Back to Journals » International Journal of Nanomedicine » Volume 13
Methyl-β-cyclodextrin quaternary ammonium chitosan conjugate: nanoparticles vs macromolecular soluble complex
Authors Piras AM ![]() , Fabiano A, Chiellini F, Zambito Y
, Fabiano A, Chiellini F, Zambito Y ![]()
Received 28 December 2017
Accepted for publication 25 January 2018
Published 24 April 2018 Volume 2018:13 Pages 2531—2541
DOI https://doi.org/10.2147/IJN.S160987
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Anna Maria Piras,1 Angela Fabiano,1 Federica Chiellini,2 Ylenia Zambito1
1Department of Pharmacy, University of Pisa, Pisa, Italy; 2BIOLab Research Group, Department of Chemistry and Industrial Chemistry, University of Pisa, UdR INSTM Pisa, Pisa, Italy
Purpose: The present study aimed to compare a novel cyclodextrin–polymer–drug complex in solution with a dispersed supramolecular nanosize system, made of the same complex, for ability to carry dexamethasone (DEX) across excised rat intestine.
Results: Methyl-β-cyclodextrin-quaternary ammonium chitosan conjugate (QA-Ch-MCD) was obtained by covalent grafting through a 10-atom spacer. The conjugate was characterized by 1H-NMR, resulting in 24.4% w/w of MCD content. Phase solubility profile analysis of the QA-Ch-MCD/DEX complex yielded an association constant of 14037 M-1, vs 4428 M-1 for the plain MCD/DEX complex. Nanoparticle (NP) dispersions resulted from ionotropic gelation of the QA-Ch-MCD/DEX complex by sodium tripolyphosphate, leading to 9.9%±1.4% drug loading efficiency. The mean diameter and zeta potential for NP were 299±32 nm (polydispersity index [PI] 0.049) and 11.5±1.1 mV, respectively. Those for QA-Ch-MCD/DEX were 2.7±0.4 nm (PI 0.048) and 6.7±0.6 mV. QA-Ch-MCD/DEX solutions and corresponding NP dispersions were compared in vitro for water-assisted transport through mucus, DEX permeation through excised rat intestine, and ex vivo mucoadhesivity. The complex showed higher mucoadhesion and lower transport rate through mucus; also, it provided faster drug permeation across excised rat intestine.
Conclusion: Carrier adhesion to mucus surface has played a most important role in favoring transepithelial permeation. Then, within the concerns of the present study, the use of NP seems not to provide any determinant advantage over using the simpler macromolecular complex.
Keywords: chitosan derivatives, cyclodextrin chitosan conjugates, mucoadhesive polymers, mucoadhesive nanoparticles, dexamethasone cyclodextrin complex
Introduction
One of the more frequently used strategies to increase the dissolution rate and hence the bioavailability of orally administered hydrophobic drugs consists in forming drug inclusion complexes with cyclodextrins. These comprise a family of hydrophilic compounds made up of 6–8 α-D-glucopyranoside units linked together by 1–4 bonds to form a thoroid.1,2 Another property of the carrier system thought to improve drug bioavailability is mucoadhesion. In fact, mucoadhesive systems for oral drug administration have raised widespread interest, thanks to their recognized potential to improve the oral bioavailability of drugs having a poor mucosal permeability.3–6 Indeed, mucoadhesion may slow down the transit of the therapeutic system down the gastrointestinal (GI) tract, thus prolonging drug contact with the absorption site and favoring the formation of high concentration gradients.7 In addition, the use of mucoadhesive nanoparticle (NP) drug systems would allow the chemical stability of active principles in the GI environment to be improved.8 Different NP systems based on cyclodextrin–chitosan conjugates have been studied, aimed at promoting absorption of encapsulated drugs by conjugating the mucoadhesive characteristics of particles with their ability to release the drug while protecting it from degradation in the GI.9
In a recent report, the solubilization ability of cyclodextrins and the properties of mucoadhesive polymers have been coupled by conjugating methyl-β-cyclodextrin (MCD) to a water-soluble mucoadhesive chitosan derivative via the hexamethylene diisocyanate (HMDI) spacer. The resulting conjugate has shown high complexing ability, mucoadhesive characteristics, and cytocompatibility.10 In that study, the water-soluble cyclodextrin–polymer complex was prepared from a quaternary ammonium chitosan derivative water-soluble irrespective of pH, previously synthesized and shown to possess mucoadhesive properties.11 This derivative was shown to be an effective enhancer of hydrophilic, hydrophobic, as well as macromolecular drug absorption across different epithelia, such as the intestinal,12,13 corneal,14 and buccal ones.15
Recently, our group has put to comparison, the ability of mucoadhesive polymeric NPs to prolong the residence of dexamethasone (DEX) phosphate or met-enkephalin in tear fluid with that of the parent non-aggregated polymers. The comparison showed that the NPs were more effective than the parent polymers in solution only if the former were able to concurrently adhere to the ocular surface and strongly interact with DEX phosphate, whereas the effect of NPs interacting weakly with the free drug was not significantly different from that for the nonaggregated polymers. Conversely, with the peptide drug in all cases the formulation based on NPs was more effective than that containing the nonaggregated polymers, probably thanks to the NP ability to protect the drug from degradation.16 Then, with drugs not liable to degradation in the GI tract, an NP dispersion cannot be assumed a priori to perform better than the simpler nonaggregated parent polymer(s) solution.
From the abovementioned considerations altogether, the need emerges to shed further light on the actual advantages/disadvantages of the cyclodextrin–polymer conjugate drug complex carrier in solution, compared to the supramolecular nanosized carrier system of the same drug complex in dispersion. The comparison concerned the carrier effect on ex vivo apparent intestinal drug permeability and mucoadhesivity. Water insoluble DEX was used as the drug, and MCD conjugated to a reduced molecular weight (MW) quaternary ammonium–chitosan polymer (QA-rCh) was used as the polymeric complexing agent for DEX, yielding a water-soluble macromolecular complex. This complex (QA-Ch-MCD/DEX) was also used to prepare a DEX-loaded nanosized carrier system.
Materials and methods
Materials
DEX was obtained from Sigma-Aldrich Co., St Louis, MO, USA. Reduced–MW chitosan (rCh) was prepared from commercial chitosan (FG90; Giusto Faravelli, Milano, Italy) according to Zambito et al.11 The viscometric MW of the resulting product, as determined after Zambito et al11 was 27.4 kDa vs a reported value of 32 kDa. The relevant quaternary ammonium–chitosan conjugate, QA-rCh60, was synthesized from rCh and characterized for chemical structure as described by Zambito et al.11 In the polymer code, the figure indicates the temperature (°C) of its synthesis. HMDI (Sigma-Aldrich Co.) was distilled under vacuum, and the fraction collected at 127°C was used. Triethylamine (TEA) (Sigma-Aldrich Co.) was distilled at room pressure at 85°C. MCD was purchased from Roquette Italia (Alessandria, Italy; MW 1,191 g/mol, degree of C6 substitution ~0.5, corresponding to ~4 methyl groups per cyclodextrin molecule). Sodium triphosphate pentabasic (TPP), dimethyl sulfoxide (DMSO), 2-diethylaminoethyl chloride, and fluorescein isothiocyanate (FITC), all from Sigma-Aldrich Co., were used as received.
Synthesis of Me-β-CD-QA-rCh60 conjugate (QA-Ch-MCD)
Accurately weighed QA-rCh60 (400 mg) was added to DMSO (8 mL), and the mixture was stirred overnight in a two neck round bottom flask under an inert atmosphere (N2). The resulting solution was added, in drops, to HMDI (720 μL) in DMSO (10 mL), leading to a solution which was heated to 70°C, mixed with TEA (65 μL), and stirred for 3 h, keeping the atmosphere inert, to complete polymer activation, after which the product was precipitated and cold washed with diethyl ether (4°C). Then, the product was redissolved in DMSO (3 mL), 2.7 g of MCD in 8 mL DMSO was added, the resulting mixture was shaken for 3 h at 70°C in the presence of TEA (150 μL), after which the solution was dripped into hot (90°C) water (about 30 mL) and was left at least 1 h under stirring. After cooling, the reaction mixture was transferred to a dialysis tubing (ethylcellulose, MWCO 12.5 kDa) and was dialyzed 3 days vs water, at the end of which the water-soluble material was separated from the dispersion by centrifugation at 20,000 rpm, 30 min, and 4°C (Sorvall, MTX, microultracentrifuge), and the supernatant was finally lyophilized.
FITC labeling of the QA-Ch-MCD derivative
The procedure previously described by Di Colo et al was followed.17 Accordingly, an FITC solution in DMSO (1 mL, 2 mg/mL) was added to an aqueous QA-Ch-MCD solution (20 mL, 2 mg/mL), and the mixture was incubated overnight at 4°C, following which it was passed through a Sephadex G-15 column, to set the labeled polymer free from unreacted FITC, and the eluate was lyophilized.
Lyophilization
A VirTis AdVantage wizard 2.0, SP Scientific freeze-dryer was used. The synthesized products were lyophilized following a manual procedure, after placing their aqueous solutions in Petri dishes. The congealing temperature was −40°C; the sublimation conditions were the following: pressure, 30–40 mTorr and temperature up to 15°C–20°C. With the different samples, the time of process would vary in the 15–17 h range.
Characterization of the QA-Ch-MCD conjugate
NMR measurements were carried out with a Bruker Avance II operating at 250.13 MHz. During data acquisition the temperature was kept at 25°C±0.1°C. For analysis, the samples were dissolved in D2O (1%–2%).
DEX complexation by QA-Ch-MCD
A DEX excess was added to QA-Ch-MCD aqueous solutions in the 0.5–20 mg/mL concentration range (1:2 DEX/QA-Ch-MCD w/w ratio). The suspensions, each contained in a screw-capped vial, were autoclave heated (20 min at 120°C). Then, they were transferred to a shaker water bath at 37°C for 2 h, subsequently kept 20 min at ambient temperature, after which the excess DEX was removed by filtration (0.45 μm). The DEX apparent solubility was determined by ultraviolet–visible (UV-VIS) technique. The intrinsic water solubility of DEX (DEX0) was determined by applying the same procedure to 2 mg/mL DEX water suspensions. The complexation K was calculated according to Equation 1 which applies to CD·Dm complexes, where CD stands for cyclodextrin and D for drug.18
| (1) |
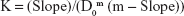
Here, D0 is the intrinsic drug solubility (in the absence of CD), and Slope is the slope of the straight line obtained by plotting the drug apparent solubility vs the corresponding CD concentration both expressed as molarity:18
With CD1·D1 complexes the complexation efficiency (CE) was calculated by the following equation:
| (2) |
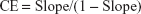
The molar ratio, on its part, was calculated as follows:
| (3) |
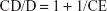
Preparation and characterization of NP from QA-Ch-MCD derivatives
Aqueous TPP (1 mL, 0.150 mg/mL) was added, under stirring at ambient temperature, to the QA-Ch-MCD/DEX complex (1.5 mL), obtained from aqueous QA-Ch-MCD (1 mg/mL), and the mixture was kept in these conditions for 30 min, after which the dispersion was centrifuged 20 min, at 8,000 rpm and 4°C. The supernatant was subjected to high-performance liquid chromatography (HPLC) analysis for the drug to calculate the DEX amount in the sediment, from the amount used in the formulation. The sediment was redispersed in water and lyophilized to determine the dry weight of NP. The DEX content in the NP dispersion was expressed as follows:
|

NP size distribution
Size distributions of QA-Ch-MCD, QA-Ch-MCD/DEX complex, and NP obtained from them were measured by a Coulter N4 PLUS (Coulter Corporation, Miami, FL, USA). The samples, dispersed in water or phosphate buffers (PB), either 1.3 M pH 7.4 PB or isotonic phosphate buffered saline (PBS) pH 6.8, were analyzed at a 90° detection angle, 20°C temperature, and 180 s analysis time. The data collected were processed in terms of mean diameter, expressed as cumulative intensity, and polydispersity index (PI).
Electrophoresis mobility measurements
Electrophoresis mobility measurements were carried out on the QA-Ch-MCD/DEX complex and on the purified NP resuspended in PB or isotonic PBS with pH 6.8. Samples were analyzed by Delsa™ Nano C (Beckman Coulter, Brea, CA, USA) in a low concentration flux cell at 25°C temperature. The Zeta potential (ζ) values were calculated by the Helmholtz–Smoluchowski equation.
Dynamic dialysis studies
These studies were carried out following already described procedures.19 Briefly, the same porous cellulose membrane (cutoff 12.5 kDa), diffusion cell, and apparatus were used at the thermostat temperature of 37°C. At t=0.5 mL of freshly prepared, medicated NP, corresponding to a DEX amount of 0.12 mg/mL, was introduced in the donor compartment of the cell. Sink conditions were maintained in the receiving phase (100 mL of PB). Drug transport across the membrane was assessed by quantifying the drug in the receiving phase by HPLC, at preset intervals. At the end of each run, the particle size in the donor phase was checked. For comparison, kinetic data were also obtained with the same membrane and procedure for the plain drug solution (0.03 mg/mL), 0.9 mg/mL of MCD solution (corresponding to 0.12 mg/mL of DEX), and QA-Ch-MCD solution (corresponding to 0.21 mg/mL of DEX).
Interrupted dialysis studies
These studies were carried out following already described procedures.20 Briefly, the dialysis was stopped after an established time from the start, and the drug fraction contained in each of the NP matrix, NP dispersion medium, and acceptor medium was determined. To determine the drug fraction in dispersion medium and calculate that in NP matrix, ultracentrifugation of donor phase followed by analysis of supernatant was carried out.
DEX quantification by HPLC
The apparatus (PerkinElmer Inc., Waltham, MA, USA) consisted of a Series 200 LC-290 AT pump, 20 μL Rheodyne injector, UV-VIS SPD–6 AV detector, Brownlee Kinetex Core-Shell C18 1.7–1.3 nm, 100A porous column (4.6×100 mm), and Turbochrom Navigator HPLC software for data integration. The mobile phase (flow rate, 1 mL/min) was 30:70 acetonitrile/aqueous acetic acid 0.35% v/v. After mixing, the mobile phase was filtered (cellulose acetate, 0.45 μm pore filter) and degassed (10 min sonication with Sonica Ultrasonic Cleaner Model 2200 ETH, Soltec). For DEX determination, the detector was set at 242 nm. A calibration curve was constructed using standard DEX solutions in the mobile phase in the concentration range of 200–10,000 ng/mL and integrating the peak at the retention time of 8.65 min (y =64.468x +8,263.9; r2=0.9996).
Fluorometric determination of FITC-labeled polymer and NP
The FITC-labeled QA-Ch-MCD/DEX complex (1 mg/mL aqueous solution) and NP (3 mg/mL aqueous dispersion) were prepared as already described, then diluted to concentrations in the range from 1.7×10−5 to 4×10−3 μg/mL. The resulting solutions/dispersions were analyzed on a fluorescence spectrometer LS 45 Perkin Elmer (excitation and emission wavelengths 485 and 530 nm, respectively). The parameters of the relevant calibration straight lines were the following: for the complex, y =648.83x −23.1 (r2, 0.997); for the NP, y =490.97x −4.5949 (r2, 0.999).
Ex vivo mucoadhesivity studies
NP mucoadhesivity was compared with that for the QA-Ch-MCD/DEX complex. Either material was FITC labeled and dispersed/dissolved in PB pH 7.4, 0.13 M (1 mg/mL). The following ex vivo procedure, taken from Pimienta et al and modified, was used. The intestinal mucosa was excised from male nonfasted Wistar rats weighing 250–300 g.21 These were treated as described in the “Guide for the care and use of laboratory animals” (NIH Publication No 92-93, revised 1985). All experiments were carried out under veterinary supervision and the protocols approved by the ethical-scientific commission of the University of Pisa. After sacrificing the rat, the first 50 cm of jejunum were removed and four 10 cm segments were cut from the excised intestine and was longitudinally opened, after which the mucosa was gently rinsed free of luminal contents with PB pH 6.8, isotonic (PBS). Each intestine segment was cut into 1 cm strips to allow its complete immersion in 1 mL of the solution/dispersion under test, under bubbling of a 95% O2/5% CO2 mixture (Oxycarb), for 4 h at 37°C. Zambito et al have shown that the intestinal mucosa would not be damaged by the abovementioned conditions.12 Then, after adding enough water to each solution/suspension to compensate for the weight loss due to evaporation, 20 μL samples, adequately diluted with PB, were analyzed for the labeled material as already described. The adsorbed mass fraction (AMF) was calculated as the percentage difference between the polymer concentration before and after incubation.
In vitro study of water-assisted transport through mucus
Purified porcine intestinal mucus was provided and transport studies across a purified mucus layer were carried out according to Fabiano et al.22 A mucus layer was realized by filling the barrel of a 1 mL plastic syringe with 350 μL of cleaned mucus. The barrel was kept, tip down, in vertical position, and the mucus was prevented from escaping through the syringe tip by an EC 0.2 μm filter. The QA-Ch-MCD/DEX complex or centrifugation-purified NP, in aqueous solution/dispersion (50 μL, 6 mg/mL), was placed on the filter before adding the mucus. The polymer used to prepare the samples was a 1:1 mixture of FITC-labeled and unlabeled QA-Ch-MCD. A bottom-to-surface PBS flow across the mucus layer (diameter, 0.6 cm) was realized by connecting the tip of the syringe to the bottom of a vertical cylindrical vessel by a flexible tubing, filling the vessel with PBS, level with the surface of the mucus layer in the syringe, and dripping PBS into the vessel so as to create a supernatant liquid phase gradually rising over the mucus layer at a rate of 1.24×10−4 cm/s corresponding to a flux of 0.446 mL/(cm2 h), without causing any phase mixing. Such a flux value was calculated from the data reported by Lennernans et al for intestinal water absorption in man.23 The system was maintained in vertical position at 37°C for 5 h, after which the supernatant was withdrawn from the mucus, the syringe was removed from the assembly, frozen, and cut into slices of 2 mm length, starting from the tip where the formulations were added. The marker in the supernatant was determined as already described. For determination of the marker in each slice, the same procedure was applied after thawing and removing the plastic debris. Then, the transport ratio (TR) was calculated as the ratio between the material amount transported across the mucus layer over 5 h and that loaded at the base of it, at the start of the experiment.
Studies of DEX and carrier permeation through excised rat intestine
These studies were carried out using the procedure from the study by Zambito et al with minor modifications.13 The DEX apparent permeability through the excised intestine was expressed as follows:
| (4) |
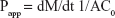
where dM/dt, the permeation flux, is the slope of the linear portion of the cumulative mass permeated vs time plot, C0 is the permeant concentration introduced in the donor phase (C0=0.2 mg/mL), supposed to be completely dissolved and constant over the whole time of linear permeation, and A is the permeation surface. At the end of each 4 h-permeation experiment, the DEX and FITC-labeled polymer and NP contents in excised intestinal tissue per unit membrane surface were evaluated by the following, already reported procedure.24 The intestinal tissue was homogenized with 1 mL of PBS using an Ultra-Turrax homogenizer. Then, the homogenate was centrifuged (5 min at 10,000 rpm), and the supernatant was withdrawn which was further treated with trichloroacetic acid 50% p/v and analyzed for FITC-labeled polymer and NP contents as described in the previous sections. For reference, this procedure was applied several times to tissue subjected to plain PB.
Results and discussion
Preparation and characterization of the QA-Ch-MCD conjugate
The oxidative depolymerization of the commercial chitosan, based on NaNO2, led to a viscometric MW reduced by a factor of 1/21.5 (27.4 kDa), in line with the value of 1/18.4 (32.0 kDa) previously obtained from the same chitosan.11 Similarly, QA-rCh60 characteristics such as the degree of substitution (DS) of pendant chains and the mean quaternary ammonium group number in each chain, n, were not significantly different from the previously reported values.11 A protocol consisting in activation, conjugation, and quenching was followed to prepare the QA-Ch-MCD conjugate, the structure of which, together with that of DEX, is shown in Figure 1. The water-soluble product was separated from the insoluble (crosslinked) material by centrifugation. The MCD conjugation to the QA-rCh60 polymer, to give QA-Ch-MCD, followed a protocol previously set up,10 where the activation step lasted 3 h. The acquisition of 1H-NMR spectra, shown in Figure 2A and B, allowed characterization of all relevant compounds, as well as the MCD and spacer conjugation fractions (Table 1). The proton unit of the quaternized chitosan was determined from the area under the signals at 4.2–2.2 ppm, corresponding to the protons of the chitosan repeating unit, but for the anomeric one, and the methylenes of the pendant chain. In addition, the following were calculated: deacetylation degree, from the diagnostic acetyl groups, at 2.0 ppm; quaternization degree, in terms of quaternary ammonium moieties per repeating unit; and mean length of pendant chains, from the signals of methyl end groups at 1.6–0.7 ppm. The interpretation of the 1H-NMR spectrum of the conjugate QA-Ch-MCD, shown in Figure 2B, has allowed calculation of the cyclodextrin-quaternized polymer conjugation degree using the signal at 2.5 ppm of the terminal methylene groups of the quaternized pendant as the reference and the signals of the cyclodextrin anomeric protons at 5.1 and 4.9 ppm as diagnostic. On account of the pendant chain DS, and the MWs of spacer, HMDI, cyclodextrin, MCD, and QArCh60 repeating unit, the weight contribution of each of the different components of the conjugate polymer synthesized was calculated. In particular, QA-Ch-MCD contains 25.4% w/w of MCD. This value has been used for the preparation and characterization of the QA-Ch-MCD/DEX complex to be described in the following sections.
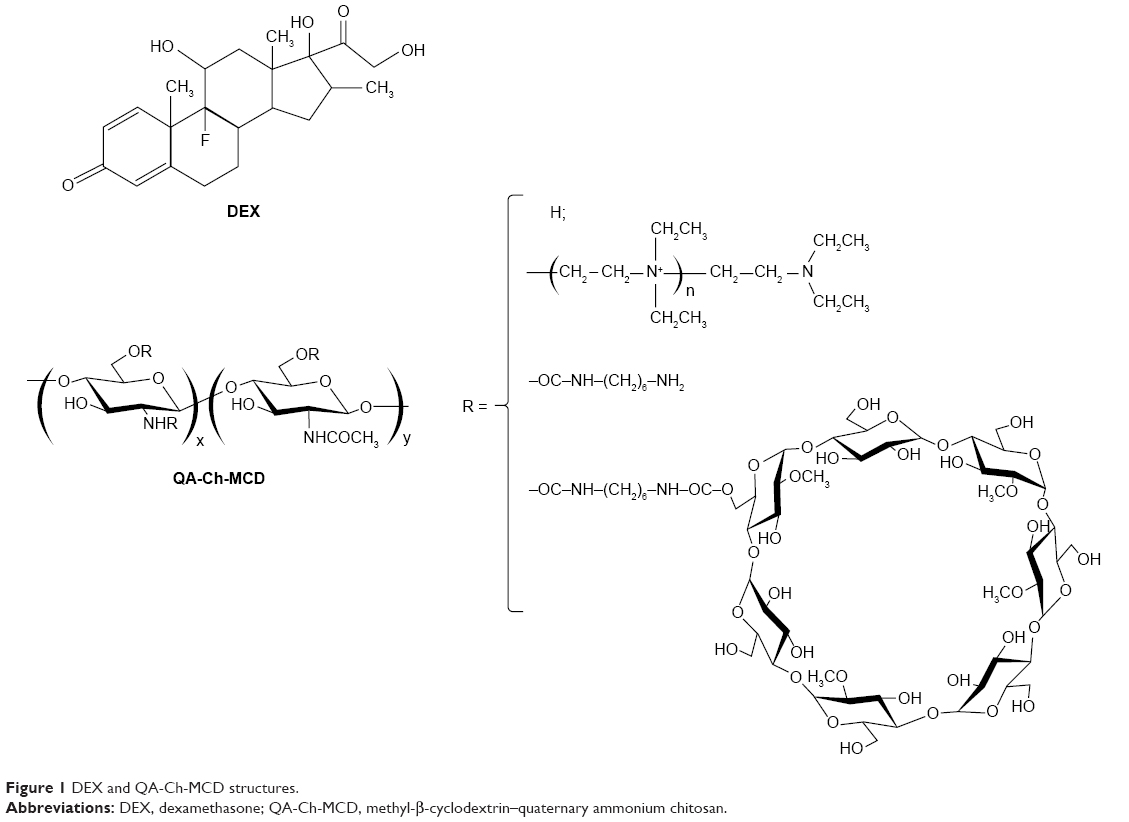 | Figure 1 DEX and QA-Ch-MCD structures. |
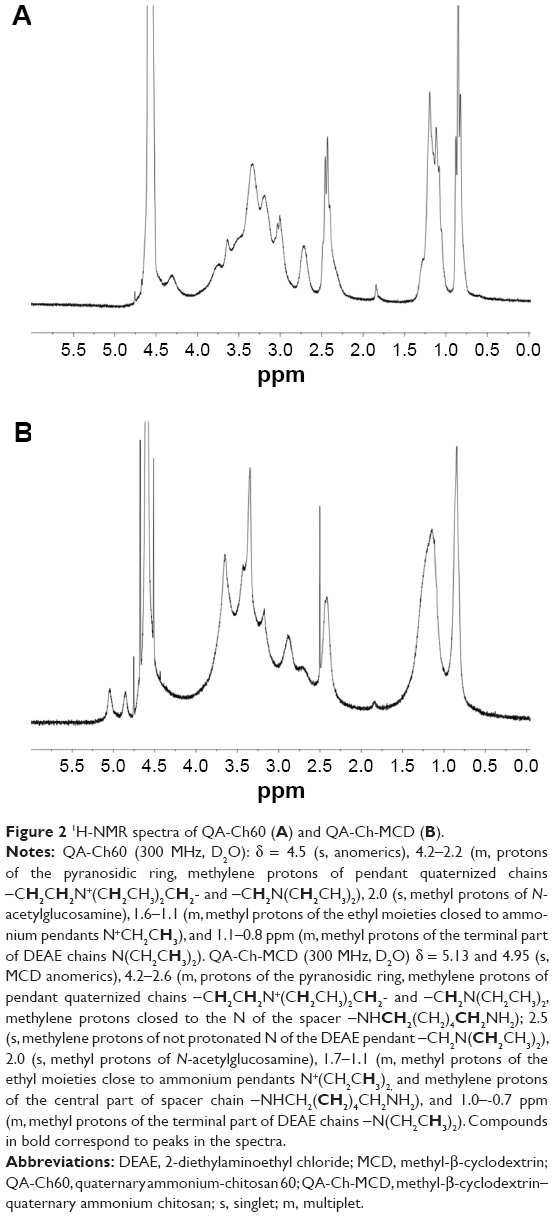 | Figure 2 1H-NMR spectra of QA-Ch60 (A) and QA-Ch-MCD (B). |
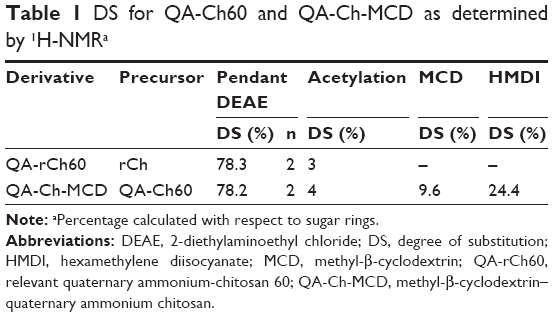 | Table 1 DS for QA-Ch60 and QA-Ch-MCD as determined by 1H-NMRa |
Preparation and characterization of the QA-Ch-MCD/DEX complex
The literature reports different complexation methods of lipophilic substances by cyclodextrins, among which are the complex formation at a controlled temperature and various equilibration times,25 the application of one or more heating–cooling cycles in autoclave,26,27 or the controlled-temperature sonication in a water bath.28 All procedures involve an undissolved excess of active principle, subsequently removed by centrifugation or filtration. In the present work, the protocol followed to prepare the QA-Ch-MCD/DEX complex involved the formation of a supersaturated solution by heating the QA-Ch-MCD/DEX mixture for 20 min at 120°C, followed by equilibration of 2 h at 37°C and 1 h at ambient temperature. The excess of insoluble drug was finally removed by filtration (0.45 μm). This last step was made possible by the low viscosity of the polymer solutions, the uppermost concentration of which was 20 mg/mL. A structurally similar chitosan derivative dealt with in a previous study by our group would not have allowed such a procedure because of a considerably higher viscosity associated with a higher molecular mass.10 Cyclodextrins are known to form inclusion complexes with lipophilic guest molecules, thus increasing the apparent solubility of the latter. A dynamic equilibrium between complexed and free forms, both water-soluble, is then established. The complex stability constant, K, is determined by studies where the apparent solubility of the guest molecule is plotted vs increasing cyclodextrin concentrations. This procedure, first published by Higuchi and Connors in 1965,29 allows the evaluation of both the complex stability constant and stoichiometry.
In Figure 3, the curve of DEX apparent solubility vs concentration of MCD covalently bound to the QA-Ch-MCD conjugate is compared with that obtained with the plain MCD as the complexing agent. In both cases, the DEX apparent solubility is proportional to the MCD concentration, as shown by the relevant AL type linear profiles. The stoichiometry of each complex can be calculated from the slope of the respective experimental straight lines (Table 2). It is shown in the table that both values are <1, which enables calculation of the CE implying a 1:1 type stoichiometry.18 The DEX intrinsic solubility (S0) in the applied conditions is 0.19 mM. From this value and the equations (1–3), the complex stability constant, K, and CE were calculated. It is observed that the QA-Ch-MCD conjugate brings about a substantial increase in both K and CE. This is reflected in a decrease in the MCD/DEX molar ratio. The higher K value for the QA-Ch-MCD/DEX than the MCD/DEX complex can be explained by DEX aspecific adsorption to the quaternary ammonium chitosan backbone and the absence of hydrophobic drug clusters.
 | Figure 3 Solubility profiles of DEX: intrinsic water solubility (DEX0), solubility in presence of MCD (MCD/DEX), and in the presence of QA-Ch-MCD (QA-Ch-MCD/DEX). |
 | Table 2 Characteristics of DEX/MCD inclusion complexes as obtained by resulting from the phase-solubility studiesa |
Preparation of NP
All conjugate polymers synthesized were studied preliminarily to pave the way for the preparation of NP via ionotropic gelation of the QA-Ch-MCD/DEX complex. In all cases, the crosslinker TPP/complex weight ratio and the volume ratio between the respective solutions were modulated. The dynamic light scattering analysis of the optimized dispersion of NP obtained from QA-Ch-MCD has shown a mean particle diameter of 299±32 nm (PI, 0.049). This value is two orders of magnitude higher than that for the QA-Ch-MCD/DEX macromolecular complex. For the NP system, EE% and LE% values of 9.9%±1.4% and 22.9%±3.3%, respectively, were assessed. In addition, the QA-Ch-MCD/DEX complex and relevant NP have been characterized for surface charge, expressed as ζ potential, in PBS or PB. As expected, both values, listed in Table 3, are positive, in line with the presence of tertiary amine and quaternary ammonium moieties. In the case of the macromolecular QA-Ch-MCD/DEX complex, the ζ value significantly changes as the pH varies from 7.4 to 6.8. This variation is correlated with a change of free amines into protonated amine moieties. The pH-dependent ζ variation is not observed with the NP. Indeed, the NP residual amine moieties are involved in the crosslinking with TPP, and the nanostructure constraint limits the chain mobility, resulting in a constant positive surface charge, due to the presence of quaternary ammonium moieties.
 | Table 3 Main characteristics of the macromolecular complex QA-Ch-MCD/DEX and its relevant NPs |
Drug release from NP
Drug release from NP was studied by a procedure based on the interrupted dialysis followed by ultracentrifugation of donor phase, as described in the “Materials and methods” section.
The results of the kinetic analysis of phases, carried out in this work for DEX loaded NP, are plotted in Figure 4. The data show a gradual drug release in the initial 4 h up to 30% of the dose, then no further release is observed in the subsequent 20 h. This indicates that ~70% drug remains irreversibly bound to the NP over 24 h.
 | Figure 4 DEX kinetics from dynamic interrupted dialysis studies on NP samples: DEX kinetics in each phase of dialysis: NP phase; NP DM; and RP. |
Drug-binding studies
Dynamic dialysis data were used to assess reversible drug interactions with the carriers in solution or the surfaces of the NP in dispersion. With mucoadhesive drug carriers, similar interactions could be correlated with a possible enhancement of drug residence time at mucus surfaces. The dynamic dialysis data obtained in this work with DEX were plotted according to the following equation:19
| (5) |
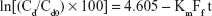
where Cd is the drug concentration in the donor phase at time t, Cd0 is the drug concentration in the donor phase at time t=0, Ff is the noninteracting to total drug ratio in the donor phase, and Km is the dialysis rate constant. The dialysis data for plain DEX, DEX-loaded NP, and DEX in the presence of MCD or the chitosan conjugate in solution, are reported as ln(Cd/Cd0)100 vs t in Figure 5. In the case of NP, a virtually time-independent fraction of total DEX mass appears, from Figure 4, to be entrapped within the NP matrix. In this case, the entrapped mass was subtracted from the total drug mass in donor compartment, to calculate the effective value of Cd, ie, the concentration of the drug portion actually variable in time. All plots of Figure 5 are significantly linear, as shown by the respective r2 values, listed in Table 4. Such a linearity indicates that in all cases Equation 5 is obeyed and Ff is actually constant with varying Cd and t at least over the time of dialysis. Also, the slopes of log-linear plots are listed in Table 4. For the plain drug, Ff=1, hence, according to Equation 5, the slope of the relative log–linear plot is equal to the dialysis rate constant, Km. From here, the drug fraction reversibly interacting with dissolved QA-Ch-MCD or MCD, or dispersed NP surface, could readily be derived from the slope of the relevant plot. The resulting values are listed in Table 4. From the data it appears that, under the experiment conditions, the reversibly interacting drug fraction ranks in the following order: MCD>QA-Ch-MCD>NP. These findings can be reconciled with the higher association constant of the QA-Ch-MCD/DEX than the MCD/DEX complex, shown in Table 2, by taking into account the lower MCD weight fraction in the former than the latter complex. This view stresses the role of inclusion complexing in DEX interactions with QA-Ch-MCD. As for the NP, in addition to the DEX fraction irreversibly bound to the carrier, it is shown in Table 4 that 30% of the residual fraction is subjected to a reversible interaction with the NP surface.
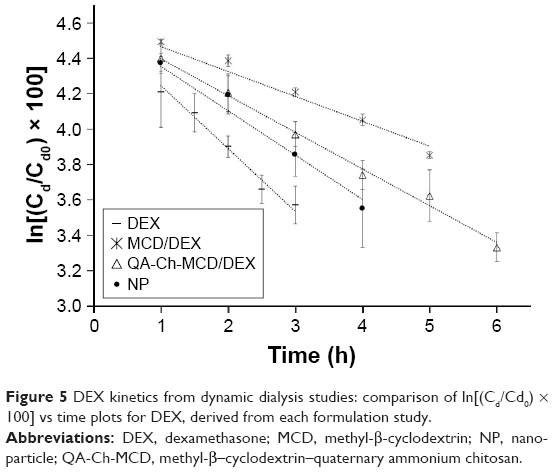 | Figure 5 DEX kinetics from dynamic dialysis studies: comparison of ln[(Cd/Cd0) × 100] vs time plots for DEX, derived from each formulation study. |
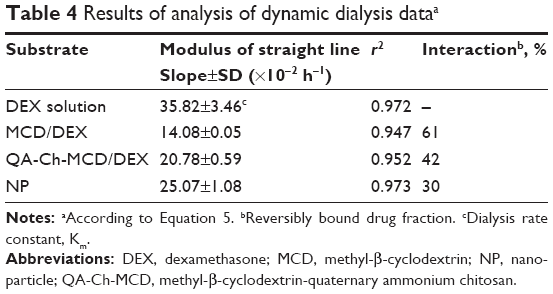 | Table 4 Results of analysis of dynamic dialysis dataa |
Ex vivo mucoadhesivity studies
These experiments have allowed the determination of the AMF value for the NP and the constituent conjugate QA-Ch-MCD. The AMF value is a measure of mucoadhesivity, indeed, the stronger is the adhesion of a material to mucus, the higher will be the fraction adsorbed on the mucus surface. The AMF values for NP (2.2%±2.0%) and QA-Ch-MCD (44.6%±1.5%) can be explained by admitting that the nonaggregated QA-Ch-MCD macromolecules in solution have more flexibility than the NP and, therefore, can give rise to stronger cooperative interactions with the mucus glycoproteins.
In vitro study of water-assisted transport of NP through mucus
The results of the relevant studies described in the “Materials and methods” are represented in Figure 6. After 5 h of experiment, the distribution of transported products over the length of the sliced mucus layer in both cases seems to decline from slice 1–11, down to comparatively low fluorescence values. Conversely, the fluorescence found in the supernatant is clearly significant in both cases, indicating that both species were transported past slice 11. From the fluorescence analysis of the supernatant, it is deduced that the NP transport through the mucus is significantly faster than that of the nonaggregated QA-Ch-MCD/DEX complex in solution. Indeed, the TR values for the NP and the nonaggregated complex are 0.35 and 0.22, respectively. These data are not in agreement with those obtained in the preceding study where it was found that the NPs were retained in the mucus gel more than the relative nonaggregated constituent polymers.22 In that case, however, the NPs were found to be more mucoadhesive than the constituent polymers. Hence, the data obtained in the present work together with those from the preceding one indicate that the transport through mucus is more influenced by the carrier mucoadhesive properties than its hydrodynamic volume.22 The latter, by the way, is shown by the data in Table 3 to be two magnitude orders greater for the NP than the nonaggregated constituent polymer.
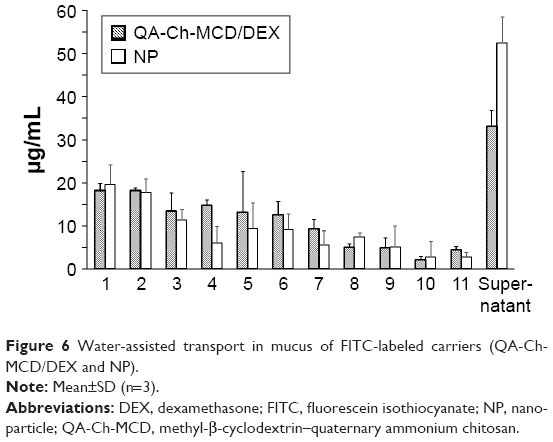 | Figure 6 Water-assisted transport in mucus of FITC-labeled carriers (QA-Ch-MCD/DEX and NP). |
Studies of DEX and carrier permeation through excised rat intestine
In these studies, the receiving phase samples withdrawn at intervals were analyzed both to determine the DEX amount permeated and monitor the possible membrane crossing by the FITC-labeled carrier, namely, QA-Ch-MCD/DEX or NP. The results obtained are shown in Table 5. The excised rat intestine was used for the studies without removing the mucus from the mucosal surface. Indeed, the presence\absence of the mucus lining in the luminal surface of the intestine is a determinant characteristic influencing the Papp of the systems studied, considering its effect of keeping the mucoadhesive carriers close to the mucus surface, thus increasing the permeant concentration in close proximity to the permeation surface. The results showed that the macromolecular complex has more ability than the NP to promote DEX permeation. Indeed, the Papp for the complexed DEX is about fivefold than that for the same drug carried by the NP. The control for Papp was calculated by assuming the DEX concentration introduced in the donor, C0, to be constant and the drug completely dissolved over the whole term of experiment. The electron paramagnetic resonance (EPR) value for NP is 1.7, which for the complex is 8.6. The Papp value for the DEX carried by the NP is shown in Table 5 to be virtually equal to that for the respective carrier. This observation agrees both with the NP tendency to retain the DEX carried, as pointed out earlier about drug release from NP, and with previous results showing that NPs of same type and size as the present ones were able to cross, unaltered, the intestinal epithelium by transcytosis.11
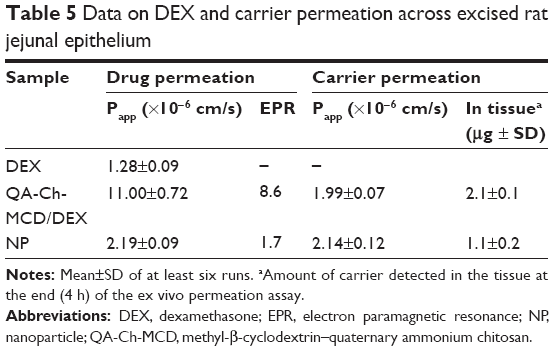 | Table 5 Data on DEX and carrier permeation across excised rat jejunal epithelium |
The comparatively high EPR value found for the macromolecular complex is mostly ascribable to the DEX apparent solubility increase produced by the complex, although some contribution from the QA-Ch-MCD conjugate cannot be ruled out. In fact, the QA-Ch-MCD parent polymer, ie, QA-Ch, was shown to be an effective intestinal absorption enhancer of hydrophobic drugs that cross the epithelium via the transcellular route.13
Macromolecular systems and NPs are generally transported via the transcellular route. It is known, in this respect, that the transport rate of particulate systems having a mean particle diameter smaller than 50 nm is higher than that of larger particles. Indeed, the particle internalization mechanisms into epithelium cells, namely, endocytosis, phagocytosis, and pinocytosis, are influenced by the carrier.30 However, the permeation data listed in Table 5 show no significant Papp difference between the two carriers. Then, the remarkable Papp difference found in DEX transport cannot be ascribed to any such difference. To rule out any NP accumulation in tissue that could reflect a decreased DEX ability to reach the receiving phase, at the end of permeation experiment, the intestinal tissue was homogenized to determine the polymer mass contained in it. No NP accumulation in tissue resulted, in fact, relevant data in Table 5 show that the tissue treated with the NP contained a smaller polymer mass than that treated with the soluble macromolecular complex. Besides, recent studies with peptides, proteins, and synthetic polymers of different nature have shown that paracellular permeation via tight junctions (hydrodynamic radius≈2–4 nm) of macromolecules (viscometric MW, Mv≈30 kg/mol, RH, 2–4 nm) is also possible and is linked to chain flexibility and conformation, irrespective of the net charge of the molecule/structure.30,31 Following this information, a contribution to the DEX permeability increase associated with complexation by QA-Ch-MCD can be thought to also result from some interaction with the tight junctions, producing a paracellular permeation enhancement.
Conclusion
The QA-Ch-MCD conjugate has successfully been prepared and characterized. The ζ potential values for the macromolecular complex and NP are similar as they are made of similar materials. However, the remarkable differences in size and molecular mobility between soluble complex and NP supramolecular structure are responsible for a higher mucoadhesivity of the former and a faster in vitro water-assisted transport across mucus of the latter. Apparently, these characteristics have compensated for each other to yield virtually equal apparent permeabilities of the drug-free carriers across the whole-thickness excised intestine. When it comes to the drug, the carrier adhesion to the mucus surface has appeared to play a most important role in favoring transepithelial permeation. Then, within the concerns of the present study, the use of NP seems not to provide any determinant advantage over using the simpler macromolecular complex. Additionally, NPs are generally preferred for the formulation of labile drugs; meanwhile, cyclodextrins are known to slow down the degradation rate of labile drugs and stabilize macromolecular drugs such as small proteins and peptides. Future investigation will then comprise the application of the developed carriers to drugs liable to degradation.
Disclosure
The authors report no conflicts of interest in this work.
References
Liu L, Zhu S. A study on the supramolecular structure of inclusion complex of β-cyclodextrin with prazosin hydrochloride. Carbohydr Polym. 2007;68(3):472–476. | ||
Marques HMC, Hadgraft J, Kellaway IW. Studies of cyclodextrin inclusion complexes I. The salbutamol–cyclodextrin complex as studied by phase solubility and DSC. Int J Pharm. 1990;63(3):259–266. | ||
Han HK, Shin HJ, Ha DH. Improved oral bioavailability of alendronate via mucoadhesive liposomal delivery system. Eur J Pharm Sci. 2012;46(5):500–507. | ||
Ramalingam P, Ko YT. Improved oral delivery of resveratrol from N-trimethyl chitosan-g-palmitic acid surface-modified solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2016;139:52–61. | ||
Millotti G, Perera G, Vigl C, Pickl K, Sinner FM, Bernkop-Schnürch A. The use of chitosan-6-mercaptonicotinic acid nanoparticles for oral peptide drug delivery. Drug Deliv. 2011;18(3):190–197. | ||
Rekha MR, Sharma CP. Synthesis and evaluation of lauryl succinyl chitosan particles towards oral insulin delivery and absorption. J Control Release. 2009;135(2):144–151. | ||
Dünnhaupt S, Barthelmes J, Hombach J, Sakloetsakun D, Arkhipova V, Bernkop-Schnürch A. Distribution of thiolated mucoadhesive nanoparticles on intestinal mucosa. Int J Pharm. 2011;408(1–2):191–199. | ||
Netsomboon K, Bernkop-Schnürch A. Mucoadhesive vs. mucopenetrating particulate drug delivery. Eur J Pharm Biopharm. 2016;98:76–89. | ||
Sivasubramanian M, Lee JY, Kim KJ, Saravanakumar G, Kang YM, Park JH. Cyclodextrin-based nanocomplexes for sustained delivery of human growth hormone. J Nanosci Nanotechnol. 2013;13(11):7306–7311. | ||
Piras AM, Zambito Y, Burgalassi S, et al. A water-soluble, mucoadhesive quaternary ammonium chitosan-methyl-β-cyclodextrin conjugate forming inclusion complexes with dexamethasone. J Mater Sci Mater Med. In press 2018. | ||
Zambito Y, Felice F, Fabiano A, Di Stefano R, Di Colo G. Mucoadhesive nanoparticles made of thiolated quaternary chitosan crosslinked with hyaluronan. Carbohydr Polym. 2013;92(1):33–39. | ||
Zambito Y, Fogli S, Zaino C, Stefanelli F, Breschi MC, Di Colo G. Synthesis, characterization and evaluation of thiolated quaternary ammonium-chitosan conjugates for enhanced intestinal drug permeation. Eur J Pharm Sci. 2009;38(2):112–120. | ||
Zambito Y, Zaino C, Uccello-Barretta G, Balzano F, Di Colo G. Improved synthesis of quaternary ammonium-chitosan conjugates (N+-Ch) for enhanced intestinal drug permeation. Eur J Pharm Sci. 2008;33(4−5):343–350. | ||
Zambito Y, Di Colo G. Thiolated quaternary ammonium-chitosan conjugates for enhanced precorneal retention, transcorneal permeation and intraocular absorption of dexamethasone. Eur J Pharm Biopharm. 2010;75(2):194–199. | ||
Zambito Y, Uccello-Barretta G, Zaino C, Balzano F, Di Colo G. Novel transmucosal absorption enhancers obtained by aminoalkylation of chitosan. Eur J Pharm Sci. 2006;29(5):460–469. | ||
Fabiano A, Mattii L, Braca A, Felice F, Di Stefano R, Zambito Y. Nanoparticles based on quaternary ammonium-chitosan conjugate: A vehicle for oral administration of antioxidants contained in red grapes. J Drug Deliv Sci Technol. 2016;32:291–297. | ||
Di Colo G, Zambito Y, Zaino C, Sansò M. Selected polysaccharide at comparison for their mucoadhesiveness and effect on precorneal residence of differentdrugs in the rabbit model. Drug Dev Ind Pharm. 2009;35(8):941–949. | ||
Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59(7):645–666. | ||
Uccello-Barretta G, Nazzi S, Balzano F, et al. Enhanced affinity of ketotifen toward tamarind seed polysaccharide in comparison with hydroxyethylcellulose and hyaluronic acid: a nuclear magnetic resonance investigation. Bioorg Med Chem. 2008;16(15):7371–7376. | ||
Zambito Y, Pedreschi E, Di Colo G. Is dialysis a reliable method for studying drug release from nanoparticulate systems? – a case study. Int J Pharm. 2012;434(1–2):28–34. | ||
Pimienta C, Chouinard F, Labib A, Lenaerts V. Effect of various poloxamer coatings on in vitro adhesion of isohexylcyanoacrylate nanospheres to rat ileal segments under liquid flow. Int J Pharm. 1992;80:1–8. | ||
Fabiano A, Zambito Y, Bernskop-Schnürch A. About the impact of water movement on the permeation behaviour of nanoparticles in mucus. Int J Pharm. 2017;517(1–2):279–285. | ||
Lennernans H, Ahrensted O, Ungell AL. Intestinal drug absorption during induced net water absorption in man; a mechanistic study using antipyrine, atenolol and enalaprilat. Br J Clin Pharmacol. 1994;37(6):589–596. | ||
Fabiano A, Brilli E, Fogli S, Beconcini D, Carpi S, Tarantino G, Zambito Y. Sucrosomial® iron absorption studied by in vitro and ex-vivo models. Eur J Pharm Sci. 2018;111:425–431. | ||
Cappello B, Iervolino M, Miro A, Chetoni P, Burgalassi S, Saettone F. Formulation and preliminary in vivo testing of Rufloxacin-Cyclodextrin ophthalmic solutions. J Incl Phenom Macrocycl Chem. 2002;44(1–4):173–176. | ||
Loftsson T, Hreinsdóttir D, Másson M. Evaluation of cyclodextrin solubilization of drugs. Int J Pharm. 2005;302(1–2):18–28. | ||
Vianna RFL, Bentleya MVLB, Ribeiro G, Carvalhoa FS, Neto AF, De Oliveira DCR, Collett JH. Formation of cyclodextrin inclusion complexes with corticosteroids: their characterization and stability. Int J Pharm. 1998;167(1–2):205–213. | ||
Maria DN, Mishra SR, Wang L, Abd-Elgawad AH, Soliman OA, El-Dahan MS, Jablonski MM. Water-soluble complex of curcumin with cyclodextrins: enhanced physical properties for ocular drug delivery. Curr Drug Deliv. 2017;14(6):875–886. | ||
Higuchi T, Connors KA. Phase-solubility techniques. In: Reilley RN, editor. Advances in Analytical Chemistry and Instrumentation. New York: Wiley-Interscience. 1965;4:117–212. | ||
Murugan K, Choonara YE, Kumar P, Bijukumar D, du Toit LC, Pillay V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int J Nanomedicine. 2015;10:2191–2206. | ||
Salamat-Miller N, Chittchang M, Mitra A, Johnston TP. A randomly coiled, high-molecular-weight polypeptide exhibits increased paracellular diffusion in vitro and in situ relative to the highly ordered α-helix conformer. Pharm Res. 2005;22:245-254. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.