PLGA and PEG; both of them are biodegradable and biocompatible materials of approved clinical application. During the prepared process, NPs based on mPEG-PLGA can form spontaneously a core–shell structure, a hydrophobic core of PLGA, and a hydrophilic shell of mPEG, fitting the specific property of MIT.45 “PEGylation” is one of the most widely used strategies to develop long-circulating NPs. The PEG-coated NPs can hide the hydrophobicity by providing a hydrophilic layer at the surface and prevent opsonins from binding to the particle surface.25 In order to avoid the disruption of PEG coating, poloxamer 188 was chosen as the emulsifier, whose hydrophilic group was poly(oxyethylene), the same as PEG.46 By this method, the DL and EE of the prepared MIT-NPs were calculated to be 2.11%±0.51% and 29.48%±7.2%, respectively. Study results revealed that the prepared formulations of MIT-NPs have realized drug-controlled release and prolonged blood circulation time, which provided optimal conditions for passive targeting.
Conclusion
In this study, MIT-NPs were prepared to improve MIT delivery to pancreatic cancer. Considering the pathologic characteristics of pancreatic cancer, the PEG-PLGA-based NPs with a small particle size have been prepared. The study demonstrated that MIT-NPs can be taken up rapidly by pancreatic cancer BxPC-3 and MIA Paca-2 cells and exert potent cytotoxicity. In vivo results suggest that MIT-NPs can improve bioavailability and therapeutic efficacy of MIT in a pancreatic carcinoma BxPC-3 xenograft model. Moreover, MIT-NPs can reduce the expression of CD47 in pancreatic cancer cells both in vitro and in vivo. It is worth doing further research to understand the relationship between MIT and CD47. In summary, the study results suggest that the mithramycin-loaded mPEG-PLGA NPs are potentially useful for cancer therapy.
Acknowledgment
This work was supported by grants from CAMS Innovation Fund for Medical Sciences (No 2016-I2M-3-013) and National Key R&D Plan (No 2016YFA0201504).
Disclosure
The authors report no conflicts of interest in this work.
References
Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. | ||
Deplanque G, Demartines N. Pancreatic cancer: are more chemotherapy and surgery needed? Lancet. 2017;389(10073):985–986. | ||
Lombo F, Menendez N, Salas JA, Mendez C. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives. Appl Microbiol Biotechnol. 2006;73(1):1–14. | ||
Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015;282(2):224–258. | ||
de Nigris F, Sica V, Herrmann J, et al. c-Myc oncoprotein: cell cycle-related events and new therapeutic challenges in cancer and cardiovascular diseases. Cell Cycle. 2003;2(4):325–328. | ||
Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42(27):8313–8324. | ||
Bond GL, Hu W, Bond EE, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119(5):591–602. | ||
Zhang M, Mathur A, Zhang Y, et al. Mithramycin represses basal and cigarette smoke-induced expression of ABCG2 and inhibits stem cell signaling in lung and esophageal cancer cells. Cancer Res. 2012;72(16):4178–4192. | ||
Tagashira M, Kitagawa T, Isonishi S, Okamoto A, Ochiai K, Ohtake Y. Mithramycin represses MDR1 gene expression in vitro, modulating multidrug resistance. Biol Pharm Bull. 2000;23(8):926–929. | ||
Lin RK, Hsu CH, Wang YC. Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer Drugs. 2007;18(10):1157–1164. | ||
Rao M, Atay SM, Shukla V, et al. Mithramycin depletes specificity protein 1 and activates p53 to mediate senescence and apoptosis of malignant pleural mesothelioma cells. Clin Cancer Res. 2016;22(5):1197–1210. | ||
Ehrenschwender M, Bittner S, Seibold K, Wajant H. XIAP-targeting drugs re-sensitize PIK3CA-mutated colorectal cancer cells for death receptor-induced apoptosis. Cell Death Dis. 2014;5:e1570. | ||
Pandiella A, Moris F, Ocana A, Nunez LE, Montero JC. Antitumoral activity of the mithralog EC-8042 in triple negative breast cancer linked to cell cycle arrest in G2. Oncotarget. 2015;6(32):32856–32867. | ||
Gopisetty G, Xu J, Sampath D, Colman H, Puduvalli VK. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene. 2013;32(26):3119–3129. | ||
Gao Y, Jia Z, Kong X, et al. Combining betulinic acid and mithramycin a effectively suppresses pancreatic cancer by inhibiting proliferation, invasion, and angiogenesis. Cancer Res. 2011;71(15):5182–5193. | ||
Choi ES, Shim JH, Jung JY, et al. Apoptotic effect of tolfenamic acid in androgen receptor-independent prostate cancer cell and xenograft tumor through specificity protein 1. Cancer Sci. 2011;102(4):742–748. | ||
Sankpal UT, Maliakal P, Bose D, Kayaleh O, Buchholz D, Basha R. Expression of specificity protein transcription factors in pancreatic cancer and their association in prognosis and therapy. Curr Med Chem. 2012;19(22):3779–3786. | ||
Jia Z, Gao Y, Wang L, et al. Combined treatment of pancreatic cancer with mithramycin A and tolfenamic acid promotes Sp1 degradation and synergistic antitumor activity. Cancer Res. 2010;70(3):1111–1119. | ||
Venkatasubbarao K, Ammanamanchi S, Brattain MG, Mimari D, Freeman JW. Reversion of transcriptional repression of Sp1 by 5 aza-2′ deoxycytidine restores TGF-beta type II receptor expression in the pancreatic cancer cell line MIA PaCa-2. Cancer Res. 2001;61(16):6239–6247. | ||
Taylor DJ, Parsons CE, Han H, Jayaraman A, Rege K. Parallel screening of FDA-approved antineoplastic drugs for identifying sensitizers of TRAIL-induced apoptosis in cancer cells. BMC Cancer. 2011;11:470. | ||
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. | ||
Graf N, Bielenberg DR, Kolishetti N, et al. α(V)β(3) integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano. 2012;6(5):4530–4539. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. | ||
Xu Q, Ensign LM, Boylan NJ, et al. Impact of surface polyethylene glycol (PEG) density on biodegradable nanoparticle transport in mucus ex vivo and distribution in vivo. ACS Nano. 2015;9(9):9217–9227. | ||
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. | ||
Li HJ, Du JZ, Liu J, et al. Smart superstructures with ultrahigh pH-sensitivity for targeting acidic tumor microenvironment: instantaneous size switching and improved tumor penetration. ACS Nano. 2016;10(7):6753–6761. | ||
Xu J, Liu XJ, Li L, et al. An engineered TIMP2-based and enediyne-integrated fusion protein for targeting MMP-14 shows potent antitumor efficacy. Oncotarget. 2015;6(28):26322–26334. | ||
Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. | ||
Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol Rev. 2017;276(1):145–164. | ||
Jia Z, Zhang J, Wei D, et al. Molecular basis of the synergistic antiangiogenic activity of bevacizumab and mithramycin A. Cancer Res. 2007;67(10):4878–4885. | ||
Nicolas M, Noe V, Jensen KB, Ciudad CJ. Cloning and characterization of the 5′-flanking region of the human transcription factor Sp1 gene. J Biol Chem. 2001;276(25):22126–22132. | ||
Choi ES, Nam JS, Jung JY, Cho NP, Cho SD. Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer. Sci Rep. 2014;4:7162. | ||
Sanchez-Arevalo Lobo VJ, Fernandez LC, Carrillo-de-Santa-Pau E, et al. c-Myc downregulation is required for preacinar to acinar maturation and pancreatic homeostasis. Gut. Epub 2017 Feb 3. | ||
Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–231. | ||
Cioffi M, Trabulo S, Hidalgo M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. 2015;21(10):2325–2337. | ||
Zhao XW, van Beek EM, Schornagel K, et al. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci U S A. 2011;108(45):18342–18347. | ||
Sick E, Jeanne A, Schneider C, Dedieu S, Takeda K, Martiny L. CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br J Pharmacol. 2012;167(7):1415–1430. | ||
Hessmann E, Patzak MS, Klein L, et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. Epub 2017 Jan 10. | ||
Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104(9):3460–3465. | ||
Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98(5):335–344. | ||
Cohen-Sela E, Teitlboim S, Chorny M, et al. Single and double emulsion manufacturing techniques of an amphiphilic drug in PLGA nanoparticles: formulations of mithramycin and bioactivity. J Pharm Sci. 2009;98(4):1452–1462. | ||
Capretto L, Mazzitelli S, Brognara E, et al. Mithramycin encapsulated in polymeric micelles by microfluidic technology as novel therapeutic protocol for beta-thalassemia. Int J Nanomedicine. 2012;7:307–324. | ||
Xu Q, Boylan NJ, Cai S, Miao B, Patel H, Hanes J. Scalable method to produce biodegradable nanoparticles that rapidly penetrate human mucus. J Control Release. 2013;170(2):279–286. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.