Back to Journals » Drug Design, Development and Therapy » Volume 14
Mini-Tablets versus Nanoparticles for Controlling the Release of Amoxicillin: In vitro/In vivo Study
Authors Gaber DA, Alhawas HS, Alfadhel FA, Abdoun SA, Alsubaiyel AM, Alsawi RM
Received 14 October 2020
Accepted for publication 6 November 2020
Published 7 December 2020 Volume 2020:14 Pages 5405—5418
DOI https://doi.org/10.2147/DDDT.S285522
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Dalia A Gaber,1,2 Hessah S Alhawas,3 Fatimah A Alfadhel,3 Siham A Abdoun,1,4 Amal M Alsubaiyel,1 Rehab M Alsawi5
1Department of Pharmaceutics, College of Pharmacy, Qassim University, Buraidah, kingdom of saudi arabia; 2Department of Quality Control & Quality Assurance, Holding Company for Biological Products and Vaccines, Cairo, Egypt; 3College of Pharmacy, Qassim University, Buraidah, kingdom of saudi arabia; 4National Medicine Quality Control Laboratory, National Medicine and Poisons Board, Sudan; 5King Faisal Specialist Hospital and Research Center, Riyadh, kingdom of saudi arabia
Correspondence: Dalia A Gaber Department of Pharmaceutics
College of pharmacy, Qassim university, Buridah, Saudi arabia
Tel +20 1001523222
Email [email protected]
Introduction: Controlling the drug release from the dosage form at a definite rate is the main challenge for a successful oral controlled-release drug delivery system. In this study, mini-tablets (MTs) and lipid/polymer nanoparticles (LPNs) of lipid polymer and chitosan in different ratios were designed to encapsulate and control the release time of Amoxicillin (AMX).
Methods: Physical characteristics and in vitro release profiles of both MT and LPN formulations were studied. Antimicrobial activity and oral pharmacokinetics of the optimum MT and LPN formulations in comparison to market tablet were studied in rats.
Results: All designed formulations of AMX as MTs and LPNs showed accepted characteristics. MT-6 (Compritol/Chitosan 1:1) showed the greatest retardation among all prepared minitablet preparations, releasing about 79.5% of AMX over 8 h. In contrast, LPN-11 (AMX: Cr 1:3/Chitosan 1 mg/mL) had the slowest drug release, revealing the sustained release of 80.9% within 8 h. The MIC of both optimized tablet formula (MT-6) and LPNs formula (LPN-11) was around two-fold lower than the control against H. pylori. The Cmax of MT-6 and LPN11 were non significantly different compared with the marketed AMX product. While the bioavailability experiment proved that the relative bioavailability of the AMX was 1.85 and 1.8 after the oral use of LPN11 and MT-6, respectively, compared to the market tablet.
Conclusion: The results verified that both controlled-release mini-tablets and lipid/polymer nanoparticles can be used for sustaining the release and hence improve the bioavailability of amoxicillin.
Keywords: lipid-polymer, nanoparticles, amoxicillin, bioavailability, mini-tablet, chitosan, sustain release
Introduction
Great steps have been taken in the management of disease through the intervention of a lot of drugs over the past years unless a drug is delivered to its target area at an optimum rate and concentration, the drug will not be optimally useful to the patient.1 Over the years there have been many drug modifications and dosage forms, with which we have tried to control the duration of drugs in the body. Moreover, the delivery of drugs must be sustained at a rate such that the disease is cured or controlled in minimum time with the lowest dose frequency and consequently fewest side effects.2 Recently, numerous polymers have been used to develop sustained release dosage forms. Hydrophilic polymers are widely used in design oral sustained-release formulations, such as xanthan gum, cellulose derivatives, alginate sodium, or Chitosan.3,4 Chitosan is a natural biodegradable cationic polysaccharide polymer obtained by deacetylation of chitin. It is used widely in the pharmaceutical industry due to its biocompatible, biodegradable, and non-toxic characteristics, as well as its adhesiveness and permeability.5,6 In this investigation, Chitosan was used as a carrier in the design of sustained-release mini-tablets and LPN for AMX. The use of lipid-based polymers showed a valuable advantage in the design of controlled release dosage forms owing to their chemical inertness, formulation versatility, better characterization, and wide compatibility with different delivery systems.5 Recently, gelucires are widely used as carriers in drug delivery systems in a particular controlled-release formulation. For example, Witepsol (Wp), Precirol (Pr), and Compritol. Compitol ATO 888 (Cr), Glyceryl monostearate (GMS) were selected in the study as a glyceride base for control the release.6 Multiple unit drug delivery systems, such as mini-tablets, proved several benefits over monolithic ones. First, Mini-tablets appreciate the benefits of adequate large size oral tablets, such as being easy to be administered, cost-effective, and can be easily protected against moisture or light.7 Mini-tablets also permit the chance to present a multiparticulate dosage form manufactured via compression, which is more efficient than extrusion-spheronization, and may avoid the use of aqueous or organic solvents.7,8 By utilizing a minitablet, modified release profiles have been demonstrated, including controlled or sustained release, chronotherapeutic, enteric or colonic release, biphasic pharmacokinetic profile, and floating bioadhesive dosage form to present a sustained pharmacokinetic profile.9,10 In addition, numerous nano-sized particle drug delivery systems such as polymeric nanoparticles, liposomes, solid lipid nanoparticles, dendrimers, etc., techniques were used to release drugs in a sustained manner.11,12 Both Polymeric and lipid-based nanoparticles have been recorded as the most effective systems.13 Liposomes are artificial fat globules, have a characteristic of one or more bilayers achieved by dispersal of either natural or synthetic lipids in water.14 Moreover, polymeric nano-carriers (PNs) were prepared via either nanoprecipitation or emulsion method by self-assembly of biodegradable copolymers with varying hydrophilicity.15,16 Recently, many studies aimed to integrate polymeric and liposomal systems provide a novel drug delivery systems termed as the lipid polymer nanoparticles (LPNs).17,18 In contrast to other nano-carriers, the LPNs offer some characteristic features, as the variety in structural components, the higher encapsulation ability, controlling in drug release, improved stability, biocompatibility, and the enhanced permeability.19,20 Amoxicillin (AMX) is one of the most widely used semisynthetic aminopenicillin (α-amino hydroxy benzylpenicillin) drugs, [see Figure 1].21 Amoxicillin is included in all current therapeutic regimens for H. pylori eradication.22–24 The most reported causes of eradication failure of H. pylori are the patient’s poor compliance with the antibiotic treatment regimen, antimicrobial resistance, and drug-related side effects. For complete eradication of H. pylori, constant concentrations of the antibiotics are required for a prolonged time.25 This can be approached by formulating an extended-release dosage form of AMX that confirms prolonged drug release.26,27 The present study was supposed to investigate the efficacy of both Mini-tablets and lipid/polymer nanoparticle systems as modern and reproducible drug delivery systems for accepted control of AMX release using both lipid polymers (Cr, GMS) and chitosan (CN) in different ratios. The formulations, with the optimal in vitro drug release, would be selected for antimicrobial activity test and pharmacokinetic study to investigate its advantage over the marketed tablet.
 |
Figure 1 Chemical structure of Amoxicillin. |
Materials and Methods
Materials
Amoxicillin (AMX) was a gift from Mash for Pharmaceutical Industries, Egypt. Compritol® 888 ATO-Cr- (glyceryl behenate NF, Gattefosse ́ s.a., Lyon, France), Glyceryl monostearate -GMS- (Loba Chemie Pvt. Ltd., Mumbai, India) was purchased from Gattefosse´ s.a., Lyon, France, poloxamer 188 (Brunsbüttel, Germany), Chitosan-CN-(50 kDa, 85%degree of deacetylation) was from Sigma-Aldrich (Sintra, Portugal). Magnesium stearate, talc, and other used chemicals were of analytical grade or equivalent and were used as received. Klavox tablets (Spimaco Co., Pharmaceutical & Chemical Industries, Al-Qassim, KSA) were used as a reference product in the study.
Preparation of AMX Minitab Tablets (AMX-MTs)
The Composition of AMX-MT formulations is presented in Table 1. The geometric dilution technique was used for mixing each formula ingredients after accurate weighing. The powder mixtures were granulated using isopropyl alcohol. The obtained masses were passed through a 1 mm mesh sieve for granulation. The prepared granules were dried at 60°C in a hot air oven. Dried granules were ground sieved through a 500 µm sieve. Granules that passed sieve (250 μm) were collected mixed with talc (1%) and magnesium stearate (2%) then compressed using a single punch tablet press machine fitted with a 4 mm diameter concave punch. Each dose equivalent to 250 mg of AMX and was encompassed 14 mini-tablets.
 |
Table 1 Composition and Physical Character of AMX-MTs Formulation, Each Dose of Tablets Contain 250 Mg of AMX |
Preparation of AMX Lipid Polymer Nanoparticles (AMX-LPNs)
AMX-LPN formulations were prepared by the solvent emulsification evaporation technique.28 LPNs compositions are expressed in Table 2. Briefly, the lipid solution in chloroform was prepared. AMX (250 mg) was dissolved in 2 mL of prepared lipid solution with a different drug: lipid ratios as shown in Table 2. Drug-loaded solution was then emulsified with 20 mL of chitosan solution (0.5, 1, or 2mg/mL) in acetic acid solution (1% w/v) containing 25 mg of poloxamer 188 surfactant under ultrasonic sonication for 5 min at 300 W ultrasonic energy, and 3-second pauses at room temperature. The organic solvent was evaporated by stirring overnight at 40°C. The prepared AMX-LPN formulations were separated from the aqueous solution by centrifugation at 10,000 rpm for 15 min, the sediment was then washed three times with deionized water and, freeze-dried until further work.
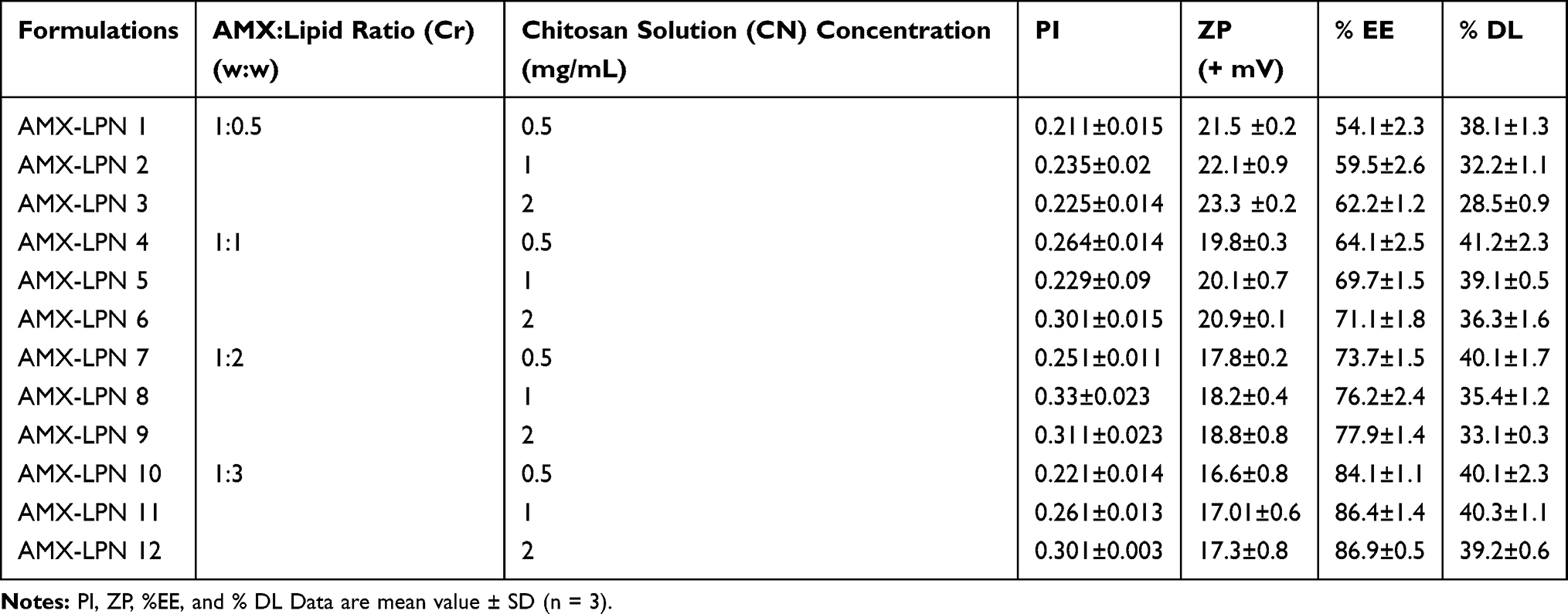 |
Table 2 Formulation Code, Drug:Lipid Ratio and Chitosan (CN) Solution Concentration (mg/mL), PI, ZP %EE and % DL of AMX-LPN Formulations |
In vitro Evaluation of AMX-MTs and AMX-LPNs Formulations
Characterization of Both Pre-Compression and Post-Compression Parameters of AMX-MTs Formulations
Powders mixtures were measured for the angle of repose, compressibility index, and Hausner’s ratio of the powder mixture. Compressed mini-tablets were evaluated for weight variation, thickness, diameter, friability, and drug content uniformity.29
Characterization of AMX-Loaded LNP Particles
Particle size (PZ), polydispersity index (PI), and zeta potential (ZP) characteristics of AMX-LPNs were defined. For particle size and zeta potential of AMX-LPN; dynamic laser light scattering (Zetasizer Ver. 5.11 Malvern) was used. Freshly prepared samples were diluted (1:100) with deionized water and sonicated for 15 minutes and measured in triplicate.
Determination of Percentage of Drug Encapsulation (% EE) and Percentage of Drug Loading (% DL) in AMX-LPNs
The percentage of drug encapsulation (% EE) and drug loading (% DL) of AMX in AMX-LPN formulations were determined. Freshly prepared samples were suspended in 10 mL distilled water and centrifuged at 10,000 rpm for 15 minutes. The supernatant was separated, diluted, and the amount of the drug was determined as reported liquid chromatography method mentioned in Pharmacopeia.29 That reversed-phase C18 column (particle size 5 mm; 250 X 4.6mm) was used for HPLC separation using a mixture (96:4v/v) of acetonitrile and phosphate buffer (pH 5) as mobile phase at a flow rate of 1.5 mL/minute with UV detection at 232 nm.30 The investigation was done at room temperature. The % EE and % DL were determined as following:
EE% = [[(Drug total- Drug free)]/Drug total] * 100
DL% = [Drug entrapped/Weight of particles] * 100
Fourier Transform Infrared (FTIR) Spectroscopic Analysis
The probable interaction between AMX, lipid, and polymer was examined. FTIR studies were performed using the FTIR spectrophotometer (Shimadzu 1800, Japan). The weighted amount of AMX and the selected sample were mixed with Potassium bromide powder and pressed into pellets.
Differential Scanning Calorimetry Studies
Thermal behavior of pure drug (AMX) and selected AMX-LPN formulations representing different drug:lipid ratios were considered using the differential scanning calorimetry method (DSC) (Mettler Toledo, Switzerland). Five milligram samples were packed in aluminum pans and were heated at a scanning rate of 10°C/min over 40–250°C temperature range under a nitrogen gas purged with a flow rate of 30 mL/min.
Surface Morphology Study of Selected Samples of AMX-LPNs
The surface morphology of the optimized AMX-LPNs formula was studied by using a transmission electron microscopy technique (TEM, Philips CM 10, Holland). To complete the TEM test, five mL suspension of the formula was pipetted on a 300 mesh copper grid coated with carbon film and allowed to sit for 15 minutes for drying. After complete drying, the sample was stained with a phospho-tungstic acid solution (2%w/v) and left for air drying at room temperature conditions.
In vitro Dissolution Studies
The release of AMX from MTs and LPNs was determined using dissolution test apparatus II (USP 24). The dissolution medium was 900 mL of 0.1 M HCl (pH 1.2), the temperature was kept at 37°C with a paddle stirrer at 50 rpm. One dose of AMX in MTs or LPN (formula was dispersed separately in a dialysis bag) was kept in the dissolution vessel. At each predetermined time interval, an exactly measured sample of the dissolution medium was withdrawn and replaced with an equal volume of a prewarmed fresh medium. The amount of AMX in withdrawn samples was measured by the HPLC method mentioned in section 2.3. All results were expressed as an average of three trials.
Mechanism and Kinetics of Drug Release
All release data were studied according to zero-order, first-order, Higuchi, Hixon–Crowell, Peppas, and Weibull kinetic equations. DDSolver, which is an add-in program for Microsoft Excel for modeling and comparison of drug release profiles was used. The model with the highest coefficient of determination (R2) was considered to be the best fitting one.31
Stability Studies
Short-term stability of optimized Formula (AMX-LPN11) was conducted. The samples were kept at 4°C, 25°C, and 40°C for 45 days. Samples were store in sealed vials and wrapped in aluminum foil and kept at a specified temperature. Samples were taken after 15, 30, and 45 days and any changes in appearance, drug content, particle size, and zeta potential were reported.
In vitro Antimicrobial Activity Assessment
MT and LPN formulations of AMX which exhibited optimal drug release features were selected for further evaluation of its antimicrobial activity. H. Pylori strain (ATCC 700392/26695) was used for assessment of antimicrobial activity of the selected formulations. Equivalent concentration of AMX from commercial tablets was suspended in 1 mL sterile water and used as a control. The bacterial susceptibility to AMX from MT, LPN, and control was performed using the agar plate diffusion method as mentioned in Clinical and Laboratory Standards Institute guidelines of the CLSI.32 The inhibition zone was then measured in mm with a slide caliper scale. In addition, the minimal inhibitory concentration test (MIC) was carried out by the microdilution broth test as described in the guidelines of the CLSI.33 MIC was described as the lowest concentration of the tested formula that inhibits the growth of bacteria compared to the control well, triphenyl tetrazolium chloride (TTC) was used as staining material. Loopfuls were taken from all wells which showed no growth, were inoculated onto Mueller Hinton agar plates using the streak plate method.34,35 All tests were completed three times and all the results were presented as mean values ± SD.
In vivo Pharmacokinetic Study of AMX in Wistar Rats
Animal Dosing and Scheme of Sampling
The in vivo study was approved by the ethical committee of bioavailability unit/Cairo University (no. N/A 161). The experiment was conducted in accordance with the reported principles of animal care reported by The European center for the validation of alternative methods (Diehl et al, 2001).36 Twenty-four male Wistar rats (190–220 g) were used in the study. Rats were divided equally and randomly into three groups (n=8). Each group was striking and housed in separate cages. Animals were having full and free access to food and water before and during the experiment. For the first group, an aliquot of AMX-PLN11 weight was suspended in deionized water and an oral dose of AMX was given to each rat via rat oral tubing. MTs with an equivalent dose of AMX were broken and delivered with an adequate volume of water to the second group. While equivalent weight of commercial tablets was also broken and given with water to the third group tablet. 0.5 mL of blood samples were collected in graduated microtainer, with lithium heparin, at zero, 0.5, 1, 2, 4, 6, 8, 12, and 24 h. Plasma samples were centrifuged (Hettich Zentrifugen, Germany) at 4000 rpm for 15 minutes and kept at −20°C till assayed.37
Chromatographic Analysis of AMX in Plasma Samples
The AMX concentration in each plasma sample was assayed according to the published Matar assay.38 After validation for the method selectivity, precision, accuracy, linearity, and stability. Briefly, AMX and cefadroxil (IS) were assayed using a C18 column (150 × 4.6 mm, 5μm) with a mobile phase (Methanol: 75 M potassium dihydrogen phosphate buffer solution) 10:90, v/v, at a flow rate of 1.5 mL/min. The assay was conducted at room temperature and a UV detector at 230 nm.
Data Calculation and Analysis
The concentrations of AMX in plasma were presented as the mean ± SD and pharmacokinetic parameters were calculated by the model-independent method. The mean maximum concentration (Cmax) of AMX and the time (Tmax) to reach Cmax were also calculated. The area under the drug concentration time curve (AUC0-24, µg mL−1 h) was calculated by the linear trapezoidal rule. The relative bioavailability (F) with the commercial product was also estimated.
Statistical Analysis of the Results
Calculation of statistical differences in in vitro data was done by SPSS Statistics 20 (Armonk, NY, USA) using one-way ANOVA with an extended LSD post hoc test for the calculation of pharmacokinetic parameters, and P value <0.05 was considered significant.
Results and Discussion
Pre-Compression Parameters of Powder Mixtures
Results showed that the angle of repose of all powder mixtures (MT1–MT12) ranged from 24.3° to 26.7° indicating an excellent flow property of all powder mixtures.39 Bulk and tapped density of the formulations powder mixture varied from 0.384 to 0.5450 gm/cm3 and from 0.532 to 0.601 gm/cm3, respectively. Hausner’s ratios and compressibility indices ranged from 1.11 to 1.20 and 19.04% to 20.98%, respectively. The flow properties results for granules are acceptable.39 The values of compressibility indices further confirmed the good compressibility of the prepared granules.22
Post-Compression Parameters for AMX-MTs
Regarding appearance, all mini-tablets formulations were yellowish-white or buff white, all showed round concave, smooth surface with no visible abrasions or cracks. The mean diameter of all MTs was 4.0± 0.0 mm while the average thickness was ranged between 3.4 and 4.5 mm. Mean hardness was in the range of 7.95±0.54 and 8.45±0.22 Kg/cm2 proving that the MTs are of adequate strength to tolerate physical abrasion.40 The friability test results showed that the loss of weight in all formulations was less than 1% which is a proof of acceptable mechanical resistance of the MTs.41 The percentage of drug content (14 mini-tablets) ranged from 98.60±0.95 to 101.24±1.02% which met the pharmacopeial requirements (90–110%).42 Since the powder mixtures were free-flowing, All the MTs were of uniform weight owing to uniformity in die fill. The mean weight of 20 MTs was 50± 0.0 mg.
Characterization of AMX-Loaded LPNs
The particle size (PS), polydispersity index (PI), and zeta potential (ZP) for all formulas were defined immediately. The results are presented in Table 2 and Figure 2. AMX-PLNs particle size was ranged between 235±34 nm and 390 ± 28 nm for AMX-PLN1 and AMX-PLN12, respectively. The results showed a direct relationship between PS and lipid/polymer concentration in the formula that the largest PS was for AMX-PLN12 which has the highest lipid and polymer content. The increase in the diameter of lipid/polymer nanoparticles by the increase of lipid and polymer concentrations was reported in many researches. Trotta M. mentioned that an increase in glyceryl monostearate (GMS) nanoparticles size was observed with the increase in GMS percentage.43 Similar observations were reported by Anwer MK et al.44 The PI values of AMX-PLNs were reached between 0.211±0.15 and 0.33±0.23 for AMX-PLN1 and AMX-PLN8, respectively, no relationship could be reported regarding lipid/polymer content and PDI of AMX-PLNs formulations, but small values showed that all formulations are monodispersed particles. The ZP values were ranged between +23.3 and +16.6 mV for AMX-PLN3 and AMX-PLN10, respectively. All formulations showed ZP less than 25 mV, so all can be considered as a stable colloidal dispersion. The magnitude of the charge was increased with the increase in the chitosan content and decreasing in the lipid content and vice versa.45 The morphology study of the optimized formula (AMX-PLN11) showed that the particles have a dense and rough spherical appearance with minimal particle aggregation-Figure 2.
 |
Figure 2 TEM image of optimized AMX-LPNs formula. |
Determination of Percentage Encapsulation Efficiency and Drug Loading of AMX-LPNs
The encapsulation efficiencies and drug loading of AMX in LPN1- LPN-12 were measured and expressed as EE% and DL%. The EE% represents the total amount of the entrapped drug, and the DL% showed the drug content in AMX in LPNs. The EE% of AMX-LPN1 and AMX-LPN12 were ranged between 54.1–86.4%, respectively, and the DL% was ranged between 28.5±0.9 and 41.2±2.3%, for AMX-LPN4 and AMX-LPN13, respectively (Table 2). A significant difference was observed in %EE and % DL were observed between formulations at p level <0.01, these variations can be explained based on the differences in the concentration of lipid and polymer. The results showed that an increase in encapsulation efficiency of AMX in LNPs was observed with the increase in lipid concentrations, which could be explained based on increasing the ability of lipids to hold the drug and decrease the drug flow to the outer medium. Also, the increase of CS content leads to a better hindrance to drug diffusion.
Differential Scanning Calorimetry (DSC) Study
DSC spectra for pure AMX and selected formulations were characterized over 40–250°C. Figure 3. The DSC of AMX showed a sharp endothermic peak at 136°C. Thermograms of the selected formulations showed no shift in the endothermic peak, but a reduction in intensity compared to pure AMX was observed due to decreasing in drug content in formulations.
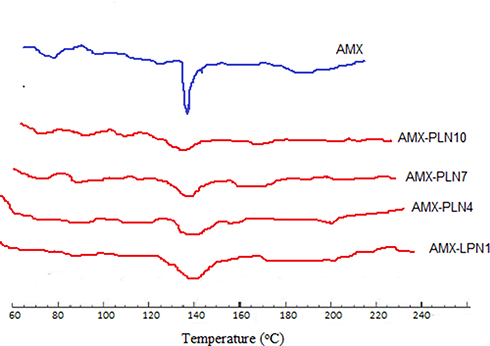 |
Figure 3 DSC thermograms of AMX, AMX-LPN1, AMX-LPN4, AMX-LPN7, and AMX-LPN10. |
FTIR Analysis Results
The FTIR analysis was used to study the compatibility of amoxicillin, with lipid and polymer in the AMX-LPNs at the four different (w/w) ratios as presented in Figure 4. The FT-IR spectrum of amoxicillin showed a band at 3475 cm−1, representing O-H, N-H stretching vibration and other representative peaks at 1767 cm−1 due to C=O stretching of β-lactamic, another peak was showed at 1680 cm−1 due to C=O stretching of amide and peaks at 1589 cm−1 showing asymmetric stretching of carboxylate.45 Spectrums of AMX-PLNs formulations revealed that there is no alternation in the main functional groups results from the incorporation of AMX into the lipid-polymer nanoparticles; thus, it confirms the stability of the drug.46
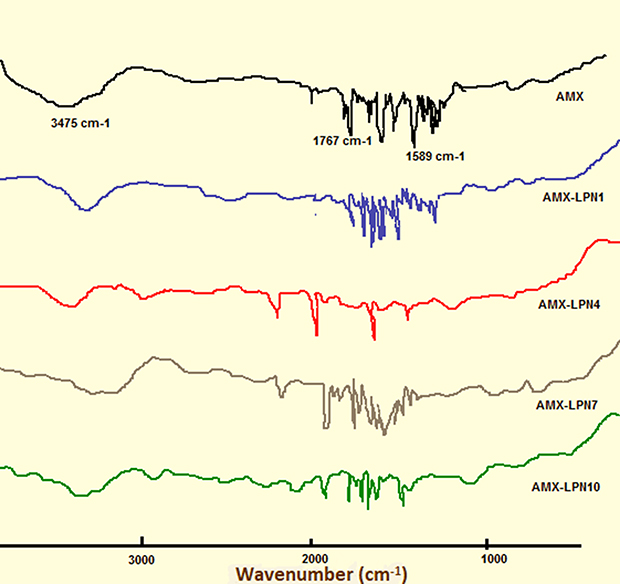 |
Figure 4 FTIR spectra of AMX, AMX-LPN1, AMX-LPN4, AMX-LPN7, and AMX-LPN10. |
In vitro Release Study
In vitro Release Study of AMX – Mini-Tablets
In vitro drug release experiments were completed to study the probability of extending the release of AMX using chitosan in different concentrations, also, to evaluate the effect of using Cr and GMS in different ratios on the delay of drug release. The in vitro dissolution profiles of AMX from MTs in 0.1 N HCl for 8 hours are shown in Figure 5A and B. All formulations (MT1–MT12) retained their integrity during the release studies, with a slow shrinking of the thickness due to polymer dissolution. It was detected that polymers undergo synchronized swelling, dissolution, and diffusion into the medium, resulting in a gradual reduction of the tablet size and strength.
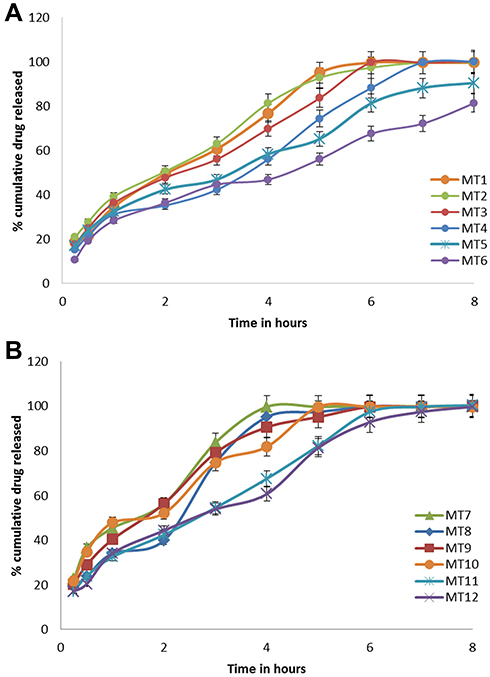 |
Figure 5 (A, B) In vitro dissolution profiles of AMX from Mini-tablets formulations (A) for MT1-MT-6, (B) for MT-7-MT-12 in 0.1N HCl. Each point is a mean value of three tablets. |
MT-1, MT-2 and MT-3 showed burst release since about 28.5%, 26.3%, and 23.2% of AMX released in 30 minutes, respectively (Figure 5A). The rapid initial burst could be interpreted on the base of the low content of Cr, the formed protective gel layer was not able to control the release of the drug,47 and about 100% of the drug was released within about 4 hours. So MT-1, MT-2, and MT-3 did not show the estimated sustained release features. Whereas, formula MT-4, MT-5, and MT-6 containing higher lipid polymer ratio released 19.1%, 17.5%, and 15.2% of AMX, respectively, in 30 min. The increase in lipid polymer content in the Mini-tablets (MT-4, MT-5, and MT-6) leads to a significant decline in the drug release (p<0.05) in the acidic medium by making thicker hydrophobic insoluble mass that acts as a better hindrance to drug diffusion and consequently, both the initial burst and release rate were decreased.48 Near inspection of tablets showed that the degree of their deformation was greater for those of higher CN content and low Cr content. That drug dissolution creates more holes in matrix structure facilitated the permeation of water into the matrix inner, helping diffusion of more drug from the Mini-tablets. These results, strength that the main reason why increasing Cr content in tablets, prolongs drug dissolution significantly. The in vitro release profiles of MT7-MT12S are shown in Figure 5B, it displays that GM-based Mini-tablets showed higher release rates compared with Cr based one. The difference in release between Cr, and GM Mini-tablets might be understood on the base of HLB value of the lipid polymers. GM has higher HLB (3.8) and consequently had a lower ability for controlling drug release, compared to Cr of lower HLB (2).44
Our study disclosed that mutual use of lipid-based polymers, namely, Cr and CN extended drug release significantly. Hydrophilic polymers depend on water absorption and gel formation, which subsequently allow drug diffusion from the matrix. When a lipid-based polymer is simultaneously present in the matrix, its lipophilic character can diminish water uptake by the matrix. Consequently, drug diffusion from tablets is reduced to yield a sustained release profile for a prolonged time. A similar observation was reported by Xiaochen Gu et al49 Considering the release pattern MT-6 was considered as the optimum formulation. The formula was selected for further in vivo pharmacokinetic studies.
In vitro Release Study Results from LPNs
Figure 6A–D showed the in vitro release patterns of AMX-PLN formulations. The dissolution rate profile of AMX from LPN1, 2, and 3 showed (Figure 6A) that rapid release of the drug was observed during 30 minutes that about 58.5±2.5%, 55.31±2.5%, and 50.20± 2.31% of the drug were released from AMX-LPN1, AMX-LPN2, and AMX-LPN3, respectively. Rapid release of the drug was shown and more than 95% of drug content was released within 6 hours. The results could be interpreted on the base of the low lipid content of these formulas and the ease of drug diffusion to the surrounding media. The rapid release pattern of the drug was also observed from LPN4, 5, and 6; that 85.1±1.5%, 80.11±2.2%, and 75.23± 1.41% of drugs were released from these formulas, respectively (Figure 6B). These results revealed that using AMX and lipid in a ratio of 1:1 was not sufficient to achieve the required hindrance of drug release to achieve a required sustain release profile of the drug. The increase in the drug: polymer ratio to 1:2 showed significant control of drug release at level p ˂ 0.5 as shown in Figure 6C, that burst release of the drug was observed due to the surface drug particles followed by slow release up to 7 h specially for AMX-LPN9 formula. The in vitro release profile of the formulations AMX-LPN10, AMX-LPN11, and AMX-LPN12 showed an accepted ability of LPNs to control the release of the drug up to 8 h (Figure 6D). The release was observed as a burst phase and a controlled release phase. In the burst phase, the release of the drug was observed 29.4±2.1%, 22.21±0.2%, and 18.13± 2.01%, for AMX-LPN10, AMX-LPN11, and AMX-LPN12, respectively. Controlling of drug release can be interpreted on the base of two factors, (a) the presence of the coating polymer that absorbs water and swells. Therefore, the drug is released through the formed pores, increasing in polymer concentration will form more narrow pores for drug flow. (b) the lipid is used for formulating nanoparticles that have a low water-absorbing ability, so it acts as a second barrier for drug release. These observations were consistent with other literature reports that reported such a biphasic release pattern for nano lipid-encapsulated drug molecules.50–52 The rate and amount of AMX released from AMX-LPN12 at all points and up to 8 hours were non significantly higher compared to AMX-LPN11. Based on the results, the AMX-LPN11 formula was selected for further investigation for its stability, in vitro antimicrobial activity, and in vivo bioavailability study. In addition, a correlation between in vitro release data and in vivo kinetics data was conducted.
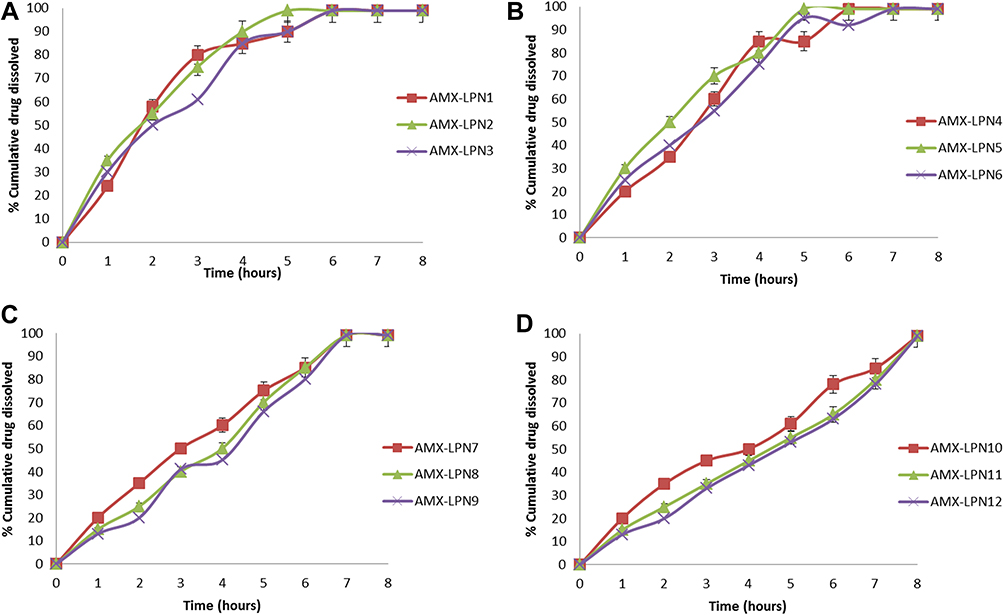 |
Figure 6 (A-D). In vitro dissolution profiles of AMX from LPNs formulations for (A) LPN1-3, (B) LPN4-6, (C) LPN7-9, (D) LPN10-12 in 0.1N HCl. Each point is a mean value of three trials. |
Study of Release Kinetics
In order to study the release kinetics of AMX from Mini-tablets and nanoparticles system, the results of in vitro release test of optimum formulations were studied using the model-dependent methods, zero-order model, first-order model, Higuchi model, Peppas model, and Hixson–Crowell model (Table 3). Conditions for choosing the most fitting model were based on the value of the coefficient of determination (R2) nearer to one.53 Concerning MT6; the highest values of R2 were after fitting into Peppas equation. Also, the value of “n” was determined to describe the release as Fickian or non-Fickian diffusion.53 The n values for MT6 (n= 0.40) have shown a Fickian diffusion mechanism of AMX from this mintablet.
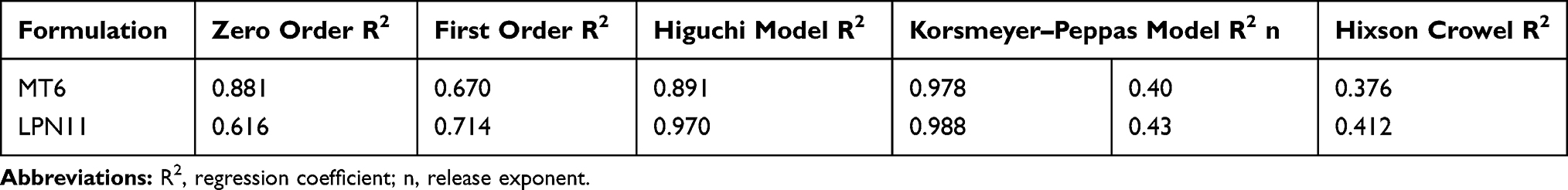 |
Table 3 Study of in vitro Release Kinetics Data for AMX from Selected Mini-Tablets and Lipid/Polymer Nanolipid Systems |
The regression coefficient (R2) of AMX-LPN11 showed that the Peppas power law had the best fit for the release data. The release exponent (n) was 0.43 for AMX-LPN11, demonstrating that the mechanism of AMX release from the LPN system is expressed by Fickian diffusion, that the release of AMX from these systems is controlled by diffusion through channels formed within the hydrogels and lipid polymers. These results are in agreement with another study stated by Zhang, L. et al.20
Stability Studies
The selected formula (AMX-LPN11) was subjected to short-term stability studies at 4°, 25°, and 40°C for 45 days. After storage under these conditions, particles were observed for changes in appearance, %EE, particle size, and zeta potential (Table 4). Storage of the samples at 4° and 25°C conditions did not result in considerable changes in the physical properties of tested samples regarding color and appearance via the visual inspection, while noticed aggregation and darkling in color was observed for samples stored at40°C. No significant change (p˂0.5) was observed in particle size when they were stored at 4° and 25°C for 45 days, but a significant increase in the size of particles at level p ˂0.5 was recorded when the particles were stored at 40°C due to aggregation. Also, there was no significant change between the zeta potential of fresh particles and particles kept at 4° and 25°C but zeta potential decreased to 9.01±1.4 after 45 days For particles stored at 40°C. This could be interpreted on the base of lipid dissolution which leads to aggregation of particles. Although the results showed accepted stability of LPNs; further longer studies are required to guarantee the stability of LPNs under different storage conditions.
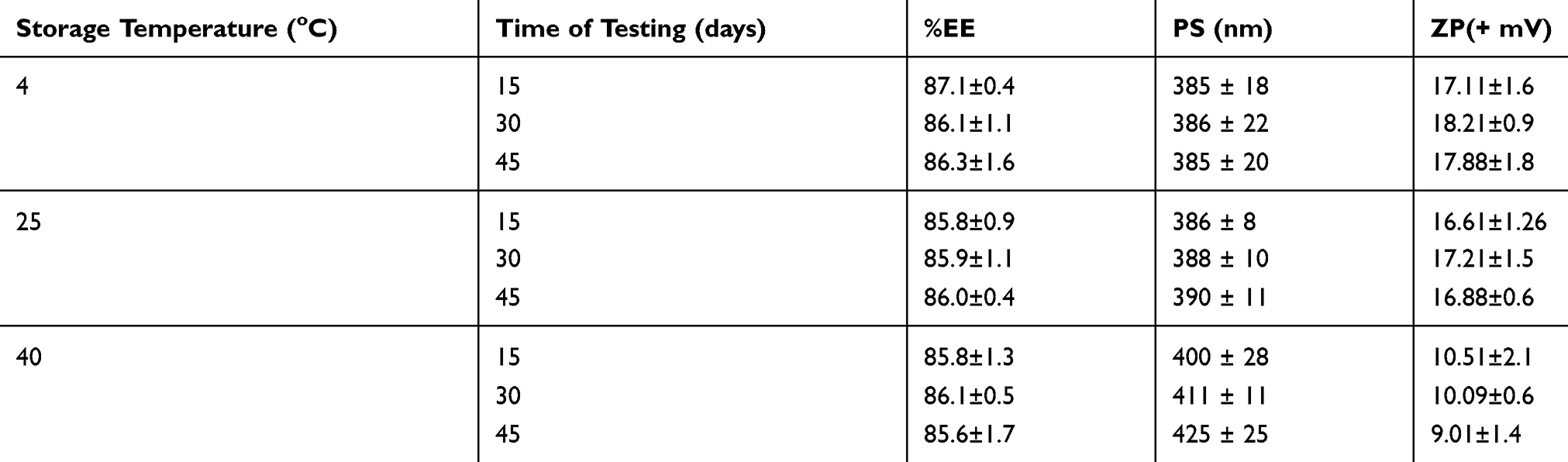 |
Table 4 %EE, PS, ZP of AMX-LPN11 Under Different Storage Conditions for 45 Days |
In vitro Antimicrobial Test Result
Table 5 showed the in vitro antibacterial activity of the optimum formula of AMX-MTs and LPNs in comparison to commercial AMX product against H. pylori. MT-6 and LPN-11 showed a significantly higher antibacterial activity compared to control as shown by their significantly wider zone of inhibition at level p < 0.001. The zones of inhibitions were 33.2±2.5 and 34.1±1.3 for MT-6 and LPN-11, respectively, compared with 28.5±1.8 for control. These results were further definite by MIC study results; the MIC of MT-6 and LPN-11 were around two-fold lower than those of control. It is approved that; designed formulations of AMX as minitabs and Lipid/polymer nanoparticles increase the ability of the drug to diffuse through the agar media and the greater the drug uptake through the bacterial cell wall, this was in good agreement with the formerly reported study by Khan, A.et al.54
 |
Table 5 Mean Results ±SD for Microbiological Tests of Selected AMX-PLN Formula |
In vivo Bioavailability Study Results
Pharmacokinetic study of the optimized Minitablet (MT-6) and lipid polymer nanoparticle (LPN11) of AMX compared with marketed tablet (Klavox® tablets 500 mg) were investigated following oral administration to rats. The plasma concentrations of AMX after oral administration of AMX-MT6, AMX-LPN11, and commercial tablets are presented in Table 6 and Figure 7. All AMX dosage forms show the same onset of action = 0.5±0.03h. Mean peak drug concentration of AMX-LPN11 Cmax (1.94±0.03 µg/mL) was higher than that of market product (1.79±0.7µg/mL) and MT-6 (1.88±0.1µg/mL). The mean time to reach the peak concentration (tmax) was equivalent, that no statistically significant difference (P>0.01) was observed amongst the tmax values of the three samples. The AUC0-24 value was 23.61±3.20, 42.49±4.0, and 43.68±4.3 (µg.h.mL−1) for commercial product, MT-6, and LPN11, respectively; recommend that LPN11 exposed both the highest rate and extent of drug absorption; however, market tablets showed the lowest rate and extent of absorption. The relative bioavailability of MT-6 and LPN11 were 1.80 and 1.85, respectively. Literature has reported that the structural integrity of the nanoparticles is an important factor for enhancing the oral absorption of the drug to its site of action.55,56 Similar results have been mentioned in another study, where the bioavailability of enoxaparin-loaded LPNs was 4.5-fold higher than the control solution.28 These results are also in good agreement with Anwer et al, results who reported a significant increase in the bioavailability of Delafloxacin-LPNs compared with the normal suspension.43
 |
Table 6 Pharmacokinetic Parameters of AMX After Oral Administration of AMX-LPN11 and Commercial Tablets in Rats |
 |
Figure 7 AMX plasma concentration (mean ± SE) time profiles in rats after oral administrations MT6, AMX-LPN11, and commercial tablets. |
Although the higher relative bioavailability of AMX from LPNs compared MTs, the simplicity, scalability, and reproducibility in manufacturing Mini-tablets, will give it higher opportunity on the industrial level.
In vitro/In vivo Correlation
In an attempted to discover the relationship between AMX plasma concentration and drug released from selected minitablet formula (MT6) and Lipid/polymer nanolipid formula (LPN11) in the dissolution medium. Plasma concentrations of drug absorbed (FA) were plotted against the fraction of drug released (FD) in vitro at the same time of 0.5, 1, 2, 3, 4, 5, 6, and 8 hours (Figure 8). The linear regression analysis showed that a statistically significant relationship (R2=0.9864) existed between the FD and FA for the Mini-tablets and was best described by the following equation: y=0.9667x −0.0267; and y= 0.9067x −0.0204 for LPN11. The slope and intercept were close to 1 and 0, respectively, indicating that the in vivo fraction absorbed could be predicted from in vitro dissolution data. Discovering a relation between the in vivo absorption and in vitro drug release from a controlled-release dosage form is an important part of the dosage form development process.57 Presented study showed a good relationship between the in vitro dissolution data and the in vivo pharmacokinetic data.
 |
Figure 8 In vitro/in vivo correlation between drug released in 0.1 N HCl (pH1.2) and plasma drug concentration in rats using MT6, LPN11, and market tablet. |
Conclusion
This study has confirmed the suitability of mixing hydrophilic polymers with lipid-based polymers to extend drug release from Mini-tablets and Lipid/polymers nanoparticles systems. According to the results, optimal formulations from both Mini-tablets and Lipid/polymers nanoparticles are able to control the release of AMX along with significant improvement in in vitro antimicrobial activity. The in vitro release profiles were modified by using different Chitosan concentrations and different ratios of lipid polymers. Analysis of the release data showed that drug release form optimal formulations followed diffusion according to fickian diffusion release mechanism. Both Mini-tablets and Lipid/polymers nanoparticles systems showed greater relative bioavailability of AMX than the reference tablets. An accepted correlation was defined between the in vitro release data and the in vivo pharmacokinetic data. These results confirmed that both controlled-released Mini-tablets and Lipid/polymers would be encouraging the delivery system for extending drugs action. Although the higher bioavailability of LPNs; the simplicity and reproducibility in manufacturing Mini-tablets will give it an advantage over the nanoparticles system.
Acknowledgments
Authors appreciate the support and help of the bioavailability unit, Cairo University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prasanna APS, Venkatasubbu GD. Sustained release of amoxicillin from hydroxyapatite nanocomposite for bone infections. Prog Biomater. 2018;7(4):289–296. doi:10.1007/s40204-018-0103-4
2. Arunachalam A, Karthikeyan M, Thulasiramaraju T. Formulation in vitro evaluation of Mebeverine HCl sustained release matrix tablets. Int J Adv Pharm Sci. 2013;4(5):1009–1020.
3. Gao P, Nie X, Zou M, et al. Recent advances in materials for extended-release antibiotic delivery system. J Antibiot. 2011;64(9):625–663. doi:10.1038/ja.2011.58
4. Hamdani J, Moës AJ, Amighi K. Development and evaluation of prolonged release pellets obtained by the melt pelletization process. Int J Pharm. 2002;245(1–2):167–177. doi:10.1016/S0378-5173(02)00348-4
5. Al Remawia M, Al-akayleh F, Salem M, Al Shami M, Badwan A. Application of excipient made from Chitosan-Xanthan as a single component for the controlled release of ambroxol tablet. J Excipients Food Chem. 2013;4:48–57.
6. Maderuelo C, Zarzuelo A, Lanao M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release. 2011;154:2–19.
7. Goole J, Deleuze P, Vanderbist F, Amighi K. New levodopa sustained-release floating Mini-tablets coated with insoluble acrylic polymer. Eur J Pharm Biopharm. 2008;68(2):310–318.
8. Roberts M, Vellucci D, Mostafa S, Miolane C, Marchaud D. Development and evaluation of sustained-release Compritol® 888 ATO matrix mini-tablets. Drug Dev Ind Pharm. 2012;38(9):1068–1076.
9. Mohamed FAA, Roberts M, Seton L, Ford JL, Levina M, Rajabi-Siahboomi AR. The influence of HPMC concentration on release of theophylline or hydrocortisone from extended release mini-tablets. Drug Dev Ind Pharm. 2013;39(8):1167–1174.
10. Biswas N, Sahoo RK, Guha A, Kuotsu K. Chronotherapeutic delivery of hydroxypropylmethylcellulose based mini-tablets: an in vitro–in vivo correlation. Int J Biol Macromol. 2014;66:179–185. doi:10.1016/j.ijbiomac.2014.02.036
11. Muller RH, Gohla S, Keck CM. State of the art of nanocrystals – special features, production, nanotoxicology aspects and intracellular delivery.Eur J Pharm Biopharm. 2011;78(1):1–9. doi:10.1016/j.ejpb.2011.01.007
12. Chokshi NV, Khatri HN, Patel MM. Formulation, optimization, and characterization of rifampicin-loaded solid lipid nanoparticles for the treatment of tuberculosis. Drug Dev Ind Pharm. 2018;44(12):1975–1989. doi:10.1080/03639045.2018.1506472
13. Kim BYS, Rutka JT, Chan WCW. Current concept: nanomedicine. N Engl J Med. 2010;363(25):2434–2443. doi:10.1056/NEJMra0912273
14. Gu L, Shi T, Sun Y, et al. Folate-modified, indocyanine green-loaded lipid-polymer hybrid nanoparticles for targeted delivery of cisplatin. J Biomater Sci Polym Ed. 2017;28(7):690–702. doi:10.1080/09205063.2017.1296347
15. Salvador-Morales C, Zhang L, Langer R, Farokhzad OC. Immunocompatibility properties of lipid–polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomaterials. 2009;30(12):2231–2240. doi:10.1016/j.biomaterials.2009.01.005
16. López-López M, Fernández-Delgado A, Moyá ML, Blanco-Arévalo D, Carrera C, de la Haba R. Optimized Preparation of Levofloxacin Loaded Polymeric Nanoparticles. Pharmaceutics. 2019;11(2):57. doi:10.3390/pharmaceutics11020057
17. Mukherjee A, Waters AK, Kalyan P, Achrol AS, Kesari S, Yenugonda VM. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: state of the art, emerging technologies, and perspectives. Int J Nanomed. 1937–1952;2019:(14).
18. Vieira ACC, Chaves LL, Pinheiro S, et al. Mucoadhesive chitosan-coated solid lipid nanoparticles for better management of tuberculosis. Int J Pharm. 2018;536(1):478–485. doi:10.1016/j.ijpharm.2017.11.071
19. Jain AS, Shah S, Nagarsenker MS, et al. Lipid Colloidal Carriers for Improvement of Anticancer Activity of Orally Delivered Quercetin: formulation, Characterization and Establishing In Vitro–In Vivo Advantage.J Biomed Nanotech. 2013;9(7):1230–1240. doi:10.1166/jbn.2013.1636
20. Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid–polymer hybrid nanoparticles: A robust drug delivery platform. ACS Nano. 2008;2;1696–1702.
21. World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019. Geneva: World Health Organization; 2019.
22. Beg S, Nayak AK, Kohli K, Swain S, Hasnain MS. Antimicrobial activity assessment of time-dependent release bilayer tablets of amoxicillin trihydrate. Brazilian J Pharm Sci. 2012;48(2):265–272. doi:10.1590/S1984-82502012000200010
23. Antos D, Schneider-Brachert W, Bästlein E, et al. 7-Day triple therapy of Helicobacter pylori infection with levofloxacin, amoxicillin, and high-dose esomeprazole in patients with known antimicrobial sensitivity. Helicobacter. 2006;11(1):39–45. doi:10.1111/j.0083-8703.2006.00375.x
24. Kaye CM, et al. The clinical pharmacokinetics of a new pharmacokinetically enhanced formulation of amoxicillin/clavulanate. Clin Ther. 2001;23(4):578–584. doi:10.1016/S0149-2918(01)80061-8
25. Sahasathian T, Kerdcholpetch T, Chanweroch A, Praphairaksit N, Suwonjandee N, Muangsin N. Ch Anwer och, A.; Praphairaksit, N.; Suwonjandee, N.; Muangsin, N. Sustained release of amoxicillin from chitosan tablets. Arch Pharm Res. 2007;30(4):526–531. doi:10.1007/BF02980229
26. Kultida S, Jatuporn P, Narong P, Nongnuj M. Sustained Release of Amoxicillin from Ethyl Cellulose-Coated Amoxicillin/Chitosan–Cyclodextrin-Based Tablets. AAPS PharmSciTech. 2011;12:1.
27. Aki H, Nakashima Y, Kawasaki Y, Niiya T. Thermodynamic evaluation of antibacterial activity for inclusion complexes of amoxicillin with cyclodextrins. J Therm Anal Calorim. 2006;85(3):685–688. doi:10.1007/s10973-006-7650-y
28. Dong W, Wang X, Liu C, et al. Chitosan based polymer-lipid hybrid nanoparticles for oral delivery of enoxaparin. International Journal of Pharmaceutics. 2018;547(1–2):499–505. doi:10.1016/j.ijpharm.2018.05.076
29. United States Pharmacopoeia-30 and National Formulary‐25. Official Compendia of Standards. Rockvile (US): United States Pharmacopoeial Convention; 2007.
30. Hadizadeh M, Toraji A. Amoxicillin-Loaded Polymeric Nanoparticles of Less than 100 nm: design, Preparation and Antimicrobial Activity Against Methicillin-Resistant Staphylococcus aureus. Iranian Journal of Science and Technology, Transactions A: Science. 2019;43(2):379–386. doi:10.1007/s40995-017-0346-2
31. Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. doi:10.1208/s12248-010-9185-1
32. CLSI. Performance Standards for Antimicrobial Susceptibility Testing, Disc Di_usion Supplemental Tables. Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2013.
33. CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard. Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2015.
34. McNulty C, Owen R, Tompkins D, et al. Helicobacter pylori susceptibility testing by disc diffusion. J Antimicrobial Chem. 2002;49(4):601–609. doi:10.1093/jac/49.4.601
35. Kahlmeter G, Brown DF, Goldstein FW, et al. European Committee on Antimicrobial Susceptibility Testing (EUCAST) Technical Notes on antimicrobial susceptibility testing.Clin Microbiol Infect. 2006;12(6):501–503. doi:10.1111/j.1469-0691.2006.01454.x
36. Diehl K-H, Hull R, Morton D, Pfister R. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21(1):15–23. doi:10.1002/jat.727
37. Radwan MA, Ela AESF, Hassan MA, El-Maraghy DA. A. el S.; Hassan, M. A.; El-Maraghy, D. A. Pharmacokinetics and analgesic effect of ketorolac floating delivery system. Drug Deliv. 2015;22(3):320–327. doi:10.3109/10717544.2014.883189
38. Matar KM. Simple and Rapid LC Method for the Determination of Amoxicillin in Plasma. Chromatographia. 2006;64(5–6):255–260. doi:10.1365/s10337-006-0021-9
39. Wilson B, Sitarambhai PH, Sajeev M, Vinothapooshan G. Design and evaluation of sustained release matrix tablets of levofloxacin for effective treatment of microbial infections. Int J Drug Deliv. 2011;3:305–314.
40. Manivannan R, Chakole V. Formulation and development of extended release floating tablet of atenolol. Int J Recent Adv Pharm Res. 2011;3:25–30.
41. Savic IM, Nikolic K, Nikolic G, Savic I, Agbaba D, Cakic M. Application of mathematical modeling for the development and optimization formulation with bioactive copper complex. Drug Dev Industrial Pharm. 2013;39(7):1–7. doi:10.3109/03639045.2012.707208
42. Garg A, Gupta M. Taste masking and formulation development & evaluation of mouth dissolving tablets of levocetirizine dihydrochloride. J Drug Deliv Ther. 2013;3:123–130.
43. Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification–diffusion technique. Int J Pharm. 2003;257(1–2):153–160. doi:10.1016/S0378-5173(03)00135-2
44. Anwer MK, Iqbal M, Muharram MM, et al. Development of Lipomer Nanoparticles for the Enhancement of Drug Release, Anti-microbial Activity and Bioavailability of Delafloxacin. Pharmaceutics. 2020;12(3):252.
45. Mohammed M, Alnafisah MS, Anwer K, et al. Chitosan surface modified PLGA nanoparticles loaded with brigatinib for the treatment of non-small cell lung cancer. J Polymer Eng. 2019;39(10):909–916. doi:10.1515/polyeng-2019-0265
46. Sonvico F, Cagnani A, Rossi A, et al. Formation of self-organized nanoparticles by lecithin/chitosan ionic interaction. Int J Pharm. 2006;324(1):67–73. doi:10.1016/j.ijpharm.2006.06.036
47. Tiwari S, Rajabi-Siahboomi A. Applications of complementary polymers in HPMC hydrophilic extended release matrices. Drug Deliv Technol. 2009;9:20–27.
48. Tatavarti A, Mehta K, Augsburger L, Hoag S. Influence of methacrylic and acrylic acid polymers on the release performance of weakly basic drugs from sustained release hydrophilic matrices. J Pharm Sci. 2014;66(9):2319–2331. doi:10.1002/jps.20129
49. Xiaochen G, Daryl J, Estelle R, Keith J. Evaluation and comparison of five matrix excipients for the controlled release of acrivastine and pseudoephedrine. Drug Dev Ind Pharm. 2004;30:1009–1017.
50. Cheow WS, Chang MW, Hadinoto K. The roles of lipid in anti-biofilm efficacy of lipid–polymer hybrid nanoparticles encapsulating antibiotics. Coll Surf a Phys Eng Asp. 2011;389:158–165.
51. Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663.
52. Zheng Y, Wang L, Lu L, Wang Q, Benicewicz B. C. pH and thermal dual-responsive nanoparticles for controlled drug delivery with high loading content. ACS Omega. 2017;2(7):3399–3405.
53. Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133.
54. Khan AA, Mudassir J, Akhtar S, Murugaiyah V, Darwis Y. Freeze-dried lopinavir-loaded nanostructured lipid carriers for enhanced cellular uptake and bioavailability: statistical optimization, in vitro and in vivo evaluations. Pharmaceutics. 2019;11:97.
55. Tahir N, Madni A, Correia A, et al. Lipid-polymer hybrid nanoparticles for controlled delivery of hydrophilic and lipophilic doxorubicin for breast cancer therapy. Int J Nanomedicine. 2019;14:4961–4974. doi:10.2147/IJN.S209325
56. Iqbal M, Ezzeldin E, Herqash RN, Anwer MK, Azam F. Development and validation of a novel UPLC-MS/MS method for quantification of delafloxacin in plasma and aqueous humour for pharmacokinetic analyses. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1138:121961.
57. Pande A, Vaidya P, Arora A. In vitro and in vivo evaluation of ethyl cellulose based floating microspheres of cefpodoxime proxetil. Int J Pharm Biomed Res. 2010;1:122–128.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.