Back to Journals » International Journal of Nanomedicine » Volume 13
Mesenchymal stem cells loaded with paclitaxel–poly(lactic-co-glycolic acid) nanoparticles for glioma-targeting therapy
Authors Wang X, Gao J, Ouyang X, Wang J, Sun X ![]() , Lv Y
, Lv Y
Received 3 March 2018
Accepted for publication 5 July 2018
Published 7 September 2018 Volume 2018:13 Pages 5231—5248
DOI https://doi.org/10.2147/IJN.S167142
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Xiaoling Wang,1,2 Jianqing Gao,2 Xumei Ouyang,1,2 Junbo Wang,1 Xiaoyi Sun,1 Yuanyuan Lv1
1Department of Pharmacy, Zhejiang University City College, 2Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, China
Background: Mesenchymal stem cells (MSCs) possess inherent tropism towards tumor cells, and so have attracted increased attention as targeted-therapy vehicles for glioma treatment.
Purpose: The objective of this study was to demonstrate the injection of MSCs loaded with paclitaxel (Ptx)-encapsulated poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles (NPs) for orthotopic glioma therapy in rats.
Methods: Ptx-PLGA NP-loaded MSC was obtained by incubating MSCs with Ptx-PLGA NPs. The drug transfer and cytotoxicity of Ptx-PLGA NP-loaded MSC against tumor cells were investigated in the transwell system. Biodistribution and antitumor activity was evaluated in the orthotopic glioma rats after contralateral injection.
Results: The optimal dose of MSC-loaded Ptx-PLGA NPs (1 pg/cell Ptx) had little effect on MSC-migration capacity, cell cycle, or multilineage-differentiation potential. Compared with Ptx-primed MSCs, Ptx-PLGA NP-primed MSCs had enhanced sustained Ptx release in the form of free Ptx and Ptx NPs. Ptx transfer from MSCs to glioma cells could induce tumor cell death in vitro. As for distribution in vivo, NP-loaded fluorescent MSCs were tracked throughout the tumor mass for 2 days after therapeutic injection. Survival was significantly longer after contralateral implantation of Ptx-PLGA NP-loaded MSCs than those injected with Ptx-primed MSCs or Ptx-PLGA NPs alone.
Conclusion: Based on timing and sufficient Ptx transfer from the MSCs to the tumor cells, Ptx-PLGA NP-loaded MSC is effective for glioma treatment. Incorporation of chemotherapeutic drug-loaded NPs into MSCs is a promising strategy for tumor-targeted therapy.
Keywords: BMSCs, contralateral injection, orthotopic glioma, drug targeting, C6 cells
Introduction
Malignant glioma is the most common form of primary brain tumor and has poor prognosis, owing to its highly proliferative, infiltrative, and invasive properties. Despite advanced treatment with surgery, radiotherapy, and chemotherapy, the median overall patient survival time of newly diagnosed glioblastoma remains at only 1–2 years.1 The inefficacy of current therapy arises mainly from inefficient delivery of therapeutic agents to the tumor site and to infiltrating tumor cells. Therefore, new strategies are needed to achieve extensive intratumoral distribution and specific tumor targeting.
Over the past few years, mesenchymal stem cells (MSCs) have attracted increased attention as cellular vehicles for cancer therapy, because of their hypoimmunogenicity, fast ex vivo expansion, and inherent tumor-tropic and migratory properties. We have previously shown that MSCs can carry therapeutic genes for tumor-targeted gene therapy.2,3 Genetically modified MSCs have been successfully engineered for the delivery of immunostimulators or toxic molecules to gliomas.4,5 Nevertheless, safety issues of viral vectors and low transfection efficiency of nonviral vectors remain significant challenges. Attempts to deliver traditional anticancer drugs via MSCs include paclitaxel (Ptx)-primed MSCs6,7 and MSCs surface attaching with nanoparticulate payloads.8,9 More recently, MSCs loaded with drug-encapsulated nanoparticles (NPs) have been made to treat refractory cancers.10,11 However, anticancer-drug cytotoxicity decreases MSC vitality and rapid drug efflux induces chemotherapeutic loss before MSC arrival at tumor sites. Previous studies using orthotopic glioma models have investigated intratumoral MSC injection, which does not demonstrate the tumor-targeted property of MSCs.12,13
Incorporating controlled-release NPs into MSCs is an alternative delivery method of targeting toxic agents to tumors. The intrinsic function of MSCs can be preserved by avoiding direct exposure through NP encapsulation. Sustained drug release from internalized NPs followed by drug excretion from MSCs may slow the payload discharge during the process of tumor-homing.6,14 Furthermore, exocytosis of intact NPs from stromal cells and then endocytosis of NPs by glioma cells would ensure that a therapeutic dose is transferred into the tumor mass.15,16
In this study, an MSC-based “Trojan horse” loaded with a Ptx reservoir of poly(D,L-lactide-co-glycolide) (PLGA) NPs was constructed. The optimal dose of Ptx-PLGA NPs carried by the MSCs with minimal influence on cell cycle, migratory activity, and differentiation was determined. The drug-transfer process between bone-marrow-derived MSCs (BMSCs) loaded with Ptx-PLGA NPs (MSCs-NPs) and glioma cells was studied. Finally, the distribution of MSC NPs within the tumor mass and their prolonged survival time was observed after MSC-NP injection into contralateral brain hemispheres in an orthotopic glioma model (Figure 1).
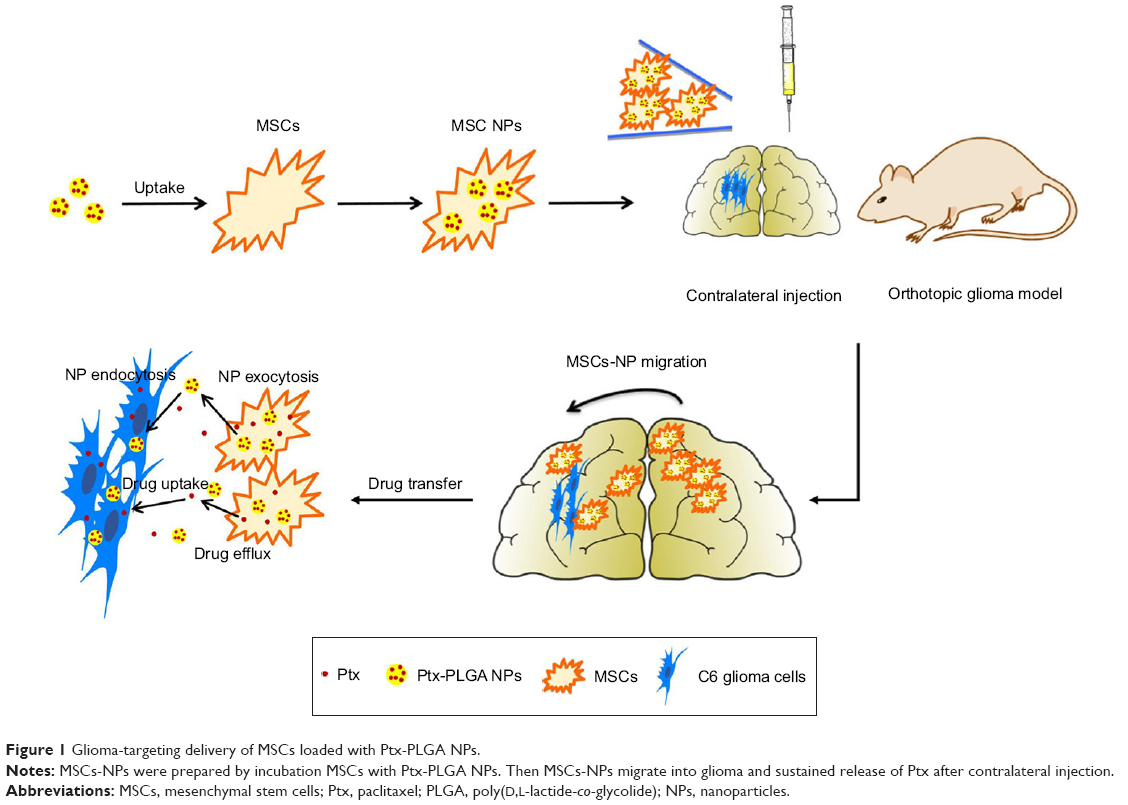 | Figure 1 Glioma-targeting delivery of MSCs loaded with Ptx-PLGA NPs. |
Methods
Materials
PLGA terminated by a carboxyl group (PLGA-COOH) or ester group (PLGA) with lactic:glycolic acid molar ratio of 50:50 (molecular weight 5 KDa) was purchased from Shandong Medical Instrumental Institute (Shandong, China), and 5(6)-carboxyfluorescein and propidium iodide (PI)/RNase were supplied by Aladdin Reagent (Shanghai, China) and Sigma-Aldrich (St Louis, MO, USA), respectively. Cell-culture medium (low-glucose DMEM and DMEM/F12) and osteogenic and adipogenic differential medium were purchased from Genom Pharmaceutical Biotechnology (Hangzhou, China) and Cyagen Biosciences (Guangzhou, China), respectively. FBS and horse serum were provided by Thermo Fisher Scientific (Waltham, MA, USA). DAPI and LysoTracker Red were supplied by Beyotime Biotechnology (Haimen, China). An ALP-assay kit was bought from Jiancheng Bioengineering Institute (Nanjing, China). C6 glioma cells were purchased from the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Science, Chinese Academy of Sciences (Shanghai, China). Male Sprague Dawley rats were purchased from Shanghai Laboratory Animal Center. Animal experiments were in accordance with Zhejiang University City College guidelines for the welfare and ethics of experimental animals, and all animal experimental procedures were performed with the approval of the Animal Experimental Ethics Committee of Zhejiang University City College.
PLGA-NP preparation and characterization
The morphology of PLGA NPs was assessed using transmission electron microscopy (JEM-1230; JEOL, Tokyo, Japan). The sample was diluted and negatively stained with phosphotungstic acid. Confocal laser-scanning microscopy (CLSM; FV1000; Olympus, Tokyo, Japan) was used to observe fluorescein–PLGA (f-PLGA) NPs dispersed in deionized water. Particle size and ζ-potential were determined by laser light scattering (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). f-PLGA was synthesized by conjugating PLGA-COOH and 5(6)-carboxyfluorescein with hexamethylenediamine. Details on method can be found in Tosi et al.17
f-PLGA NPs were prepared by the modified emulsion-solvent evaporation method.18 Briefly, 100 mg f-PLGA was added to 5 mL CH2Cl2, then 25 mL deionized water containing 1% polyvinyl alcohol was added to the organic phase. The mixture was sonicated for 2 minutes, and then stirred for another 4 hours to evaporate CH2Cl2. NPs were collected and washed by centrifugation at 12,000 rpm/min for 10 minutes (Microfuge 22R; Beckman, USA). Ptx-PLGA NPs were prepared in the same way as f-PLGA NPs, except that 5 mg Ptx and 100 mg PLGA were added to the organic phase. The obtained nanosuspension was filtered (pore size 0.8 μm) to remove the unencapsulated Ptx. NPs were washed three times by centrifugation.
For measurement of encapsulation efficiency and drug-loading rate, NPs were dissolved in acetonitrile. The supernatant was centrifuged and analyzed by HPLC (Alliance 2690; Waters, Milford, MA, USA). Detection conditions were: Diamonsil C18 column (250×4.6 mm, 5 μm); flow rate 1 mL/min, the ultraviolet-detector wavelength 227 nm. The mobile phase was acetonitrile:water (55:45). Linearity was good: 0.5–50 μg/mL (y=44,326x+27706, R2=0.9934).
In vitro release of Ptx from Ptx-PLGA NPs
In vitro release of Ptx from Ptx-PLGA NPs was investigated using dialysis. NPs loaded with 80 μg Ptx were suspended in PBS (pH 7.4) and placed in a dialysis bag (molecular weight cutoff 14 kDa), which was placed in a receiving compartment filled with 15 mL PBS containing 0.1% Tween 80. Samples were then incubated at 37°C, 100 rpm/min. Dialysate (0.5 mL) was collected at indicated time intervals, while an equivalent volume of fresh medium was added to maintain the sink conditions. The collected dialysate was analyzed by HPLC. Detection conditions were the same as previously mentioned.
Cell culture
BMSCs were isolated from the bone shaft of femurs of 3-week-old male Sprague Dawley rats as per our previously reported method.3 BMSCs were cultured with modified low-sugar DMEM containing 10% FBS, penicillin, and streptomycin. Second- to fifth-passage cells were used for all experiments. C6 glioma cells were cultured in DMEM/F12 supplemented with 15% horse serum, 2.5% FBS, penicillin, and streptomycin. All cells were cultured in a humidified incubator (95% air, 5% CO2) at 37°C.
Cytotoxicity of Ptx-PLGA NPs and MSC NPs
The cytotoxicity of Ptx-PLGA NPs and Ptx toward MSCs and C6 cells was determined by MTT assay. MSCs (5×103 cells/well) and C6 cells (104 cells/well) were seeded in 96-well plates and incubated overnight. Then, Ptx and Ptx-PLGA NPs were added at concentrations of 0.5–1,500 ng/mL to MSCs and 1–100 ng/mL to C6 cells. The cytotoxicity of blank PLGA NPs was also tested. After 72 hours, cells were washed and treated with MTT solution for 4 hours. The crystals formed were dissolved by dimethyl sulfoxide. Absorbance at 570 nm was measured by a microplate reader (BioTek, Winooski, VT, USA). In vitro antitumor effects of MSCs-NPs against C6 cells were evaluated by a transwell system (8 μm pore size; Costar; Corning, NY, USA). MSC NPs were added to the upper chamber, while C6 cells were seeded in the lower one. After 3 days, the viability of C6 cells was analyzed by MTT.
Uptake kinetics and intracellular distribution of f-PLGA NPs in MSCs
Internalization kinetics of f-PLGA NPs into MSCs was studied by incubating MSCs with 5 μg/mL f-PLGA NPs for various periods. Cells were washed with PBS and then lysed with Triton X-100. Intracellular levels of f-PLGA NPs were determined by fluorescence spectrophotometry (RF-5301PC; Shimadzu, Kyoto, Japan) with excitation wavelength 492 nm and emission wavelength 517 nm. After incubation for 8 hours, MSCs were washed and stained with LysoTracker and DAPI to observe the intracellular distribution of f-PLGA NPs by CLSM.
In vitro migration assays
The effect of Ptx-PLGA NPs on the tumor-tropism capacity of MSCs was conducted using 24-well transwell plates. MSC NPs were constructed by incubating MSCs with different concentrations of Ptx-PLGA NPs (5, 8, and 10 ng/mL Ptx-equivalent) for 8 hours. Then, MSC NPs were seeded in the upper chamber at 3×104 cells per well and C6 cells (DMEM containing 0.5% serum) placed into the lower well at 105 cells per well. Untreated MSCs served as the control. At 1, 3, and 5 days after seeding, cells that had not migrated through the pores were removed with cotton swabs. Then, cells on the lower surface of the membrane were fixed with methanol and stained with 0.1% crystal violet for 30 minutes. Cells that had migrated were observed and counted in three randomly selected microscopic fields (400×) using an Olympus CKX41microscope. The migration rate in vitro was calculated: counts (sample)/counts (control) ×100%.
Flow-cytometry analysis
To measure the cell cycle of MSC NPs, the MSCs were treated with Ptx-PLGA NPs for 8 hours at three Ptx concentrations (5, 8, and 10 ng/mL Ptx-equivalent). Then, cells were washed and added to Ptx-free culture medium. Cells were collected at 0, 3, and 5 days after drug withdrawal, respectively, and then fixed with ethanol. Samples were stained with PI/RNase. Fluorescence intensity was analyzed in the FL3 channel (FACSCalibur; BD Biosciences, San Jose, CA, USA).
Differentiation of MSC NPs
Osteogenic and adipogenic differentiation of MSC NPs were performed. MSCs (3×104 cells/well) in 24-well plates were exposed to 8 ng/mL Ptx-PLGA NPs for 8 hours, and then cells were washed and cultured in drug-free osteogenic or adipogenic differential medium for another 21 days. Early status of osteogenic differentiation was assessed by determination of extracellular ALP production. ALP activity was determined on days 3, 6, 9, and 12 after drug uptake with an ALP-assay kit. Late status of osteogenic differentiation was assessed by alizarin red staining to detect calcium-nodule deposits. For adipogenic differentiation, cells were fixed with 4% paraformaldehyde and stained with oil red O for observation. Fractions of positive cells were calculated by counting cells in 20 fields in each group.
Drug released from MSC NPs
MSCs (106 cells) were treated with Ptx-PLGA NPs or Ptx solution (6 mg/mL, dissolved in Cremophor EL–ethanol 1:1) for 8 hours (8 ng/mL). Then, cells were washed and incubated with drug-fresh medium. At predetermined time points, cells were trypsinized and mixed with methanol. After sonication for 10 minutes, samples were centrifuged (6,000 rpm, 5 minutes). The supernatant was air-dried and redissolved in methanol. Intracellular Ptx was quantitatively analyzed by HPLC. For investigation of NP exocytosis from MSC NPs, MSCs were treated with 5 μg/mL f-PLGA NPs for 8 hours. After incubation, MSCs were washed and recultured in NP-free medium. In the following days (0, 1, 2, and 3), cells were lysed and fluorescence intensity determined.
Drug transferred from MSC NPs to C6 cells
To investigate Ptx transfer from MSC NPs to tumor cells, MSCs (3×104 cells) were incubated with Ptx-PLGA NPs (8 ng/mL) for 8 hours. Then, MSC NPs were collected and seeded in the upper chamber of the 24-well transwell, while the lower well was seeded with 2×105 C6 cells. At the indicated time points, sets of C6 cells (four wells per set) were trypsinized and mixed with methanol. Samples were sonicated, centrifuged, air-dried, and redissolved in 80 μL methanol. Intracellular Ptx was analyzed quantitatively by HPLC. Linearity was good: 0.15–2.00 μg/mL (y=116,400x+13,099, R2=0.9969).
In vivo studies
Intratumoral distribution of implanted MSC NPs
Developing an orthotropic brain-tumor model was carried out as reported previously.19 Briefly, male Sprague Dawley rats weighing 180–200 g were anesthetized by an intraperitoneal injection of pentobarbital sodium (60 mg/kg). Then, rats were injected stereotactically (Stoelting, Wood Dale, IL, USA) with 106 C6 glioma cells in 10 μL PBS into the left forebrain (3 mm lateral, 1 mm anterior to bregma, 5 mm depth from the skull surface). Seven days later, 2×105 therapeutic MSCs (MSCs incubated with 8 ng/mL Ptx-PLGA NPs for 8 hours) stained with CM-Dil were injected contralaterally into the right forebrain (3 mm lateral, 1 mm anterior to bregma, 5 mm depth from the skull surface). After 2 days, frozen specimens of glioma and contralateral brain tissue were collected (transverse sections) and the nuclei stained with DAPI (Figure S1). The distribution of MSCs was observed by CLSM. Fluorescence intensity (FI) of CM-Dil-stained MSC NPs in the glioma area and contralateral normal tissue from six rats was measured by ImageJ software. The migration rate in vivo was calculated: FIglioma/(FIglioma + FInormal) ×100%.
Antitumor effect in vivo
The orthotropic brain-tumor model was first established. Seven days after tumor inoculation, rats were then randomly assigned to five groups (n=6) receiving a contralateral injection of saline, MSCs, Ptx-PLGA NPs, MSCs-Ptx, or MSC NPs. The injection volume was 10 μL in all five groups. Ptx dose was 1 μg/kg in drug-containing groups. MSC-Ptx and MSC NPs were obtained according to the method in the “Drug released from MSC NPs” section. The Ptx dose was adjusted by the results of drug content determined in that part. MSC NPs (2×105) and MSCs-Ptx (2.2×105) were injected. The survival study lasted for 2 months, and the rats were killed when they had lost >15% of their body weight. On day 7 after therapeutic injection, tumor masses were stained with H&E for histopathology evaluation.
Statistical analysis
Data are presented as means ± SD, and comparison among the different groups was analyzed by one-way ANOVA followed by Student-Newman-Keuls, Dunnett’s, or two-tailed Student’s t-test, assuming equal variance in two groups. The probability of survival was estimated by the Kaplan–Meier method and compared with the log-rank test in GraphPad Prism 6.0. The level of statistical significance in all analyses was set at P<0.05.
Results
Physicochemical properties of f-PLGA NPs and Ptx-PLGA NPs
To study PLGA-NP exocytosis from MSCs, f-PLGA was synthesized to prepare f-PLGA NPs (Figure S2). The structure of f-PLGA and its intermediates was confirmed by 1H nuclear magnetic resonance spectra (Figures S3–S5). The extent of f-PLGA derivatization was 21.6 μmoL fluorescein/g of polymer, determined by integration of the fluorescein and methyl-group protons of the PLGA. f-PLGA-NP fluorescence is presented in Figure S6. Fluorescence intensity was high enough for PLGA-NP endocytosis and exocytosis kinetic study in MSCs. Particle size, polydispersity index, and ζ-potential of PLGA NPs, f-PLGA NPs, and Ptx-PLGA NPs are presented in Table 1. The physicochemical properties of f-PLGA NPs were found to be similar to those of Ptx-PLGA NPs, supporting the use of f-PLGA NPs as a model particle to mimic Ptx-PLGA NPs. The encapsulation efficiency and drug-loading rate of Ptx-PLGA NPs were 86.87%±3.63% and 4.34%±0.18%, respectively. Transmission electron microscopy (Figure 2A) and size distribution (Figure 2B) indicated that Ptx-PLGA NPs were spherical and uniformly distributed. Figure 2C presents a slightly negative charge of Ptx-PLGA NPs. In vitro release of Ptx from Ptx-PLGA NPs is shown in Figure 2D, illustrating sustained release lasting beyond 5 days.
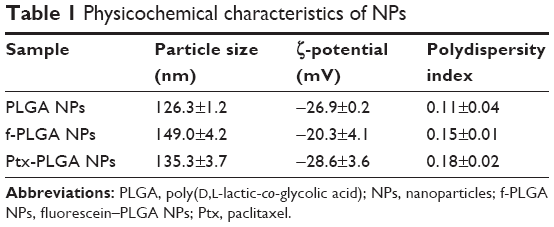 | Table 1 Physicochemical characteristics of NPs |
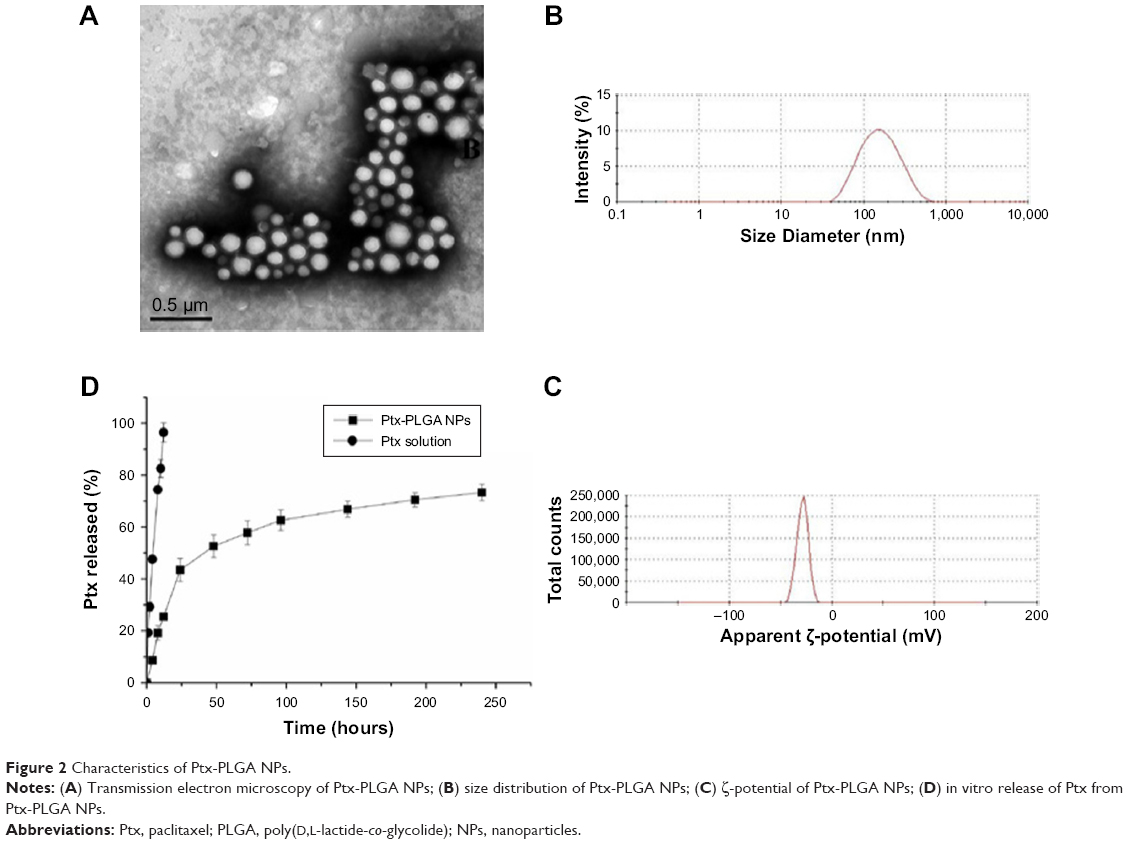 | Figure 2 Characteristics of Ptx-PLGA NPs. |
Cytotoxicity of Ptx-PLGA NPs against MSCs and C6 cells
A significant difference in Ptx sensitivity was observed between MSCs and C6 cells. MSC and C6-cell viability following exposure to Ptx-PLGA NPs is shown in Figure 3A. Significant dose-dependent cytotoxicity was observed for Ptx <100 ng/mL in C6 cells. Conversely, MSCs exposed to wide-ranging doses of Ptx-PLGA NPs (up to ~1,500 ng/mL Ptx) maintained relatively high levels of viability. The IC50 of Ptx-PLGA NPs was significantly higher in MSCs (1,632±427 ng/mL) than C6 cells (3.37±0.35 ng/mL) (Figure 3B). This difference is crucial for loading and releasing sufficient amounts of Ptx from MSCs to target sensitive glioma cells. Encapsulation of Ptx into PLGA NPs enabled MSCs to be protected from direct exposure to the antimicrotubule agent. Sustained drug release from internalized Ptx-PLGA NPs contributed to the relatively low intracellular Ptx levels observed in MSCs. As such, higher tolerance to Ptx-PLGA NPs than free Ptx was observed in MSCs (Figure 3B). Cytotoxicity was absent from the control PLGA-NP group throughout the administered concentration range. To enable a higher dose of Ptx to be loaded into the MSCs while maintaining cell vitality, MSCs loaded with Ptx-PLGA NPs were constructed and their tumor tropism and antitumor activity investigated further.
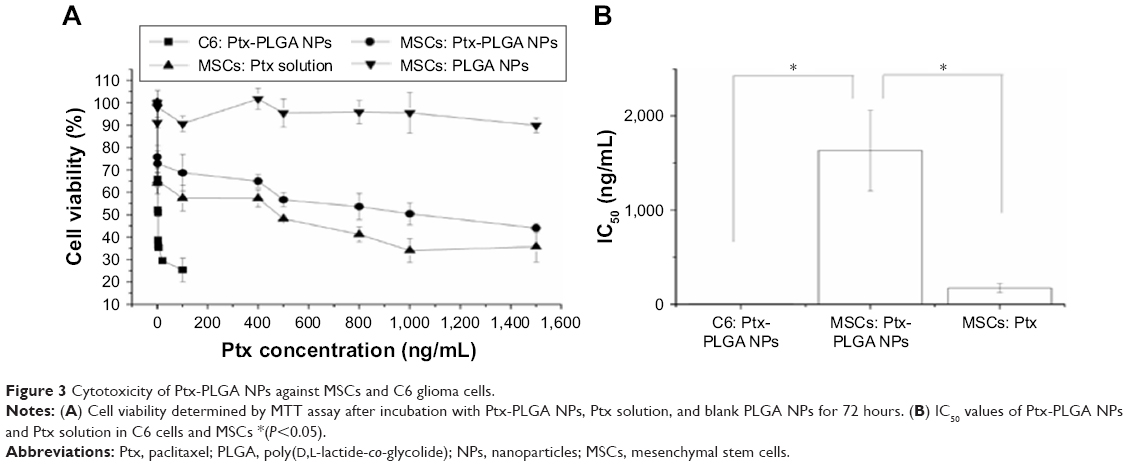 | Figure 3 Cytotoxicity of Ptx-PLGA NPs against MSCs and C6 glioma cells. |
Uptake kinetics and intracellular distribution of f-PLGA NPs in MSCs
The internalization kinetics of f-PLGA NPs into MSCs was investigated to determine incubation time. Cellular uptake of NPs increased with time, and reached a plateau at 8 hours (Figure 4B). As a result, incubation time was fixed at 8 hours for MSC-NP preparation. f-PLGA-NP intracellular distribution was also observed. NPs were colocalized with the lysosome tracker (Figure 4A).
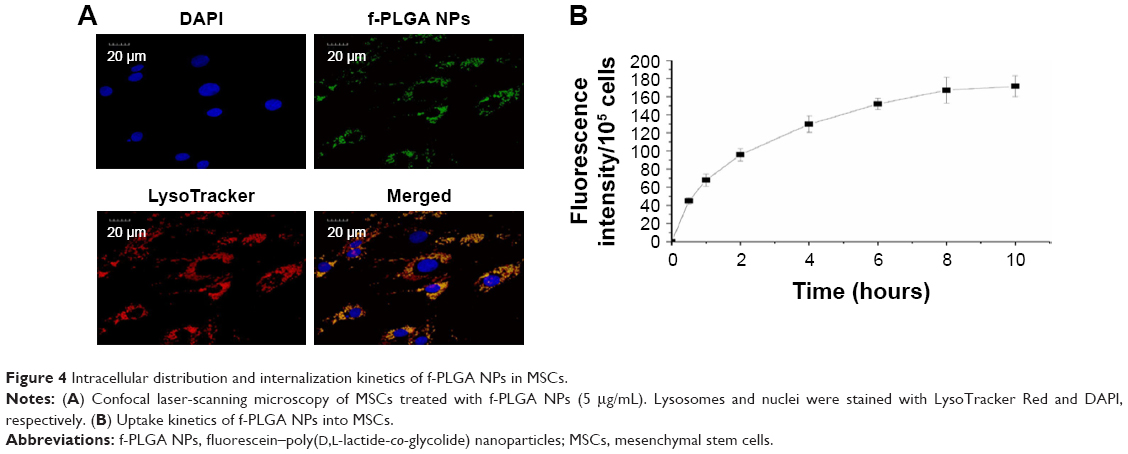 | Figure 4 Intracellular distribution and internalization kinetics of f-PLGA NPs in MSCs. |
In vitro tumor tropism of MSC NPs
A major challenge in the development of MSC NPs is the preservation of MSC migration for the delivery of toxic agents to tumor areas. Using a transwell migration assay, we showed that Ptx-PLGA-NP incubation decreased the number of migratory MSCs by about 50%. Surprisingly, these MSCs recovered from migration inhibition. Representative images of migratory MSCs loaded with 8 ng/mL Ptx-PLGA NPs are shown in Figure 5A. Restoration of the tumor-tropism trait was found to be both dose- and time-dependent (Figure 5B). No significant difference was observed between the number of migratory MSCs 3 days after drug loading for low-concentration Ptx-PLGA NPs (5 ng/mL) or unloaded MSCs. These results demonstrated almost full recovery of MSC-migration capacity. However, in the high-dose group (10 ng/mL), about 30% of MSCs were unable to migrate toward the C6 cells up to day 5. In the mid-dose group (8 ng/mL), 70% and 85% of MSCs migrated through the membrane pores on days 3 and 5, respectively.
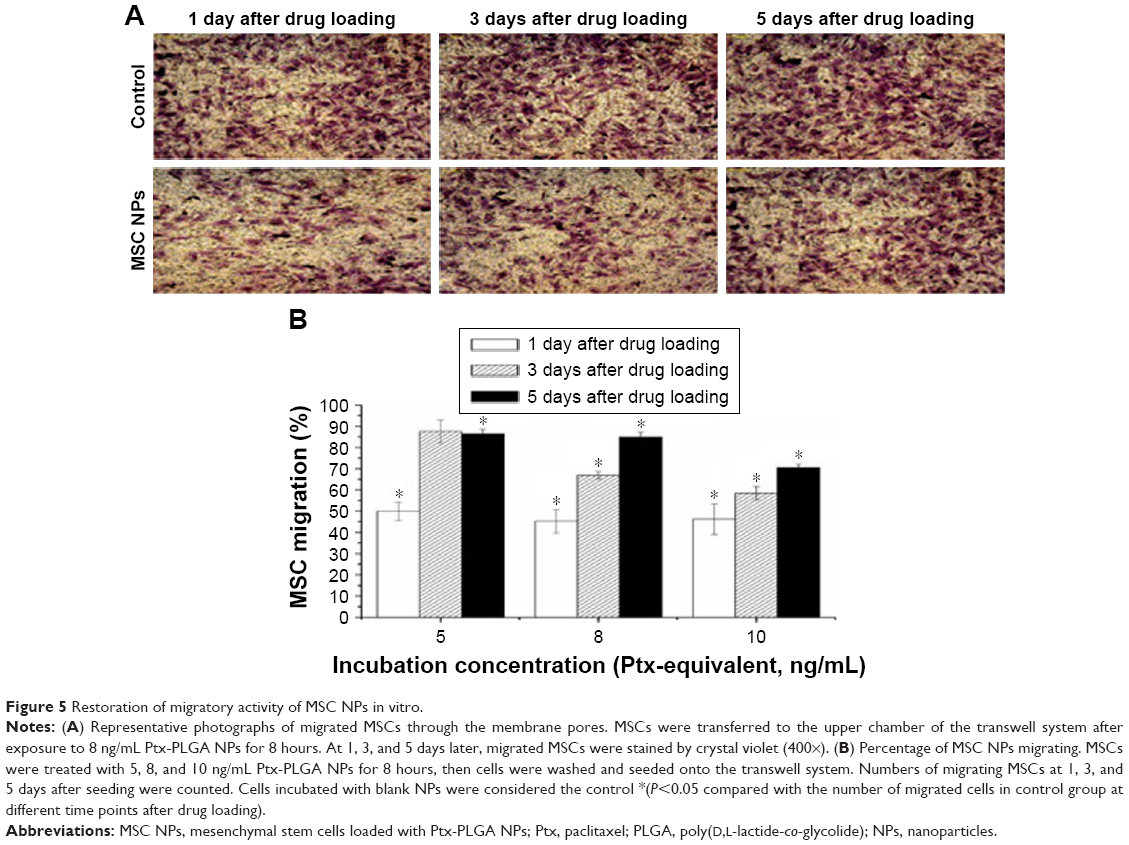 | Figure 5 Restoration of migratory activity of MSC NPs in vitro. |
Cell-cycle analysis of MSC NPs
To determine the effect of Ptx NPs on MSC division, cell-cycle analysis of MSC NPs was carried out by flow cytometry. Unloaded MSCs treated with PI were used as a control. Ptx exerts its cytotoxic effects by inducing tubular polymerization, resulting in unstable microtubules that interfere with mitotic spindle function and block cells in the G2/M phase of the cell cycle.20 Consequently, the number of MSCs in the G2/M phase was increased significantly after loading with Ptx-PLGA NPs (Figure 6B). Similarly to the migration-assay results, cell cycle phases of MSCs loaded with 8 ng/mL Ptx-PLGA NPs had recovered after a further 5 days of incubation (Figure 6A). Restoration of MSCs from G2/M arrest was also dose- and time-dependent. Compared with untreated MSCs, no significant differences were observed in percentages of cells in the G2/M phase at 5 days after drug loading for either the low (5 ng/mL) or middle (8 ng/mL) doses. However, the high dose (10 ng/mL) caused continuous G2/M-phase blocking (Figure 6B). Given these results, MSCs were incubated in subsequent experiments with 8 ng/mL Ptx-PLGA NPs for 8 hours to achieve high Ptx loading, undisturbed cell-cycle progression, and efficient tumor tropism. According to the level of Ptx released from the MSC NPs (“Drug transferred from MSC NPs to C6 glioma cells” section), the intracellular dose was calculated to be about 1 pg/cell (Ptx-equivalent).
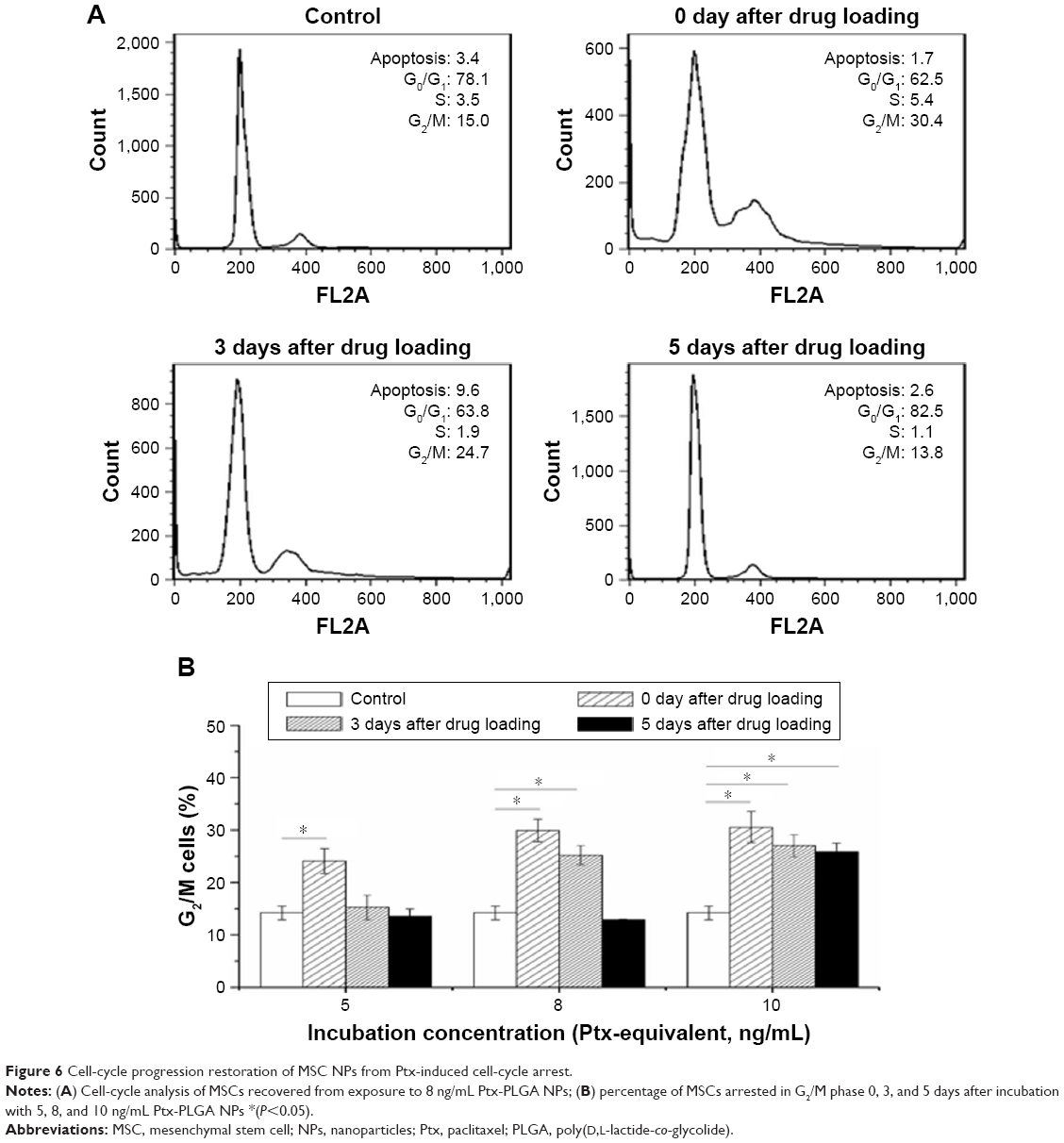 | Figure 6 Cell-cycle progression restoration of MSC NPs from Ptx-induced cell-cycle arrest. |
Osteogenic and adipogenic differentiation of MSC NPs
To examine whether internalized Ptx-PLGA NPs (8 ng/mL) would interfere with BMSC differentiation, MSC NPs were treated with inducing agents for osteogenic and adipogenic differentiation. Images were obtained 21 days after induction. There were no significant differences in either early (ALP production) or late (calcium deposition) osteogenic markers (Figure 7A) or in accumulation of lipid vacuoles (Figure 7B [red]) between treated and control BMSCs. Based on these findings, exposure of BMSCs to 8 ng/mL Ptx-PLGA NPs did not affect osteogenic or adipogenic differentiation.
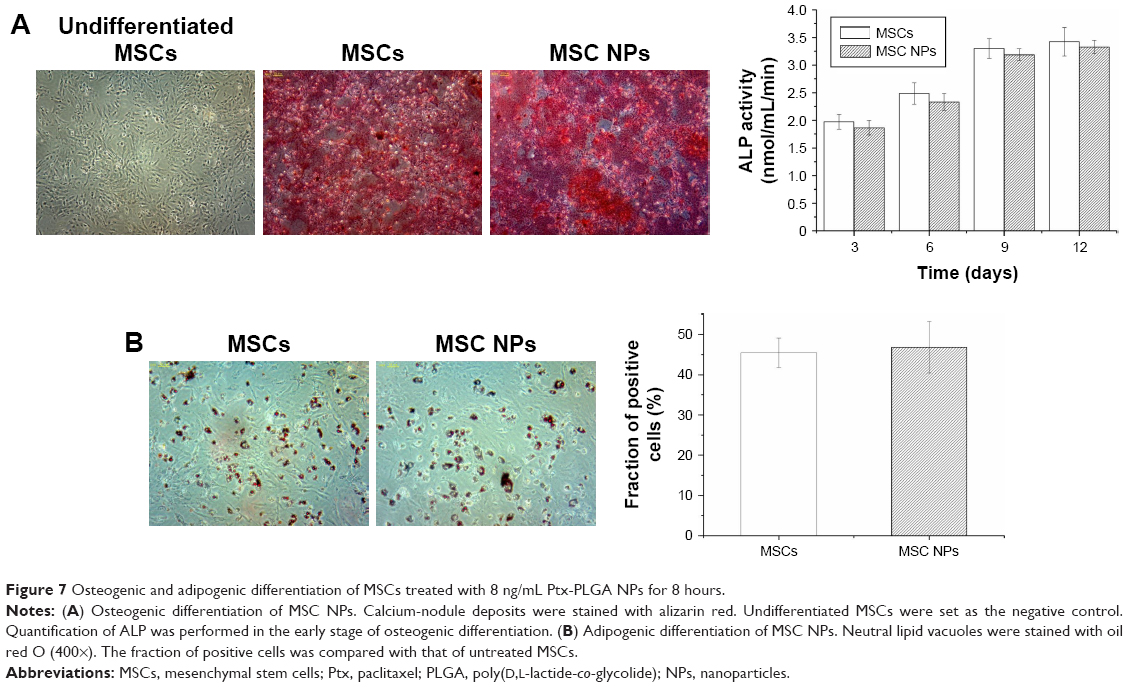 | Figure 7 Osteogenic and adipogenic differentiation of MSCs treated with 8 ng/mL Ptx-PLGA NPs for 8 hours. |
Drug transferred from MSC NPs to C6 glioma cells
Following NP loading into MSCs, efficient drug transfer to rat tumors was confirmed. Time-dependent export of Ptx by MSCs was evaluated by collecting cell lysates after drug withdrawal at different intervals (Figure 8B). A representative graph of HPLC results for intracellular Ptx is shown in Figure S7. MSCs primed with Ptx (MSC Ptx) were used as the control. At initiation of drug excretion, a higher intracellular Ptx level was measured in MSC NPs compared with MSC Ptx, indicating enhanced uptake of Ptx NPs. Both MSC Ptx and MSC NPs displayed an initially fast and then slow excretion process. During the process of MSC migration toward the tumor, sustained Ptx release increased from MSC NPs. Approximately 25% and 40% of intracellular Ptx, corresponding to 0.23±0.02 ng/cell and 0.38±0.02 ng/cell, was released from MSC Ptx and MSC NPs, respectively. Drug export from the MSC-Ptx group was almost completed in the first 6 hours. Ptx/cell (0.21 ng) was pumped rapidly out, and the intracellular drug was maintained at a stable level. No further intracellular drug reduction was observed from MSC NPs after sustained release 3 days later. Compared with MSC Ptx, enhanced drug transfer and reduced drug loss before MSC arrival at the tumor site was achieved by the incorporation of Ptx NPs into the MSCs.
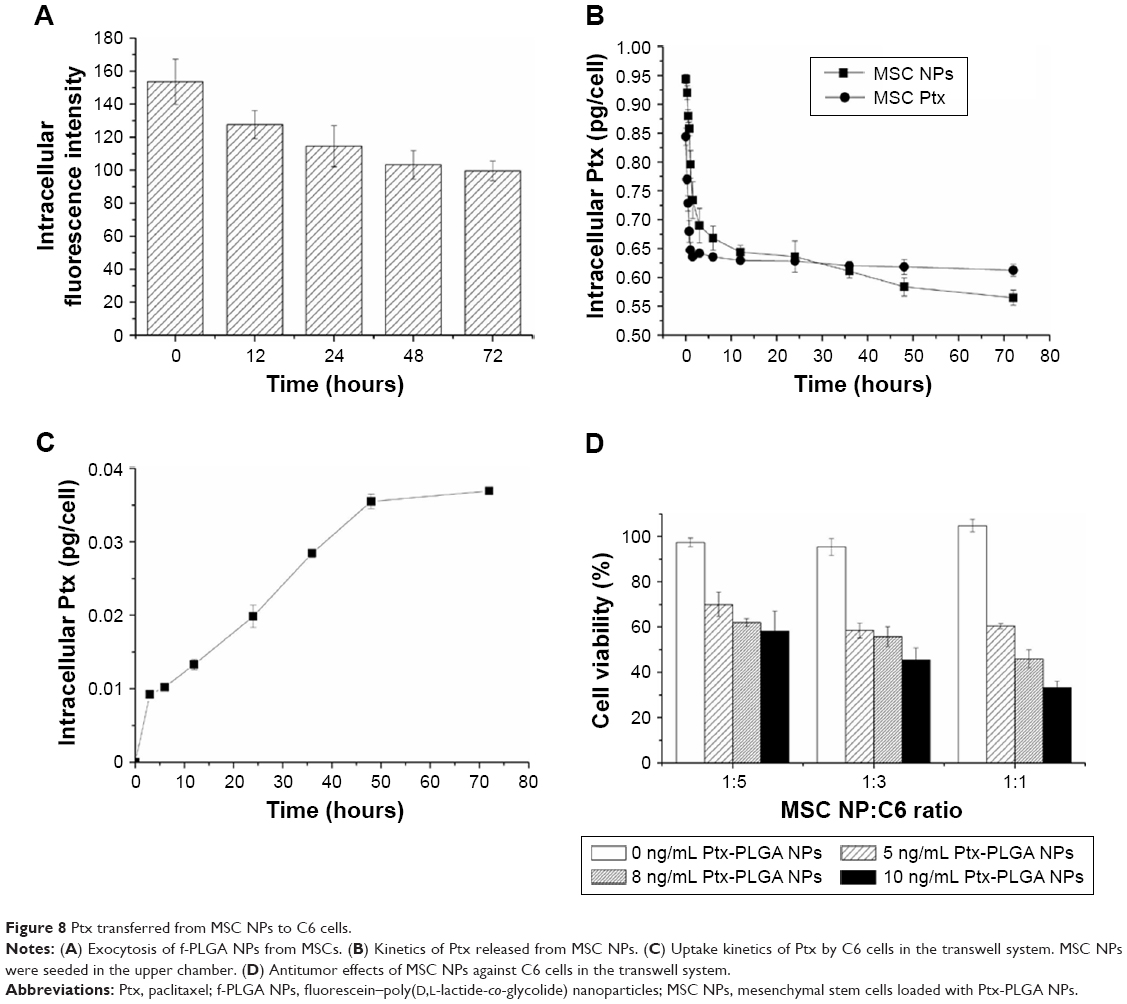 | Figure 8 Ptx transferred from MSC NPs to C6 cells. |
To determine the form of Ptx released (free or encapsulated), PLGA NP export from MSC NPs was investigated. This was achieved by synthesizing fluorescein-labeled PLGA to prepare f-PLGA NPs to demonstrate the exocytosis process of PLGA NPs in MSCs (Figure 8A). NP exocytosis was time-dependent, with 35% of internalized NPs being eliminated in 3 days. These findings support both free Ptx being released intracellularly and unreleased Ptx in PLGA NPs being exported from MSC NPs.
Figure 8C demonstrates the transfer of Ptx from MSC NPs to the lower chamber in the transwell system. In order to maintain relatively high viability of C6 cells during the kinetic study, the number of C6 cells was twice that seeded in the cytotoxicity study of MSC NPs towards C6 in the transwell system. The Ptx-uptake process was time-dependent and lasted for 48 hours. Efficient Ptx transfer was also confirmed by MSC-NP cytotoxicity against C6 cells. We found that antitumor activity was dose- and cell number-dependent (Figure 8D). The transwell system allowed passage of Ptx and Ptx-PLGA NPs, but not of MSCs. A higher ratio of MSC NPs to C6 cells and higher drug loading of MSCs resulted in increased Ptx export for transfer into the tumor cells. MSC NPs (1 pg/cell Ptx) decreased C6-cell survival by 40%–50%. Almost 100% of C6 cells survived, demonstrating that untreated MSCs had no effect on tumor cells.
Intratumoral distribution of implanted MSC NPs
CM-Dil was used as a red fluorescent probe to track the in vivo distribution of MSC NPs in a rat model. C6 glioma cells were injected into the left hemisphere of the rat brain, and 7 days later the right-brain hemisphere was injected with CM-Dil-stained MSC NPs. Two days after MSC NP injection, fluorescent MSC NPs were observed to be distributed in clusters (Figure 9B) at the site of MSC injection. Drug-loaded CM-Dil-MSCs were observed surrounding the tumor periphery, and were distributed throughout the tumor mass (Figure 9A), confirming retention of the migratory ability of MSC NPs toward C6 glioma tumors in vivo. These results were in accordance with the restoration of migration observed in vitro. Fluorescence intensity of CM-Dil-stained MSC NPs in the glioma area and contralateral normal tissues was 18,138±1,944 and 22,763±2,679, respectively, measured by ImageJ software. The drug-loaded MSC-migration rate toward gliomas in vivo was calculated to be 44.4%±5.4%. This clear in vivo tropism of MSC NPs suggested successful construction of a tumor-targeted Ptx-delivery system.
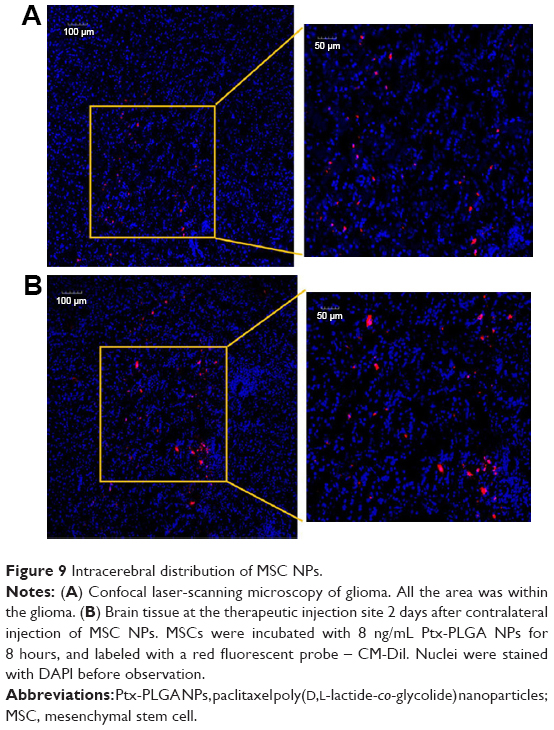 | Figure 9 Intracerebral distribution of MSC NPs. |
Antitumor efficacy in vivo
The antiglioma effect was investigated in an orthotopic C6 glioma-bearing rats. Ptx-PLGA NPs, MSC-Ptx, and MSC NPs were injected contralaterally at the same dose. On day 12 postinjection, rat status and neurological functions were recorded. Rats in the saline group injected with MSCs had poor ambulation, owing to hemiplegia. These rats were unable to move with pain stimulus and displayed ulcerations and bleeding around the eyes. Although the rats injected with Ptx-PLGA NPs and MSC Ptx had healthier mental status and moved in response to pain stimuli, they ambulated with notable paresis. Interestingly, rats in the MSC-NP group had no obvious abnormalities in consciousness or motor response and ambulated well, moving spontaneously when picked up by their tails.
Rat-survival evaluation is presented in Figure 10A as a Kaplan–Meier plot. Median and mean survival time were calculated by the log-rank test (Figure 10B). There was no significant difference in mean survival time between the MSC and saline groups. Furthermore, MSCs had no curative or stimulatory effect on glioma growth. Administration of Ptx significantly prolonged life span over the control groups. Rat-survival time was comparable between the Ptx-PLGA-NP and MSC-Ptx groups. MSC-NP treatment dramatically prolonged survival time in tumor-bearing rats compared with the Ptx-PLGA-NP and MSC-Ptx groups, with medians of 35.5, 24.5, 22.0, 13.5, and 14.5 days for MSC NPs, MSC Ptx, Ptx-PLGA NPs, MSCs, and saline, respectively.
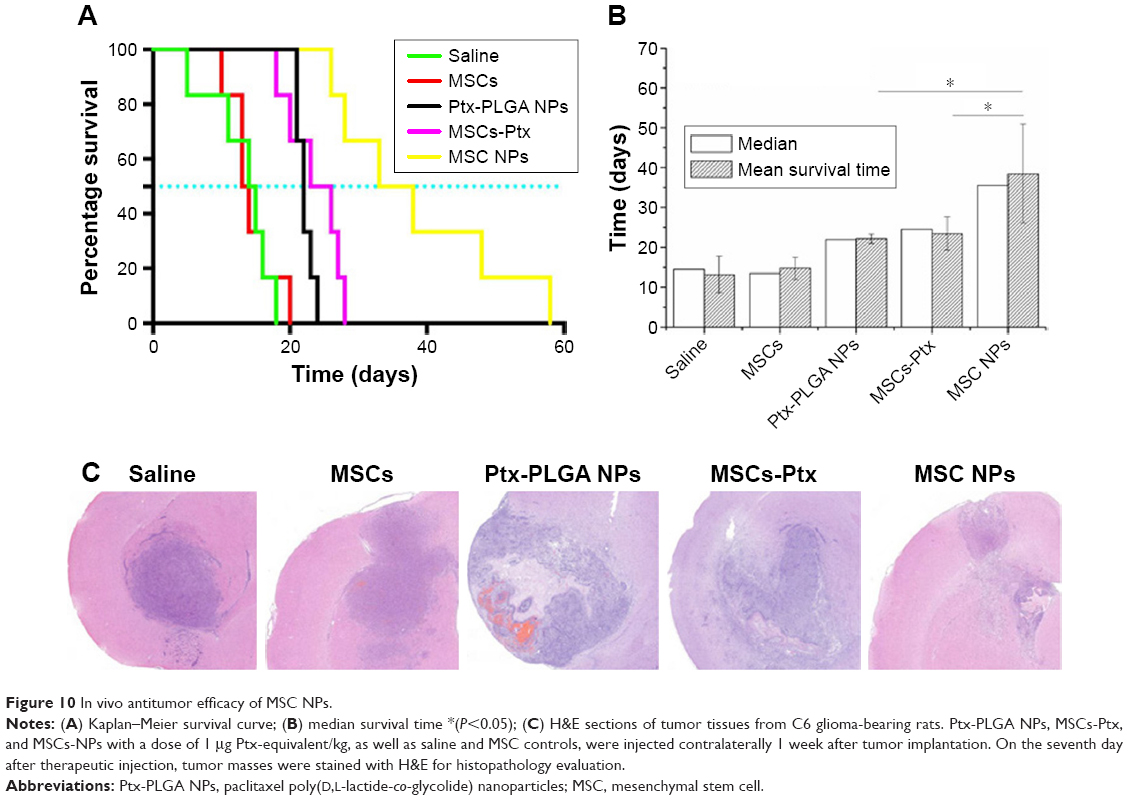 | Figure 10 In vivo antitumor efficacy of MSC NPs. |
Seven days after treatment, tumor tissue was evaluated by H&E staining (Figure 10C). There were no significant differences between the control and MSC groups, both of which exhibited large tracts of tumor tissue in the brain with amplified hyperchromatic nuclei. No necrosis or obvious apoptosis was observed in the tumor mass, and the tumor border was clear. However, tumors that received Ptx-PLGA NPs, MSC Ptx, or MSC NPs showed significant cell destruction and extensively damaged areas, with shrunken nuclei and obvious necrosis or apoptosis. The most significant reduction in glioma area was observed in the MSC-NP group. These data clearly indicated that MSC NPs had an effective antitumor effect in vivo.
Discussion
MSCs are attracted to TNFα, TGFβ, and other cytokines secreted within the tumor microenvironment, causing them to home toward infiltrated/disseminated tumor cells.21 Several studies have shown that MSCs have excellent tumor-homing activity toward intracranial neoplasms.22,23 Therefore, combining chemotherapeutic agents with MSCs to treat glioma is a promising therapeutic strategy. Ptx is a commonly used chemotherapeutic compound that acts as a potent microtubule toxin. It has exhibited potent cytotoxicity against malignant gliomas by intratumoral convection-enhanced delivery in Phase I/II clinical studies and combination therapy with topotecan and filgrastim or fractionated stereotactic radiotherapy in clinical trials.24–26 Recent in vitro studies have shown that after MSC uptake, the active form of Ptx can be released from MSC Ptx via the Pgp system or via membrane microvesicles, giving MSCs their strong antitumor activity.14,27 However, cell dysfunction after Ptx priming, mechanical filtration of circulating MSCs, and the blood–brain barrier make convenient systematic injection methods challenging, including injections into the tail vein, femoral vein, and carotid artery. This is because few MSCs engraft at the brain-tumor site.28 Until now, effective antiglioma-targeting chemotherapeutically integrated MSCs have only been achieved via direct intratumoral injection.12,13 Successful contralateral and intrathecal injections have only been reported using genetically engineered MSCs to preserve their tumor-homing ability.19,29 This study shows for the first time the effectiveness of MSC NPs injected contralaterally into an orthotopic glioma model.
MSC-NP construction requires careful optimization to acquire an appropriate dose at which MSC functions are maintained. Migration assays have demonstrated the effect of different chemotherapeutic doses on MSC function; however, the dynamic recovery process has not been accurately reported.11,12 The optimal Ptx-PLGA-NP-incubation concentration was determined to be 8 ng/mL (equivalent to 1 pg/cell intracellular Ptx), owing to almost complete restoration of migratory activity and cell-cycle patterns from transient disturbance after replacement with drug-free medium. Drug toxicity on MSC osteogenic and adipogenic differentiation was also assessed by stimulation with differentiation factors and genes.30 Our results showed that Ptx-PLGA NP loading did not substantially interfere with multilineage differentiation. Compared with C6 glioma cells, MSCs were more tolerant toward Ptx, which could be explained by the nonproliferative fibroblastic state adopted by the MSCs.31 With regard to the IC50 of Ptx-NPs on C6 cells, differences were observed between our results and those reported by Xu et al.32 The lower IC50 might have contributed to the encapsulation of Ptx into PLGA NPs and differences in incubation time.
Internalization of Ptx-PLGA NPs reduced Ptx toxicity toward MSCs (Figure 3), enabling relatively high drug loading into MSCs. With a pathway to export intracellular Ptx, Ptx released may exert antitumor activity at low concentration. The active form of Ptx and its antitumor effect has been investigated in MSC Ptx.6,27 One study reported that about 60% of Ptx was released from human gingival MSCs in 48 hours and the microtubule-bound Ptx remained in the cells.33 In the present study, we found that about 25% of Ptx was released from MSC Ptx. This relatively lower level might reflect different origins of the MSCs. For MSC Ptx, insufficient and quick-drug-release demand on increasing intracellular Ptx loading likely led to the loss of tumor tropism. However, the export of Ptx from MSC NPs was likely enhanced by the exocytosis of Ptx-PLGA NPs and efflux of free Ptx by Pgp or exosomes.
Unmodified NPs are mainly internalized into MSCs via the clathrin-dependent pathway, which ends up in lysosome entrapment.34,35 This was confirmed by our intracellular distribution results. Exocytosis of inorganic and polymeric NPs exists widely in many cell types, including stem cells, tumor cells, hepatocytes, fibroblasts, and epithelial cells. This process is closely related to the repair of cell membranes, antiautophagy, and maintenance of cell homeostasis.36 Endocytosed NPs colocalized to lysosomes mainly undergo lysosomal exocytosis by fusion lysosomes to plasma membrane.37 Although the dynamic efflux of a fluorescent probe encapsulated in PLGA NPs has been reported, exocytosis of PLGA NPs has not been studied.38 We thus synthesized a fluorescent PLGA polymer to prepare f-PLGA NPs. The efflux of Ptx by Pgp or secretion by exosomes is often measured in minutes, sometimes in hours.14,39 These processes were much faster than the exocytosis of PLGA NPs (Figure 8A), consistent with the exocytosis results of dysfunctional NPs, whereby exocytosis often finishes after several days.15,16 In addition to the controllable release of PLGA NPs in a time-dependent manner, slow and enhanced drug transfer was achieved between MSCs and tumor cells. Exported NPs with unreleased Ptx and free Ptx were reinternalized into C6 cells to induce cell death. Efficient drug transfer was observed and confirmed using the transwell system (Figure 8C and D).
In vivo antitumor evaluation was implemented in rats bearing intracranial C6 glioma cells. The role of MSCs in cancer development remains controversial: MSCs may promote or suppress tumor progression.40 In our previous study, we demonstrated that intravenous injection of 2×105 BMSCs into C57BL6 mice caused no remarkable abnormality in lung cells.2 In the present study, we found that there were no significant differences in survival time or tumor regions between those injected with unloaded MSCs and saline, which indicated that contralateral implantation of 2×105 BMSCs contributes neither aggregative nor inhibitory effects on brain tumors (Figure 10). The safety of MSCs in C6 glioma treatment was reported by Amano et al.41 Furthermore, human MSCs have been reported to inhibit tumor growth in orthotopic glioblastoma xenografts. The discrepancies between these results might be to differences in MSC origins, dose of MSC injection, and tumor models used.28
Although the intracellular Ptx level was much lower than that reported in the MSC-Ptx system, the observed antiglioma effect demonstrated that MSC loading with low-dose Ptx is potent.6,10 Low-dose Ptx loading preserved the homing property of MSCs and enabled MSC NPs to arrive at the tumor area in the contralateral hemisphere within 2 days. This is an obviously shorter time frame than the 2–5 migration days with MSC injection, which could be explained by migration over time to the brain from predominant entrapment in the lung vasculature42 and the shorter migration distance between the two hemispheres.43 Ptx-loaded fluorescent MSCs migrated and penetrated the glioma to offload Ptx in the manner previously described. Meanwhile, MSC-NP aggregation at the injection site had the potential to regain tumor tropism after recovery for 3–5 days (Figure 4B). The enhanced antitumor activity achieved in rats administered with MSC NPs could be explained by efficient loading of Ptx-NPs in MSCs, cell-function maintenance of MSC NPs, and sustained and efficient drug transfer from MSC NPs to tumor cells.
Conclusion
We successfully established a tumor-targeting vector combining MSCs with chemotherapeutically encapsulated NPs for glioma therapy. The NPs were taken in readily by the MSCs and toxicity reduced. An optimal antitumor effect was achieved by contralateral implantation of this delivery system into orthotopic glioma rats, because MSC NPs infiltrated from normal brain parenchyma, then migrated toward gliomas and efficiently unloaded the drug in a sustained way. The use of MSCs as cellular carriers provides a novel approach for the delivery of nanoparticulate drugs to gliomas, thus representing a promising therapeutic strategy for targeted chemotherapy.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81402872, 81620108028) and the Natural Science Foundation of Zhejiang Province of China (LY17H160002, LY17B020002, LY14C090001).
Disclosure
The authors report no conflicts of interest in this work.
References
Chang L, Su J, Jia X, Ren H. Treating malignant glioma in Chinese patients: update on temozolomide. Onco Targets Ther. 2014;7:235–244. | ||
Zhang TY, Huang B, Yuan ZY, et al. Gene recombinant bone marrow mesenchymal stem cells as a tumor-targeted suicide gene delivery vehicle in pulmonary metastasis therapy using non-viral transfection. Nanomedicine. 2014;10(1):257–267. | ||
Zhang TY, Huang B, Wu HB, et al. Synergistic effects of co-administration of suicide gene expressing mesenchymal stem cells and prodrug-encapsulated liposome on aggressive lung melanoma metastases in mice. J Control Release. 2015;209:260–271. | ||
Choi SA, Lee YE, Kwak PA, et al. Clinically applicable human adipose tissue-derived mesenchymal stem cells delivering therapeutic genes to brainstem gliomas. Cancer Gene Ther. 2015;22(6):302–311. | ||
Bexell D, Svensson A, Bengzon J. Stem cell-based therapy for malignant glioma. Cancer Treat Rev. 2013;39(4):358–365. | ||
Pessina A, Bonomi A, Coccè V, et al. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS One. 2011;6(12):e28321. | ||
Bonomi A, Silini A, Vertua E, et al. Human amniotic mesenchymal stromal cells (hAMSCs) as potential vehicles for drug delivery in cancer therapy: an in vitro study. Stem Cell Res Ther. 2015;6(1):1–10. | ||
Cheng H, Kastrup CJ, Ramanathan R, et al. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano. 2010;4(2):625–631. | ||
Li L, Guan Y, Liu H, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011;5(9):7462–7470. | ||
Dai T, Yang E, Sun Y, et al. Preparation and drug release mechanism of CTS-TAX-NP-MSCs drug delivery system. Int J Pharm. 2013;456(1):186–194. | ||
Zhao Y, Tang S, Guo J, et al. Targeted delivery of doxorubicin by nano-loaded mesenchymal stem cells for lung melanoma metastases therapy. Sci Rep. 2017;7:44758. | ||
Clavreul A, Montagu A, Lainé AL, et al. Targeting and treatment of glioblastomas with human mesenchymal stem cells carrying ferrociphenol lipid nanocapsules. Int J Nanomedicine. 2015;10:1259–1271. | ||
Zhang X, Yao S, Liu C, Jiang Y. Tumor tropic delivery of doxorubicin-polymer conjugates using mesenchymal stem cells for glioma therapy. Biomaterials. 2015;39:269–281. | ||
Pascucci L, Coccè V, Bonomi A, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262–270. | ||
Fang CY, Vaijayanthimala V, Cheng CA, et al. The exocytosis of fluorescent nanodiamond and its use as a long-term cell tracker. Small. 2011;7(23):3363–3370. | ||
Sakhtianchi R, Minchin RF, Lee KB, et al. Exocytosis of nanoparticles from cells: role in cellular retention and toxicity. Adv Colloid Interface Sci. 2013;201–202:18–29. | ||
Tosi G, Rivasi F, Gandolfi F, et al. Conjugated poly(D,L-lactide-co-glycolide) for the preparation of in vivo detectable nanoparticles. Biomaterials. 2005;26(19):4189–4195. | ||
Gomezgaete C, Tsapis N, Besnard M, et al. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int J Pharm. 2007;331(2):153–159. | ||
Gu C, Li S, Tokuyama T, et al. Therapeutic effect of genetically engineered mesenchymal stem cells in rat experimental leptomeningeal glioma model. Cancer Lett. 2010;291(2):256–262. | ||
Costa MA, Simon DI. Molecular basis of restenosis and drug-eluting stents. Circulation. 2005;111(17):2257–2273. | ||
Hu YL, Fu YH, Tabata Y, Gao JQ. Mesenchymal stem cells: a promising targeted-delivery vehicle in cancer gene therapy. J Control Release. 2010;147(2):154–162. | ||
Menon LG, Pratt J, Yang HW, et al. Imaging of human mesenchymal stromal cells: homing to human brain tumors. J Neurooncol. 2012;107(2):257–267. | ||
Huang X, Zhang F, Wang Y, et al. Design considerations of iron-based nanoclusters for noninvasive tracking of mesenchymal stem cell homing. ACS Nano. 2014;8(5):4403–4414. | ||
Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100(3):472–479. | ||
Pipas JM, Meyer LP, Rhodes CH, et al. A phase II trial of paclitaxel and topotecan with filgrastim in patients with recurrent or refractory glioblastoma multiforme or anaplastic astrocytoma. J Neurooncol. 2005;71(3):301–305. | ||
Ashamalla H, Zaki B, Mokhtar B, et al. Fractionated stereotactic radiotherapy boost and weekly paclitaxel in malignant gliomas clinical and pharmacokinetics results. Technol Cancer Res Treat. 2007;6(3):169–175. | ||
Bonomi A, Ghezzi E, Pascucci L, et al. Effect of canine mesenchymal stromal cells loaded with paclitaxel on growth of canine glioma and human glioblastoma cell lines. Vet J. 2017;223:41–47. | ||
Pacioni S, D’Alessandris QG, Giannetti S, et al. Human mesenchymal stromal cells inhibit tumor growth in orthotopic glioblastoma xenografts. Stem Cell Res Ther. 2017;8(1):53. | ||
Kinoshita Y, Kamitani H, Mamun MH, et al. A gene delivery system with a human artificial chromosome vector based on migration of mesenchymal stem cells towards human glioblastoma HTB14 cells. Neurol Res. 2010;32(4):429–437. | ||
Ranjbarvaziri S, Kiani S, Akhlaghi A, et al. Quantum dot labeling using positive charged peptides in human hematopoetic [sic] and mesenchymal stem cells. Biomaterials. 2011;32(22):5195–5205. | ||
Bosco DB, Kenworthy R, Zorio DA, et al. Human mesenchymal stem cells are resistant to paclitaxel by adopting a non-proliferative fibroblastic state. PLoS One. 2015;10(6):e0128511. | ||
Xu Y, Shen M, Li Y, et al. The synergic antitumor effects of paclitaxel and temozolomide co-loaded in mPEG-PLGA nanoparticles on glioblastoma cells. Oncotarget. 2016;7(15):20890–20901. | ||
Coccè V, Farronato D, Brini AT, et al. Drug loaded gingival mesenchymal stromal cells (GinPa-MSCs) inhibit in vitro proliferation of oral squamous cell carcinoma. Sci Rep. 2017;7:9376. | ||
Saulite L, Dapkute D, Pleiko K, et al. Nano-engineered skin mesenchymal stem cells: potential vehicles for tumour-targeted quantum-dot delivery. Beilstein J Nanotechnol. 2017;8:1218–1230. | ||
Shi H, He X, Yuan Y, et al. Nanoparticle-based biocompatible and long-life marker for lysosome labeling and tracking. Anal Chem. 2010;82(6):2213–2220. | ||
Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014;9 Suppl 1:51–63. | ||
Yanes RE, Tarn D, Hwang AA, et al. Involvement of lysosomal exocytosis in the excretion of mesoporous silica nanoparticles and enhancement of the drug delivery effect by exocytosis inhibition. Small. 2013;9(5):697–704. | ||
Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D,L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20(2):212–220. | ||
Jang SH, Wientjes MG, Au JL. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J Pharmacol Exp Ther. 2001;298(3):1236–1242. | ||
Yagi H, Kitagawa Y. The role of mesenchymal stem cells in cancer development. Front Genet. 2013;4:261. | ||
Amano S, Gu C, Koizumi S, et al. Tumoricidal bystander effect in the suicide gene therapy using mesenchymal stem cells does not injure normal brain tissues. Cancer Lett. 2011;306(1):99–105. | ||
Kim SM, Jeong CH, Woo JS, et al. In vivo near-infrared imaging for the tracking of systemically delivered mesenchymal stem cells: tropism for brain tumors and biodistribution. Int J Nanomedicine. 2015;11:13–23. | ||
Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97(23):12846–12851. |
Supplementary materials
 | Figure S1 C6 implantation and MSC-NP therapeutic injection (cross-section). |
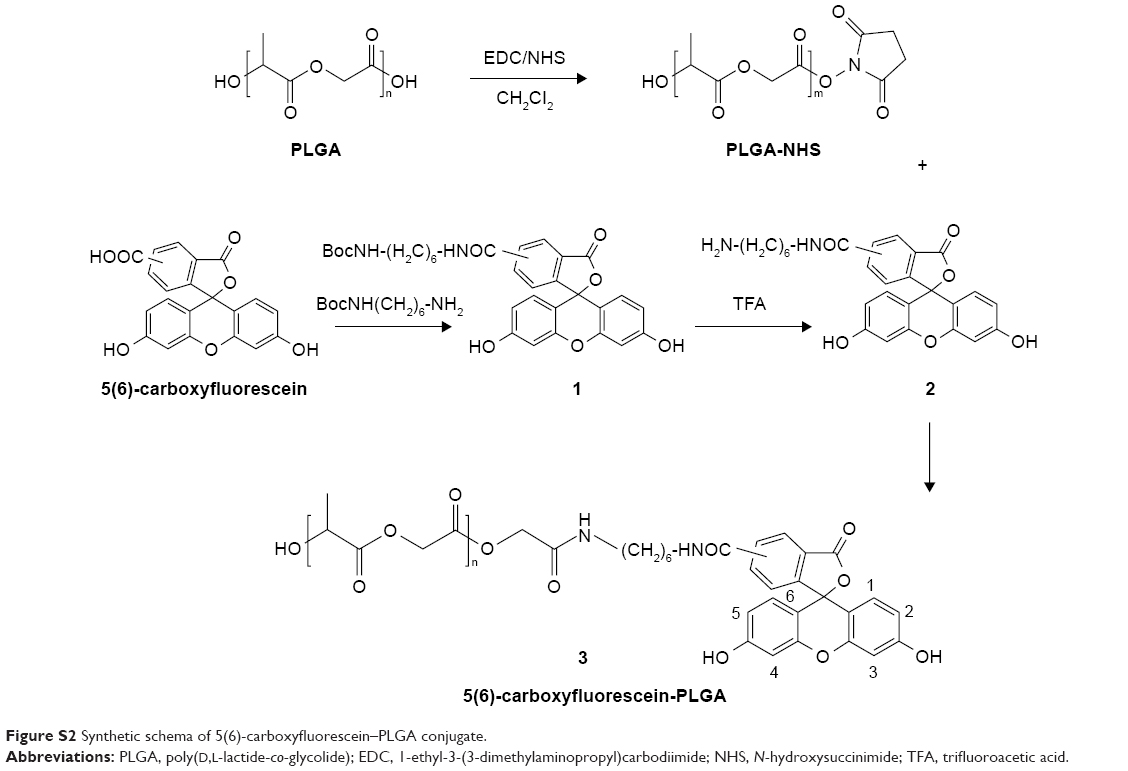 | Figure S2 Synthetic schema of 5(6)-carboxyfluorescein–PLGA conjugate. |
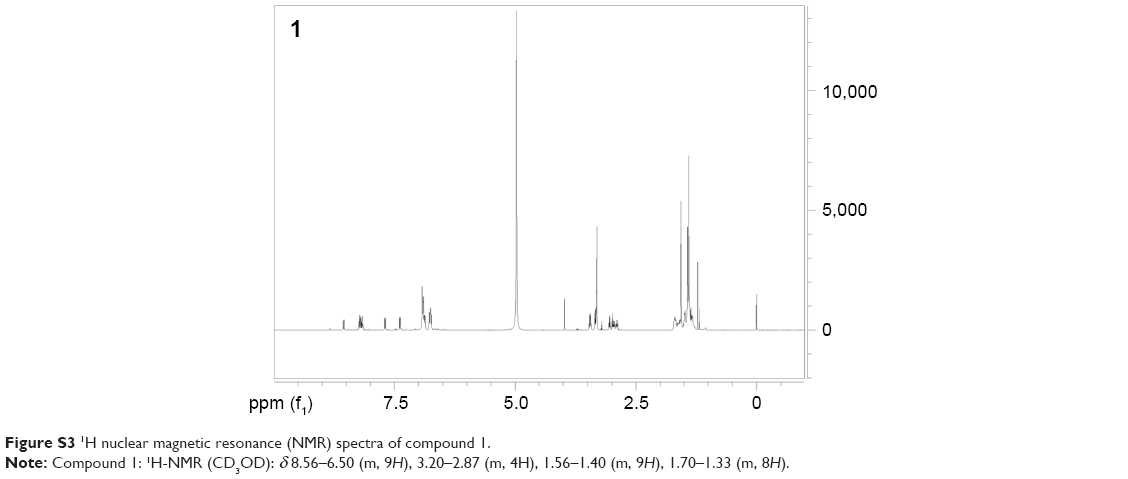 | Figure S3 1H nuclear magnetic resonance (NMR) spectra of compound 1. |
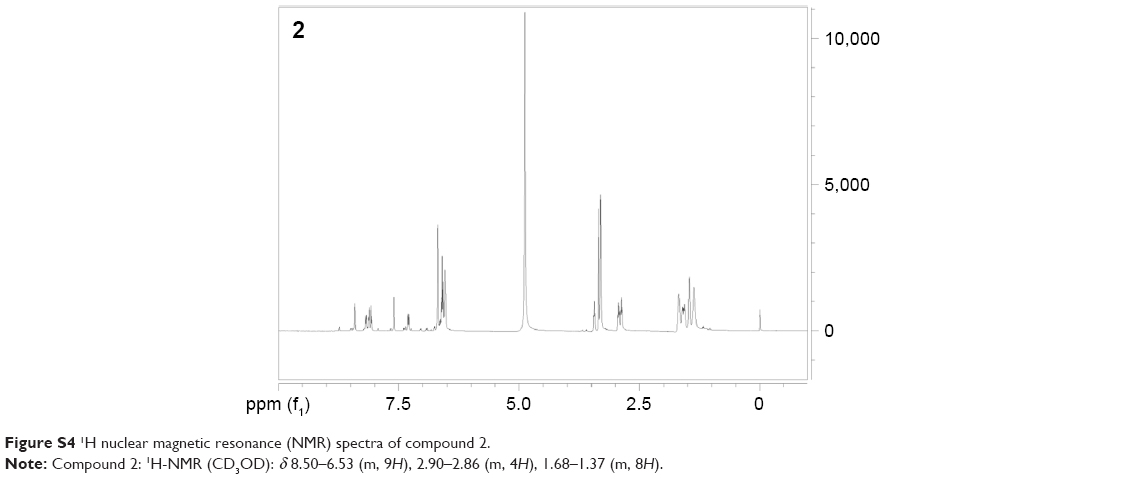 | Figure S4 1H nuclear magnetic resonance (NMR) spectra of compound 2. |
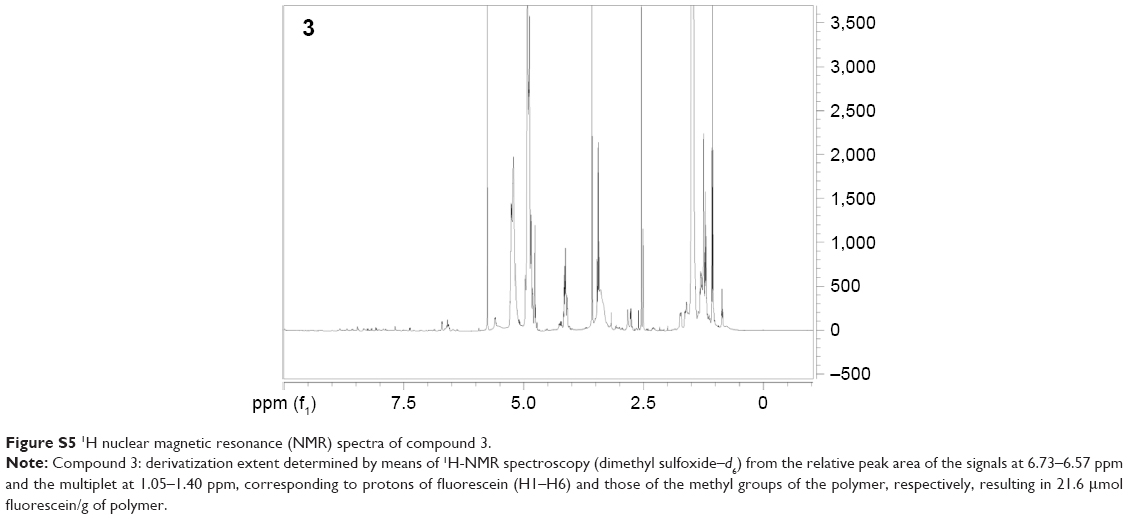 | Figure S5 1H nuclear magnetic resonance (NMR) spectra of compound 3. |
 | Figure S6 Fluorescence of f-PLGA NPs. |
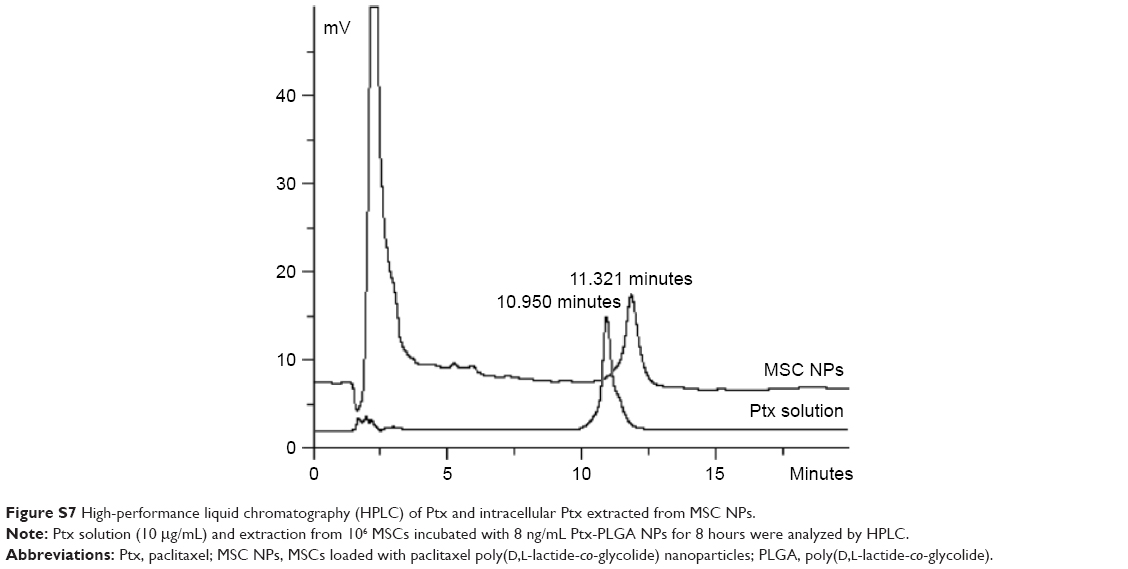 | Figure S7 High-performance liquid chromatography (HPLC) of Ptx and intracellular Ptx extracted from MSC NPs. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.