Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Microvascular pericytes in healthy and diseased kidneys
Received 21 October 2013
Accepted for publication 22 November 2013
Published 17 January 2014 Volume 2014:7 Pages 39—48
DOI https://doi.org/10.2147/IJNRD.S37892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Szu-Yu Pan,1,2 Yu-Ting Chang,3 Shuei-Liong Lin1,3
1Renal Division, Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan; 2Department of Internal Medicine, National Taiwan University Hospital, Yun-Lin Branch, Yun-Lin, Taiwan; 3Graduate Institute of Physiology, College of Medicine, National Taiwan University, Taipei, Taiwan
Abstract: Pericytes are interstitial mesenchymal cells found in many major organs. In the kidney, microvascular pericytes are defined anatomically as extensively branched, collagen-producing cells in close contact with endothelial cells. Although many molecular markers have been proposed, none of them can identify the pericytes with satisfactory specificity or sensitivity. The roles of microvascular pericytes in kidneys were poorly understood in the past. Recently, by using genetic lineage tracing to label collagen-producing cells or mesenchymal cells, the elusive characteristics of the pericytes have been illuminated. The purpose of this article is to review recent advances in the understanding of microvascular pericytes in the kidneys. In healthy kidney, the pericytes are found to take part in the maintenance of microvascular stability. Detachment of the pericytes from the microvasculature and loss of the close contact with endothelial cells have been observed during renal insult. Renal microvascular pericytes have been shown to be the major source of scar-forming myofibroblasts in fibrogenic kidney disease. Targeting the crosstalk between pericytes and neighboring endothelial cells or tubular epithelial cells may inhibit the pericyte-myofibroblast transition, prevent peritubular capillary rarefaction, and attenuate renal fibrosis. In addition, renal pericytes deserve attention for their potential to produce erythropoietin in healthy kidneys as pericytes stand in the front line, sensing the change of oxygenation and hemoglobin concentration. Further delineation of the mechanisms underlying the reduced erythropoietin production occurring during pericyte-myofibroblast transition may be promising for the development of new treatment strategies for anemia in chronic kidney disease.
Keywords: endothelial cell, fibroblast, chronic kidney disease, myofibroblast, renal fibrosis
Introduction
The pericytes have gained much attention in various organ systems in recent times. In the kidney, the pericytes have been shown to take the central role with regard to fibrogenesis, microvascular stability, and erythropoietin (EPO) production.1,2 Our knowledge of the pericytes has expanded markedly in recent years thanks largely to the advances of genetic lineage–tracing techniques.
Pericytes and perivascular fibroblasts have been traditionally defined by their morphologic and anatomic characteristics.3 They are both highly branched interstitial cells of mesenchymal origin with the ability to generate collagen. These cells are named pericytes when they are embedded within the microvascular basement membrane and are in close contact with endothelial cells. When these cells are not in direct contact with endothelial cells, they are called perivascular fibroblasts. According to a previous work, the term pericytes refers collectively to a heterogeneous population of interstitial fibroblasts with similar morphology, located at the perivascular area.4
This review focuses on the recent studies of pericytes in healthy and diseased kidneys.
Characteristics of renal pericytes
The identification of renal pericytes remains a difficult task. An approach that combines the consideration of genetic lineage tracing, morphological and anatomical criteria, and the expression of multiple surface markers may be the best available solution for this challenge.
Morphological and anatomic characteristics of renal pericytes revealed by genetic lineage tracing
In our previous study, the morphologic characteristics of renal pericytes were depicted.1 By using collagen, type 1, alpha 1-green fluorescent protein, transgenic (Col1a1-GFPTg) mice expressing enhanced GFP under the regulation of Col1a1 promoter and enhancers, we labeled the cells producing Col1a1.1 These Col1a1-GFP+ cells in the kidney included the pericytes, perivascular fibroblasts, and glomerular podocytes, while the mesangial cells and vascular smooth muscle cells did not show green fluorescence (Figure 1). We then bred forkhead box d1 (Foxd1Cre/+) knock-in mice with tdTomato reporter mice, and the fate of Foxd1-derived mesenchymal cells of the kidneys was mapped in the Foxd1Cre/+; Rosa26fstdTomato/+ mice (Figure 2A). In developmental nephrogenesis, Foxd1 is activated in the metanephric mesenchymal progenitor cells fated to become the kidney stromal cells. The Foxd1-derived cells are fated to become pericytes, perivascular fibroblasts, vascular smooth muscle cells, and mesangial cells.5 In fact, when Foxd1-tdTomato+ cells were compared with Col1a1-GFP+ pericytes, they were found to be identical.6 The pericytes are located in the subendothelial area, in close contact with the endothelial cells (Figure 1). A single pericyte has numerous extended processes, and different processes may be in contact with different endothelial cells. The cell processes of the pericytes and the cytoplasm of the endothelial cells are configured in a “peg and socket” arrangement, which supports an intimate crosstalk between the pericytes and the endothelial cells. The ratio of pericytes to endothelial cells is ~0.40.1 Perivascular fibroblasts are located near arterioles and venules, without direct contact with the endothelial cells. The ratio of perivascular fibroblasts to vascular smooth muscle cells is ~0.33.
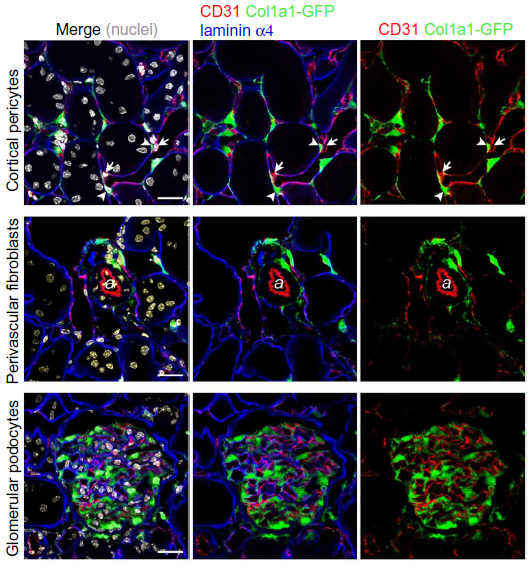 | Figure 1 Characterization of microvascular pericytes and perivascular fibroblasts in normal adult kidney of Col1a1-GFPTg mice. |
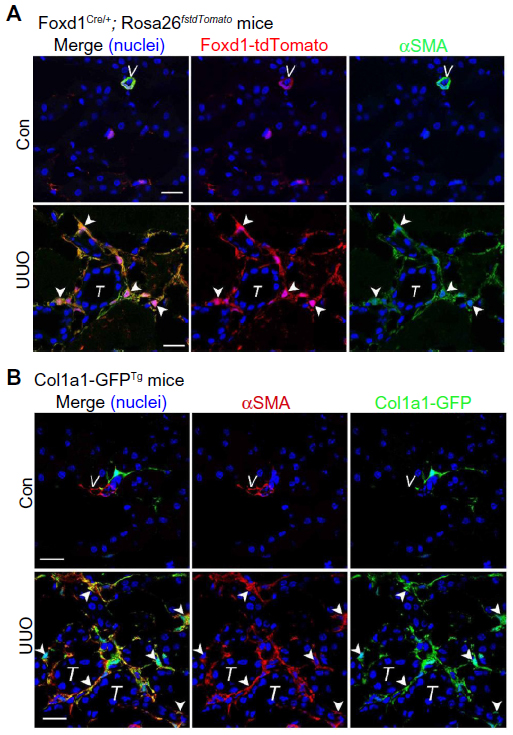 | Figure 2 Renal pericytes are the origin of scar-producing myofibroblasts in fibrotic kidney. |
Molecular markers of renal pericytes
In addition to the morphologic and structural criteria, many molecular markers have been used to identify the pericytes. However, the expressions of surface markers in pericytes are determined, not only by the organ system of interest but also, by the specific development stages of the pericytes. Moreover, some of these markers are expressed nonspecifically in cells other than the pericytes. For example, α-smooth muscle actin (αSMA) (a contractile protein present in smooth muscle lineages and myofibroblasts), often used as a marker for the pericytes in the retina,7 has been shown to have a low level of expression in renal pericytes in adult healthy kidneys (Figure 2B).1 Neuron-glial antigen 2 (NG2) (a chondroitin sulfate proteoglycan) is also minimally expressed on quiescent adult renal pericytes.1 However, both αSMA and NG2 are expressed in the pericytes of neonatal kidney, and their expressions are lost as the kidney matures from day 12 after birth (Table 1).1,8 When a renal insult, such as a unilateral ureteral obstruction (UUO) is applied, the pericytes are activated, and NG2 is again highly expressed on these activated renal pericytes.1,9 Other pericyte markers, such as platelet-derived growth factor (PDGF) receptor β (PDGFRβ), desmin, vimentin, and 5' ectonucleotidase (also known as cluster of differentiation [CD]73), when used alone, also fail to label renal pericytes unequivocally and specifically.
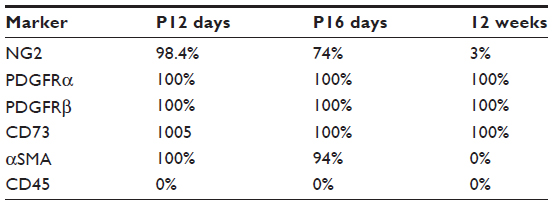 | Table 1 The proportion of Col1a1-GFP+ pericytes concurrently expressed the markers in normal kidney |
The identity of EPO-producing resident fibroblasts and renal pericytes
Since Col1a1-GFP+ microvascular pericytes are the major collagen-producing cells in the interstitium of the healthy kidney, we think the perivascular mesenchymal cells studied by the other independent groups are probably pericytes.10–14 A study reported by Asada et al mapped mesenchymal cells in early development and identified a population of mesenchymal cells expressing a myelin protein P0, which completed migration from the neural crest to the kidney metanephric mesenchyme by embryonic day 13.5.11 These cells populate the nephrogenic interstitial mesenchyme zone of the metanephric mesenchyme, which is identical to the Foxd1+ mesenchymal progenitor area, before migrating into the kidney stroma. The investigators showed that these cells became the PDGFRβ+, CD73+ mesenchymal cells of the adult kidney. Although the authors named them as resident fibroblasts of the adult kidney, rather than pericytes, they showed that these cells attached to capillaries and could generate EPO. It is very likely, therefore, that the researchers mapped the same cells as the pericytes, providing strong new evidence that mesenchymal cells may arise, before nephrogenesis commences, as neural crest progenitors.
Hence adult kidney pericytes are located in the subendothelial area, derived from Foxd1-expressing progenitor cells, and are positive for Col1a1-GFP, PDGFRβ, and CD73, and negative for αSMA and CD45.1,5,8,9
Roles of microvascular pericytes in kidneys
We have gained some insights into the roles of renal pericytes in renal fibrosis, microvascular stability, and EPO production. Through transition into scar-producing myofibroblasts, activated renal pericytes contribute to renal fibrosis. To the contrary, quiescent renal pericytes maintain microvascular stability in coordination with endothelial cells. In addition, the inadequate EPO production by renal pericytes after their transition into myofibroblasts may explain, in part, the anemia encountered in patients with chronic kidney disease (CKD).
Renal fibrosis
Renal fibrosis is the common pathway of various kinds of kidney injury. The severity of renal fibrosis is highly associated with the impairment of renal function. Microscopically, renal fibrosis is related to extracellular matrix (ECM) deposition, cellular infiltration by inflammatory cells and myofibroblasts, tubular atrophy, and capillary rarefaction.15 The deposited ECM consists mainly of type I collagen. Previous study has unequivocally identified myofibroblast as the key effector in producing the pathogenic collagen.16 However, the source of the myofibroblast remains a great debate.
Origin of myofibroblasts
An earlier study using protein S100A4 (also known as fibroblast-specific protein-1) as a marker of fibroblasts suggested epithelial-mesenchymal transition (EMT) and bone marrow-derived circulating cells as the major source of renal myofibroblasts.17 The ablation of S100A4+ cells is associated with the amelioration of renal fibrosis. However, subsequent studies have revealed that S100A4 might better serve as a marker of macrophages and that the effect of ablating S100A4+ cells might be related to the elimination of macrophages.1,18,19 In our studies using genetic lineage tracing mouse models, achieved by labeling collagen-producing cells or stromal cells with fluorescent protein or β galactosidase, along with the analysis of surface markers, morphological, and structural characteristics, we have identified that the majority of myofibroblasts originate from resident renal microvascular pericytes (Figure 2).1,5,6 Renal microvascular pericytes, derived from Foxd1-expressing progenitors during kidney development, differentiate to myofibroblasts during renal fibrogenic injury.5,6 In contrast, renal cells with a fate marker activated by Six2 (a transcription factor expressed transiently during development, in cells of the metanephric mesenchyme destined to become only renal epithelial cells) or homeobox B7 (HoxB7) (a transcription factor expressed in cells of the ureteric bud) do not show any evidence of differentiation into myofibroblasts during renal fibrogenic injury.5 Different independent studies have shown the same conclusion that an EMT of injured tubular epithelial cells does not exist during renal fibrosis.13,20,21
According to the aforementioned findings, it is plausible that pericytes are the major source of renal myofibroblasts. Apart from the kidney, pericytes have also been shown to be the source of scar-forming myofibroblasts in other organ systems, such as the central nervous system, the skin, skeletal muscle, lung, intestine, and the liver.22-25
The contribution of circulating fibrocytes to the active renal myofibroblast population is also an issue of dispute. Studies performed in the kidney and other organs have reported variable results regarding the proportion of active myofibroblasts derived from circulating fibrocytes.26,27 In order to dissect the contribution of bone marrow-derived fibrocytes to renal myofibroblasts, we performed transplantation of bone marrow from Col1a1-GFPTg mice to wild type C57BL/6 mice after sublethal irradiation.1 The result revealed only very few Col1a1-GFP+ cells (indicating bone marrow-derived fibrocytes) appearing at the perivascular area after UUO or ischemia-reperfusion injury surgery (less than 0.1% of total myofibroblast population).
Recently, LeBleu et al used multiple genetically engineered mice to track and determine the origin and function of myofibroblasts in kidney fibrosis.28 The authors suggested that nearly 50% of renal myofibroblasts derive from local resident myofibroblasts, through proliferation. The researchers found that nonproliferating myofibroblasts were derived from bone marrow (35%), the endothelial-to-mesenchymal transition (EndoMT) (10%), and from the EMT (5%). This study relied heavily on transgenic mouse model with short promoter-driven Cre, which is more likely to produce a mosaic, weak, and aberrant expression pattern compared with the longer promoter.29 The results, supporting EMT-, EndoMT-, and bone marrow-derived myofibroblasts, relied on red fluorescent protein (αSMA-RFP)Tg mice, which were generated using a very short promoter of actin, alpha 2 (Acta2) to drive the reporter RFP. The faithfulness of αSMA-RFP in reporting the αSMA+ cells was questionable because the aberrant expression of RFP driven by the short αSMA promoter was seen in healthy renal epithelial tubules in all mice and in cells negative for fluorescein isothiocyanate (FITC)-labeled αSMA.28 They further demonstrated that the ablation of proliferating NG2+ or PDGFRβ+ cells did not greatly alter the fibrosis. It is not clear why so few αSMA+ cells concurrently expressed PDGFRβ compared with other published data that would suggest that a greater number should have been positive.30 The use of BALB/c mice in the study, which are less prone to fibrosis compared with C57BL/6 mice, may in part, account for the small number of αSMA+ cells that concurrently expressed PDGFRβ.31,32 However, it is more likely that the cause was the use of the mice with short promoter-driven Cre. Thus these provocative findings of this study are not definitive and should be interpreted with caution because of inherent limitations in the genetic tagging methods used to identify the myofibroblasts and because of the use of short promoter transgenic mice which do not efficiently identify cells of interest.
To sum up, the majority of studies have identified renal microvascular pericytes as the origin of renal myofibroblasts. The contribution of circulating fibrocytes, EMT, and EndoMT, though not completely excluded, is still unclear.
Interruption of pericyte-myofibroblast transition to attenuate renal fibrosis
The clinical burden of CKD calls for an effective way to halt the progression of renal fibrosis. However, so far, the treatment options for fighting irreversible renal fibrosis have been scarce and limited. Since renal myofibroblasts are responsible for the fibrogenesis in the kidney and myofibroblasts are mainly derived from renal pericytes, the interruption of the pericyte-myofibroblast transition stands a good chance of halting the development of renal fibrosis. Multiple growth factors have been implicated in the crosstalk between renal pericytes and their neighboring endothelial cells or tubular epithelial cells. Among them, PDGF, vascular endothelial growth factor (VEGF)-A, and transforming growth factor (TGF)-β are under the most intensive investigation.
The PDGF system consists of four ligands (PDGF-A, -B, -C, and -D) and two receptors (PDGFRα and PDGFRβ).30 This system regulates a variety of biological events, including cell proliferation, cell migration, cell activation, inflammation, tissue permeability, and ECM deposition. PDGFR has tyrosine kinase activity and can be autophosphorylated after ligand binding. In the kidney, PDGF is expressed mainly in endothelial cells, glomeruli, and tubular epithelial cells, whereas PDGFR is found in pericytes, vascular smooth muscle cells, and glomerular mesangial cells.30 We examined the role of the PDGF system in renal fibrosis, in Col1a1-GFPTg reporter mice following UUO or ischemia-reperfusion injury surgery.33 Renal pericyte-myofibroblast transition was found to be associated with increased expression of both PDGF and PDGFR. In the injured renal interstitium, the increased expression of PDGF was found in the tubular epithelial cells, endothelial cells, and macrophages, whereas the PDGFR was expressed exclusively in the renal pericytes and myofibroblasts. A PDGFR blockade with anti-PDGFR antibody or the PDGFR tyrosine kinase inhibitor imatinib attenuated the pericyte-myofibroblast transition and subsequent renal fibrosis. Notably, PDGFR blockade was also associated with a reduced infiltration of macrophages.
The VEGF-A system is known to regulate physiological and pathological angiogenesis.34,35 This system comprises multiple isoforms, such as VEGF120, VEGF164, and VEGF188, based on alternative exon splicing.36–38 Receptors for VEGF-A, including VEGFR1, VEGFR2, VEGFR3, and neuropilin have been identified in endothelial cells. In the normal kidney, VEGF-A has been detected mainly in the glomerular podocytes, tubular epithelial cells, and pericytes, while VEGFR is found in endothelial cells.38 Eremina et al have highlighted the crosstalk between the glomerular podocytes (secreting VEGF-A) and glomerular endothelial cells (expressing VEGFR).39 The association of VEGF-A blockade and thrombotic microangiopathy has also been depicted in both clinical and experimental conditions. However in a transgenic mouse model with VEGF-A overexpression by renal tubular epithelial cells, a high level of VEGF-A was found to stimulate fibroblast proliferation and transition into myofibroblasts.40 The effect of VEGF-A on fibroblasts is possibly mediated by endothelial cells indirectly. We previously showed that VEGFR2 blockade following fibrogenic injuries induced by UUO or ischemia-reperfusion injury resulted in an attenuation of pericyte-myofibroblast transition and renal fibrosis.38 Notably, VEGFR2 blockade also reduced the expression of PDGF-B and macrophage recruitment. Thus it is reasonable to consider that VEGF-A promotes renal fibrosis through endothelial activation and subsequent induction of PDGF, TGF-β1, and inflammation. It should be noted that, in terms of the VEGF-A system, the peritubular capillary compartment of the kidney is quite different from the glomerular capillary compartment. A blockade of glomerular VEGF-A results in thrombotic microangiopathy, while the blockade of interstitial VEGF-A attenuates the injury related renal fibrosis. The glomerulus is known as a vascular bed with high shear stresses and a very high turnover of glomerular endothelial cells. It is likely that unlike the peritubular capillaries, the VEGF-A signaling from podocytes to glomerular endothelial cells is active physiologically and that a chronic angiogenic response is normal. A complete loss of the crosstalk with the endothelium in the glomerulus in physiological conditions destabilizes the glomerular endothelium. By contrast, the peritubular capillaries are nonangiogenic in physiological conditions, and injury-stimulated angiogenesis is at first adaptive but subsequently, maladaptive. Moreover, unlike a genetic deletion of VEGF-A, a pharmacological blockade attenuates VEGF-A signaling but does not completely ablate it. Clearly, the distinction between VEGF-A signaling in the glomerulus and the peritubular capillaries requires further study because it may be that in the short term, a VEGFR2 blockade is beneficial during acute injury, whereas chronic blockade becomes deleterious.
TGF-β is another growth factor that is important in renal fibrosis.41,42 There are three isoforms of TGF-β, namely, TGF-β1, TGF-β2, and TGF-β3. Three different TGF-β receptors - type I, II, and III - have been identified. The binding of TGF-β to TGF-β receptor type II (TGFβRII) recruits and transphosphorylates TGFβRI. The activated TGFβRI regulates downstream fibrosis and inflammation through Smad protein signals. In the normal kidney, TGF-β1 is detected mainly in the glomerular mesangial cells, glomerular epithelial cells, and distal tubular cells. After UUO injury, the expression of TGF-β1 is remarkably increased, predominantly in the proximal tubular epithelial cells and, to a lesser extent, in the interstitial macrophages.43 The role of the TGF-β1/Smad pathway in the pathogenesis of renal fibrosis has been depicted in previous studies.20,44 To further clarify the effect of TGF-β1 on pericyte-myofibroblast transition, we created a TGF-β blockade after a UUO injury in Col1a1-GFPTg reporter mice.9 Blunting of the pericyte-myofibroblast transition was evident after the TGF-β blockade, resulting in an attenuation of renal fibrosis. Instead of mesenchymal transition, the injured tubular epithelial cells were arrested in cell cycle G2/M and an increased production of profibrotic cytokines was seen in the UUO kidney. TGF-β1 has been shown to be one of the factors leading to a G2/M cell cycle arrest in injured tubular epithelial cells.9 In accordance with our finding, Yang et al also showed a causal role of epithelial cell cycle G2/M arrest in renal fibrosis.45 Interestingly, TGF-β1 has not been shown to induce the proliferation of renal pericytes. Instead, TGF-β1 drives pericyte-myofibroblast transition.9
In addition to the roles of PDGF, TGF-β, and VEGF-A in mediating the interaction between pericytes and neighboring endothelial cells or tubular epithelial cells during renal fibrosis, the role of Wnt signaling in myofibroblasts and their precursor pericytes has been studied recently.46,47 In healthy kidneys, pericytes have active Wnt/β-catenin signaling responses, which are markedly upregulated following fibrogenic kidney injury.46,47 Pericytes have been shown to express the Frizzled cell-surface receptors and the low-density lipoprotein receptor protein 5 and 6 (LRP5 and LRP6) coreceptors for Wnt ligand responsiveness. Biochemically LRP6 interacts closely with PDGFRβ and TGF-βR1 at the cell membrane. The dominant-negative mutated forms of LRP6 inhibit PDGF-induced fibroblast cell proliferation.47 Furthermore, Dickkopf-related protein 1 (Dkk1), which inhibits Wnt/β-catenin signaling through binding of the Wnt LRP5/6 coreceptors, has been shown to block the proliferation of myofibroblasts cultured in vitro, to block pericyte proliferation in response to PDGF, pericyte migration, and gene activation, and to block cytoskeletal reorganization in response to TGF-β or connective tissue growth factor (CTGF). Dkk1 acts predominantly through LRP6 and inhibits PDGF-, TGF-β–, and the CTGF-activated mitogen-activated protein kinase (MAPK) and c-Jun amino-terminal kinase (JNK) signaling cascades, largely independent of inhibition of downstream β-catenin signaling. Dkk1 has also been shown to effectively inhibit pericyte-myofibroblast transition in vivo in response to fibrogenic kidney injury, resulting in the attenuation of fibrogenesis, capillary rarefaction, and inflammation.47
Taken together, PDGF, VEGF-A, TGF-β, and the Wnt signaling pathways are all important in renal pericyte-myofibroblast transition (Figure 3). Blocking these signaling pathways results in the attenuation of the pericyte-myofibroblast transition and subsequent fibrosis, after renal insult.
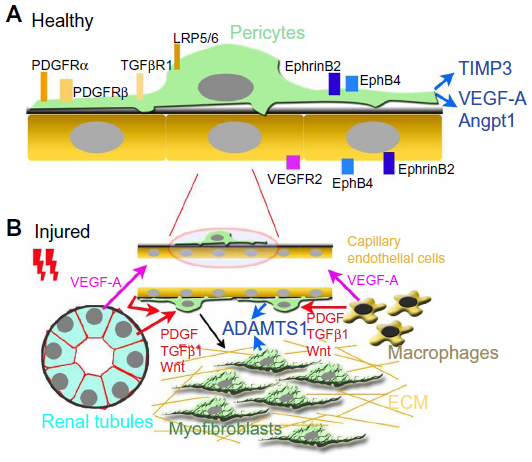 | Figure 3 Microvascular pericytes in healthy and injured kidneys. |
Microvascular stability
Given the close anatomical relation between the pericyte and the endothelial cell, a role of pericytes in the maintenance of proper endothelial function and microvascular stability is not hard to imagine. In the central nervous system, the role of pericytes in regulating microvascular stability is clear. Using a pericyte-deficient mouse model, Lindahl et al demonstrated that the absence of microvascular pericytes in the brain was associated with the formation of microaneurysms and spontaneous hemorrhage.48 Armulik et al also found that pericytes regulate the brain blood-brain barrier, in a set of adult, viable pericyte-deficient mouse mutants.49 Further, the pericytes have been shown to play an important role in the blood-retina barrier, and the dropout of the pericytes is an important feature of diabetic retinopathy.50
In the kidney, with regard to the role that pericytes play in microvascular stability, our understanding is only just beginning. In order to investigate the mechanisms underlying capillary rarefaction during kidney injury, we examined the relation of microvascular pericytes and endothelial cells in a kidney injury model of UUO.38 At day 1 following kidney injury onset, the detachment of pericytes and their migration away from the capillaries was demonstrated. By day 2, the detached pericyte population had expanded in parallel with the early angiogenic response, which manifested as an increased capillary density of the kidney microvasculature. The angiogenic response persisted through day 7. However, when the kidney insult persisted, significant rarefaction of the microvasculature replaced the increased capillary density by day 10, suggesting that the endothelial proliferation was initially effective but later deleterious. In order to clarify the significance of VEGF-A and PDGF in renal pericyte detachment and the transition to myofibroblasts, we administered soluble ectodomains of PDGFRβ (sPDGFRβ) or VEGFR2 (sVEGFR2) to block the signal transduction through the PDGFRβ and VEGFR2 in pericytes and endothelial cells, respectively. With the blockade of either PDGFRβ or VEGFR2, the detachment of pericytes was attenuated by day 1, as was, by day 2, the expansion of pericyte population and the transition to myofibroblasts. As for the early angiogenic response seen at day 2 of injury, the VEGFR2 blockade completely abolished this response, while the PDGFRβ blockade partially attenuated it. Most importantly, either the PDGFRβ or the VEGFR2 blockade was capable of reducing capillary rarefaction on day 14 after the UUO. This study underscores the significance of microvascular pericyte detachment and myofibroblast transition to the development of capillary rarefaction, implicating both PDGFRβ and VEGFR2 as important mechanisms. In line with our study, Greenberg et al demonstrated that VEGF-A promoted pericyte detachment and vessel destabilization in a PDGFRβ-dependent manner.51 VEGFR2 blockade, in the condition of both VEGF-A and PDGF exposure, restores angiogenesis. Moreover, the switching of the VEGF-A isoform, from VEGF164 to VEGF120 and VEGF188, has been observed after renal injury.38 VEGF164 is the predominant VEGF-A isoform expressed in renal pericytes, while renal myofibroblasts express predominantly VEGF120 and VEGF188.38 In the retina, the overexpression of VEGF120 and VEGF188 has been linked to abnormal arteriolar and venular patterning.52 In tumor neovascularization, VEGF120 and the highly ECM-bound VEGF188 are responsible for dysfunctional angiogenesis, which results in impaired tumor growth and vascular instability.37 It is reasonable that the isoform switch of VEGF-A after kidney insult is linked to renal pericyte-myofibroblast transition and that the predominance of dysangiogenic VEGF120 and VEGF188 contribute to renal microvasculature rarefaction.
Recently, using a three-dimensional (3D) gel assay, the ability of renal microvascular pericytes, but not myofibroblasts, to stabilize capillary tubes (possibly through the production of VEGF-A, angiopoietin 1, and tissue inhibitor of metalloproteinase 3 [TIMP3]) was demonstrated.53 Microarray analyses also showed that renal pericytes rapidly activate the expression of a disintegrin and metalloproteinase with thrombospondin motif 1 (ADAMTS1) and downregulate its inhibitor, TIMP3, in response to UUO insult.53 Further, TIMP3-deficient mice were found to have a spontaneous microvascular phenotype in the kidney, resulting from overactivated pericytes, and were more susceptible to injury-stimulated microvascular rarefaction, with an exuberant fibrotic response.53
Bidirectional signaling by the ephrin B4 receptor (EphB4) and ephrinB2 ligand is critical for angiogenesis during embryonic development in mice.54 Although ephrinB2 is a membrane-bound ligand, it has an intracellular domain that also possesses intrinsic signaling capacity, called “reverse signaling.” Very recently, Kida et al demonstrated that ephrinB2 reverse signaling was activated in the kidney after injury.55 In mice lacking the PDZ (post synaptic density protein [PSD95], drosophila disc large tumor suppressor [Dlg1], and zonula occludens-1 protein [zo-1]) intracellular signaling domain of ephrinB2, kidney injury led to impaired angiogenesis, increased peritubular capillary rarefaction, and renal fibrosis. The primary renal pericytes from mice lacking ephrinB2 intracellular signaling migrated more than did the wild-type pericytes and were less able to stabilize capillary tubes in 3D culture and to stimulate synthesis of the capillary basement membrane. The primary renal microvascular endothelial cells from mice lacking ephrinB2 intracellular signaling also showed less migration and proliferation in response to VEGF-A than did the wild-type endothelial cells. These results indicate that bidirectional signaling by the EphB4 and ephrinB2 in renal microvascular pericytes and endothelial cells is important, to limit pericyte-myofibroblast transition and renal fibrosis.
Taking all these studies together, the role of renal microvascular pericytes in the stabilization of the microvasculature is evident (Figure 3A). It is plausible that the transition of pericyte to myofibroblast is associated with pericyte detachment and microvasculature rarefaction (Figure 3B). The accumulation of dysangiogenic VEGF-A isoforms, the upregulation of ADAMTS1, and the downregulation of TIMP3 may be the mechanisms responsible for pathological vascular remodeling. PDGFRβ and VEGFR2 blockade are shown to ameliorate the pathological pericyte-myofibroblast transition and subsequent microvasculature rarefaction.
EPO production
In normal subjects, the production of erythrocytes is regulated by the hormone EPO, through its effect on bone marrow erythroid progenitor cells. Anemia and hypoxia stimulate the production of EPO through the activation of hypoxia-inducible factor (HIF)-2α.56,57 However, in patients with advanced CKD, the level of EPO fails to increase appropriately despite remarkable anemia.58,59 The causes of the relative EPO deficiency may be related to the dysfunction of the renal pericytes.
Renal pericytes as the major EPO-producing cells
The major organ responsible for EPO synthesis in adult life is the kidney. The cell types responsible for EPO production have recently been confirmed to be the peritubular fibroblasts.11,14,60,61 Using transgenic mice with GFP under the control of mouse EPO promoter, the renal EPO-producing cells (REPCs) were identified to be the peritubular cells in the renal cortex and outer medulla.60 Morphologically, the REPCs display a unique stellar shape, with multiple extending processes. Structurally, these cells are located between the vascular endothelial cells and the tubular epithelial cells. When examining the surface markers expressed, the REPCs were found to be positive for PDGFRβ, CD73, microtubule-associated protein 2, and neurofilament light polypeptide, but they were negative for CD31. Based on the similar morphological and structural characteristics and the surface markers expressed, these REPCs share much in common with the renal pericytes identified in our studies.1,5,6,9,33,38,53 A study by Yanagita et al42 used P0-Cre to label renal fibroblasts and revealed that the P0-Cre lineage-derived renal fibroblasts possessed both the ability to produce EPO and the potential to differentiate into scar-forming myofibroblasts. Most importantly, when these fibroblasts differentiated into myofibroblasts after kidney injury, the ability to produce EPO was reduced. As mentioned, the P0-Cre lineage-derived cells populate the nephrogenic interstitial mesenchyme zone of the metanephric mesenchyme, which is identical to the Foxd1+ mesenchymal progenitor area, before migrating into the kidney stroma. Therefore Foxd1-derived, Col1a1-GFP+ pericytes enwrapping the endothelia of capillaries deserve attention for their potential to produce EPO in healthy kidneys because pericytes stand in the front line, sensing the change of oxygenation and hemoglobin concentration.
The mechanisms underlying the inadequate EPO production in CKD
Why the production of EPO by the diseased kidney is inadequately low is obscure. Both a disturbed oxygen-sensing mechanism and a destroyed production capacity for EPO have been suggested. The most important oxygen-sensing system is probably the HIF system. HIF is a DNA-binding transcription factor activated under hypoxic conditions. A recent Phase I clinical trial studied the ability of an orally active inhibitor of the prolyl-hydroxylase domain enzyme, FG-2216, to stabilize HIF independently of oxygen availability in hemodialysis (HD) patients and healthy volunteers.62 FG-2216 was found to increase plasma EPO levels 30.8-fold in HD patients with kidneys, 14.5-fold in anephric HD patients, and 12.7-fold in healthy volunteers. These data demonstrate, not only that pharmacologic manipulation of the HIF system can stimulate endogenous EPO production but also, that there is no loss of REPCs in fibrotic kidneys. Hence the question emerges as to why the response to anemic stimulation by the REPCs in fibrotic kidneys decreases or becomes inadequate. Although treatments against fibrosis and inflammation, with tamoxifen and dexamethasone, respectively, can restore the expression of EPO transcripts in fibrotic kidneys,11,14 the cause of the loss of EPO production in REPCs in fibrotic kidneys is still obscure. Recently, Bechtel et al demonstrated that perpetuation of renal fibrosis in kidney is caused by the hypermethylation of rat sarcoma (Ras) protein activator like 1 (RASAL1), which encodes Ras oncoprotein inhibitor.63 Additionally, they found that the epigenetic change was mediated by DNA methyltransferase 1, which is upregulated in most of the fibroblasts in fibrotic kidney. Accordingly, we hypothesize that the insufficient EPO production might be caused by the increased DNA methyltransferase when pericytes transdifferentiate into myofibroblasts in the fibrotic kidney, contributing to the hypermethylation of the EPO regulatory sequence and the silencing of the EPO gene.64,65 Further studies should be conducted to confirm the causal relationship between methylation and expression of the EPO gene during pericyte-myofibroblast transition, using an animal model of kidney fibrosis. If possible, DNA demethylating agents might be a choice to revive EPO production in CKD patients. The more clearly the mechanism underlying inadequate EPO production is identified, the closer we get to an avenue to the renaissance of EPO production in diseased kidneys.
Conclusion
In the quiescent state, the renal microvascular pericytes maintain the stability of the microvasculature of healthy kidneys. Upon renal insult, the development of the pericyte-myofibroblast transition parallels pericyte detachment, pericyte proliferation, and myofibroblast differentiation. The myofibroblasts lose the ability to maintain the stability of microvasculature during renal fibrosis, while they are the major effector cells responsible for the deposition of pathological fibrogenic ECM. The renal pericytes express receptors for PDGF, TGF-β, and for Wnt ligands. Crosstalk between pericytes and their neighboring endothelial cells and tubular epithelial cells is critical for the phenotypic change of pericytes, upon fibrogenic renal injury. Blockade either of PDGF, VEGF-A, TGF-β, or Wnt signaling has been proven to attenuate the pericyte-myofibroblast transition, resulting in reduced capillary rarefaction, inflammatory infiltration, and renal fibrosis. Further delineation of the mechanisms underlying the reduced EPO production occurring during pericyte-myofibroblast transition will be promising for the development of new treatment strategies for anemia in CKD.
Acknowledgments
The Lin laboratory is supported by National Science Council (NSC) grants (101-2321-B-002-060, 101-2314-B-002-084, 102-2628-B-002-015, and 102-2321-B002-045) and the Mrs Hsiu-Chin Lee Kidney Research Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173(6):1617–1627. | |
Chang FC, Chou YH, Chen YT, Lin SL. Novel insights into pericyte-myofibroblast transition and therapeutic targets in renal fibrosis. J Formos Med Assoc. 2012;111(11):589–598. | |
Díaz-Flores L, Gutiérrez R, Madrid JF, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24(7):909–969. | |
Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130(2):247–262. | |
Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. | |
Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304(7):C591–C603. | |
Wilkinson-Berka JL, Babic S, De Gooyer T, et al. Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am J Pathol. 2004;164(4):1263–1273. | |
Rojas A, Chang FC, Lin SL, Duffield JS. The role played by perivascular cells in kidney interstitial injury. Clin Nephrol. 2012;77(5):400–408. | |
Wu CF, Chiang WC, Lai CF, et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. 2013;182(1):118–131. | |
Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130(1):141–155. | |
Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121(10):3981–3990. | |
Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am J Pathol. 2005;167(5):1193–1205. | |
Bielesz B, Sirin Y, Si H, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120(11):4040–4054. | |
Souma T, Yamazaki S, Moriguchi T, et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol. 2013;24(10):1599–1616. | |
Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6(11):643–656. | |
Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229(2):298–309. | |
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–350. | |
Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005;67(6):2488–2493. | |
Österreicher CH, Penz-Österreicher M, Grivennikov SI, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108(1):308–313. | |
Koesters R, Kaissling B, Lehir M, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177(2):632–643. | |
Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol. 2010;176(4):1767–1778. | |
Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333(6039):238–242. | |
Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18(8):1262–1270. | |
Hung C, Linn G, Chow YH, et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(7):820–830. | |
Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(24):9448–9453. | |
Direkze NC, Forbes SJ, Brittan M, et al. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells. 2003;21(5):514–520. | |
Duffield JS, Park KM, Hsiao LL, et al. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest. 2005;115(7):1743–1755. | |
LeBleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. | |
Le YZ, Zheng L, Zheng W, et al. Mouse opsin promoter-directed Cre recombinase expression in transgenic mice. Mol Vis. 2006;12:389–398. | |
Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19(1):12–23. | |
Eddy AA, LÓpez-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol. 2012;27(8):1233–1247. | |
Eddy AA. The origin of scar-forming kidney myofibroblasts. Nat Med. 2013;19(8):964–966. | |
Chen YT, Chang FC, Wu CF, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80(11):1170–1181. | |
Darland DC, Massingham LJ, Smith SR, Piek E, Saint-Geniez M, D’Amore PA. Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol. 2003;264(1):275–288. | |
Evensen L, Micklem DR, Blois A, et al. Mural cell associated VEGF is required for organotypic vessel formation. PLoS One. 2009;4(6):e5798. | |
Perrin RM, Konopatskaya O, Qiu Y, Harper S, Bates DO, Churchill AJ. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia. 2005;48(11):2422–2427. | |
Grunstein J, Masbad JJ, Hickey R, Giordano F, Johnson RS. Isoforms of vascular endothelial growth factor act in a coordinate fashion To recruit and expand tumor vasculature. Mol Cell Biol. 2000;20(19):7282–7291. | |
Lin SL, Chang FC, Schrimpf C, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178(2):911–923. | |
Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. | |
Hakroush S, Moeller MJ, Theilig F, et al. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am J Pathol. 2009;175(5):1883–1895. | |
Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350–1358. | |
Yanagita M. Inhibitors/antagonists of TGF-β system in kidney fibrosis. Nephrol Dial Transplant. 2012;27(10):3686–3691. | |
Fukuda K, Yoshitomi K, Yanagida T, Tokumoto M, Hirakata H. Quantification of TGF-beta1 mRNA along rat nephron in obstructive nephropathy. Am J Physiol Renal Physiol. 2001;281(3):F513–F521. | |
Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112(10):1486–1494. | |
Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543, 1p following 143. | |
DiRocco DP, Kobayashi A, Taketo MM, McMahon AP, Humphreys BD. Wnt4/β-catenin signaling in medullary kidney myofibroblasts. J Am Soc Nephrol. 2013;24(9):1399–1412. | |
Ren S, Johnson BG, Kida Y, et al. LRP-6 is a coreceptor for multiple fibrogenic signaling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci U S A. 2013;110(4):1440–1445. | |
Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–245. | |
Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–561. | |
Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012;366(13):1227–1239. | |
Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456(7223):809–813. | |
Stalmans I, Ng YS, Rohan R, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109(3):327–336. | |
Schrimpf C, Xin C, Campanholle G, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23(5):868–883. | |
Gerety SS, Wang HU, Chen ZF, Anderson DJ. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol Cell. 1999;4(3):403–414. | |
Kida Y, Ieronimakis N, Schrimpf C, Reyes M, Duffield JS. EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol. 2013;24(4):559–572. | |
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104(7):2301–2306. | |
Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27(1):41–53. | |
Fehr T, Ammann P, Garzoni D, et al. Interpretation of erythropoietin levels in patients with various degrees of renal insufficiency and anemia. Kidney Int. 2004;66(3):1206–1211. | |
Hung SC, Lin YP, Tarng DC. Erythropoiesis-stimulating agents in chronic kidney disease: What have we learned in 25 years? J Formos Med Assoc. Epub September 30, 2013. | |
Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood. 2008;111(10):5223–5232. | |
Paliege A, Rosenberger C, Bondke A, et al. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010;77(4):312–318. | |
Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. 2010;21(12):2151–2156. | |
Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. | |
Chang YT, Pan SY, Lin SL. Seeking for a way to revive erythropoietin production in chronic kidney disease. J Formos Med Assoc. 2013;112(11):657–658. | |
Pan SY, Chang YT, Lin SL. The role of hypoxia-inducible factors in renal fibrosis. J Formos Med Assoc. 2013;112(10):587–588. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.