Back to Journals » Medical Devices: Evidence and Research » Volume 7
Methodological choices for the clinical development of medical devices
Authors Bernard A, Vaneau M, Fournel I, Galmiche H, Nony P, Dubernard JM
Received 10 March 2014
Accepted for publication 16 April 2014
Published 23 September 2014 Volume 2014:7 Pages 325—334
DOI https://doi.org/10.2147/MDER.S63869
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Alain Bernard,1 Michel Vaneau,2 Isabelle Fournel,3 Hubert Galmiche,2 Patrice Nony,4,5 Jean Michel Dubernard6
1Department of Thoracic Surgery CHU Bocage, Dijon, France; 2Department for Assessment of Medical Devices, HAS (French National Authority of Health), Saint-Denis La Plaine, France; 3Centre of Epidemiology of the Populations, Burgundy University, Dijon, France; 4Department of Clinical Pharmacology, Lyon University CNRS, Lyon, France; 5Laboratory of Biometry and Biology, CNRS, Lyon, France; 6HAS Board (French National Authority of Health), Saint-Denis La Plaine, France
Abstract: Clinical evidence available for the assessment of medical devices (MDs) is frequently insufficient. New MDs should be subjected to high quality clinical studies to demonstrate their benefit to patients. The randomized controlled trial (RCT) is the study design reaching the highest level of evidence in order to demonstrate the efficacy of a new MD. However, the clinical context of some MDs makes it difficult to carry out a conventional RCT. The objectives of this review are to present problems related to conducting conventional RCTs and to identify other experimental designs, their limitations, and their applications. A systematic literature search was conducted for the period January 2000 to July 2012 by searching medical bibliographic databases. Problems related to conducting conventional RCTs of MDs were identified: timing the assessment, eligible population and recruitment, acceptability, blinding, choice of comparator group, and learning curve. Other types of experimental designs have been described. Zelen's design trials and randomized consent design trials facilitate the recruitment of patients, but can cause ethical problems to arise. Expertise-based RCTs involve randomization to a team that specializes in a given intervention. Sometimes, the feasibility of an expertise-based randomized trial may be greater than that of a conventional trial. Cross-over trials reduce the number of patients, but are not applicable when a learning curve is required. Sequential trials have the advantage of allowing a trial to be stopped early depending on the results of first inclusions, but they require an independent committee. Bayesian methods combine existing information with information from the ongoing trial. These methods are particularly useful in situations where the number of subjects is small. The disadvantage is the risk of including erroneous prior information. Other types of experimental designs exist when conventional trials cannot always be applied to the clinical development of MDs.
Keywords: medical device, randomized controlled trials, assessment, clinical development
Introduction
Clinical data for the assessment of medical devices (MDs) are often inadequate or limited.1 This view is shared at the European level as shown by recent publications.2,3 New MDs, which are health products to treat patients, should be subjected to high quality clinical studies to demonstrate their benefit. Taken alone, the technical performance of new MDs does not guarantee the clinical efficacy and its benefit for the patient.
The randomized controlled trial (RCT) is the study offering the highest level of evidence in order to demonstrate the efficacy of a new MD4 and in most cases, conducting an RCT is possible. However, the clinical context can make the achievement of a conventional RCT in parallel groups impossible. To overcome the issues, other experimental designs have been proposed to demonstrate the efficacy of new MDs. The methods described in the following document do however have their limitations and should be reserved for unique situations where it is considered impossible to conduct a conventional RCT. Use of these alternative methods should be scientifically backed up and justified.
In view of the shortcomings frequently observed in assessments of the efficacy of non-pharmacological treatments, the National Committee for the Assessment of Medical Devices and Health Technologies (CNEDiMTS) wished to identify a set of methods and conditions that will allow high-quality clinical assessment, particularly when conventional RCTs cannot be performed. The CNEDiMTS has produced this document for manufacturers, research organizations, and project developers. It aims to provide an up-to-date overview of comparative methods that can be used to evaluate the potential clinical benefit of a new MD or health technology, and to describe possible research designs.
This document focuses on aspects of the clinical efficacy assessment for a new MD or a new health technology from development onwards, following feasibility studies. It aims to identify the methods and conditions that allow a high-quality clinical assessment of an MD to be made. The objectives of this review are to present problems related to conducting conventional RCTs and to identify other experimental designs, their limitations, and their applications.
Methods
A systematic literature search was conducted for the period January 2000 to July 2012 by searching medical bibliographic databases: Medline (National Library of Medicine, USA), the Cochrane Library (Wiley Interscience, USA), BDSP (French Public Health Database), National registers, Websites that publish guidelines, technological assessment reports or economic assessment reports, Websites of learned societies competent in the field studied, and specialist sources, particularly epidemiology and economics sources. The search was limited to publications in English and French. Monitoring continued until 27 June 2013. Technological assessments, guidelines, consensus conferences, meta-analyses, systematic reviews, RCTs and other controlled trials, comparative studies, and cohort studies were sought. The search was completed by the bibliographies of experts consulted and by manufacturers’ data. The search strategy for the Medline bibliographic database was constructed by using either terms from the thesaurus (Medline MeSH descriptors) or free-text terms (from the title or abstract) for each subject. These were combined with terms describing the types of study. The results of the search strategy are reported in a flow chart (Figure 1).
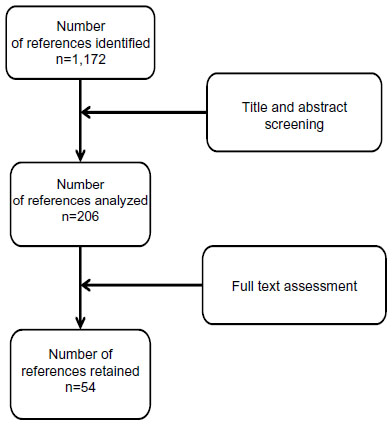 | Figure 1 Flow diagram of references evaluated for inclusion or exclusion. |
Methodological problems
Some methodological principles that are intrinsic to randomized trials of pharmacological treatments may be more difficult to apply when assessing MDs and health technologies. Problems related to conducting conventional RCTs of MDs are detailed below.
Timing the assessment
Choosing the most appropriate time in its life cycle to clinically assess an MD is one issue to consider. MDs usually undergo changes after they have been launched, which aim to improve them. Therefore, a study carried out too early may not reflect the true performance of the MD if it does not sufficiently take into account the period needed to learn the technique. In this case, an unfavorable assessment may reflect a poorly mastered technique rather than a genuinely ineffective technique.4
On the other hand, an assessment conducted too late is responsible for MDs or health technologies being used without any proof of efficacy. An assessment should take place before they are widely distributed. In fact, once an MD or technology is widely distributed, it is difficult to get doctors to adhere to a study protocol,5 because a technique already used is often empirically considered to be effective.
Over time, professionals will change the situations in which the MD is used. These developments may invalidate the initial assessment.5
Eligible population and recruitment
The small size of the eligible population is also a particular feature of studies of MDs. Indeed, the target population may be far less sizeable for MDs than for most drug treatments,5 possibly only involving a few hundred patients in some cases.6 In this situation, a conventional parallel-group trial may be more difficult to implement due to its complexity and cost.
The selection of the population studied is important.6 If the selection of eligible patients is too strict, the risk/benefit ratio for the device will be optimized, but the study’s external validity will be more limited. On the other hand, a broader selection can facilitate recruitment and make it easier to generalize from the results, but may fail to delineate the population most likely to benefit from the new treatment.
Acceptability
The acceptability of the study to patients plays an important role when assessing treatments. Obtaining patient consent is a prerequisite for conducting a clinical trial. When patients are informed before giving their consent, they should be provided with clear, documented, and reliable information. If patients will not consent, the feasibility of the study is called into question. Where there are reasons to believe that the risk/benefit ratio differs between the treatments, both patients and surgeons may prefer a specific intervention and refuse to take part in the trial.
Some patients prefer to choose their treatment, and refuse to be randomized.6 These issues can disrupt patient recruitment and make randomization difficult. Whether or not the use of the technique is widespread may also be a source of difficulty when convincing patients to take part in a clinical trial.
Questions of acceptability may additionally be raised by surgeons, if they are absolutely convinced that the technique they normally use is the best strategy.7 A cross-sectional survey showed that 58% of orthopedic surgeons prefer to participate in expertise-based controlled trials, versus 17% for conventional RCTs.7 In addition, there is improved acceptability because surgeons only perform the procedure that they are used to carrying out, which they prefer, and in which they are “experts”.7 Overall, surgeons are less reluctant to take part in an expertise-based clinical trial than a conventional trial.7
Blinding
Blinding is an important element in clinical trials, because it can reduce measurement bias related to the doctor’s or patient’s subjectivity. A crucial element of blinding is that it must be impossible to distinguish between the treatments compared. The patient, doctor administering the treatment, endpoint committee and/or statistician may be blinded. This is particularly harmful given that the doctor’s influence is also more marked than in pharmacological trials.8 Open-label trials overestimate the therapeutic effect by 14% in comparison with double-blind trials.9
Blinding is more often impossible in non-pharmacological studies,10 for ethical or practical reasons.4 In these situations, in order to evaluate the efficacy of a non-pharmacological treatment as objectively as possible, alternatives have been developed. Boutron et al have summarized the different blinding methods used in non-pharmacological trials.11 Blinding may be complete, partial, or only apply to the assessment of endpoints.
Choice of control or comparator group
The choice of the control or comparator group is crucial in non-pharmacological trials. This problem does not apply to studies where the treatment evaluated is added to the standard treatment, which is used alone in the control group. According to the eleventh directive of the Council for International Organizations of Medical Sciences regarding biomedical research in human beings, the use of a placebo (or an inactive treatment) may be considered ethically acceptable in the following circumstances:
- where there is no effective treatment or procedure;
- where abstaining from a treatment or procedure with known efficacy will lead at worst to temporary discomfort or a delay in relieving symptoms;
- where comparison with an effective treatment or procedure would not provide scientifically reliable results, and administering a placebo does not add any significant risk of irreversible damage.12,13
From an ethical point of view, it is difficult to offer patients an invasive sham procedure.14 In fact, the more invasive the procedure, the harder it is to justify exposing patients in the control group to risks that may be substantial without any expected benefit.15 An important counter-argument to this point of view is that ethical considerations also apply to treatments received by future patients, in that the widespread use of an unassessed treatment is not ethical. This suggests that it may be important to make participants aware of the overall benefit of a study.
In particular, it has been suggested that it is not ethical to administer a placebo in place of a standard treatment with demonstrated efficacy,13 or indeed an “invasive placebo” that does not help to strengthen the demonstration. The decision to use this placebo (or sham) surgery was widely controversial for several reasons. Firstly, it was of no benefit to patients whereas it did entail risks (relating to anesthesia),16 and secondly, alternatives did exist.17
A literature review has summarized the main surgical placebos used in non-pharmacological trials.11 For surgery and technological interventions, different methods have been reported depending on the procedure. Thus, patients may be under general anesthetic, or a surgical drape may be used to conceal the procedure. In some cases, the procedure is simulated by making an incision similar to that made in the treated group, or by injecting a placebo. In practice, so-called placebo surgery is virtually impossible and limited to cases where there is no suitable comparator and where it involves little risk.18 It is also important to standardize preoperative care (patients or equipment in the same position), perioperative care (duration of procedure, instruments, manipulation or care), and postoperative care. In other studies, the surgeon who performed the procedure is not involved in patient follow-up. Boutron et al have also reported the different placebos possible when using MDs: placebo prostheses, hidden MDs, identical but inactive MDs, active devices made ineffective, or use of similar equipment.11
Learning curve
A particular feature of health technologies using MDs is that the operator’s experience has an impact on the results of the technique.4 Different levels of experience may lead to different levels of performance when carrying out interventions. A lack of experience may influence the result of the study, penalizing the new treatment tested.19
Therefore, the learning curve for operators must be taken into account when assessing surgical or interventional techniques. During the development of a new MD, provision must be made for training and learning plans. In fact, the surgeons’ knowledge and skill are variability factors6 and they should be taken into account, for example with a breaking-in period.20
An assessment performed too early risks reflecting complications related to learning the new procedure. During surgical interventions, the impact of learning may be assessed through indicators such as the duration of the procedure or the volume of blood lost. The variability of the treatment effect according to the operator’s level of expertise should also be assessed.
Therefore, an assessment of a new technology versus a control risks being unbalanced in favor of the control treatment, because of the operator’s experience.18 The study should therefore incorporate the effect of learning, for example by recording training and experience.18 From a pragmatic point of view, this learning phase must be taken into account in the trial so that any benefit provided by the device or health technology can be evaluated accurately.
Methods used to overcome the problems identified
Compensating for the lack of blinding
When it is impossible to blind health care professionals, a blind assessment of the endpoint should be planned. This guarantees a neutral assessment of the endpoint. In this situation, the assessment is performed by assessors independent of the study who are blinded as to the treatment received. This assessment may also be centralized in the case of laboratory tests, radiological investigations, or excerpts from clinical examination (videos, photos, recorded interviews). For surgical interventions, patient blinding can be achieved if the patient is under general anesthetic or masked with a drape.11
In some cases, an adjudication committee independent of the investigators is formed to check the endpoint. Blinding as to the study’s hypotheses (or partial blinding) is also described as an alternative.11 When blinding or alternatives to blinding are impossible, it is important to choose the most objective efficacy endpoint possible and for a blind assessment of this endpoint to be made (for example by an independent expert committee).
Other types of experimental designs
A trial with two parallel groups (or arms) should be considered before any other experimental design in any case where a comparative clinical assessment is needed. In such trials, the study treatment is compared with a control treatment by using two groups of patients formed by contemporaneous randomization and followed up in parallel. Other types of experimental designs have been described, in particular for surgery, a field with similar difficulties to those encountered in MD assessment.
Zelen’s design or randomized consent design trial
Zelen’s design (Table 1) involves randomizing patients without first obtaining their informed consent. Only patients randomized to the new treatment group must sign an informed consent form. Patients who refuse will be given the standard treatment.21 Three variants of Zelen’s design have been proposed as clinically indicated.22 The advantage of this experimental design is to facilitate the inclusion of patients by reducing refusals.23,24 Furthermore, it is easier to recruit patients even when they have a strong preference for one treatment over another.25 Nonetheless, several drawbacks have been reported for this type of experimental design. The first is ethical because only patients who provide informed consent receive the new treatment. Zelen’s design may also dilute the effect of the new treatment with the risk of not being able to conclude whether the randomized patients refused the new treatment in favor of the therapeutic reference.21 Selection bias is possible with the risk of under-representation of patients with a poor prognosis in the experimental group as patients at high risk may refuse the new treatment in favor of the therapeutic reference.21 To date, few trials of this type have been proposed in France for ethical reasons. This experimental design is particularly useful when patients receiving the standard treatment do not require additional visits and when death is the only endpoint.25 A trial comparing radiofrequency ablation to surgery or the treatment of small hepatocellular carcinoma used Zelen’s design.26 The authors chose this experimental design to prevent the withdrawals of patients to be treated by radiofrequency ablation while surgery was the standard treatment.26 According to the flow chart, four patients refused to participate in the trial.26 After randomization, the number of patients refusing the proposed treatment was well balanced, four in the radiofrequency group and four in the surgery group.26 In this trial, 168 patients (84 in each group) were included. The characteristics of patients were similar in both treatment groups.
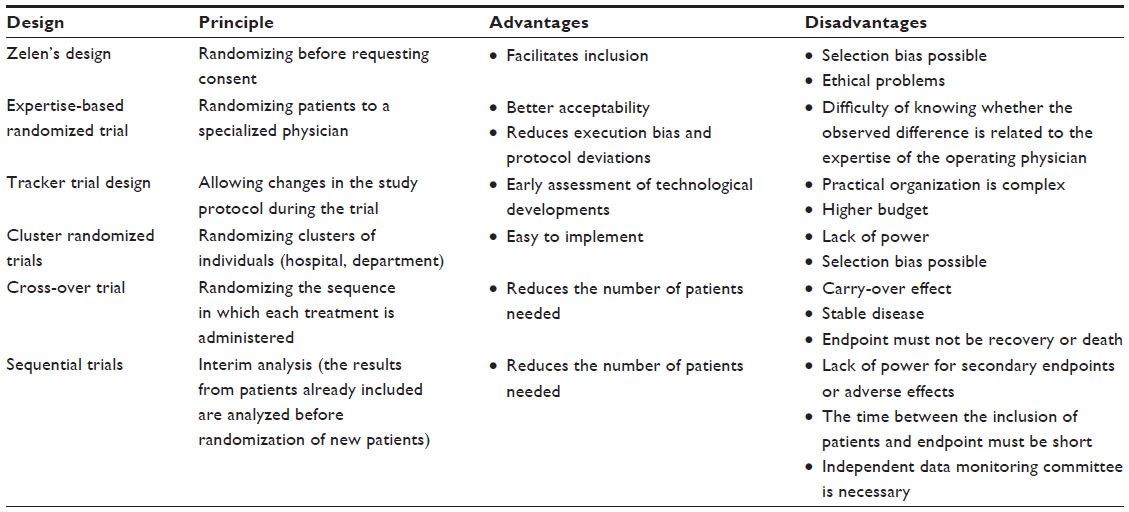 | Table 1 Different experimental designs |
Expertise-based RCTs
Unlike a conventional clinical trial, where patients are randomized to receive either intervention A or intervention B provided by the same team, an expertise-based randomized controlled design (Table 1) involves randomizing patients to a surgeon or team that specializes in a given intervention.27 In this case, the physician is deemed to master the procedure. This type of design requires at least one expert in each randomized intervention at each center.27 More initial pre-inclusion consultations should be carried out by a neutral person to determine the eligibility of patients.27 This design requires stratification by surgeon and by center.27 Among the advantages, this method limits execution bias related to the absence of blinding.28 In fact, as each physician only performs the procedure that he or she specializes in, the risk of differences between patients regarding the procedure and related factors is lower than in conventional trials, where physicians may follow patients in different groups in different ways. This experimental design could reduce deviations from the protocol27 and improve feasibility, as physicians do not need to be trained in both procedures.27 Last but not least, this approach has the advantage of acceptability because physicians only perform the procedure that they are used to.27,28 The disadvantage of this expertise-based experimental design lies in the fact that it can sometimes be difficult to tell whether the superiority of one technique over another is real, or whether it is related to the expertise of the physician performing it.27 This type of experimental design is interesting when a learning curve is required to master the technique, especially as the level of expertise can affect the result. In a multicenter trial that compared arthroscopy with open surgery for the treatment of rotator cuff tears,28,29 at the same center, the surgeon performing the open surgery was associated with a surgeon performing arthroscopic surgery. The objective of this trial was to minimize the impact of expertise.
Tracker trial designs
This type of trial was developed so that technological changes or improvements in the procedure could be taken into account during the trial (Table 1).30 Changes in the trial are authorized and taken into account in the statistical analysis.30 The main advantage is to achieve a very early assessment of a new MD or new technology before making it widely available.18 This design has several disadvantages: the methods are more sophisticated than in conventional trials and the practical organization is difficult with an impact on the budget of the study.30 To date, there are few examples of this type of study in the literature.
Cluster randomized trials
Cluster randomized trials involve randomizing clusters of individuals (by center, hospital, department) (Table 1). Each center is randomly assigned one of two treatments.31 This experimental design is easy to implement in terms of logistics and may be of interest to compare strategies. The main disadvantage is a lack of power31 as recruitment in the clusters in terms of numbers and characteristics of patients may vary. Sometimes, some clusters may remain empty when the person responsible does not include patients.32 These situations jeopardize the comparability of groups and lead to selection bias.33 The application of this type of study is limited to the clinical development of MDs.
Cross-over trials
In contrast with parallel group trials, where each patient receives only one treatment, cross-over trials involve randomizing the sequence in which each treatment is administered (Table 1). Each patient therefore receives both treatments.34 Cross-over experimental design has the advantage of reducing the number of patients needed.34 The main disadvantage concerns the risk of the so-called “carry-over” effect. This effect results from the fact that the first treatment administered may have a remaining effect during the second period. Cross-over trials are not suitable when the endpoint studied is recovery or death, or when there is a learning curve.34 In a cross-over trial, the disease must be stable. This type of trial may have a benefit in dermatology, cosmetology, and in certain implantable MDs such as neuro-stimulation devices. In 2013, a cross-over trial was reported comparing subthalamic and pallidal deep brain stimulation for dystonia.35 Each patient was randomly selected to undergo initial bilateral stimulation of either the subthalamic nucleus or the globus pallidus internus for 6 months, followed by bilateral stimulation of the other nucleus for another 6 months. For this trial, only 12 patients were necessary.35
Sequential trials
Sequential trials are one technique that can be used for the interim analysis of RCTs (Table 1). During the study, results obtained with patients already included are analyzed before the randomization of new patients. Several methods have been described.36–40 These methods have the advantage of allowing a trial to be stopped earlier.41 The average number of patients required is also lower than in conventional trials.42,43 However, they have a number of constraints: there is a single outcome endpoint and the time between the inclusion of patients and measurement of the endpoint must be short. Stopping the trial early may lead to insufficient power for the endpoints or may influence measurements of the frequency of adverse effects. An independent data monitoring committee should be set up to decide whether the trial should continue or be stopped.42 It is advisable to have regular follow-up with good-quality inclusions to avoid delays in updating data. These techniques are of particular interest for rare diseases or in pediatrics. Hamilton et al44 reported sequential design to reduce the time required to bring a prosthetic heart valve to market, helping to ensure that patients and clinicians have access to the latest devices in less time.44 Clinical trials involving prosthetic heart valves are typically based on a set of objective performance criteria for certain late adverse event rates. The objective performance criteria were defined under the guidance of the US Food and Drug Administration. This sequential design alternative allows for the possibility of stopping the trial early.
Adaptive randomized trials
The objective is to maximize the total number of patients receiving the best treatment (Table 2). Thus, at the start of the trial patients are randomized with a ratio 1:1 in each arm, but as the results start to show a difference between the treatments, this ratio is modified in favor of the group receiving the treatment that seems to be more effective.43 These trials are based on achieving interim analyses. The experimental design can be changed depending on the results obtained during the interim analysis.45 Possible adjustments relate to the re-evaluation of the number of patients required for the endpoint and the addition or deletion of interim analyses.42 The adaptive trial may be constructed in two or more stages.45 During a two-stage trial, at first, a sample of size N1 is selected. Depending on the results, the study can be interrupted. It is necessary to establish an independent data monitoring committee that will decide on the adaptations, whether to adapt the number of patients required, and whether to proceed or not with new inclusions.42,45 An adaptive trial implies greater logistical constraints,45 and the outcome endpoint should be simple and unique.41 Adaptive trials allow for greater flexibility and a reduction in the number of patients required. However, the internal validity of these trials has been called into question, and they could have the same disadvantage as historical controlled trials.36,41 They have been judged no more interesting than sequential trials according to some authors.41,45,46 This type of trial could be interesting in the context of techniques requiring a learning curve or new MDs.
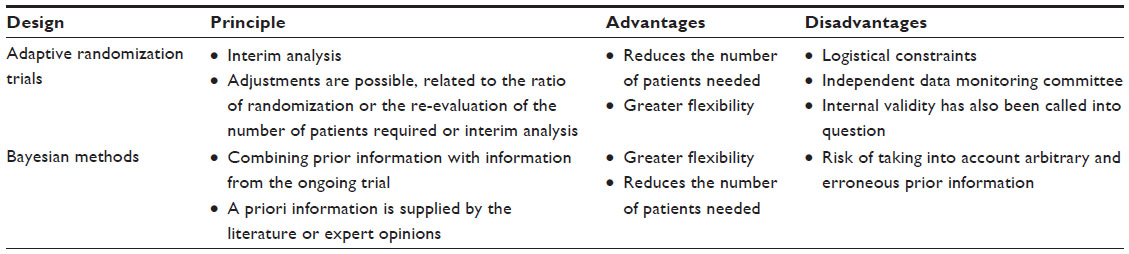 | Table 2 Adaptative methods |
Bayesian methods
Bayesian methods can be used as alternatives to conventional sequential methods (Table 2).47 In a conventional RCT, data from previous studies are used when the trial is being designed. Subsequently, only information collected during the trial is used. By contrast, the Bayesian approach combines prior information with information from the ongoing trial.47 It uses existing data, which may be obtained from the literature. Some publications argue that it is even possible to use expert opinions by modelling them. From Bayes’ theorem, posterior probabilities are estimated using data from the ongoing trial, which are conditional on prior information.47 The Bayesian estimation provides not a confidence interval but a credible interval based on the posterior distribution.47 Unlike the so-called “frequentist” approach, there are no statistical tests but results with a 95% credible interval. These methods provide some flexibility and can be used in the test by performing adaptive interim analysis.48 The use of Bayesian methods is recommended for the clinical evaluation of MDs.48–51 The advantage of these methods lies in the reduction of the number of patients required, which is particularly interesting for small target populations. The main disadvantage is that arbitrary and erroneous prior information can be taken into account, which can substantially influence the final result. This would be similar to including in a meta-analysis a large biased study in favor of the treatment, which would fully compensate for the results of a small unbiased study. In 2010, an RCT using Bayesian methods compared antiarrhythmic drugs with radiofrequency ablation for the treatment of paroxysmal atrial fibrillation.52 The maximum number of subjects required was estimated at 230 patients. Bayesian interim analyses were planned for 150, 175, and 200 patients. The trial was stopped at 150 patients because the Bayesian interim analysis showed a predictive probability of success with radiofrequency ablation of 99.9% above the threshold set by the protocol.
Comparative non-randomized observational studies
This type of study should be used under circumstances where it is impossible to conduct an RCT. The choice of an observational study should remain the exception. Observational studies offer lower-quality evidence than RCTs.
Prospective comparative non-randomized studies involve comparing the incidence of an endpoint in a group receiving procedure A with that in a group not receiving procedure A. The distribution of patients in each group is not determined by the investigator. It is simply observed data. This type of study does not guarantee that the two groups will be comparable on other variables, especially variables that have not been measured, and cannot conclude causality in terms of the superiority of one treatment over another. The only authorized conclusion is that a difference between the two treatments is observed. To reduce the risk of confounding bias, the propensity score was developed.53 The propensity score is defined as the conditional probability of a patient receiving treatment A rather than treatment B given his/her baseline characteristics (propensity to receive one treatment according to characteristics). The objective is to balance the distribution of covariates (age, sex, comorbidities, etc) between the two groups so as to neutralize confounding factors.53,54 The comparison between the two groups is made within the same propensity score category.53 Once established, the propensity score can be used for purposes of matching, stratifying, adjusting, or weighting.55,56 The propensity score can only be calculated for collected variables57 with the risk of getting non-comparable groups,57 and a sensitivity analysis can identify potential hidden bias.58 A systematic review that compared the results of RCTs with those of observational studies using propensity score on the various treatments during acute coronary syndromes has been published.59 Overall, observational studies report a greater amount of effect for long-term mortality than RCTs. It should be noted that only one RCT among ten reported significant results while nine out of ten observational studies reported statistically significant results.
Discussion
This review of the different existing methodologies shows that favorable conditions exist for the clinical development of new MDs. Any medical progress is inconceivable without comparison with existing treatment. This is the only way to confirm that the new MD improves the health of patients compared with conventional treatment. Conducting a comparative trial should become the norm for all new high-risk MDs. Obviously, a company is interested in carrying out a comparative trial only if it considers that its MD can contribute to medical progress. Conducting a high quality trial is a constraint, but ultimately it is an investment for the future and carries the value of insurance by showing that the new MD can be a more effective treatment alternative. Contrary to what one may read, a comparative trial does not constitute a barrier to innovation. The demonstration from a clinical study of interest to patients enhances an innovative MD. Actually a clinical trial is costly, but this is not an obstacle to innovation. This is an investment for the future for patients. The different experimental designs adapted to small target populations, technological changes, and learning curves are available.
Depending on the circumstances, one or more experimental designs can be offered to assist developers or manufacturers. They depend on the characteristics of the MD and the acceptability to patients or physicians. When an MD requires rapid technological change, in early clinical development, several experimental designs (Table 3) such as adaptive randomization trials or Bayesian methods can be proposed. When the target population is small, Bayesian methods are appropriate as are cross-over trials providing that the disease studied is stable and that the outcome endpoint can be repeated. Finally, a sequential trial may be considered. When the MD carries potentially serious adverse effects, sequential trials allow the study to be stopped early.
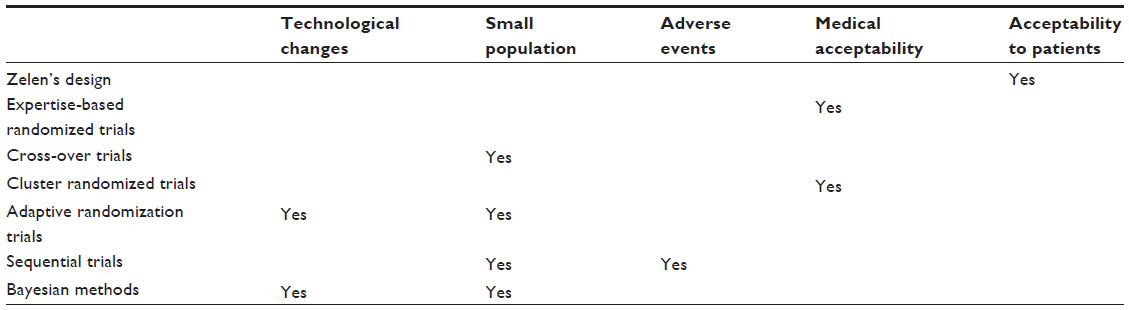 | Table 3 Experimental design according to characteristics of the medical devices |
Medical eligibility is essential for the success of a trial and especially when the technique influences the result or when the techniques are very different or when a learning curve is required. Expertise-based randomized trials are appropriate to overcome such problems (Table 3). Under some circumstances, cluster randomized trials could be used.
Problems concerning patient acceptability may arise when the comparator is an invasive technique or the technique is already widely known. In these circumstances, Zelen’s design is suitable to promote patient acceptability (Table 3).
Following this review of different experimental designs, the role of observational studies in the clinical development of a new high-risk MD is very small. Observational studies cannot guarantee the absence of bias even when the methodology is rigorous. Interpretations of this type in demonstrating the superiority of the new MD compared with the reference treatment are more limited. Observational studies may be proposed only when an RCT is not possible. However, an observational study is still better than no study at all to demonstrate the efficacy of a new MD.
All of the methods exist to carry out a clinical evaluation of a new MD. However, the support and training of a developer or company is necessary. These methods are far less well-known than conventional trials. The implementation of a trial is a constraint in terms of organization and costs. But it is worth considering spending a little time at the beginning of clinical development in order to achieve a high quality trial soundly demonstrating efficacy. This investment will make it possible to evaluate the new MD and to demonstrate medical progress. Ultimately, the public authorities will be less reluctant to cover the costs of such MDs. A new paradigm may be considered if the company carries out a clinical trial of quality with the highest level of evidence.
In conclusion, a conventional RCT is applicable in many situations. The diversity and heterogeneity of situations mean that these conventional trials cannot always be applied to the clinical development of some high-risk MDs. To meet these challenges, this paper proposes a toolkit to allow everyone to find an answer to his/her problem.
Acknowledgments
We are grateful to Catherine Denis and Michele Morin-Surocca, head of medical devices assessment department, HAS (French National Authority of Health). We are also grateful to Sophie Stamenkovic for reading this manuscript (HAS [French National Authority of Health]).
Authors’ contributions
MV contributed toward analysis and interpretation of literature; IF contributed toward the acquisitions, analysis, and interpretation of literature; HG contributed toward drafting the work and revising it; PN revised it critically for important intellectual content; JMD revised it critically for important intellectual content; AB had the original idea for this article and gave final approval of the version to be published. All authors read and approved the submitted manuscript. All authors contributed toward data analysis, drafting, and revising the manuscript.
Disclosure
The authors declare that they have no competing interests in this work.
References
Activity report of the National committee for the evaluation of medical devices and health technologies. Available from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2012-10/ra-cnedimts_2011.pdf. | |
Stordeur S, Vinck I, Neyt M, Van Brabandt H, Hulstaert F. [Introduction of innovative high-risk medical devices in Europe: are clinical efficacy and safety guaranteed?]. Rev Epidemiol Sante Publique. 2013;61(2):105–110. French. | |
Storz-Pfennig P, Schmedders M, Dettloff M. Trials are needed before new devices are used in routine practice in Europe. As the EU debates new legislation to regulate medical devices. BMJ. 2013;346:f1646. | |
Vinck I, Neyt M, Thiry N, et al. An evaluation procedure for the emerging medical technologies. Belgian health care knowledge centre (KCE) Bruxelles. KCE Reports 44. 2006. | |
Bernard A. Le développement clinique des nouveaux dispositifs médicaux [Clinical development of new medical devices]. e-memoires de l’académie nationale de chirurgie. 2012;11(4):27–31. French. | |
Bernard A, Vicaut E. Medical devices. First party: what type of study is needed? Medicine. 2008;4:464–467. | |
Bednarska E, Bryant D, Devereaux PJ; Expertise-Based Working Group. Orthopaedic surgeons prefer to participate in expertise-based randomized trials. Clin Orthop Relat Res. 2008;466(7):1734–1744. | |
Boutron I, Tubach F, Giraudeau B, Ravaud P. Methodological differences in clinical trials evaluating nonpharmacological and pharmacological treatments of hip and knee osteoarthritis. JAMA. 2003;290(8):1062–1070. | |
Juni P, Altmann DG, Egger M. Assessing the quality of controlled clinical trials. Systematic reviews in health care. BMJ. 2001;323(7303):42–46. | |
Boutron I, Moher D, Tugwell P, et al. A checklist to evaluate a report of nonpharmacological trial (CLEAR NPT) was developed using consensus. J Clin Epidemiol. 2005;58(12):1233–1240. | |
Boutron I, Guittet L, Estellat C, Moher D, Hrobjartsson A, Ravaud P. Reporting methods of blinding in randomised trials assessing nonpharmacological treatments. PLos Med. 2007;4(2):e61. | |
Kinnaert P. Factual surgery: ethics and methodology. Rev Med Brux. 2006;27:451–458. | |
Kinnaer P. [Placebo and placebo effect (second part): ethical aspects]. Rev Med Brux. 2007;28(1):39–44. French. | |
Ravaud P. Assessment of nonpharmacological treatments. 4ème conference d’epidemiologie Clinique; May 26–28; 2010; Paris. | |
Li H, Yue LQ. Statistical and regulatory issues in nonrandomized medical device clinical studies. J Biopharm Stat. 2008;18(1):20–30. | |
Macklin R. The ethical problems with sham surgery in clinical research. N Engl J Med. 1999;341(13):992–996. | |
Macklin R. Placebo surgery in trials of therapy for parkinson’s disease. N Engl J Med. 2000;342(5):355. | |
Ergina PL, Cook JA, Blazeby JM, et al. Challenges in evaluating surgical innovation. Lancet. 2009;374(9695):1097–1104. | |
Rudicel S, Esdaile J. The randomized clinical trial in orthopaedics: obligation or option? J Bone Joint Surg Am. 1985;67-A(8):1284–1293. | |
Campbell G. Statistics in the world of medical devices: the contrast with pharmaceuticals. J Biopharm Stat. 2008;18(1):4–19. | |
Zelen M. A new design for randomized clinical trials. N Engl J Med. 1979;300(22):1242–1245. | |
Zelen M. Innovations in the design of clinical trials in breast cancer. Breast Cancer Res Treat. 1983;3(2):137–142. | |
Zelen M. Strategy and alternate randomized designs in cancer clinical trials. Cancer Treat Rep. 1982;66(5):1095–1100. | |
Zelen M. Randomized consent designs for clinical trials: an update. Stat Med. 1990;9(6):645–656. | |
Horwitz RI, Feinstein AR. Advantages and drawbacks of the Zelen design for randomized clinical trials. J Clin Pharmacol. 1980;20(7):425–427. | |
Feng K, Yan J, Li X, et al. A randomized controlled trial of radiofrequency ablation and surgical resection in the treatment of small hepatocellular carcinoma. J Hepatol. 2012;57(54):794–802. | |
Devereaux PJ, Bhandari M, Clarke M, et al. Need for expertise based randomized controlled trials. BMJ. 2005;330(7482):88–91. | |
Mastracci TM, Clase CM, Devereaux PJ, Cina CS. Open versus endovascular repair of abdominal aortic aneurysm: a suvey of Canadian vascular surgeons. Can J Surg. 2008;51(2):142–148. | |
MacDermid JC, Holtby R, Razmjou H, et al. All-arthroscopic versus mini-open repair of small or moderate-sized rotator cuff tears: a protocol for a randomized trial [NCT00128076]. BMC Musculoskelet Disord. 2006;7:25. | |
Lilford RJ, Braunholz DA, Greenhalgh R, Edwards SJ. Trials and fast changing technologies: the case for tracker studies. BMJ. 2000; 320(7226):43–46. | |
Giraudeau B, Ravaud P. Preventing bias in cluster randomized trials. PLos Med. 2009;6(5):e1000065. | |
Campbell MJ. Cluster randomized trials in general practice research. Stat Methods Med Res. 2000;9(2):81–94. | |
Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV. Meta-analyses involving cross-over trials: methodological issues. Int J Epidemiol. 2002;31(1):140–149. | |
Hills M, Armitage P. The two-period cross-over clinical trial. Br J Clin Pharmacol. 1979;8(1):7–20. | |
Schejerling L, Hjermind L, Jespersen B, et al. A randomized double-blind crossover trial comparing subthalamic and pallidal deep brain stimulation for dystonia. J Neurosurg. 2013;119(6):1537–1545. | |
Armitage P. Sequential Medical Trials. 2nd ed. New York: Wiley; 1975. | |
Pocock SJ. Group sequential methods in the design and analysis of clinical trials. Biometrika. 1977;64(2):191–199. | |
O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35(3):549–556. | |
Peto R, Pike MC, Armitage P, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br J Cancer. 1976;34(6):585–612. | |
Falissard B, Lellouch J. A new procedure for group sequential analysis in clinical trials. Biometrics. 1992;48(2):373–388. | |
Clayton DG. Ethically optimised designs. Br J Clin Pharmacol. 1982; 13(4):469–480. | |
Van der Lee JH, Wesseling J, Tanck MW, Offringa M. Efficient ways exist to obtain the optimal sample size in clinical trials in rare disease. J Clin Epidemiol. 2008;61(4):324–330. | |
Sebille V, Bellissant E. Comparison of four sequential methods allowing for early stopping of comparative clinical trials. Clin Sci (Lond). 2000;98(5):569–578. | |
Hamilton C, Lu M, Lewis S, Anderson W. Sequential design for clinical trials evaluating a prosthetic heart valve. Ann Thorac Surg. 2012;93(4):1162–1166. | |
Bauer P, Brannath W. The advantages and disadvantages of adaptive designs for clinical trials. Drug Discov Today. 2004;9(8):351–357. | |
Simon R. Adaptive treatment assignment methods and clinical trials. Biometrics. 1977;33(4):743–749. | |
Lellouch J, Schwartz D. Therapeutic trial: individual or collective ethics. Rev Inst Int Stat. 1971;39:127–136. | |
Guidance for the use of bayesian statistics in medical device clinical trial. Center for devices and radiological health, center for biologics evaluation and research. Silver Spring, Rockville: CDRH, CBER 2006. Available from: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationGuidance/GuidanceDocumentation/ucmo071121.pdf. Accessed August 31, 2014. | |
Berry SM, Carlin BP, Lee JJ, Muller P. Bayesian Adaptive Methods for Clinical Trials. Ed Chapman and Hall/CRC Biostatistics series; 2011. | |
Campbell G. Bayesian statistics in medical devices: innovation sparked by the FDA. J Biopharm Stat. 2011;21(5):871–887. | |
Pibouleau L, Chevret S. Bayesian statistical method was underused despite its advantages in the assessment of implantable medical devices. J Clin Epidemiol. 2011;64(3):270–279. | |
Wilber DJ, Pappone C, Neuzil P, et al. Comparison of antiarrhythmic drug therapy and radiofrequency catheter ablation in patients with paroxysmal atrial fibrillation. A randomized controlled trial. JAMA. 2010;303(4):333–340. | |
Rosenbaum PR, Rubin DB. Reducing bias in observational studies using subclassification on the propensity score. J Am Stat Assoc. 1984;79:516–524. | |
Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41–55. | |
Yue LQ. Statistical and regulatory issues with the application of propensity score analysis to nonrandomized medical device clinical studies. J Biopharm Stat. 2007;17(1):1–13. | |
Stuart EA. Matching methods for causal inference: a review and a look. Stat Sci. 2010;25(1):1–21. | |
Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424. | |
Rosenbaum PR. Discussing hidden bias in observational studies. Ann Intern Med. 1991;115(11):901–905. | |
Dahabreh IJ, Sheldrick RC, Paulus JK, et al. Do observational studies using propensity score methods agree with randomized trials? A systematic comparison of studies on acute coronary syndromes. Eur Heart J. 2012;33(15):1893–1901. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.