Back to Journals » International Journal of Nanomedicine » Volume 14
Mesoporous silica nanoparticles as a delivery system for improving antiangiogenic therapy
Authors Sun JG, Jiang Q ![]() , Zhang XP, Shan K, Liu B, Zhao C, Yan B
, Zhang XP, Shan K, Liu B, Zhao C, Yan B ![]()
Received 21 November 2018
Accepted for publication 25 January 2019
Published 25 February 2019 Volume 2019:14 Pages 1489—1501
DOI https://doi.org/10.2147/IJN.S195504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Jian-Guo Sun,1–3,* Qin Jiang,4,* Xiao-Pei Zhang,4,* Kun Shan,1 Bai-Hui Liu,5 Chen Zhao,1 Biao Yan1,2
1Eye Institute, Eye & ENT Hospital, Shanghai Medical College, Fudan University, Shanghai, China; 2NHC Key Laboratory of Myopia, Fudan University, Key Laboratory of Myopia, Chinese Academy of Medical Sciences, Shanghai, China; 3Shanghai Key Laboratory of Visual Impairment and Restoration, Fudan University, Shanghai, China; 4The Affiliated Eye Hospital, Nanjing Medical University, Nanjing, China; 5Department of Pediatric Surgery, Children’s Hospital of Fudan University, Shanghai, China
*These authors contributed equally to this work
Purpose: Antiangiogenic drugs usually have short-acting efficacy and poor treatment compliance. The purpose of this study was to determine whether mesoporous silica nanoparticles (MSNs) could be utilized as a nanodrug delivery system for improving antiangiogenic therapy.
Materials and methods: MSN-encapsulated bevacizumab nanoparticles were prepared by the nanocasting strategy and characterized by Fourier transform infrared, transmission electron microscopy, and Brunauer–Emmett–Teller method. Encapsulation efficiency and drug loading efficiency of MSN-encapsulated bevacizumab nanoparticles were calculated. The pharmacokinetics, cytotoxicity, and tissue toxicity were evaluated in vitro and in vivo. The antiangiogenic effects of MSN-bevacizumab nanoparticles were evaluated in vitro and in vivo.
Results: MSN encapsulation could prolong the residency of bevacizumab in vitreous/aqueous humor and maintain the long-lasting drug concentration. MSN-encapsulated bevacizumab nanoparticles did not show any obvious cytotoxicity and tissue toxicity. MSN-encapsulated bevacizumab nanoparticles were more effective than bevacizumab in suppressing vascular endothelial growth factor-induced endothelial cell proliferation, migration, and tube formation in vitro. MSN-encapsulated bevacizumab nanoparticles showed sustained inhibitory effects on corneal neovascularization and retinal neovascularization in vivo.
Conclusion: This study provides a novel strategy of encapsulating bevacizumab to protect and deliver it, which could increase the time between administration and formulation shelf-life. MSN-encapsulated bevacizumab is a promising drug delivery alternative of antiangiogenic therapy.
Keywords: mesoporous silica nanoparticle, bevacizumab, ocular angiogenesis, antiangiogenic therapy
Introduction
Angiogenesis is the process through which new blood vessels form from the pre-existing vessels. It plays important roles in several physiological and pathological processes, including embryonic development, wound healing, tumor growth, ischemic, inflammatory, infectious, and immune disorders.1,2 Angiogenesis is usually controlled by the balance of proangiogenic factors (such as vascular endothelial growth factor [VEGF], basic fibroblast growth factor, platelet-derived growth factor) and antiangiogenic factors (such as angiostatin, angiopoietin-2, and endostatin).3 Currently, VEGF is usually recognized as the prominent cytokine during the angiogenic cascade. Many anti-VEGF drugs have been developed, such as bevacizumab, pegaptanib, ranibizumab, and aflibercept.4,5
Bevacizumab (Avastin®; Genentech, Inc., South San Francisco, CA, USA) is a recombinant humanized monoclonal anti-VEGF antibody. Bevacizumab binds to VEGF that keeps VEGF away from VEGF receptor, thereby preventing the formation of new blood vessels. It has been used for treating colon cancer, lung cancer, glioblastoma, renal cell carcinoma, and ocular vascular diseases.6,7 However, bevacizumab may lead to rare but serious neurologic disorders, such as headache, confusion, fainting, and vision problems.8,9 Moreover, bevacizumab usually has a short half-life in vivo, and repetitive injections are required to maintain drug efficiency.10 Therefore, a novel strategy for bevacizumab administration should be developed for improving antiangiogenic therapy.
The advent of nanotechnology has revolutionized the way of drug administration.11 The application of mesoporous silica nanoparticles (MSNs) in drug delivery has attracted much attention due to the favorable chemical property, thermal stability, satisfying biocompatibility, and improved bioavailability.12 MSNs are solid materials, which contain a great number of mesopores. The mesoporous structure of silica facilitates effective drug loading and subsequent controlled release.13 MSNs have been widely used in several biomedical fields, such as drug delivery, diagnosis, biosensing, and cellular uptake. In this study, we determined whether MSNs could be used as a drug delivery system for antiangiogenic therapy.
The retina and its clinical disorders provide an important system to investigate the mechanism of angiogenic cascade.14 VEGF is responsible for ocular neovascular pathology.15 Currently, bevacizumab has been approved for treating neovascular ocular diseases, including diabetic retinopathy, retinopathy of prematurity, and neovascular glaucoma.16 However, the cost and frequent intravitreal injection leads to poor patient compliance and ocular side effects such as endophthalmitis, increased intraocular pressure, hemorrhage, corneal epithelial defect, and retinal detachment.17 Thus, it is required to develop a novel drug delivery method to improve the efficacy and safety of bevacizumab. In this study, bevacizumab was encapsulated into MSN nanoparticles, and the profiling of bevacizumab release from MSN-bevacizumab nanoparticles and the long-acting efficacy for antiangiogenic activity in vitro and in vivo were determined.
Materials and methods
Chemicals
Tetraethyl orthosilicate (TEOS), cetyltrimethylammonium chloride (CTAC), triethanolamine (TEA), 3-aminopropyltriethoxysilane (3-aminopropyl) triethoxysilane (APTES), and mPEG-succinimidyl carboxymethyl ester (mPEG-NHS, MW5K) were purchased from Sigma-Aldrich St Louis, MO, USA. Bevacizumab (Avastin) was the product of Genentech Inc. Cyclohexane, anhydrous ethanol, isopropanol, and ammonium nitrate (NH4NO3) were purchased from Shanghai Chemical Corp. (Shanghai, China). All other chemicals used were of analytical grade and used without further purification.
Preparation and characterization of MSN-encapsulated bevacizumab nanoparticles
Synthesis of MSNs
The uniform MSNs were synthesized by the soft template method as previously reported.18 In brief, CTAC (1.5 g) and TEA (0.045 g) were dissolved in 15 mL deionized water in a 50 mL round bottom flask and stirred gently at 65°C in an oil bath for 1 hour. Then, 1 mL TEOS dispersed in 4 mL cyclohexane was carefully added to CTAC–TEA mixture and then stirred for additional 24 hours with continuous mechanical stirring (150 rpm). MSNs were collected by centrifugation at 4,000 rpm, washed by ethanol and deionized water to remove the residual reactants, extracted with 0.6 wt % NH4NO3 ethanol solution at 60°C for 12 hours to remove CTAC and TEA, followed by washing with anhydrous ethanol for three times. The extracted MSN products were lyophilized to obtain nanoparticle powder.
Surface modification of MSNs
Surface modification of MSNs consisted of two steps: −NH2-modification and PEG-modification. The −NH2-modification of MSNs was performed according to a simple surface alkylation process.19 In brief, 12 mg MSNs was dispersed in 30 mL anhydrous ethanol with 20 μL APTES and then stirred at 70°C for 3 hours. The −NH2-modified MSNs (MSN-NH2) were obtained by centrifugation at 4,000 rpm, washed with anhydrous alcohol, dried at 110°C for 3 hours, and vacuum dried for 24 hours. The MSN-NH2 products were dispersed in 3 mL 2-(N-morpholino)ethanesulfonic acid (MES) buffer, and then 0.5 mg mPEG-NHS dissolved in 1 mL MES buffer was added. The mixture dispersion was stirred at room temperature for 2 hours. PEG-modified MSNs (MSN-PEG-NH2) were collected by centrifugation at 4,000 rpm and washed with deionized water to remove the unreacted mPEG-NHS.20 The MSN-PEG-NH2 products were lyophilized to obtain the final nanocarriers.
Preparation of MSN-encapsulated bevacizumab nanoparticles
MSN-encapsulated bevacizumab nanoparticles were prepared by encapsulating bevacizumab into the surface-functionalized MSNs via the nanocasting strategy. Briefly, surface-functionalized MSNs (10 mg) were mixed with bevacizumab solution (10 mL, 1 mg/mL) under stirring (500 rpm) for 6 hours. Then, MSN-encapsulated bevacizumab nanoparticles were collected by centrifugation.
Characterization of MSN-encapsulated bevacizumab nanoparticles
The morphology, particle size, and particle size distribution of MSNs were observed by transmission electron microscopy (200 kV; JEOL 2011 microscope; JEOL, Tokyo, Japan). Zeta potential of nanoparticles was detected in deionized water by the Zetasizer (Malvern Instruments, Malvern, UK). The Brunauer–Emmett–Teller method was used to calculate the specific surface areas using adsorption data in a relative pressure (P/P0) ranging from 0.04 to 0.1. The Barrett–Joyner–Halenda method was used to calculate the pore size distribution and pore volume, which was derived from the adsorption branches of the isotherms. Fourier transform infrared (FTIR) was used to confirm the surface modifications of MSNs by −NH2 and Polyethylene glycol (PEG) groups and drug loading, respectively. FTIR measurements were performed on a Nicolet 6700 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) using KBr disk at a resolution of 4 cm−1 in the frequency interval of 500–4,000 cm−1.
Encapsulation efficiency and drug loading efficiency were calculated by the following equations. BEV indicates bevacizumab.
|
|

|
|

|
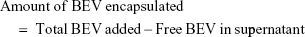
Pharmacokinetic study of bevacizumab release from MSN-encapsulated bevacizumab nanoparticles
MSN-bevacizumab (containing 0.01 mg bevacizumab) and bevacizumab (0.01 mg, control group) were injected to the vitreous of C57B/L mice, respectively. About 6 hours and 1, 3, 7, 21, 30, and 45 days after injection, the animals were sacrificed (n=6). The amount of bevacizumab in the vitreous and aqueous humor was determined by ELISA.
The maximum concentration of bevacizumab (Cmax) and the time required to reach the maximum concentration (tmax) were obtained from the bevacizumab concentration vs time curves. The rate constant (K) was calculated by log-linear regression of the last data points (terminal portion) of bevacizumab concentration vs time curve. The half-life of bevacizumab was calculated using the following equation: t1/2=0.693/K. The total body clearance (CL) was calculated as: CL = dose/area under the curve (AUC). The mean residence time (MRT) was calculated as: MRT = area under the first moment concentration–time curve0–∞/AUC0–∞. The area under the bevacizumab concentration vs time curve was calculated by trapezoidal rule.
In vitro release of bevacizumab from MSN-encapsulated bevacizumab nanoparticles
To determine the in vitro release of bevacizumab, bevacizumab-MSN or MSN-encapsulated bevacizumab nanoparticles (containing 1 mg bevacizumab) were filled into a dialysis bag (6 mm [MD10], molecular weight 300 kD) and 40 μL of PBS was added to disperse the powders to reduce error due to the delay of mass exchange and penetration. The dialysis bag with samples was distributed in 5 mL PBS (containing 0.1% sodium azide) and was placed in an incubator water bath at 37°C with rotational shaking at 45 rpm. At specific time intervals (3 and 9 hours and 1, 2, 4, 7, 14, 21, and 28 days), 0.5 mL of leaching liquor was withdrawn, and then 0.5 mL of fresh PBS was added to the solution to keep the sink condition. The same amount of pure bevacizumab powder (1 mg) was also subjected to the above-mentioned steps. The withdrawn media were stored at −20°C and concentrated by freeze drying and dissolving till analysis of the amount of released bevacizumab. The bevacizumab in the release media was quantified by measuring the absorbance of bevacizumab at 269 nm using a spectrophotometer (NanoDrop 2000 Spectrophotometer; Thermo Fisher Scientific). The drug release curves of bevacizumab, bevacizumab-MSN, and MSN-encapsulated bevacizumab nanoparticles in terms of cumulative amount were then plotted against time.
Alkaline burn-induced corneal neovascularization (CNV) model
Eight-week-old male C57BL/6J mice were anesthetized with ketamine (80 mg/kg) and xylazine (4 mg/kg), and 0.5% proparacaine hydrochloride was used for topical anesthesia. Corneal center was touched with a 3 mm diameter filter paper (soaked with 1 mol/L NaOH) for 30 seconds and irrigated with sterile saline solution. Subconjunctival injection of bevacizumab was performed immediately after alkaline burn using a 33-gauge needle under an operating microscope. The length of CNV was analyzed by ImageJ.
Oxygen-induced retinopathy (OIR) model
The newborn mice at postnatal day 7 (P7) and their nursing mothers were exposed to 75% oxygen in a hyperoxic chamber for 5 days until P12 and then returned to normoxia (21% oxygen) for 5 days. Bevacizumab or MSN-encapsulated bevacizumab nanoparticles were injected into the vitreous at P13. Pups were euthanized at P17 and their retinas were dissected for Isolectin B4 (IB4) staining.
IB4 staining
The eyes of OIR pups were collected and fixed in 4% paraformaldehyde for 30 minutes at room temperature. The retinas were carefully dissected, flat-mounted, and permeabilized with 0.1% Triton X-100 (cat. no A110694; Sangon Biotech). The retinas were blocked with 5% BSA (cat. no B2064; Sigma-Aldrich) for 30 minutes and then incubated with the fluorescence antibody-conjugated IB4 at 4°C overnight. Retinal neovascularization was observed using a fluorescence microscope (Olympus IX-73).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from the ScienCell Research Laboratories (Carlsbad, CA, USA). They were maintained in an incubator at 37°C with 95% humidity and 5% CO2. The culture medium was composed of DMEM/F-12 (1:1) medium, 1% endothelial cell growth supplement, 5% fetal bovine serum (FBS; Thermo Fisher Scientific), and 1% penicillin/streptomycin solution. Passage 4–6 cells were used for cell phenotype evaluation.
MTT assay
HUVECs were cultured in 96-well plates at a density of 1×104 cells per well. After the required treatment, they were washed with PBS for three times and then incubated with MTT (0.5 mg/mL) for 3 hours. Dimethyl sulfoxide solution was used to dissolve formazan crystals. MTT absorbance was detected at 570 nm wavelength by a microplate reader (Molecular Devices).
Cell proliferation assay
Cell proliferation was detected by Ki67 staining. After the required treatment, HUVECs were fixed in 4% formaldehyde for 15 minutes, permeabilized with 0.1% Triton X-100 for 15 minutes, and blocked with 5% BSA for 30 minutes. These cells were incubated with Ki67 antibody (1:200; Abcam) at 4°C overnight and then stained with the fluorescent secondary antibody. Cells were stained with Hoechst 33342 to show nuclei and observed using a fluorescence microscope (Olympus IX-73).
Cell migration assay
Cell migration was detected by Transwell migration assay. Briefly, HUVECs were seeded into 24-well Transwell inserts (8 mm diameter, 0.4 μm pore size polycarbonate filters; Corning Costar Corporation) at a density of 1×105/insert and cultured in the complete media to allow cell migration. The migrated cells on the bottom surface were fixed with 4% formaldehyde and stained with 0.5% crystal violet (Beyotime Biotechnology) for 30 minutes. The non-invaded cells were removed using a cotton swab. The number of migrated cells was counted using a bright-field microscope.
Tube formation assay
A layer of 50 μL growth factor-reduced Matrigel (cat. no 356234; BD Biosciences) was made on the bottom of 96-well plates. After the required treatment, HUVECs were seeded on the top of the Matrigel at a density of 1×104 cells/well. The plates were incubated at 37°C in 5% CO2 and 95% humidity for 6 hours. The tube-like structures were observed using a bright-field microscope.
Statistical analysis
All data are presented as mean ± standard error of the mean. Student’s t-test was used to detect significant difference between two groups. One-way ANOVA with Bonferroni’s comparison test was used for multiple comparisons. P<0.05 was considered statistically significant.
Ethics statement
The animals were housed according to the Guide of the Care and Use of Laboratory Animals (published by the National Institutes of Health). Animal experiments were approved by the Animal Care and Use Committee of Nanjing Medical University and performed according to the guidelines of the Statement for the Use of Animals in Ophthalmic and Vision Research established by the Association for Research in Vision and Ophthalmology.
Results
Preparation and characterization of MSN-encapsulated bevacizumab nanoparticles
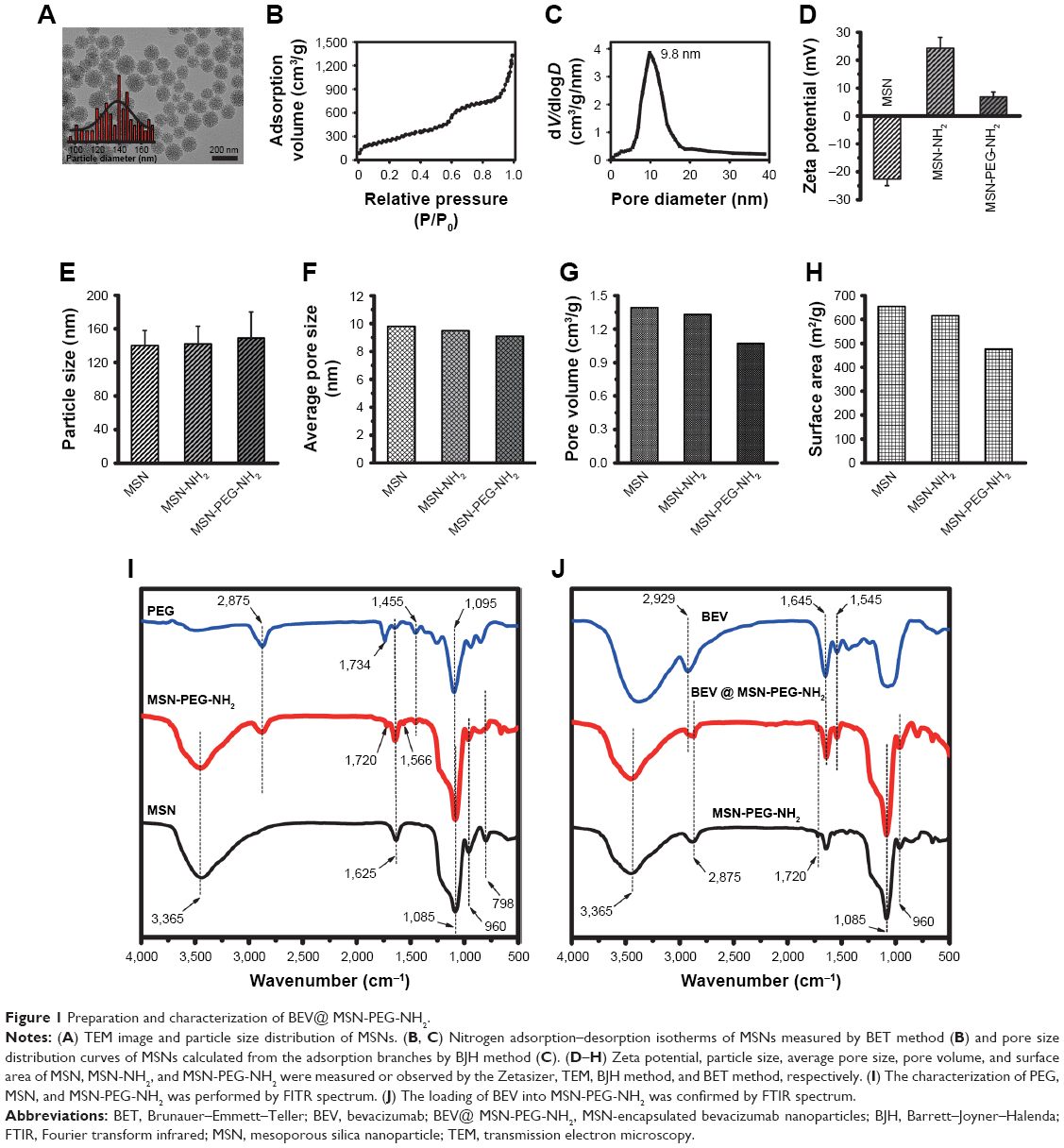
MSNs were prepared by the soft template method. MSNs were discrete spheres and had a narrow particle size distribution with an average diameter of 140±18 nm (Figure 1A). The nanoparticles had plenty of regular and center-radial pore channels. Nitrogen adsorption–desorption plot showed that MSNs were the type IV isotherms characterized by ordered mesoporous materials (Figure 1B). Nitrogen adsorption measurements revealed that the average pore size of MSNs was 9.8 nm (Figure 1C).
 | Figure 1 Preparation and characterization of BEV@ MSN-PEG-NH2. |
MSNs were then functionally modified with −NH2 and PEG groups. The success of surface functionalization was confirmed by zeta potential measurement and infrared spectroscopy. The zeta potential of MSNs was −22.6±2.3 mV. After the functionalization of MSNs with APTES, the surface had a charge reversal from negative to positive (+24.3±3.9 mV) due to the full dominance of protonated −NH3+ group on the surface. PEG modification led to decreased zeta potential to +6.9±1.7 mV (Figure 1D). NH2 and PEG modification led to a slight increase in the particle size of MSNs (Figure 1E), but a slight decrease in average pore size (Figure 1F), pore volume (Figure 1G), and surface area (Figure 1H).
Infrared spectroscopy showed the features of PEG, MSN, and MSN-PEG-NH2 (Figure 1I). The black curve indicated the characteristic peaks of MSNs, including two weak bands at 798 cm−1 (Si−O stretching of Si−O−Si groups) and 960 cm−1 (Si−O stretching of Si−OH groups) and a broad peak in the region of 1,050–1,200 cm−1 (highest point at 1,085 cm−1, Si−O−Si bending). After the modification of MSNs by −NH2 and PEG (red curve), some new peaks appeared, including a peak at 1,720 cm−1 (the bending vibrations of C=O group in −NHCO−) and three characteristic peaks at 1,095 cm−1 (−C−O−C stretching), 1,455 cm−1 (−CH2 bending), and 2,875 cm−1 (−CH2 stretching), which correspond to the PEG chain (blue curve). The peak at 1,566 cm−1 responded to −NH stretching vibration of NH2 in APTES. It should be noted that the peak at 1,734 cm−1 in the PEG spectrum was the fingerprint of N-succinimidyl ester in the mPEG-NHS. The peak at 3,365 cm−1 could be assigned to −NH stretching combined with the peak −OH from water. Comparing with the curves of MSNs and MSN-PEG-NH2, the characteristic peaks at 1,720 cm−1 (−NHCO−), 1,095 cm−1 (C−O−C), and 2,875 cm−1 (−CH2) demonstrated that PEG was chemically grafted to the MSN surface through the reaction of −NHS and NH2.
After surface modification by −NH2 and PEG, bevacizumab was loaded into MSN-PEG-NH2 to obtain MSN-encapsulated bevacizumab nanoparticles. As shown in Figure 1J, the blue curve in FTIR spectrum characterized bevacizumab, which had three characteristic peaks: 2,929 cm−1 (−CH stretch), 1,645 cm−l (Amide I), and 1,545 cm−l (Amide II). After bevacizumab loading, the above three peaks were found in the spectrum of MSN-encapsulated bevacizumab nanoparticles (red curve), which confirm that bevacizumab was uploaded into MSN-PEG-NH2. In addition, a peak at 1,625 cm−1 (bending vibration of H2O) and a broad band at 3,000–4,000 cm−1 (stretching vibration of OH) could be found in all spectra, which show the presence of condensed atmospheric moisture in all samples.
We also calculated the Encapsulation efficiency (%) (EE%) and Loading efficiency (%) (LE%) of bevacizumab-MSN and MSN-encapsulated bevacizumab nanoparticles. According to the above Equations 1 and 2, EE% for bevacizumab-MSN and MSN-encapsulated bevacizumab nanoparticles was 70.4% and 79.2%, respectively. LE% for bevacizumab-MSN and MSN-encapsulated bevacizumab nanoparticles was 72.1% and 85.3%, respectively.
In vitro and in vivo release of bevacizumab from MSN-encapsulated bevacizumab nanoparticles
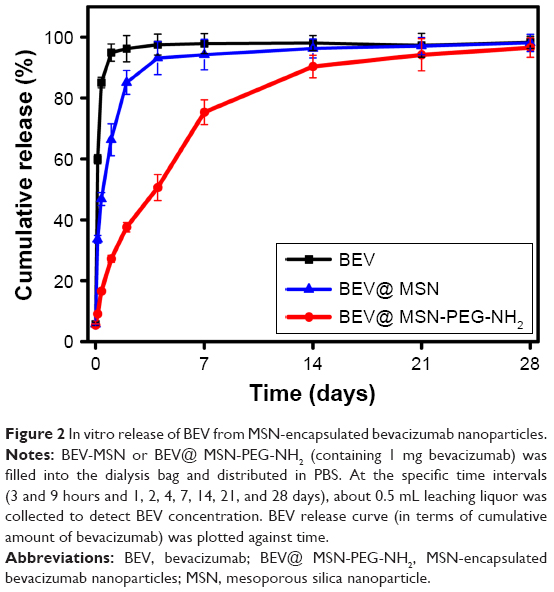
We investigated the release profile of bevacizumab from bevacizumab-MSN and MSN-encapsulated bevacizumab nanoparticles in vitro. Bevacizumab experienced an initial burst release, sustained release, and a slow release from bevacizumab-MSN and MSN-encapsulated bevacizumab nanoparticles. More than 35% of bevacizumab was released from bevacizumab-MSN in the first 3 hours, and about 35% of bevacizumab was released in the next 24 hours, followed by a slow release up to 7 days. By contrast, the release of bevacizumab from MSN-encapsulated bevacizumab nanoparticles was much slower. More than 35% of bevacizumab was released in the first 2 days, but with a burst release in the first 1–10 hours. About 35% of bevacizumab was released in the next 5 days (sustained release) with a subsequent slow release up to 28 days (Figure 2).
 | Figure 2 In vitro release of BEV from MSN-encapsulated bevacizumab nanoparticles. |
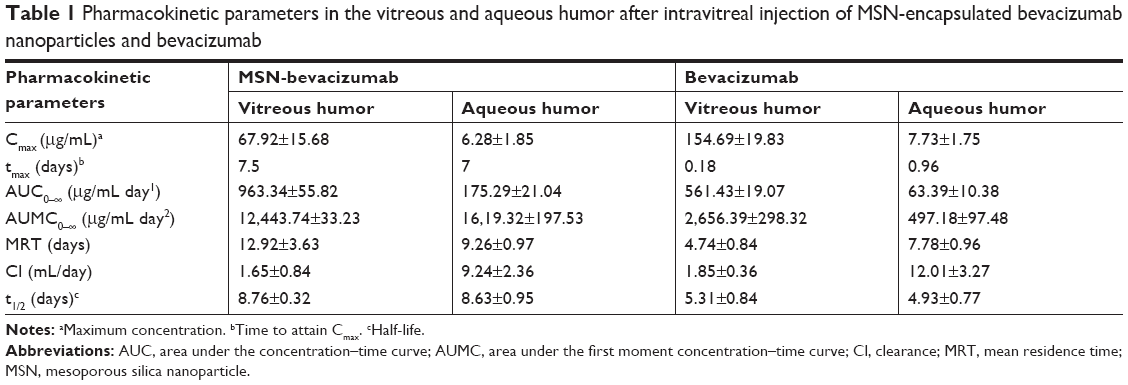
We investigated the in vivo release of bevacizumab. The amount of free bevacizumab in the vitreous and aqueous humor were plotted against the time after intravitreal injection of MSN-encapsulated bevacizumab nanoparticles. We calculated the pharmacokinetic parameters of bevacizumab in the vitreous and aqueous humor by the non-compartmental method (Table 1). Maximum bevacizumab concentration (Cmax) was observed about 7 days after injection of MSN-encapsulated bevacizumab nanoparticles in the vitreous and aqueous humor. By contrast, Cmax was detected immediately after bevacizumab injection. The MRT in the injected group of MSN-encapsulated bevacizumab nanoparticles was significantly greater than that in bevacizumab-injected group both in vitreous and aqueous humor. Accordingly, the half-life of MSN-encapsulated bevacizumab nanoparticles was significantly greater than that of bevacizumab.
 | Table 1 Pharmacokinetic parameters in the vitreous and aqueous humor after intravitreal injection of MSN-encapsulated bevacizumab nanoparticles and bevacizumab |
In vivo and in vitro toxicity assessment of MSN-encapsulated bevacizumab nanoparticles
The mouse retinas received an intravitreal injection of bevacizumab, MSN, MSN-encapsulated bevacizumab nanoparticles, or PBS (Ctrl) for 30 days. H&E staining revealed that there was no significant change in the histological structure of retinas or choroids among different groups. No necrosis, hemorrhage, leukocytic infiltration, or granulation tissue formation was observed in different groups (Figure 3A). Dark-adapted electroretinography (ERG) results revealed that the amplitudes of a-wave and b-wave increased as the light intensity increased. However, we did not detect significant difference in the amplitude of a-wave or b-wave among different groups at a certain light intensity (Figure 3B and C). Collectively, both histologic and ERG experiments suggest that MSN-encapsulated bevacizumab nanoparticles have no tissue toxicity in vivo.
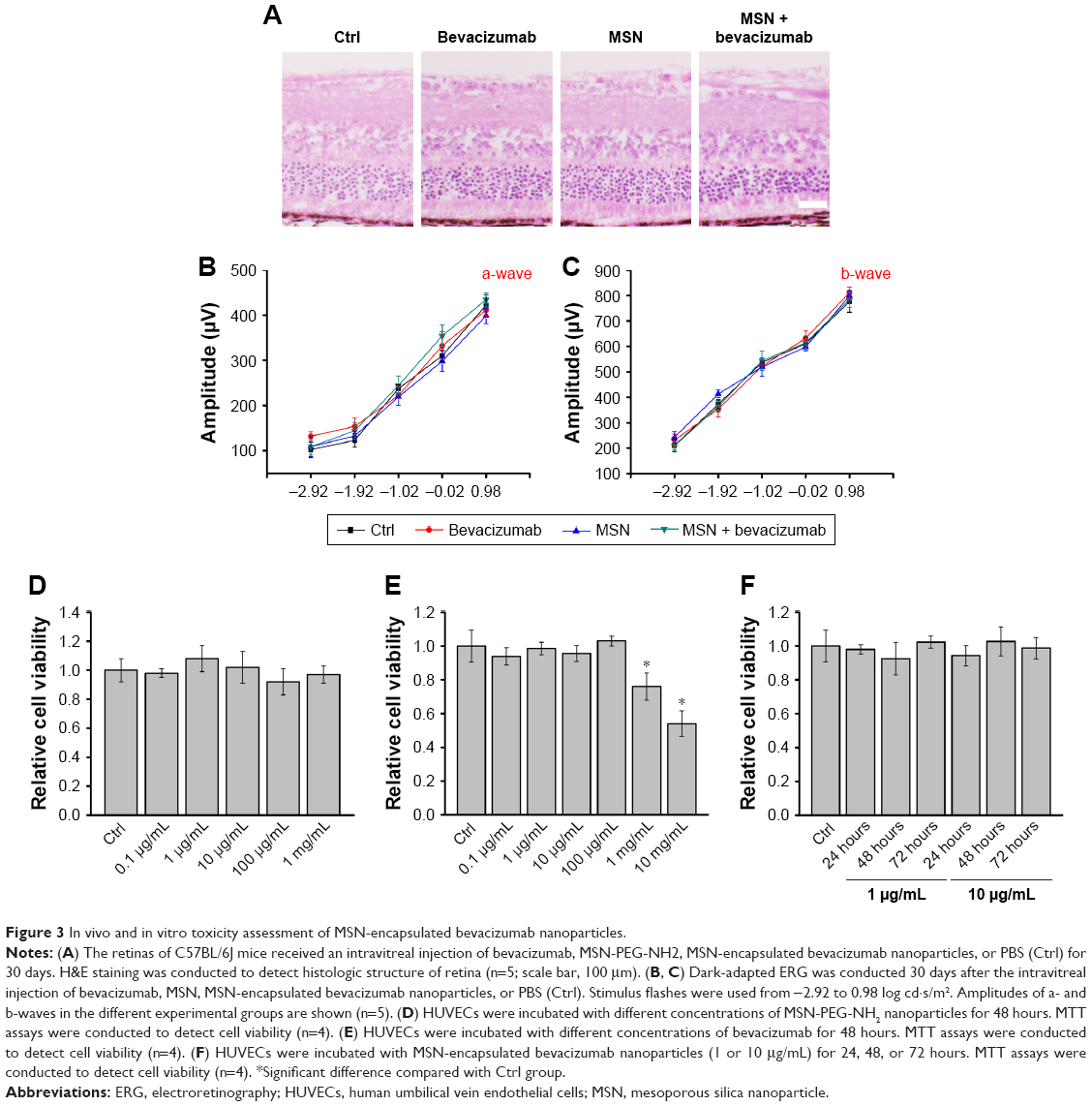 | Figure 3 In vivo and in vitro toxicity assessment of MSN-encapsulated bevacizumab nanoparticles. |
We next investigated whether MSN-encapsulated bevacizumab nanoparticles had endothelial cell toxicity. MTT assays showed that MSNs did not show any cytotoxic effect on endothelial cell viability at the tested concentrations (Figure 3D). Bevacizumab did not show any cytotoxic effects on endothelial cell viability, except in extremely high concentrations (1 and 10 mg/mL; Figure 3E). HUVECs were then incubated with or without MSN-encapsulated bevacizumab nanoparticles for 24, 48, and 72 hours. We also did not detect any cytotoxic effect at the tested time points (Figure 3F). Taken together, the above-mentioned evidence suggests that MSN, bevacizumab, or MSN-encapsulated bevacizumab nanoparticles have no cytotoxicity on endothelial cells.
MSN-encapsulated bevacizumab nanoparticles inhibit VEGF-mediated endothelial angiogenic function in vitro
VEGF controls endothelial angiogenic function during vascular growth. It is a key regulator of endothelial cell functions, such as proliferation, migration, and tube formation.21 We thus determined whether MSN-encapsulated bevacizumab nanoparticles affected VEGF-mediated endothelial angiogenic functions in vitro. MTT assays showed that MSN-encapsulated bevacizumab nanoparticles or bevacizumab administration significantly inhibited VEGF-mediated increase in endothelial cell viability. MSN-encapsulated bevacizumab nanoparticles were more effective than bevacizumab in inhibiting cell viability (Figure 4A). Ki67 staining showed that MSN-encapsulated bevacizumab nanoparticles had greater efficiency than bevacizumab in inhibiting endothelial cell proliferation, especially after 48 hours of culture (Figure 4B). Matrigel tube formation and Transwell migration assays showed that MSN-encapsulated bevacizumab nanoparticles were more effective than bevacizumab in inhibiting VEGF-mediated tube formation and migration ability of endothelial cells, especially after 48 or 72 hours of culture (Figure 4C and D).
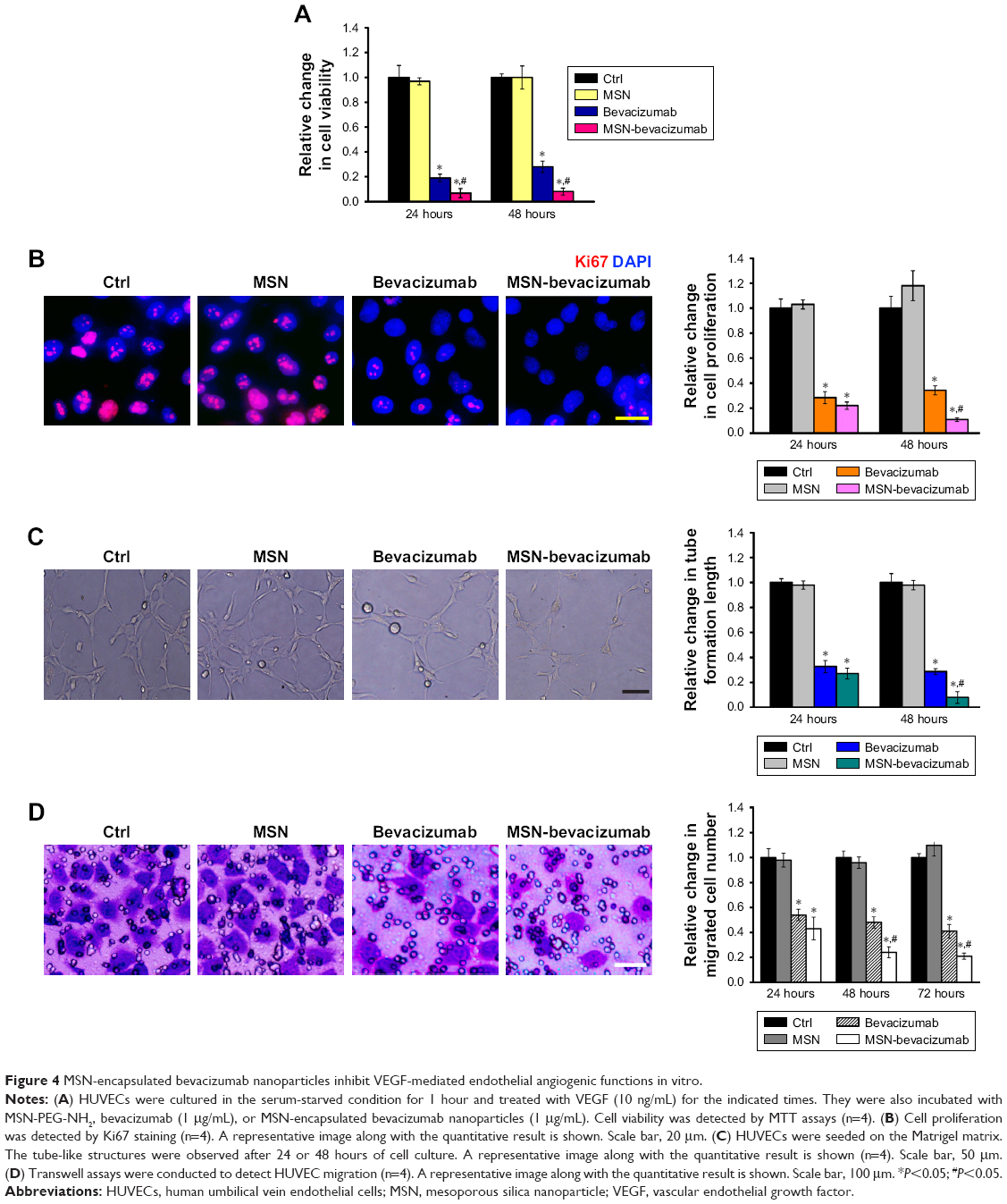 | Figure 4 MSN-encapsulated bevacizumab nanoparticles inhibit VEGF-mediated endothelial angiogenic functions in vitro. |
Subconjunctival injection of MSN-encapsulated bevacizumab nanoparticles inhibits CNV in vivo
Both VEGF and the VEGF receptors are present in higher concentrations than in normal corneas with CNV.22 Subconjunctival injection of bevacizumab significantly inhibited CNV in vivo. We then determined the effects of subconjunctival injection of MSN-encapsulated bevacizumab on CNV. We did not observe obvious corneal epithelial side effects, such as corneal epithelial defect, corneal ulcer, and conjunctival necrosis, in alkali-injured mouse corneas. The area of neovascularization decreased 2, 10, or 14 days after treatment with MSN-encapsulated bevacizumab nanoparticles or bevacizumab. CNV was almost completely absent in MSN-encapsulated bevacizumab group after 14 days of treatment. MSN-encapsulated bevacizumab nanoparticles could enhance the inhibitory effects of bevacizumab on CNV, especially at 14 days after alkali injury, as shown by decreased vascular length (Figure 5A). The representative CNV images are shown at 14 days after alkali injury (Figure 5B).
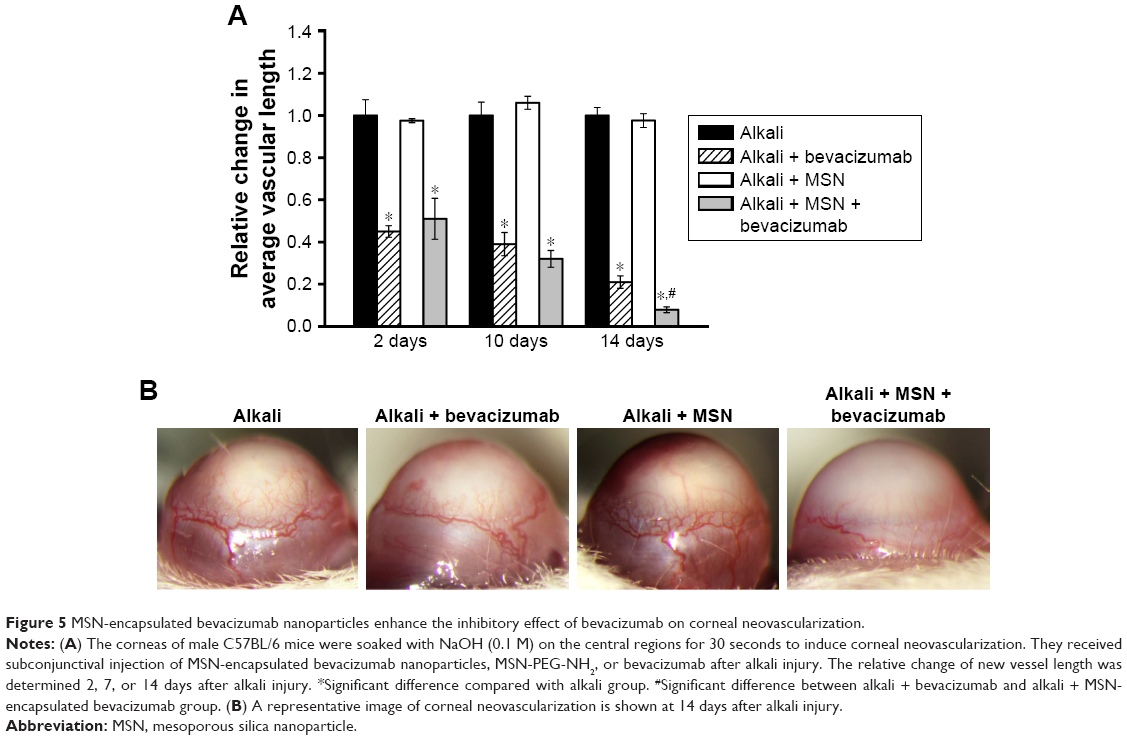 | Figure 5 MSN-encapsulated bevacizumab nanoparticles enhance the inhibitory effect of bevacizumab on corneal neovascularization. |
Intravitreal injection of MSN-encapsulated bevacizumab nanoparticles inhibits retinal neovascularization in vivo
Ischemia-induced proliferative retinal neovascularization is usually associated with increased VEGF expression.23 We determined whether MSN-encapsulated bevacizumab could enhance the anti-VEGF effects of bevacizumab on retinal neovascularization in vivo. Retinal neovascularization was induced in C57BL/6 mice by exposing 7-day-old mice to 75% oxygen for 5 days, followed by exposure for 5 days to room air. Compared with PBS-injected group or MSN nanoparticle-injected group, MSN-encapsulated bevacizumab and bevacizumab injection significantly attenuated the area of retinal neovascularization (Figure 6A). Moreover, the area of retinal neovascularization in the group injected with MSN-encapsulated bevacizumab nanoparticles was significantly smaller than that in bevacizumab-injected group (Figure 6B).
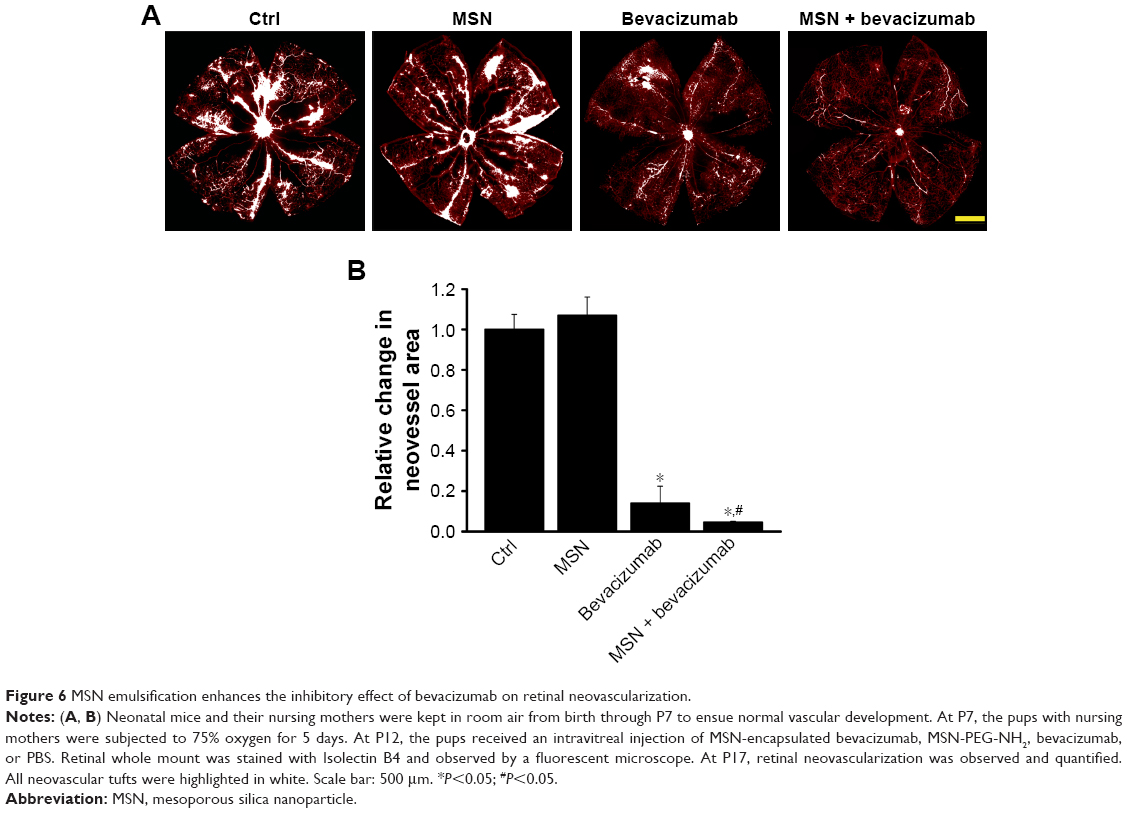 | Figure 6 MSN emulsification enhances the inhibitory effect of bevacizumab on retinal neovascularization. |
Discussion
Bevacizumab is a humanized recombinant antibody against VEGF. It can block VEGF and its receptors, thus inhibiting the formation of new blood vessels and reducing vascular permeability. Several laboratory and clinical studies have supported the safety and efficacy of intravitreal bevacizumab.6,7 However, anti-VEGF strategy for neovascular diseases is usually limited by the short half-life of bevacizumab. In this study, we provide a novel drug delivery system in which bevacizumab was encapsulated within MSN nanoparticles. MSN-encapsulated bevacizumab nanoparticles could increase the time between administration and formulation shelf-life and showed sustained inhibitory effects on corneal and retinal neovascularization.
MSNs are composed of a honeycomb-like porous structure that can encapsulate a great number of bioactive molecules.24 Previous studies have revealed that MSNs are capable of carrying imaging agents such as fluorescein, Texas Red, and rhodamine B. MSN carrier system has been used for controlled release of drugs such as vancomycin, ibuprofen, erythromycin, and alendronate.25,26 In this study, we showed that MSN encapsulation could prolong the residency of bevacizumab in vitreous and aqueous humor and maintain long-lasting drug concentration. We speculate that the unique properties such as surface area, large pore volume, and good chemical and thermal stability make MSNs suitable for drug delivery and release.27 Moreover, MSNs or MSN-encapsulated bevacizumab nanoparticles have no toxicity effects on endothelial cell function in vitro and mouse retina function in vivo. MSNs could also be efficiently internalized by animal cells.28 We envision that MSNs could serve as a drug carrier for controlled release of bevacizumab in the endothelial cells, which would in turn improve the antiangiogenic effects of bevacizumab. This study also expands the scope of MSNs for biotechnological and biomedical applications.
After bevacizumab is encapsulated into MSN nanoparticles, the primary concern is whether MSN-encapsulated bevacizumab nanoparticles still have the bioactivity. Abnormal endothelial cell proliferation, migration, and tube formation usually occurs in angiogenic cascade.29 MSN-encapsulated bevacizumab nanoparticles are more effective than bevacizumab in suppressing HUVEC proliferation, migration, and tube formation, providing the evidence that the bioactivity of bevacizumab is well kept after MSN encapsulation. Moreover, MSN-encapsulated bevacizumab nanoparticles show long-acting antiangiogenic effects on endothelial cells due to the sustained release of bevacizumab. We thus conclude that the bioactivity of MSN-encapsulated bevacizumab nanoparticles on antiangiogenic effects is perfectly preserved and greatly improved.
Regardless of the source, CNV is a major complication in transplant rejection, infection, and injury. The treatments for CNV mainly include photodynamic therapy, photocoagulation, corticosteroids, and nonsteroidal anti-inflammatory eye drops.30,31 Subconjunctival bevacizumab was found to be a promising method for suppressing CNV. However, CNV is far from complete due to the reason that the dosage and duration of bevacizumab treatment are insufficient to antagonize VEGF activity.32 In previous studies, nanomedicine had been used for treating ocular surface diseases by providing much safer, less-invasive, and cheaper treatment options.33 Natamycin-encapsulated licithan/chitosan nanoparticles prolong ocular exposure to the drug and decrease the clearance, compared to commercially available suspensions.34 Silica nanoparticles are shown as a novel antiangiogenic agent for inhibiting CNV following chemical burn.35 We found that subconjunctival injection of MSN-encapsulated bevacizumab nanoparticles almost completely inhibited CNV. Bevacizumab is continuously released from MSN-encapsulated bevacizumab nanoparticles in a controlled manner, which might continuously antagonize VEGF activity. MSN nanocarriers can also interact with the unique chemical composition of the cornea, allowing for longer residence and greater resistance to ocular clearance.
Ischemia-induced proliferative angiogenesis is a common pathological process in several retinal diseases such as retinopathy of prematurity and proliferative diabetic retinopathy.36 We found that MSN-encapsulated bevacizumab nanoparticles could produce long-lasting drug concentrations in the vitreous and aqueous humor. MSN-bevacizumab obviously inhibits oxygen-induced retinal angiogenesis. The area of neovessel formation in MSN-encapsulated bevacizumab nanoparticle-injected group was obviously smaller than that in bevacizumab-injected group. Thus, MSN encapsulation is also a promising drug delivery system for intraocular neovascular diseases.
Conclusion
This study provides a novel strategy to obtain MSN-encapsulated bevacizumab nanoparticles to protect and deliver bevacizumab. MSN encapsulation could prolong the residency of bevacizumab in vitreous/aqueous humor and maintain the long-lasting drug concentration. MSN-encapsulated bevacizumab nanoparticles have no obvious cytotoxicity in vitro and tissue toxicity in vivo. MSN-encapsulated bevacizumab nanoparticles are more effective than bevacizumab in suppressing VEGF-induced endothelial cell proliferation, migration, and tube formation in vitro. MSN-encapsulated bevacizumab nanoparticles show sustained inhibitory effects on CNV and retinal neovascularization in vivo. MSN-encapsulated bevacizumab nanoparticles enhance the efficiency of bevacizumab for treating neovascular diseases. MSNs would become a promising drug delivery system for improving antiangiogenic therapy.
Acknowledgment
This work was generously supported by the grants from the National Natural Science Foundation of China (Grant No.81770945 to B.Y., and Grant No. 81570859 to Q.J.), and the grant from the Shanghai Youth Talent Support Program (to B.Y.).
Disclosure
The authors report no conflicts of interest in this work.
References
Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–660. | ||
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–974. | ||
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. | ||
Ferrara N, Gerber H-P, Lecouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–676. | ||
Klettner A, Roider J. Comparison of bevacizumab, ranibizumab, and pegaptanib in vitro: efficiency and possible additional pathways. Invest Ophthalmol Vis Sci. 2008;49(10):4523–4527. | ||
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. | ||
Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7(4):335–345. | ||
Mailliez A, Baldini C, Van JT, Servent V, Mallet Y, Bonneterre J. Nasal septum perforation: a side effect of bevacizumab chemotherapy in breast cancer patients. Br J Cancer. 2010;103(6):772–775. | ||
Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70(10):779–787. | ||
Rodrigues EB, Farah ME, Maia M, et al. Therapeutic monoclonal antibodies in ophthalmology. Prog Retin Eye Res. 2009;28(2):117–144. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Slowing II, Trewyn BG, Giri S, Lin VSY. Mesoporous silica nanoparticles for drug delivery and biosensing applications. Adv Funct Mater. 2007;17(8):1225–1236. | ||
Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24(12):1504–1534. | ||
Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438(7070):960–966. | ||
Pieramici DJ, Rabena MD. Anti-VEGF therapy: comparison of current and future agents. Eye. 2008;22(10):1330–1336. | ||
Lynch SS, Cheng CM. Bevacizumab for neovascular ocular diseases. Ann Pharmacother. 2007;41(4):614–625. | ||
Andreoli CM, Miller JW. Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Curr Opin Ophthalmol. 2007;18(6):502–508. | ||
Shen D, Yang J, Li X, et al. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 2014;14(2):923–932. | ||
Dai Z, Yu X, Hong J, Liu X, Sun J, Sun X. Development of a novel CsA-PLGA drug delivery system based on a glaucoma drainage device for the prevention of postoperative fibrosis. Mater Sci Eng C Mater Biol Appl. 2016;66:206–214. | ||
Cruz LJ, Tacken PJ, Fokkink R, Figdor CG. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials. 2011;32(28):6791–6803. | ||
Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Bio. 2016;17(10):611–625. | ||
Roshandel D, Eslani M, Baradaran-Rafii A, et al. Current and upcoming therapies for corneal neovascularization. Ocul Surf. 2018;22(6):761–767. | ||
Chen J, Smith LEH. Retinopathy of prematurity. Angiogenesis. 2007;10(2):133–140. | ||
Vallet-Regí M. Ordered mesoporous materials in the context of drug delivery systems and bone tissue engineering. Chemistry. 2006;12(23):5934–5943. | ||
Doadrio JC, Sousa EMB, Izquierdo-Barba I, Doadrio AL, Perez-Pariente J, Vallet-Regí M. Functionalization of mesoporous materials with long alkyl chains as a strategy for controlling drug delivery pattern. J Mater Chem. 2006;16(5):462–466. | ||
Balas F, Manzano M, Horcajada P, Vallet-Regí M. Confinement and controlled release of bisphosphonates on ordered mesoporous silica-based materials. J Am Chem Soc. 2006;128(25):8116–8117. | ||
Takahashi H, Li B, Sasaki T, Miyazaki C, Kajino T, Inagaki S. Catalytic activity in organic solvents and stability of immobilized enzymes depend on the pore size and surface characteristics of mesoporous silica. Chem Mater. 2000;12(11):3301–3305. | ||
Weng Y, Liu J, Jin S, Guo W, Liang X, Hu Z. Nanotechnology-based strategies for treatment of ocular disease. Acta Pharm Sin B. 2017;7(3):281–291. | ||
Liu C, Yao MD, Li CP, et al. Silencing of circular RNA-ZNF609 ameliorates vascular endothelial dysfunction. Theranostics. 2017;7(11):2863–2877. | ||
Chang JH, Gabison EE, Kato T, Azar DT. Corneal neovascularization. Curr Opin Ophthalmol. 2001;12(4):242–249. | ||
Gupta D, Illingworth C. Treatments for corneal neovascularization: a review. Cornea. 2011;30(8):927–938. | ||
Kim SW, Ha BJ, Kim EK, Tchah H, Kim TI. The effect of topical bevacizumab on corneal neovascularization. Ophthalmology. 2008;115(6):e33–e38. | ||
Park K. Nanotechnology: what it can do for drug delivery. J Control Release. 2007;120(1–2):1–3. | ||
Bhatta RS, Chandasana H, Chhonker YS, et al. Mucoadhesive nanoparticles for prolonged ocular delivery of natamycin: in vitro and pharmacokinetics studies. Int J Pharm. 2012;432(1–2):105–112. | ||
Mohammadpour M, Jabbarvand M, Hashemi H, Delrish E. Prophylactic effect of topical silica nanoparticles as a novel antineovascularization agent for inhibiting corneal neovascularization following chemical burn. Adv Biomed Res. 2015;4:124. | ||
Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009;4(11):1565–1573. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.