Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Mesoporous magnesium silicate-incorporated poly(ε-caprolactone)-poly(ethylene glycol)- poly(ε-caprolactone) bioactive composite beneficial to osteoblast behaviors
Authors Niu Y, Dong W, Guo H, Deng Y, Guo L, An X, He D, Wei J, Li M, Wang H, Chen J, Shen H, Boccaccini B, Yao X
Received 11 December 2013
Accepted for publication 7 February 2014
Published 27 May 2014 Volume 2014:9(1) Pages 2665—2675
DOI https://doi.org/10.2147/IJN.S59040
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Yunfei Niu,1,* Wei Dong,1,* Han Guo,2 Yuhu Deng,3 Lieping Guo,1 Xiaofei An,1 Dawei He,1 Jie Wei,3 Ming Li1
1Department of Orthopedic Surgery, Changhai Hospital, Second Military Medical University, Shanghai, People's Republic of China; 2Shanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, People's Republic of China; 3Key Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai, People's Republic of China
*These authors contributed equally to this work
Abstract: Mesoporous magnesium silicate (m-MS) and poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) (PCL-PEG-PCL) composite (m-MPC) was synthesized by solvent casting method. The results suggest that the mechanical properties of compressive strength and elastic modulus, as well as hydrophilicity, of the m-MPC increased with increase of m-MS content in the composites. In addition, the weight loss of the m-MPC improved significantly with the increase of m-MS content during composite soaking in phosphate-buffered saline for 10 weeks, indicating that incorporation of m-MS into PCL-PEG-PCL could enhance the degradability of the m-MPC. Moreover, the m-MPC with 40 w% m-MS could induce a dense and continuous apatite layer on its surface after soaking in simulated body fluid for 5 days, which was better than m-MPC 20 w% m-MS, exhibiting excellent in vitro bioactivity. In cell cultural experiments, the results showed that the attachment and viability ratio of MG63 cells on m-MPC increased significantly with the increase of m-MS content, showing that the addition of m-MS into PCL-PEG-PCL could promote cell attachment and proliferation. The results suggest that the incorporation of m-MS into PCL-PEG-PCL could produce bioactive composites with improved hydrophilicity, degradability, bioactivity, and cytocompatibility.
Keywords: PCL-PEG-PCL, degradation, cytocompatibility, cell attachment, cell proliferation
Introduction
Mesoporous material with pore size ranging from 2–50 nm has tunable pore size, which, together with its large specific surface area and pore volume, should enable it to be used in a variety of biomedical and biotechnological applications, such as drug delivery and enzyme immobilization.1,2 As the mesoporous material has unique structural characteristics, it is reasonable to assume that mesoporous material may possess special physico-chemical and biological performances. The use of mesoporous material in bone tissue regeneration has been proposed because of its large specific surface area and pore volume, which may improve its bioactivity and allow it to be loaded with osteogenic agents and promote new bone formation.3–5
The biocompatible and bioactive mesoporous material will favor cellular growth and bone regeneration, which is very useful for building macroporous devices to be applied in bone tissue engineering.6,7 Poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) (PCL-PEG-PCL), a linear polyester copolymer composed of a hydrophobic PCL block and a hydrophilic PEG block, possesses good biocompatibility and suitable degradability.8 PCL and PEG are both US Food and Drug Administration-approved biomaterials and they have been widely used in the biomedical field. PCL-PEG-PCL copolymer was used as drug-loaded nanoparticles or thermosensitive hydrogel, and their drug delivery behaviors were extensively investigated.9,10
Studies have focused on the bioactive composites created by combining biodegradable polymers and bioactive inorganic materials such as bioglass, calcium phosphate, and silicate.11,12 However, to our knowledge, there has been no report about the preparation of mesoporous magnesium silicate (m-MS) and PCL-PEG-PCL bioactive composite for use as bone repair material. It is expected that the biological performance of polymer-based bioactive composite should be improved if m-MS were incorporated into PCL-PEG-PCL. Therefore, in this study, m-MS/PCL-PEG-PCL bioactive composite (m-MPC) was prepared, and the effects of m-MS content on hydrophilicity, degradability, and apatite formation in vitro and cell attachment and proliferation on the bioactive composite were investigated.
Materials and methods
Preparation of m-MS and PCL-PEG-PCL
Six grams of EO20PO70EO20 (polyethylene oxide)20(polypropylene oxide)70(polyethylene oxide)20 (EO20PO70EO20 [P123]) was dissolved in 240 mL of 2 M hydrochloric acid and 60 mL of water while stirring at 37°C in water bath until the solution became clear. Seventeen grams of tetraethyl orthosilicate and 19.28 g of Mg(NO3)2 · 4H2O was then added into the solution. The mixture was stirred at room temperature for 24 hours, and the resulting product was dried at 50°C for 24 hours. After that, the product was calcined at 600°C for 3 hours and the final product of m-MS powder was obtained. Nitrogen adsorption–desorption isotherms of m-MS were obtained on a TriStar 3000 porosimeter (Micromeritics, Norcross, GA USA) at 77 K under a continuous adsorption condition. Specific surface area and pore size were calculated by Brunauer–Emmett–Teller and Barrett–Joyner–Halenda methods, respectively. The m-MS was characterized by using scanning electron microscopy ([SEM] S-3400N; Hitachi, Tokyo, Japan); transmission electron microscopy ([TEM] JEM2010; JEOL, Tokyo, Japan); wide angle X-ray diffraction ([XRD] D/max 2550 V; Rigaku, Tokyo, Japan); and Fourier transform infrared spectroscopy ([FTIR] 6700; Thermo Scientific, Waltham, MA, USA).
PCL-PEG-PCL with both hydrophobic PCL and hydrophilic PEG was synthesized as the raw material for fabricating composite. A three-necked flask was purged by nitrogen and heated to 90°C. Then, PEG methyl ether and a catalyst, stannous 2-ethylhexanoate, were introduced to the flask for 30 minutes. Then, CL (caprolactone) was added to the flask to start the ring-opening polymerization at 120°C for 24 hours. In the purification process, the product was first dissolved in dichloromethane and then precipitated in a mixture of n-hexane and ethyl ether at a volumetric ratio of 3:7. This step was done several times to purify the product (PCL-PEG-PCL). The molecular weight (Mw) of the block copolymer (PCL-PEG-PCL) was 66,000 g/mol, which was determined by a gel permeation chromatography test.13
Preparation of m-MS/ PCL-PEG-PCL composite
PCL-PEG-PCL was dissolved into chloroform at a concentration of 10% (w/v) and m-MS powder was added to produce composites with m-MS content of 20 w%, 40 w%, and 60 w%. The mixture was stirred continuously for 2 hours, and the mixture was cast into 12×12×2 mm PTEF (polytetrafluoroethylene) molds. The obtained m-MPC was air-dried in a fume hood for 24 hours and subsequently vacuum-dried at 50°C for 24 hours to remove any remaining solvent. The 12×12×2 mm PCL-PEG-PCL samples were synthesized as a control in the same way. The surface morphology of the composite was examined by SEM. The mechanical properties of compressive strength and elastic modulus of the composite samples (Φ6×12 mm) were measured at room temperature using a compression test at a constant displacement rate of 2 mm/minute, and a Bionix 858.20 servohydraulic testing system (MTS Systems Corp, Eden Prairie, MN, USA) was used to conduct tests.
Hydrophilicity and degradability
The water contact angle of the m-MPC samples (size 12×12×2 mm) with 0 w%, 20 w%, and 40 w% m-MS content was determined by using a DropMeter A-200 contact angle system (MAIST Vision Inspection and Measurement Ltd, Co, Ningbo City, People’s Republic of China). The in vitro degradation behavior of m-MPC was determined by testing the weight loss ratio of the samples with 0 w%, 20 w%, and 40 w% m-MS (size 12×12×2 mm) after soaking in phosphate-buffered saline (PBS) separately, and the buffer was refreshed every 3 days. After soaking, the specimens were removed from the liquid, rinsed with distilled water, and dried in a vacuum oven for 12 hours. Weight loss was computed according to the following equation:
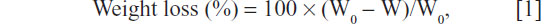
where W0 is the starting dry weight and W is the dry weight at time t.
In vitro bioactivity
The in vitro bioactivity of m-MPC with 0 w%, 20 w%, and 40 w% m-MS was evaluated by examining the apatite formation on their surfaces into simulated body fluid (SBF). SBF was prepared by dissolving reagent-grade NaCl, NaHCO3, KCl, K2HPO4 · 3H2O, MgCl2 · 6H2O, CaCl2, and Na2SO4 in deionized water; the ion concentrations were similar to those in human blood plasma.14 The solution was buffered at pH 7.4 with tris(hydroxymethyl) aminomethane ([CH2OH]3CNH2) and 1 M hydrochloric acid at 37°C.
The 12×12×2 mm m-MPC samples were then soaked in 40 mL SBF at 37°C for 1, 2, 3, 4, and 5 days without shaking, with a change of solution during soaking for each sample. After soaking, specimens were removed from the SBF solution, gently rinsed with deionized water, and dried at room temperature. Field-emission SEM (FE-SEM S-4300SE) and energy-dispersive X-ray spectrometry (EDS) (Hitachi) were used to monitor the surface morphology and composition of these samples. At each time point (6, 12, 24, 48, and 96 hours), the samples were taken out and the ion concentrations of calcium, phosphorus, magnesium, and silicon in the SBF solution were measured using inductively coupled plasma atomic emission spectroscopy (ICP-AES; IRIS 1000, Thermo Scientific).
Cell attachment
The m-MPC with 0 w%, 20 w%, and 40 w% m-MS samples (size 12×12×2 mm) were used to examine the cell adhesion on the samples (tissue culture plate [TCP] used as a control). MG63 cells with a density of 2×104 were seeded on the samples located on 24-well tissue culture plates. MG63 osteoblast-like cells (Cambrex Corporation, East Rutherford, NJ, USA) were cultured in α-Modified Eagle’s Medium (Life Technologies, Carlsbad, CA, USA) containing 10% fetal calf serum (Life Technologies), 100 μg/mL streptomycin (AMRESCO, Solon, OH, USA) and 100 μg/mL penicillin (Amresco) at 37°C in a humidified atmosphere of 5% (v/v) CO2. The cell-seed samples were cultured for 4 hours, and then the culture medium was removed. Thereafter, the residual cultured medium and unattached cells were removed by washing with PBS three times. After the attached cells on the samples were digested by trypsin, the adherent cells were counted with a hemocytometer (Shanghai All-Time Commercial Co., Ltd, Shanghai, People’s Republic of China), and the cell attachment efficiency was determined by counting the number of cells remaining in the wells.
Cell proliferation
The m-MPC with 0 w%, 20 w%, and 40 w% m-MS samples (size 12×12×2 mm) were sonicated in ethanol and sterilized using ultraviolet light. Cell proliferation was evaluated after seeding MG63 cells with a density of 2.5×104 on the samples (TCP used as a control), followed by incubation for 1, 4, and 7 days, with the medium replaced every day. Cell proliferation on the substrate was assessed quantitatively using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, sample-cell constructs were placed in culture medium containing MTT and incubated in a humidified atmosphere at 37°C for 4 hours. After the supernatants were removed, dimethyl sulfoxide (Sinopharm Foreign Trade Co., Ltd, Shanghai, People’s Republic of China) was added to each well to completely dissolve the MTT reagent. The optical density (OD) of each well was measured at 590 nm using a microplate reader (ELx800™; BioTek, Winooski, VT, USA) at a reference wavelength of 620 nm, and the results are reported as OD units.
Cell morphology
Approximately 50 mL of culture medium containing 5×104 MG63 cells was seeded on the top of the m-MPC that had previously been placed in 24-well culture plates. Cells were allowed to attach to substrates for 4 hours, and then 1 mL of fresh culture medium was added to each well. Cells were incubated for different lengths of time in a humidified atmosphere at 37°C and 5% CO2. After the various culture times, the samples were washed twice with PBS solution and fixed with 4% formalin in PBS (pH 7.4) for 20 minutes. The samples were subsequently washed twice with PBS solution and dehydrated in a series of graded ethanol (50%, 60%, 70%, 80%, 90%, and 100% v/v) for 3 minutes each time. Finally, samples were air-dried in a desiccator overnight. Cell morphology on the samples was observed by SEM.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance with post hoc tests. All results are expressed as the mean ± standard deviation of six samples. Difference was considered statistically significant at P<0.05.
Results
Microstructure and morphology of m-MS and m-MPC
The N2 adsorption–desorption isotherms and pore size distribution of m-MS are shown in Figure 1A and B, respectively. The results indicated that the m-MS displayed a very narrow pore size distribution with a pore size of approximately 3 nm, a relatively high specific surface area of 428.0 m2/g, and a single-point adsorption total pore volume of 0.40 cm3/g.
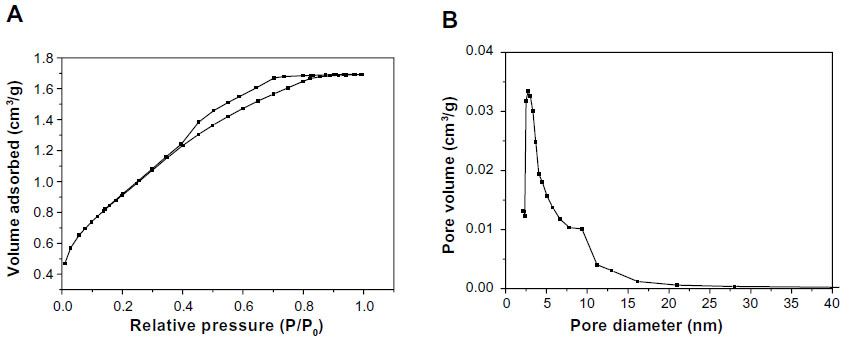 | Figure 1 N2 adsorption–desorption isotherms (A) and pore size distribution (B) of mesoporous magnesium silicate. |
Figure 2 presents the SEM and TEM images of the morphology and microstructure of the m-MS powder samples. It can be seen that the m-MS particles gathered into irregular ball-like particles (Figure 2A). The TEM image shows that the sphere-like m-MS particles had a uniform mesoporous structure with a pore size of approximately 3 nm (mesoporous diameter) (Figure 2B).
 | Figure 2 Scanning electron microscope (A) and transmission electron microscope (B) images of mesoporous magnesium silicate. |
Figure 3A shows that the fabricated m-MS could be well identified as pure magnesium silicate phase according to the Joint Committee on Powder Diffraction Standards, and no other phases were observed. FTIR was applied to further evaluate the surface properties of materials. Figure 3B presents the FTIR spectra of the m-MS. The peak of 461 cm−1 was assigned to the Mg-O vibrations, the intense peaks between 963 and 1,078 cm−1 were assigned to the absorption peaks for silicate, and the peaks at 803, 1,641, 3,448 cm−1 were assigned to the silicate bending mode.
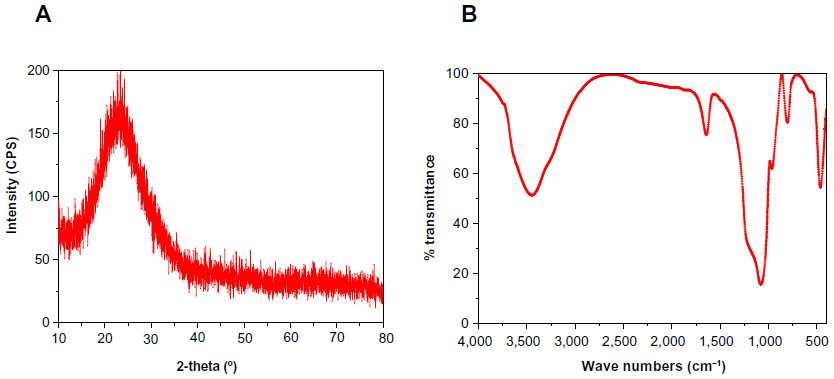 | Figure 3 X-ray diffraction (A) and Fourier transform infrared spectroscopy (B) of mesoporous magnesium silicate. |
Figure 4 shows the surface morphology of SEM images of PCL-PEG-PCL and m-MPC with 20 w% and 40 w% m-MS. It was found that the m-MS was entrapped by the PCL-PEG-PCL polymer matrix for the m-MPC with 20 w%, while it was difficult to distinguish m-MS particles and PCL-PEG-PCL for the m-MPC with 40 w%. It can be seen that the surface of the m-MPC with 40 w% m-MS had more m-MS particles than m-MPC with 20 w% m-MS.
 | Figure 4 Scanning electron microscope images of PCL-PEG-PCL (A) and m-MPC with 20 w% (B) and 40 w% (C) m-MS. |
Mechanical properties and hydrophilicity of m-MPC
The mechanical properties of compressive strength and elastic modulus of composites increased with the increasing m-MS content (P<0.05) (Table 1). The composite containing 40 w% m-MS resulted in the highest compressive strength (48 MPa) and elastic modulus (548 MPa), indicating that the addition of m-MS into PCL-PEG-PCL improved the mechanical properties. The highest m-MS content (60 w%) yielded a lower compressive strength (34 MPa) and elastic modulus (350 MPa) than did the composite with 40 w% m-MS (P<0.05), indicating that there was a critical concentration beyond which additional m-MS diminished the mechanical properties of the composite.
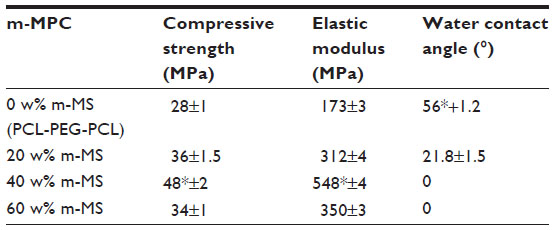 | Table 1 Mechanical properties and water contact angle of the composites with different m-MS contents |
Table 1 shows the results of the measurements of water contact angles of the composite surfaces. The results revealed that the water contact angles on the m-MPC reduced significantly with increasing m-MS content, indicating improved hydrophilicity. The results show that the water contact angle of PCL-PEG-PCL was 56°; after adding 20 w% m-MS, the composite was 21.8°, and after adding 40 w% MS into the polymer, the composite contact angle was 0, indicating that the addition of m-MS into PCL-PEG-PCL improved the hydrophilicity of m-MPC.
Degradability of m-MPC in PBS solution
The in vitro degradation behavior of m-MPC was determined by testing weight loss ratio after soaking in PBS. Figure 5 presents the weight loss ratio of the m-MPC with 0, 20 w%, and 40 w% m-MS after immersion in PBS as a function of incubation time. The results revealed that the weight loss ratio of all the samples increased with incubation time, and the weight loss ratios of m-MPC increased with the increase of the m-MS content. It was found that the weight loss ratio of m-MPC with 40 w% m-MS was 75.6 w%, while m-MPC with 20 w% m-MS was 50.3 w% and PCL-PEG-PCL was 26.7 w% at the end of 10 weeks. The results show that the m-MPC with high m-MS content had a faster degradation rate, and that m-MS not only degraded completely in m-MPC, but also promoted the degradation of PCL-PEG-PCL after 10 weeks.
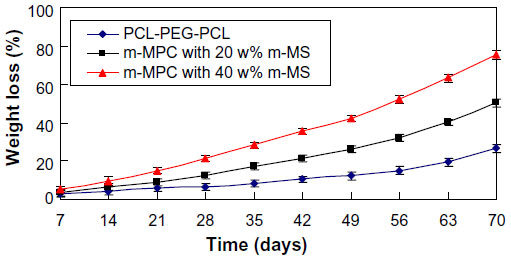 | Figure 5 Weight loss ratio of PCL-PEG-PCL and m-MPC with 20 w% and 40 w% m-MS after immersion in phosphate-buffered saline over time. |
Apatite formation in SBF
After soaking in SBF for 5 days, it was found that spherical apatite granules were formed on the surfaces of m-MPC with 20 w% and 40 w% m-MS. No apatite formation was found on the PCL-PEG-PCL surface (Figure 6A). Only a few spherical apatite granules were found on the m-MPC surface with 20 w% m-MS (Figure 6B), while spherical granules were found in a densely packed apatite layer on m-MPC with 40 w% m-MS (Figure 6C). The EDS spectra were used to examine the apatite formation on the surface of m-MPC with 20 w% and 40 w% m-MS after soaking in SBF for 5 days (Figure 6D). Calcium and phosphorous peaks were detected and the calcium/phosphorous ratios of the apatites on the m-MPC with 20 w% and 40 w% m-MS were found to be 1.5 and 1.52, respectively, which were both less than the calcium/phosphorous ratio of 1.67 in hydroxyapatite. This indicates that the apatite formation on m-MPC was calcium deficient.
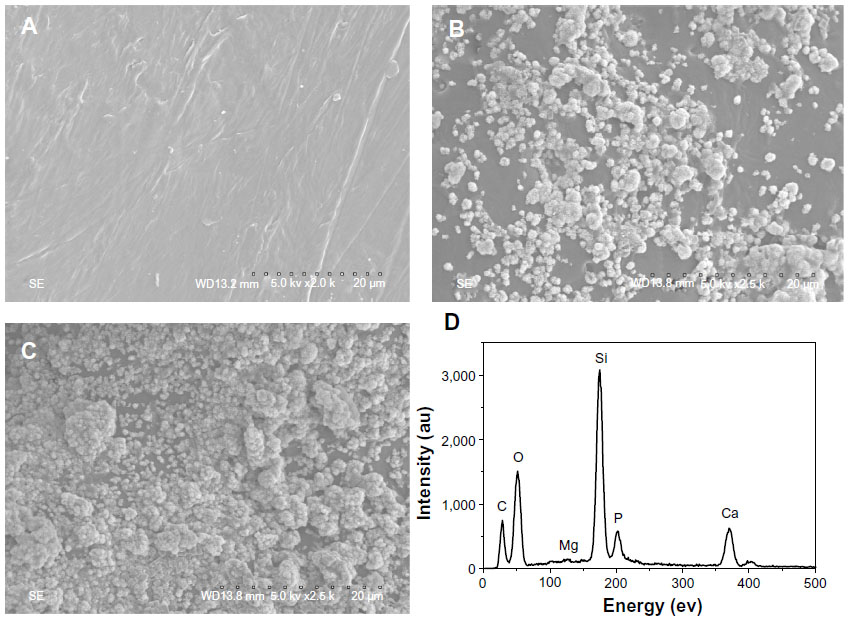 | Figure 6 Scanning electron microscope images of PCL-PEG-PCL (A) and m-MPC with 20 w% (B) and 40 w% (C) m-MS after being soaked in simulated body fluid for 5 days, and EDS of m-MPC with 40 w% m-MS after being soaked in simulated body fluid for 5 days (D). |
Changes of ion concentrations in SBF solution
We examined the changes in the concentrations of calcium, phosphorus, magnesium, and silicon after soaking the m-MPC with 40 w% m-MS in SBF solution for various periods of time (Figure 7). Magnesium and silicon concentrations increased rapidly up to 6 hours in the specimens, and, thereafter, continued to increase at a slower rate up to 96 hours. In contrast to the increasing magnesium and silicon concentrations, calcium and phosphorous ion concentrations decreased gradually throughout the soaking period.
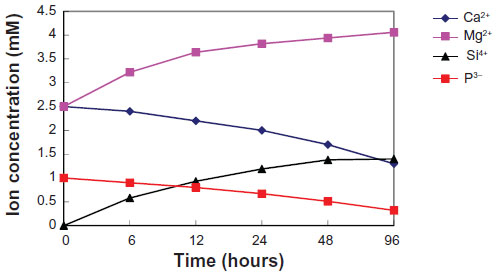 | Figure 7 Changes of calcium, silicon, magnesium, and phosphorus concentrations in m-MPC with 40 w% m-MS after soaking in simulated body fluid. |
Cell attachment
Cell attachment was assessed using MG63 cells cultured on m-MPC with 0 w%, 20 w%, and 40 w% m-MS, and TCP was used as a control. The results of cell attachment efficiency are profiled in Figure 8. In a period of 4 hours, it was found that the cell attachment ratio of the m-MPC with 40 w% m-MS (131%) was significantly higher than that of m-MPC with 20 w% m-MS (114%). Moreover, the cell attachment ratio of the PCL-PEG-PCL was only 91%, which was lower than that of the TCP control (100%). The results show improved cell attachment relative to the m-MPC with higher m-MS content, suggesting that the addition of m-MS into PCL-PEG-PCL facilitated cell adhesion.
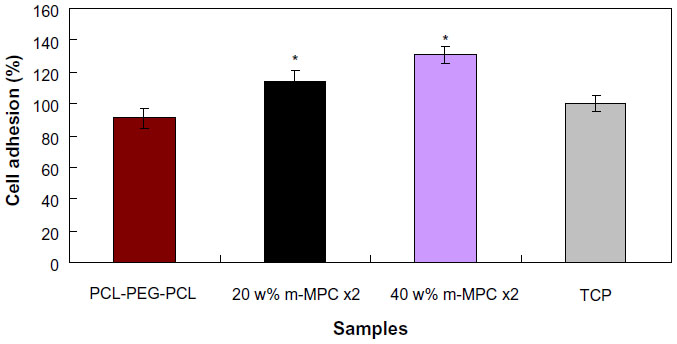 | Figure 8 MG63 cell adhesion on m-MPC with 20 w% and 40 w% m-MS, and on PCL-PEG-PCL and TCP (controls). |
Cell proliferation
Proliferation of MG63 cells cultured on the m-MPC was assessed using the MTT assay because OD values could provide an indication of cell growth. Figure 9 reveals that the OD value for all the samples increased with time, and no significant differences were found among the samples at day 1. The OD values for m-MPC with 20 w% and 40 w% m-MS were significantly higher than that of PCL-PEG-PCL at 4 and 7 days (P<0.05); no significant differences appeared between m-MPC with 20 w% and 40 w% m-MS at 4 and 7 days. The results indicate that cell growth and viability in m-MPC were superior to those in PCL-PEG-PCL, suggesting that m-MPC facilitates cell growth and can promote cell viability.
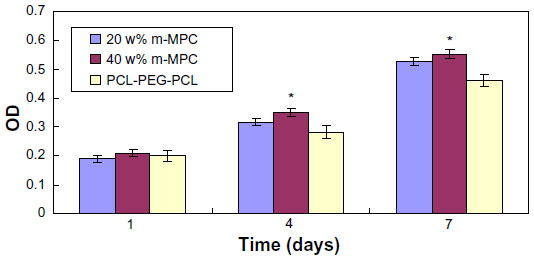 | Figure 9 Proliferation of MG63 cells on m-MPC with 20 w% and 40 w% m-MS and on PCL-PEG-PCL (control). |
Cell morphology
Figure 10 presents SEM images showing the morphology of MG63 cells cultured on the m-MPC with 0 w%, 20 w%, and 40 w% m-MS, respectively, for 4 days. At 4 days, the MG63 cells extended and spread better on the m-MPC surfaces with 20 w% and 40 w% m-MS than on the PCL-PEG-PCL surfaces.
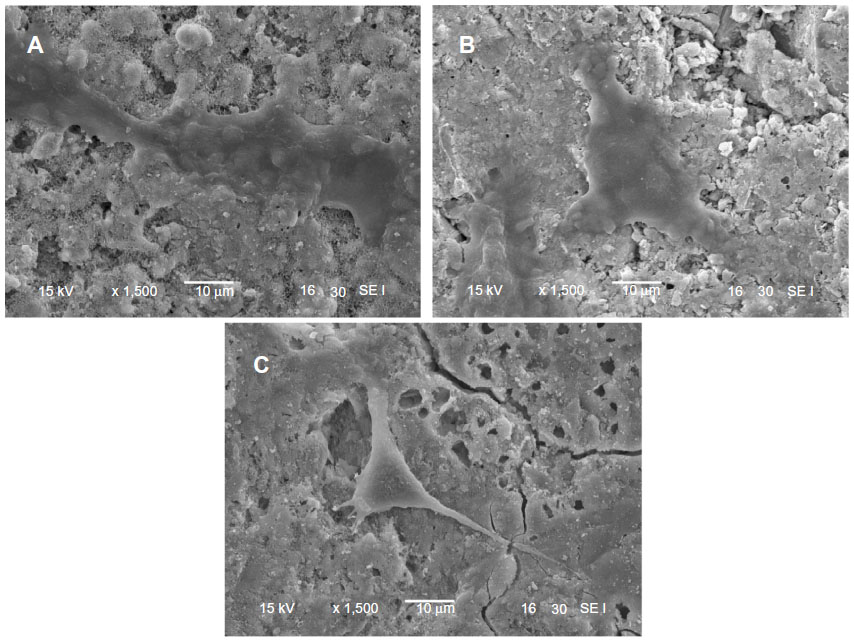 | Figure 10 Scanning electron micrscope images of MG63 cells cultured on m-MPC with 20 w% (A) and 40 w% (B) m-MS and on PCL-PEG-PCL (C) for 4 days. |
Discussion
Over the past few years, considerable efforts have been made in studying inorganic/organic bioactive composites for bone regeneration.15,16 Mesoporous material with large specific surface area and pore volume may have enhanced bioactivity, be conducive to the cellular growth, and promote new bone formation, and it has also been proposed to be used in mesoporous/macroporous scaffolds that can be used in bone tissue engineering.17,18 In this study, m-MS with a specific surface area of 428.0 m2/g and pore volume of 0.40 cm3/g was prepared, and its composites with PCL-PEG-PCL were fabricated.
The results showed that the mechanical properties of compressive strength and elastic modulus of the m-MPC increased significantly with the increase of the m-MS in PCL-PEG-PCL. In the composite system, the mechanical properties of the compressive strength and elastic modulus of m-MPC with 40 w% m-MS were significantly higher than those of m-MPC with 20 w% and 60 w% m-MS, indicating that a weight ratio of approximately 40 w% is optimal. Composites containing high m-MS content, for example 60 w%, may result in a dramatic reduction in the structural integrity and mechanical strength of the composite. The results suggest that the content of m-MS in PCL-PEG-PCL had obvious effects on the mechanical properties of the composite.
It is well known that surface property greatly affects the bioperformance of the implanted biomaterial. The hydrophilicity of the biomaterial is an important factor for cell adhesion, and thus improved surface hydrophilicity is beneficial to the interactions between biomaterials and cells.19 For the m-MPC, the results showed that the water contact angle of the m-MPC decreased from 56° to 0° with the increase of m-MS from 0 w% to 40 w% in the composite, indicating that the m-MS content had significant effects on the hydrophilicity of the m-MPC. The remarkable improvement in hydrophilicity of the m-MPC through incorporation of m-MS into PCL-PEG-PCL suggests that the addition of inorganic bioactive material into polymer is a good way to enhance the hydrophilicity.
The in vitro degradation behavior was determined by testing weight loss ratio of the m-MPC after soaking in PBS.20 The results showed that the weight loss ratio of the m-MPC with different m-MS content in the PBS increased with the incubation time, and that the weight loss ratio of the m-MPC with 40 w% m-MS was significantly higher than that of either m-MPC with 20 w% and PCL-PEG-PCL. At the end of 10 weeks, the m-MPC with 20 w% and that with 40 w% m-MS lost 50.3 w% and 75.6 w%, respectively, of their initial weight, while PCL-PEG-PCL lost 26.7 w%. Clearly, the m-MS degraded completely in both m-MPC with 20 w% and that with 40 w% m-MS, and the degradation ratio of PCL-PEG-PCL in each m-MPC was 30.3 w% and 35.6 w%, respectively, suggesting that m-MS with a huge specific surface area and pore volume promoted the degradation of PCL-PEG-PCL in the two composites. The results suggest that incorporation of m-MS into PCL-PEG-PCL could regulate the degradability of m-MPC.
Bioactive materials can bond to living bone tissue through the apatite layer formed on their surfaces when in contact with body fluid, and the apatite formation on the implant surface plays an essential role in the formation of the tissue–biomaterial interface.21–23 In this study, the m-MS as filler was added into PCL-PEG-PCL to form bioactive m-MPC, which could form the apatite layer on the composite surfaces upon soaking in SBF. In addition, the apatite formation rate on m-MPC with 40 w% m-MS was significantly faster than that on m-MPC with 20 w% m-MS, indicating that the m-MPC with high m-MS content possessed better in vitro bioactivity. The results suggest that the m-MPC had excellent bioactivity when m-MS was added into PCL-PEG-PCL, which could form bone-bonding with bone tissue when implanted in vivo. However, apatite could not form on the surface of PCL-PEG-PCL after soaking in SBF for 5 days, indicating that PCL-PEG-PCL was not a bioactive material.
After the m-MPC was immersed in SBF, the magnesium and silicon ion concentrations in SBF increased with time, which was attributed to the dissolution of magnesium and silicon ions from m-MPC. However, the calcium and phosphorous ion concentrations in SBF gradually decreased with time, which was attributed to calcium and phosphorous ion precipitation forming amorphous calcium phosphate and the subsequent formation of apatite by incorporating OH− ions from the SBF, which provided an indirect indication that the apatite precipitation reaction had occurred.
The EDS results showed that the calcium/phosphorous molar ratios of the apatite formation on the m-MPC with 20 w% and 40 w% m-MS were found to be 1.5 and 1.52, respectively, indicating that the formed apatite was calcium deficient. The mechanism of apatite formation on m-MPC in SBF was Mg2+ release from m-MPC surface and ionic interchange of Mg2+ for 2H+, resulting in the formation of an amorphous silica layer on the m-MPC surface and providing a favorable site for apatite nucleation. The dissolution of m-MS increased the ionic activity of apatite in SBF, thereby promoting the nucleation of apatite. Apatite formation on the implant surface may promote cell adhesion, proliferation, and function, which would build a strong bond between the implant and the surrounding bone tissue.24,25
Bioactive biomaterials need to interact actively with cells and stimulate cell growth.26 In the present study, the results showed that MG63 cells adhered better to m-MPC with 40 w% m-MS than to that with 20 w% m-MS and PCL-PEG-PCL within the first 4 hours of culture due to the presence of m-MS. The m-MS exposed on the m-MPC surfaces might have had additional special surface properties that promoted cell attachment. It could be concluded that the composite surface features (m-MS on the m-MPC surfaces) were responsible for promoting cell attachment. In addition, the MG63 cells were able to grow on the m-MPC, and the cell viability on the m-MPC was significantly higher than that on PCL-PEG-PCL. The results revealed that m-MPC with m-MS could stimulate MG63 cell viability. The superior ability of MG63 cell responses (such as attachment, spreading, and viability) on the m-MPC was probably associated with differing material surface features between the m-MPC and PCL-PEG-PCL.
Cellular responses to the biomaterial, such as attachment, spreading, and proliferation, not only depend on the surface morphology of the biomaterial, but also its chemical composition.27 The chemical compositions of the biomaterials might play crucial roles in determining cell behaviors, which influence the quantity of ions released from the materials and consequent cell–material interaction.28 A previous study has confirmed that the ion dissolution products containing silicon and magnesium from bioactive glasses and ceramics could stimulate osteoblast proliferation and differentiation.29 In this study, magnesium and silicon ions were able to be released from the m-MPC. Moreover, MTT assay showed that the m-MPC had obviously stimulated cell proliferation. It can be suggested that the improved cell responses might have resulted from the release of magnesium and silicon ions from m-MPC.
Cytocompatibility is very closely associated with cell behavior, such as cell spread on the substrate surface.27 In this study, the results revealed that the MG63 cells with normal phenotype extended and spread well on the composite surface and intimately attached to substrate at 4 days of culture, indicating that the composite had no negative effects on cell morphology and viability. The process of cell spread on the composite surface was influenced by the underlying substrate. Therefore, the results suggested that the composite surface was favorable for cell attachment, growth, and spread, disclosing good cytocompatibility. In short, enhancing m-MPC bioactivity by inducing apatite formation on its surface, which is beneficial to osteoblast proliferation and promotes new bone formation when implanted in vivo, therefore accelerates the healing process.
Conclusion
m-MPC was synthesized by solvent casting method. The addition of m-MS into PCL-PEG-PCL could improve the mechanical properties and hydrophilicity of the m-MPC, which increased with the increase of m-MS content in the m-MPC. Moreover, addition of m-MS into PCL-PEG-PCL could improve the degradability of the m-MPC, and the degradation ratio of the m-MPC increased with the increase of m-MS content. The m-MPC had good in vitro bioactivity because the apatite layer could form on the surface after immersion in SBF, and the m-MS content had obvious effects on the apatite formation rate of the composites. The m-MPC could enhance the cell attachment, and attachment rate of the cells increased with the increase of the m-MS content in the m-MPC. The viability ratio of MG63 cells on the m-MPC was significantly higher than that on PCL-PEG-PCL, demonstrating that m-MPC with m-MS in the polymer could stimulate cell viability. The results demonstrate that m-MPC had good in vitro bioactivity and cytocompatibility. The bioactive m-MPC shows promise in the biomedical field as a candidate for use as a new-generation biomaterial that promotes bone regeneration.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No 81000799, 31100680, and 81271705), Nano special program of Science and Technology Development of Shanghai (No 12nm0500400), and Key Medical Program of Science and Technology Development of Shanghai (No 12441903600).
Disclosure
The authors report no conflicts of interest in this work.
References
He Q, Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26:391–411. | |
He QJ, Shi JL. Mesoporous silica nanoparticle based nano drug delivery systems: synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility. J Mater Chem. 2011;21:5845–5855. | |
Vallet-Regí M. Ordered mesoporous materials in the context of drug delivery systems and bone tissue engineering. Chemistry. 2006;12:5934–5943. | |
Vallet-Regí M, Izquierdo-Barba I, Colilla M. Structure and functionalization of mesoporous bioceramics for bone tissue regeneration and local drug delivery. Philos Trans A Math Phys Eng Sci. 2012;370:1400–1421. | |
Zhu M, Zhang JH, Zhou YH, et al. Preparation and characterization of magnetic mesoporous bioactive glass/carbon composite scaffolds. Journal Chem. 2013;2013:893479. | |
Yan X, Yu C, Zhou X, Tang J, Zhao D. Highly ordered mesoporous bioactive glasses with superior in vitro bone-forming bioactivities. Angew Chem Int Ed Engl. 2004;43:5980–5984. | |
Wu C, Zhou Y, Fan W, et al. Hypoxia-mimicking mesoporous bioactive glass scaffolds with controllable cobalt ion release for bone tissue engineering. Biomaterials. 2012;33:2076–2085. | |
Liu CB, Gong CY, Huang MJ, et al. Thermoreversible gel-sol behavior of biodegradable PCL-PEG-PCL triblock copolymer in aqueous solutions. J Biomed Mater Res B Appl Biomater. 2008;84:165–175. | |
Wang W, Deng L, Liu S, et al. Adjustable degradation and drug release of a thermosensitive hydrogel based on a pendant cyclic ether modified poly(ε-caprolactone) and poly(ethylene glycol) co-polymer. Acta Biomater. 2012;8:3963–3973. | |
Gong CY, Dong PW, Shi S, et al. Thermosensitive PEG-PCL-PEG hydrogel controlled drug delivery system: sol-gel-sol transition and in vitro drug release study. J Pharm Sci. 2009;98:3707–3717. | |
Li X, Shi JL, Dong X, Zhang L, Zeng H. A mesoporous bioactive glass/polycaprolactone composite scaffold and its bioactivity behavior. J Biomed Mater Res A. 2008;84:84–91. | |
He QJ, Huang ZL, Liu Y, Chen W, Xu T. Template-directed one-step synthesis of flowerlike porous carbonated hydroxyapatite spheres. Mater Lett. 2007;61:141–143. | |
Fu SZ, Wang XH, Guo G, et al. Preparation and characterization of nano-hydroxyapatite/poly(epsilon-caprolactone)-poly(ethylene glycol)-poly(epsilon-caprolactone) composite fibers for tissue engineering. J Phys Chem. 2010;114:18372–18378. | |
Kokubo T, Matsushita T, Takadama H, Kizuki T. Development of bioactive materials based on surface chemistry. J Eur Ceram Soc. 2009;29:1267–1274. | |
Yun HS, Kim SE, Hyun YT, Heo SJ, Shin JW. Three-dimensional mesoporous – giantporous inorganic/organic composite scaffolds for tissue engineering. Chem Mat. 2007;19:6363–6366. | |
Su J, Cao L, Yu B, et al. Composite scaffolds of mesoporous bioactive glass and polyamide for bone repair. Int J Nanomedicine. 2012;7:2547–2555. | |
Miller DC, Thapa A, Haberstroh KM, Webster TJ. Endothelial and vascular smooth muscle cell function on poly(lactic-co-glycolic acid) with nano-structured surface features. Biomaterials. 2004;25:53–61. | |
Parent LR, Robinson DB, Woehl TJ, et al. Direct in situ observation of nanoparticle synthesis in a liquid crystal surfactant template. ACS Nano. 2012;6:3589–3596. | |
Holmes CA, Tabrizian M. Enhanced MC3T3 preosteoblast viability and adhesion on polyelectrolyte multilayer films composed of glycol-modified chitosan and hyaluronic acid. J Biomed Mater Res A. Epub November 29, 2011. | |
Huang W, Lv X, Liu C, et al. The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem. 2012;287:26245–26253. | |
Lu X, Leng Y. Theoretical analysis of calcium phosphate precipitation in simulated body fluid. Biomaterials. 2005;26:1097–1108. | |
Azami M, Moosavifar MJ, Baheiraei N, Moztarzadeh F, Ai J. Preparation of a biomimetic nanocomposite scaffold for bone tissue engineering via mineralization of gelatin hydrogel and study of mineral transformation in simulated body fluid. J Biomed Mater Res A. 2012;100:1347–1355. | |
Wu CT, Fan W, Chang J. Functional mesoporous bioactive glass nanospheres: synthesis, high loading efficiency, controllable delivery of doxorubicin and inhibitory effect on bone cancer cells. J Mater Chem B. 2013;1:2710–2718. | |
Wang J, Layrolle P, Stigter M, de Groot K. Biomimetic and electrolytic calcium phosphate coatings on titanium alloy: physicochemical characteristics and cell attachment. Biomaterials. 2004;25:583–592. | |
Oyane A, Wang X, Sogo Y, Ito A, Tsurushima H. Calcium phosphate composite layers for surface-mediated gene transfer. Acta Biomater. 2012;8:2034–2046. | |
Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23:47–55. | |
Zinelis S, Thomas A, Syres K, Silikas N, Eliades G. Surface characterization of zirconia dental implants. Dent Mater. 2010;26:295–305. | |
Prodanov L, Lamers E, Domanski M, Luttge R, Jansen JA, Walboomers XF. The effect of nanometric surface texture on bone contact to titanium implants in rabbit tibia. Biomaterials. 2013;34:2920–2927. | |
Varanasi VG, Saiz E, Loomer PM, et al. Enhanced osteocalcin expression by osteoblast-like cells (MC3T3-E1) exposed to bioactive coating glass (SiO2-CaO-P2O5-MgO-K2O-Na2O system) ions. Acta Biomater. 2009;5:3536–3547. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.