Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 6
Melatonergic drugs in development
Authors Carocci A, Catalano A, Sinicropi MS
Received 7 May 2014
Accepted for publication 17 June 2014
Published 18 September 2014 Volume 2014:6 Pages 127—137
DOI https://doi.org/10.2147/CPAA.S36600
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Alessia Carocci,1 Alessia Catalano,1 Maria Stefania Sinicropi2
1Department of Pharmacy–Drug Sciences, University of Bari Aldo Moro, Bari, 2Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Cosenza, Italy
Abstract: Melatonin (N-acetyl-5-methoxytryptamine) is widely known as "the darkness hormone". It is a major chronobiological regulator involved in circadian phasing and sleep-wake cycle in humans. Numerous other functions, including cyto/neuroprotection, immune modulation, and energy metabolism have been ascribed to melatonin. A variety of studies have revealed a role for melatonin and its receptors in different pathophysiological conditions. However, the suitability of melatonin as a drug is limited because of its short half-life, poor oral bioavailability, and ubiquitous action. Due to the therapeutic potential of melatonin in a wide variety of clinical conditions, the development of new agents able to interact selectively with melatonin receptors has become an area of great interest during the last decade. Therefore, the field of melatonergic receptor agonists comprises a great number of structurally different chemical entities, which range from indolic to nonindolic compounds. Melatonergic agonists are suitable for sleep disturbances, neuropsychiatric disorders related to circadian dysphasing, and metabolic diseases associated with insulin resistance. The results of preclinical studies on animal models show that melatonin receptor agonists can be considered promising agents for the treatment of central nervous system-related pathologies. An overview of recent advances in the field of investigational melatonergic drugs will be presented in this review.
Keywords: MT1/MT2 ligands, circadian rhythms, melatonin
Introduction
Melatonin (MLT), N-acetyl-5-methoxytryptamine (Figure 1), a product of tryptophan metabolism, is the main secretory product of the pineal gland.1 Its release follows a marked circadian rhythm, governed by the central circadian pacemaker in the suprachiasmatic nucleus (SCN) of the hypothalamus, the highest levels occurring during the period of darkness.2 While MLT concentrations found in circulating plasma and cerebrospinal fluid are almost exclusively of pineal origin, MLT seemed to be also produced in other tissues, such as retina,3,4 lymphocytes,5 and gastrointestinal tract,6,7 but the papers claiming this had no convincing sequel. Tryptophan serves as the precursor for the biosynthesis of MLT. It is converted into serotonin via 5-hydroxytryptophan. Serotonin (5-HT) is then acetylated to form N-acetylserotonin by arylalkylamine N-acetyltransferase, which has also been called “the timezyme”, since N-acetylation of serotonin is usually regarded as the rate-limiting step of MLT formation, and determinant of its circadian rhythm.8 N-acetylserotonin is then converted to MLT by hydroxyindole-O-methyltransferase. In the mammalian pineal gland, MLT biosynthesis is synchronized to the light/dark cycle by the SCN, which receives its input from the retinohypothalamic tract. Special photoreceptive retinal ganglion cells, containing melanopsin as a photopigment, are involved in the projection from retina.9 Fibers from the SCN pass through a circuitous route, involving the paraventricular nucleus of the hypothalamus, then proceed to innervate the pineal gland, as postganglionic sympathetic fibers. Norepinephrine released from these fibers binds to postsynaptic adrenoceptors, whose activation induces an increase in cyclic adenosine-3′,5′-monophosphate (cAMP) accumulation and a subsequent activation of N-acetyltransferase.10 MLT regulates and modulates a wide variety of physiological functions. Besides the well-known chronobiotic and sleep-inducing properties,11 many other physiological effects have been ascribed to MLT, such as modulation of the cardiovascular system,12 enhancement of immunological functioning,13 and influence on hormone secretion and metabolism.14 Other functions of MLT described in the literature include anti-inflammatory,15 pain modulator,16 analgesic,17 and neuroprotective,18,19 as well as free-radical scavenging and antioxidant properties.20,21 More recently, a role for MLT and melatonin receptors has been found in cancer protection,22,23 glucose regulation,24 bone physiology,25 and neurodegenerative disorders.26 Moreover, many research findings provide scientific evidence for the protective role of MLT in a number of oxidative stress-related diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD); the protective actions of MLT are attributable to its direct and indirect antioxidative properties. With respect to PD, a beneficial effect of melatonin in preclinical and cell culture models has been reported.27 Melatonin treatment has been reported to prevent neuronal dopamine loss or dopamine transporter downregulation induced by rotenone, 6-hydroxydopamine, or 1-methyl-4-phenylpyridinium. MLT exerts its actions via multiple mechanisms. Many of its physiological actions are mediated through G protein-coupled receptors, expressed in a wide variety of tissues. Membrane-bound melatonin receptors (MT1 and MT2) have been identified and cloned from several tissues in the body.28,29 They are localized in different areas of the central nervous system as well as in peripheral tissues.30,31 These receptors belong to the superfamily of G protein-coupled receptors that contain the typical 7-transmembrane domain.32 Activation of melatonin receptors, in general leads, to a decrease in cAMP concentration. The exact physiological roles of MT1 and MT2 receptors have not been not well-defined yet. However, MT1 receptors seem to be involved in the sleep-promoting effects of melatonin,33,34 while MT2 receptors appear to play a major role in the resynchronizing activity of MLT,35,36 and seem to be involved in the pathophysiology and pharmacology of sleep disorders, anxiety, depression, AD, and pain.37 As regards vasal activity, MT1 receptors seem to be involved in mediating vasoconstriction,38 and MT2 in vasodilation. Moreover, a low-affinity MLT binding site, termed MT3, has been characterized as a MLT-sensitive form of the human enzyme, quinone reductase 2.39 Many in vitro and in vivo experimental models have contributed to demonstrate the role of MLT as an efficient radical scavenger against several reactive oxygen species; for example, the hydroxyl radical, the peroxynitrite anion, the superoxide anion, and singlet oxygen.40 MLT has also been shown to enhance the production and the activity of several antioxidant enzymes, including superoxide dismutase, glutathione peroxidase, glutathione reductase, catalase, and glucose-6-phosphate dehydrogenase.41,42 Finally, MLT has been shown to bind directly to calmodulin in vitro; furthermore, its ability to inhibit Ca2+/calmodulin-dependent kinase II at physiological concentrations has been reported.43
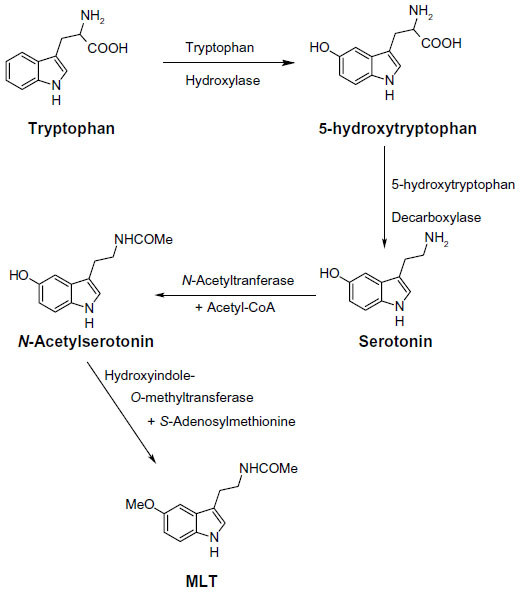 | Figure 1 Biosynthetic pathway of melatonin (MLT). |
The effects of MLT on the Plasmodium cell cycle were first described by Hotta et al in a work that demonstrated that the synchronization of parasite development is lost in pinealectomized mice, but restored when MLT is administered.44 The effects of MLT in Plasmodium falciparum were extensively evaluated in several works, which revealed a complex signaling pathway.45 MLT seems to have a central role in the control of parasite replication and the establishment of parasitemia. Therefore, targeting and blocking this hormone pathway can contribute to the discovery of new antimalarial drugs.
Melatonergic drugs in clinical and preclinical practice
As MLT exhibits both hypnotic and chronobiotic properties, it has been used for the treatment of age-related insomnia, as well as of other primary and secondary insomnia.46,47 A recent consensus of the British Association for Psychopharmacology, on evidence-based treatment of insomnia, parasomnia, and circadian rhythm sleep disorders, concluded that MLT is the first-choice treatment when a hypnotic is indicated in patients >55 years.48 MLT has also been used successfully for the treatment of sleep problems related to perturbations of the circadian timekeeping system, like those caused by jetlag, shift work disorder, and delayed sleep phase syndrome.49,50 Unfortunately, a substitution therapy is not easily achieved with MLT. The major obstacle for the purposes of treatment is its rapid elimination from the circulation. Its half-life in the blood varies from less than 30 minutes to (maximally) about 45 minutes. Various factors contribute to its relatively poor bioavailability, such as uptake, hepatic first-pass metabolism; also, perhaps, transient loading to the gastrointestinal tract.51,52 Therefore, if only a chronobiotic action is desired (ie, induction of phase shifts to readjust the circadian oscillator system), MLT may be fully effective.53 However, if the restitution of a complete nocturnal peak would be desired, the clinical outcome with the rapidly-metabolized, natural hormone remains relatively poor. The development of either prolonged-release formulations of the natural hormone, or melatonergic drugs of longer half-life, represents two different solutions to this problem. In 2007, a prolonged-release formulation of MLT (Circadin®, Neurim Pharmaceuticals, Tel-Aviv, Israel) was approved in Europe by the European Medicines Agency (EMEA) for the treatment of insomnia in patients aged >55 years.54 It was licensed on the basis of the combination of improvement of sleep quality and next-day feeling. Although this formulation improves several sleep parameters by statistical measures, its efficacy is below that of conventional hypnotics. However, it has the advantage of being devoid of the typical side effects of benzodiazepines, such as next-day hangover, withdrawal effects, and dependence liability. Another obstacle for the clinical use of MLT is the lack of selectivity for the different biological targets, including MT1 and MT2 receptors. These difficulties should be overcome with the development of selective melatonergic ligands, with improved properties. Numerous patents claiming the use of melatonergic agonists, mostly for the treatment of sleep disorders, have been filed in the past decade. Melatonergic drugs now used in therapy (ramelteon55 and agomelatine56), as well as the most advanced melatonergic ligands under clinical evaluation (tasimelteon57 and TIK-30158) (Figure 2 ), will be described below.
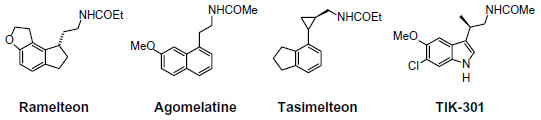 | Figure 2 Structures of melatonergic ligands in clinical and preclinical practice. |
Ramelteon
Ramelteon (Rozerem®; TAK-375, Takeda Pharmaceutical Co, Osaka, Japan) (Figure 2) was the first melatonergic agonist approved for human use. In 2005, it was licensed in the USA by the US Food and Drug Administration (FDA) for the treatment of insomnia. It is a tricyclic, synthetic analog of MLT with the chemical name of (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno[5,4-b]furan-8-yl)-ethyl]propionamide. In vitro binding studies have shown that ramelteon’s affinity for MT1 (Ki=0.014 nM) and MT2 (Ki=0.112 nM) receptors is 3 to 16 times greater than for MLT.59 The selectivity of ramelteon for MT1 has been found to be greater than for MT2, thus suggesting that it targets sleep onset more specifically than MLT itself does. Ramelteon does not bind with substantial affinity to quinone reductase 2, and seems to have no influence on calmodulin-dependent actions, as is known of MLT. It has negligible affinities to the majority of serotonin receptors, except for a rather moderate binding to 5-HT1A (pKi=5.25), and to gamma-aminobutyric acid, dopamine, norepinephrine, acetylcholine, and opiate receptors.60 When compared to MLT, ramelteon has a higher lipophilicity. Therefore, it is more easily taken up and retained by tissues. Ramelteon is usually administered by the oral route at a dose of 8 mg, or 4 mg in elderly subjects, and is rapidly absorbed by the gastrointestinal tract, with an absorption rate, under fasting conditions, of about 84%. It is characterized by a half-life of 0.83–1.93 hours, which is indeed longer than that of MLT (20–30 minutes).61 Ramelteon undergoes extensive liver metabolism via oxidation to hydroxyl and carbonyl groups, and is then conjugated with glucuronide.62 Cytochrome P450 isoenzyme CYP1A2 is the major hepatic enzyme involved in ramelteon metabolism. The major metabolite is a product of ω-1 hydroxylation in the propionamide side chain. Although the activity of the major metabolite is 30-fold lower than that of ramelteon, its serum levels are 20 to 100-fold higher than those of the parent drug, suggesting that it may significantly contribute to the overall clinical effect of ramelteon.63 Like MLT, ramelteon exerts its direct sleep-inducing effects via MT1 receptors located in the SCN, while inducing phase shifting mainly via MT2 receptors. In all clinical studies undertaken so far to evaluate the efficacy and safety of ramelteon in various doses ranging from 4–32 mg/day in patients with chronic insomnia, the drug reduced sleep onset latency and increased sleep duration.64,65 Besides acting as a sedative-hypnotic drug, ramelteon also exhibited chronobiotic properties.66 Interestingly, impairment of MLT rhythm may play a role in the development of sleep disturbances and delirium in critically ill patients, and it seems that MLT replenishment could decrease the incidence of these phenomena.67 Evidence of the association between MLT and delirium is limited.68 Studies of the effects of ramelteon in delirium have been recently reported.69
Agomelatine
Another approved melatonergic drug is agomelatine (Valdoxan®; S20098, Servier Laboratories, Neuilly-sur-Seine, France) (Figure 2), the naphtalenic analogue of MLT, which has been licensed in Europe by EMEA for the treatment of major depressive episodes in adults. Agomelatine is a potent MT1 and MT2 agonist (with pKi values of 10.21 and 9.57, respectively), which displays antagonist behavior at 5-HT2C serotonin receptors (pKi=6.68).56 It does not show any significant affinities for muscarinic, histaminergic, adrenergic, or dopaminergic receptor subtypes.70 The blockade of 5-HT2C receptors has been interpreted to explain the direct antidepressant actions of agomelatine.56 This action should be distinguished from its indirect melatonergic actions related to adjustments of circadian rhythms, which are effective in subtypes of depression having an etiology of circadian dysfunction. A synergistic interaction of melatonergic and 5-HT2C-signaling mechanisms has been assumed, to explain the direct antidepressant action of this drug.71 The stimulation of both melatonergic receptors and the blockade of 5-HT2C seem to be essential for the antidepressant effect of agomelatine.72 It has been hypothesized that, through this dual action, agomelatine may promote and maintain sleep at night, and help to maintain alertness during daytime. Agomelatine given before sleep would have an immediate sleep-promoting melatonergic effect, which would prevail over its potentially antihypnotic 5-HT2C-antagonism. In contrast, during the day, the drug antagonism towards 5-HT2C receptors would predominate over the melatonergic action, thus having an alerting action.73 Agomelatine has a short half-life (~2 hours) in humans. It is absorbed rapidly by the oral route and is metabolized in the liver by cytochrome P450 isoenzymes CYPA1, CYPA2, and CYP2C9. 3-Hydroxy-, 3-hydroxy-7-methoxy-, and 7-desmethyl-agomelatine (S20098) were identified as the three metabolites of agomelatine.74,75 Agomelatine has been also shown to display robust anxiolytic properties, which seem to be superior to those of MLT and which may, again, be explained by a concerted action of melatonergic stimulation and 5-HT2C-inhibition.76 In a recent study, agomelatine has been used successfully for treating patients with migraine attacks. Agomelatine administered to patients suffering from migraine attacks (in doses of 25 mg/day for a duration of 6 months) decreased both the frequency and duration of migraine attacks; thus, it reduced the intensity of pain in migraine patients. Moreover, it also reduced significantly the severity of depression and normalized sleep disturbances.77
Melatonin ligands under clinical evaluation
Melatonergic agonists
The two nonselective MT1/MT2 agonists tasimelteon and TIK-301 are the most advanced melatonergic drug candidates undergoing clinical trials for the treatment of sleep disorders.
Tasimelteon (VEC-162, BMS-214778, MA-1) (Figure 2) is an orally bioavailable melatonergic drug that has been clinically tested, to phase III, by Vanda Pharmaceuticals Inc (Washington, DC, USA), under license from Bristol-Myers Squibb Co (New York, USA).78,79 It has a slightly lower affinity to MT1 (pKi=9.45) and a moderately higher affinity than MLT to MT2 (pKi=9.80). No substantial interactions with receptors for other neurotransmitters or neuromodulators, or with other melatonin binding sites have been observed or reported. In some pre-clinical and exploratory trials, the compound has been also tested for antidepressive effects, although no antagonist properties at 5-HT2C receptors are obvious. In humans, time to maximum plasma concentration (Tmax) values varied from 1.9 to 3.0 hours, with considerable interindividual deviations. Tasimelteon is extensively metabolized by CYP1A2 and CYP3A4 enzymes. It is metabolized by oxidation, resulting in the opening of the dihydrofuran ring, followed by further oxidation to carboxylic acid. The major phase II metabolic route is glucuronidation. In the phase III study, tasimelteon improved sleep latency, sleep efficiency, and wake after sleep onset.69 Recently, in May 2013, Vanda Pharmaceuticals submitted a new drug application to the FDA, for tasimelteon, for the treatment of non-24-hour sleep–wake disorder in subjects suffering total blindness.80 It is the first FDA-approved medication for this orphan indication. Tasimelteon is recommended for use at a dosage of 20 mg/day, taken before bedtime (at the same time every night), without food.81 Vanda Pharmaceuticals intends to launch the drug under the brand name Hetlioz™ in the second quarter of 2014. The same company has also evaluated the use of tasimelteon for the treatment of insomnia in phase II and III studies, while it discontinued development of tasimelteon for the treatment of major depressive disorders in January 2013, after top-line results from a phase III study failed to meet the primary endpoint.
TIK-301 (LY 156735; β-methyl-6-chloromelatonin) (Figure 2) is a melatonergic agonist originally developed by Eli Lilly and Co (Indianapolis, IN, USA) that is now sublicensed to Tikvah Therapeutics Inc. (Altanta, GA, USA).82 TIK-301 has received an orphan drug designation by FDA for the treatment of sleep disorders in blind individuals.83 It displays a high affinity towards MT1 and MT2 receptors; its affinity for MT1 receptors (pKi=10.09) is similar to that of melatonin, and even higher for MT2 (pKi=10.38).84 The preferential binding to MT2 is a typical feature of 6-chlorinated melatonin derivatives, and is also observed with 6-chloromelatonin.54 The half-life of TIK-301 has been reported to be in the range of 1 hour. This only moderately extended action is somewhat surprising, because TIK-301 carries a chlorine atom at position 6 of the indolic moiety, which prevents 6-hydroxylation (ie, degradation) via the major catabolic route of MLT.85 When tested for its soporific properties, the outcome has been relatively moderate, despite some statistically demonstrable effects. TIK-301 has been reported to also combine properties as a melatonergic agonist and serotonergic antagonist, at both receptor subtypes 5-HT2C and 5-HT2B, opening new perspectives for its possible antidepressant action.86
Melatonergic antagonists
MLT receptor antagonists have only been evaluated in pre-clinical studies; for example, luzindole (N-0774; 2-benzyl-N-acetyltryptamine) for its antidepressant-like effect,87 S22153 in circadian rhythm entrainment experiments,88 and ML-23 in the treatment of Parkinson’s disease.89
Melatonergic ligands in development
Although the functions of MLT in the body are numerous and extensive, its role in entraining circadian rhythms to the light-dark cycle is probably the best known. Probably for this reason, the effort of most laboratories has been focused upon developing melatonergic drugs for the treatment of circadian pathologies (ie, sleep disturbances or seasonal affective disorders). Thus, during the past decade, a great number of structurally different MLT receptor ligands, which range from simple indole derivatives and their bioisosteres to phenylalkyl amides, and constrained melatoninergic agents, have been reported in the literature.90–92 According to the guidelines of the International Union of Basic and Clinical Pharmacology, a selective ligand should display at least 50–100 times greater binding affinity and/or potency for one receptor subtype, relative to the other. Applying these criteria, compounds with moderate, approximately ten to 50-fold selectivity and agents showing just two to ten-fold preference for one of the melatonin receptor subtypes are considered to be nonselective. Below, the most recent and interesting melatonergic ligands will be briefly reported (Figure 3).
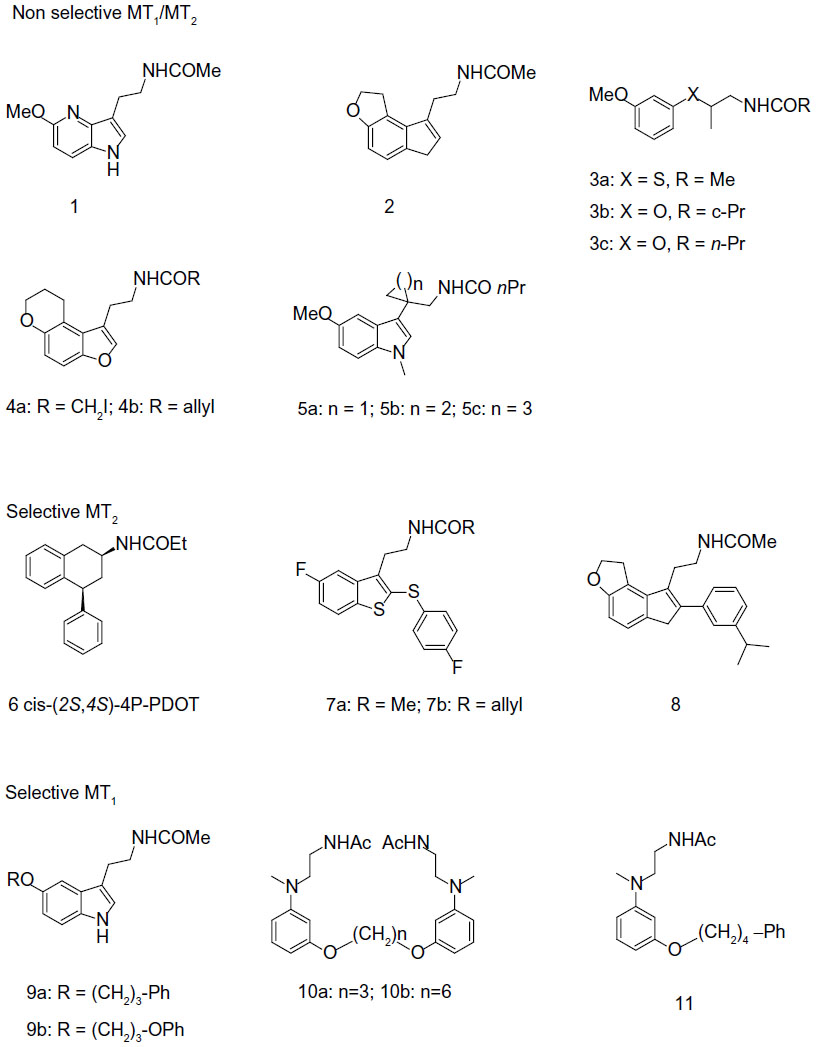 | Figure 3 Structures of melatonergic ligands in development. |
Nonselective MT1/MT2 melatonergic ligands
Most of the melatonergic ligands are not able to distinguish between MT1 and MT2 receptors, or they show just a preference in binding affinity for one of the subtypes (eg, maximally 50-fold selectivity). Their structures range from bioisosteric MLT analogs, obtained by exchange of the indole nucleus with others (mostly heteroaromatic rings), to ring-opened derivatives and conformationally constrained analogues. Recently, bioisosteric relationships between the indole nucleus and both 4-azaindole and indene nuclei have been demonstrated. Actually, 4-azamelatonin (Figure 3-1) has shown identical MT1 (Ki=0.24 nM) and MT2 (Ki=0.36 nM) binding affinities to MLT, for receptors expressed in HEK-293 cells,93 while indene (Figure 3-2) showed indeed five to ten times higher affinities than MLT, in both receptor subtypes, expressed in CHO cells.94 Moreover, regarding bioisosteric substitutions of the indole nucleus, a series of chiral phenoxyalkyl and phenylthioalkyl amides have been always reported.95 This study provided compounds with nanomolar affinity for both MLT receptor subtypes, expressed in NIH3T3 cells. The (S)-enantiomers behave as eutomers at both MT1 and MT2 receptors. The highest stereoselectivity was observed for the thioanalog shown in Figure 3-3a (Ki distomer/Ki eutomer ratio: 457, for MT1 receptors). Of particular interest, the (S)-compound shown in Figure 3-3b displayed binding affinity for MT1 receptors (pKi=9.14), close to that of MLT, behaving as a full agonist in [35S]GTPγS binding assay. Recently, new series of tricyclic analogues, obtained by the insertion of the methoxy group into a ring, have been reported as MLT ligands.96 Some of the newly synthesized compounds showed higher binding affinity at both receptors than MLT itself. Compounds 4a and 4b in Figure 3 were the most interesting derivatives. Functional activity studies showed full agonist activity of these compounds with half maximal effective concentration in the nanomolar range. MLT ligands with the N-acylaminoethyl side chain partly incorporated into a ring have been frequently reported, as in compounds 5a and 5c in Figure 3.97
Selective MT2 melatonergic ligands
Numerous efforts to develop selective MT1 or MT2 ligands have been also reported. Selectivity towards MT2 is much easier to accomplish than for MT1, and many series of MT2-selective ligands have been studied. 4-P-PDOT (4-phenyl-2-propionamidotetralin) is a well-known, highly MT2-selective antagonist, displaying 330 times greater affinity for MT2 (Ki=1.5 nM) than for MT1 (Ki=501 nM), in receptors expressed in COS-7 cells.98 4-P-PDOT exists in the form of four stereoisomers, a pair of cis and a pair of trans enantiomers. Only recently, all four single stereoisomers of 4-P-PDOT have been separated99 and pharmacologically evaluated at receptors expressed in NIH3T3 cells, in terms of their binding affinities and intrinsic activities (GTPγS assay).100 The eutomer for (+/−)-cis-4-P-PDOT is the (+)-(2S,4S) enantiomer shown in Figure 3-6, displaying ~170-fold greater affinity for MT2 than for MT1 receptors, and 15% and 45% intrinsic activity at MT1 and MT2, relative to MLT. The (−)-(2R,4R) mirror image shows ~3 times less agonist potency. A few years ago, a series of benzothiophene analogs was evaluated at receptors expressed in CHO cells.101 Acetamide (Figure 3-7a) and allyl carboxamide (Figure 3-7b) displayed 65.5- and 220-fold selectivity towards MT2 receptors, respectively. Both ligands were demonstrated to be MT1/MT2 partial agonists. A high selectivity against MT2 was obtained by modifying the compound shown in Figure 3-2, to obtain the compound shown in Figure 3-8, which showed a 1,200-fold MT2 selectivity, when evaluated at MLT receptors expressed in CHO cells.94
Selective MT1 melatonergic ligands
Development of MT1-selective ligands is still a challenge, and only a few examples of selective compounds have been reported so far. Preferential binding to MT1 receptors is, at maximum, ~100 times higher for MT1 than for MT2. A ~10-fold selectivity towards MT1 receptors was found for the compounds shown in Figure 3-9a and 9b.102 Compound 9b behaves as an MT1/MT2 agonist in a cAMP assay, at receptors expressed in CHO cells. Recently, a series of dimeric ligands has been designed. The most interesting compounds were those shown as 10a and 10b (Figure 3).103 In particular, analogue 11b exhibited the highest MT1 affinity (3.4 nM) and was 54 times more selective for MT1 than for MT2 receptors. Both compounds behave as partial MT1/MT2 agonists, as assessed through [35S]GTPγS binding analysis. A structurally-related series of N-(anilinoalkyl)amides, bearing 3-arylalkyloxy substituents, was also examined.104 The highest selectivity was found for compound 11 (Figure 3), which was 78-fold more selective towards MT1 (MT1: Ki=1.17 nM; MT2: Ki=91.2 nM), acting as a partial agonist at both receptor subtypes.
Investigational melatonergic drugs
Melatonin receptor agonists offer promising possibilities for treating neurological and sleep disorders related to circadian dysfunction, and other different disorders, such as various subtypes of depression and anxia, and metabolic diseases. This paragraph summarizes progress in the development of melatonergic drugs (Figure 4). However, it should be noted that investigational melatonergic drugs have been mainly characterized with regard to MT1 and MT2 receptors, whereas data on interference with other signaling pathways and on other melatonin binding sites are mostly missing or have, at least, not been disclosed. To date, it is difficult to judge whether an absence of effects mediated by binding sites different from the MLT membrane receptors represents a disadvantage, or the advantage of higher drug specificity, relative to MLT. There is increasing evidence that MLT plays an important role in modulating learning and memory processing.105 For example, MLT facilitates memory performance in the novel object recognition (NOR) task and water-maze task in rats.106,107 Neu-P11 is an MT1/MT2 receptor agonist and a serotonin 5-HT1A/1D receptor agonist, recently developed for the treatment of insomnia.108 Neu-P11 has been shown to promote sleep, improve insulin sensitivity, and exert antidepressant and anxiolytic activities in rodent models.109–111 Recent studies have demonstrated that Neu-P11 enhances memory performance in the NOR task in rats, and improves neuronal and cognitive impairments in a rat model of AD (induced by intrahippocampal Aβ(1–42) injection), thus proposing Neu-P11 as a novel agent for the treatment of AD.112 Recently, a new 1-N substituted melatonin analog, M3C has been proposed for the treatment of anxiety.113 It has been demonstrated to be effective as an anxiolytic-like agent in pinealectomized rats, at doses lower than any other melatonin analogues previously reported. Data suggest that the anxiolytic properties of M3C may be mediated by MT1 and MT2 receptors. Evidence of the oncostatic properties of MLT in hormone-dependent breast cancer is stimulating research for synthetic molecules useful in breast cancer therapy. In the last few years, two nonselective benzofuran MT1/MT2 melatonin receptor agonists (S23219-1 and S23478-1) have been tested for their in vitro and in vivo antitumoral activity, and it has been determined that these compounds are more effective than MLT in inhibiting proliferation and promoting apoptosis of MCF-7 cells, as well as in inducing regression of N-nitroso-N-methylurea- (NMU) induced rat mammary tumors.114 The moderately MT2-selective partial agonist UCM765 has been reported to promote non-rapid eye movement sleep (NREMS) in rats and mice.115 This effect is nullified by the pharmacological blockage or genetic deletion of MT2 receptors. Remarkably, the effects of this ligand on sleep are different from those of nonselective MT1/MT2 agonists. For example, the structurally-related MT1/MT2 agonist UCM793 slightly decrease sleep onset, without effect on NREMS maintenance, similar to ramelteon, indicating that dual MT1/MT2 agonistic activity accounts for the effect on sleep onset, whereas selectivity for MT2 receptors has an additional effect on NREMS maintenance. Because of the selective promotion of NREMS, MT2 receptors could become an interesting future target for the treatment of sleep disorders. Preliminary data were also reported concerning anxiolytic effects of UCM765 in rats.116 Recent studies were aimed at understanding how MLT influences the quality of the three vigilance states of sleep in adult Sprague-Dawley male rats.117 The effects of MLT on 24-hour vigilance states were studied and compared with those produced by UCM793 and a selective MT2 receptor partial agonist (UCM924). UCM924 has the same binding profile as the MT2 receptor, but, unlike UCM765, it is more metabolically stable, thus showing a longer half-life. In particular, UCM924 has the free phenyl ring of UCM765 protected by a fluorine, and the methoxy group is bioisosterically replaced with bromine. The authors demonstrated that the three compounds do not increase the quantity of sleep, but differently influence the quality of the three vigilance states. Studies also demonstrated that selective activation of MT2 receptors increases NREMS duration more than do MLT and nonselective MT1/MT2 receptors. Thus, selective MT2 receptor agonists have high potential to be further developed as novel hypnotic agents. Recently, new melatonin-related compounds were developed to modulate the human malaria parasite cell cycle and block the parasite’s development, acting as antimalarials.118 These novel indole derivatives showed very significant antimalarial activity, opening good perspectives for the development of new drugs with novel mechanisms of action. Recently, a new MLT agonist (Figure 3-3c), displaying potent antioxidant activity and strong cytoprotective activity against H2O2-induced cytotoxicity, has been reported.119 The same compound has been shown to perform as a Ca2+/calmodulin-dependent kinase II (CaMKII) inhibitor. This compound, which presents both of these characteristics, should be a valid candidate for further investigations. In fact, both antioxidant and CaMKII-inhibiting properties might prove beneficial in neurodegenerative disorders. Thus, it should be very useful to obtain an MLT analogue that shows antioxidant capability and, at the same time, inhibits CaMKII, since this is, in turn, an important source of oxidative stress.
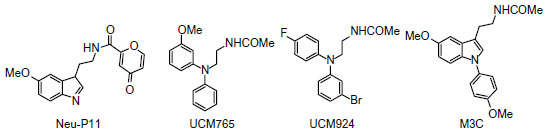 | Figure 4 Structures of investigational melatonergic drugs. |
Conclusion
Recent advances in the field of melatonergic drugs have been reported in this review, focusing on agents in therapy, as well as new MT1/MT2 ligands in development. Many of the current melatonergic drugs used in therapy or in clinical development, indicated for the treatment of depression (agomelatine) and sleep disorders (ramelteon, tasimelteon, TIK-301), have dual action; for example, agomelatine for MT1/MT2 and 5-HT2C, TIK-301 for MT1/MT2, and 5-HT2C/5-HT2B. Therefore, drug therapies targeted at multiple receptors seem to be preferable to those selective for one type of receptor. Most researchers have pursued high affinity for a single therapeutic target, combined with high selectivity, believing that this will maximize efficacy, minimize side effects, and give the drug a therapeutic window. However, this view has been challenged in recent years, with the introduction of the concept of polypharmacology.120 It has been found that activity at multiple therapeutic targets can result in superior efficacy and safety, giving opportunities for the discovery of better drugs.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
Lerner AB, Case JD, Takahashi Y, Lee TH, Mori W. Isolation of melatonin, the pineal factor that lightens melanocytes. J Am Chem Soc. 1958;80:2587–2587. | |
Reiter RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev. 1991;12(2):151–180. | |
Cardinali DP, Rosner JM. Metabolism of serotonin by the rat retina in vitro. J Neurochem. 1971;18(9):1769–1770. | |
Tosini G, Menaker M. The clock in the mouse retina: melatonin synthesis and photoreceptor degeneration. Brain Res. 1998;789(2):221–228. | |
Carrillo-Vico A, Calvo JR, Abreu P, et al. Evidence of melatonin synthesis by human lymphocytes and its physiological significance: possible role as intracrine, autocrine and/or paracrine substance. FASEB J. 2004;18(3):537–539. | |
Raikhlin NT, Kvetnoy IM. Melatonin and enterochromaffine cells. Acta Histochem. 1976;55(1):19–24. | |
Bubenik GA. Gastrointestinal melatonin: localization, function, and clinical relevance. Dig Dis Sci. 2002;47(10):2336–2348. | |
Klein DC. Arylalkylamine N-acetyltransferase: “the Timezyme”. J Biol Chem. 2007;282(7):4233–4237. | |
Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(5557):1070–1073. | |
Sugden D. Melatonin biosynthesis in the mammalian pineal gland. Experientia. 1989;45(10):922–932. | |
Pevet P, Bothorel B, Slotten H, Saboureau M. The chronobiotic properties of melatonin. Cell Tissue Res. 2002;309(1):183–191. | |
Sewerynek E. Melatonin and the cardiovascular system. Neuro Endocrinol Lett. 2002;23 Suppl 1:79–83. | |
Carrillo-Vico A, Reiter RJ, Lardone PJ, et al. The modulatory role of melatonin on immune responsiveness. Curr Opin Investig Drugs. 2006;7(5):423–431. | |
Barrenetxe J, Delagrange P, Martinexz JA. Physiological and metabolic functions of melatonin. J Physiol Biochem. 2004;60(1):61–72. | |
Genovese T1, Di Paola R, Mazzon E, Muià C, Caputi AP, Cuzzocrea S. Melatonin limits lung injury in bleomycin treated mice. J Pineal Res. 2005;38(2):198–208. | |
Peres MF. Melatonin, the pineal gland and their implications for headache disorders. Chephalalgia. 2005;25(6):403–411. | |
Srinivasan V, Pandi-Perumal SR, Spence DW, et al. Potential use of melatonergic drugs in analgesia: mechanisms of action. Brain Res Bull. 2010;81(4–5):362–371. | |
Srinivasan V, Pandi-Perumal SR, Cardinali DP, Poeggeler B, Hardeland R. Melatonin in Alzheimer’s disease and other neurodegenerative disorders. Behav Brain Funct. 2006;2:15. | |
Medeiros CA, Carvalhedo de Bruin PF, Lopes LA, Magalhães MC, de Lourdes Seabra M, de Bruin VM. Effect of exogenous melatonin on sleep and motor dysfunction in Parkinson’s disease. A randomized, double blind, placebo-controlled study. J Neurol. 2007;254(4): 459–464. | |
Sofic E, Rimpapa Z, Kundurovic Z, et al. Antioxidant capacity of the neurohormone melatonin. J Neural Transm. 2005;112(3):349–358. | |
Reiter RJ, Paredes SD, Manchester LC, Tan DX. Reducing oxidative/nitrosative stress: a newly-discovered genre for melatonin. Crit Rev Biochem Mol Biol. 2009;44(4):175–200. | |
Grant SG, Melan MA, Latimer JJ, Witt-Enderby PA. Melatonin and breast cancer: cellular mechanisms, clinical studies and future perspectives. Expert Rev Mol Med. 2009;11:e5. | |
Davis VL, Dodda BR, Witt-Enderby PA. The prevention and treatment of breast cancer using melatonin. In: Watson, R, editor. Melatonin in the Promotion of Health. 2nd ed. Abingdon, UK: Taylor and Francis; 2011. p. 271–286. | |
Karamitri A1, Renault N, Clement N, Guillaume JL, Jockers R. Minireview: Toward the establishment of a link between melatonin and glucose homeostasis: association of melatonin MT2 receptor variants with type 2 diabetes. Mol Endocrinol. 2013;27(8):1217–1233. | |
Witt-Enderby PA, Clafshenkel WP, Kotlarczyk MP, Sethi S. Melatonin in bone health. In: Watson, R, editor. Melatonin in the Promotion of Health. 2nd ed. Abingdon, UK: Taylor and Francis; 2011. p. 261–270. | |
Maria S, Witt-Enderby PA. Melatonin effects on bone: potential use for the prevention and treatment for osteopenia, osteoporosis, and periodontal disease and for use in bone-grafting procedures. J Pineal Res. 2014;56(2):115–125. | |
Carocci A, Sinicropi MS, Catalano A, Lauria G, Genchi G. Melatonin in Parkinson’s Disease. In: Rana QA, editor. A Synopsis of Parkinson’s Disease. Intech; 2014. | |
Reppert SM, Weaver DR, Godson C. Melatonin receptors step into the light: Cloning and classification of subtypes. Trends Pharmacol Sci. 1996;17(3):100–102. | |
Von Gall C, Stehle JH, Weaver DR. Mammalian melatonin receptors: molecular biology and signal transduction. Cell Tissue Res. 2002;309(1):151–162. | |
Blask DE, Sauer LA, Dauchy RT. Melatonin as a chronobiotic/anticancer agent: cellular, biochemical, and molecular mechanisms of action and their implications for circadian-based cancer therapy. Curr Top Med Chem. 2002;2(2):113–132. | |
Reppert SM, Godson C, Mahle CD, Weaver DR, Slaugenhaupt SA, Gusella JF. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci U S A. 1995;92(19):8734–8738. | |
Reppert SM. Melatonin receptors: molecular biology of a new family of G protein-coupled receptors. J Biol Rhythms. 1997;12(6):528–531. | |
Dubocovich ML, Yun K, Al-Ghoul WM, Benloucif S, Masana MI. Selective MT2 melatonin receptor antagonists block melatonin-mediated phase advances of circadian rhythms. FASEB J. 1998;12(12): 1211–1220. | |
Liu C, Weaver DR, Jin X, et al. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1997;19(1):91–102. | |
Hunt AE, Al-Ghoul WM, Gillette MU, Dubocovich ML. Activation of MT(2) melatonin receptors in rat suprachiasmatic nucleus phase advances the circadian clock. Am J Physiol Cell Physiol. 2001;280(1): C110–C118. | |
Dubocovich ML, Markowska M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine. 2005;27(2):101–110. | |
Comai S, Gobbi B. Unveiling the role of melatonin MT2 receptors in sleep, anxiety and other neuropsychiatric diseases: a novel target in psychopharmacology. J Psych Neurosci. 2014;39(1):6–21. | |
Doolen S, Krause DN, Dubocovich ML, Duckles SP. Melatonin mediates two distinct responses in vascular smooth muscle. Eur J Pharmacol. 1998;345(1):67–69. | |
Nosjean O, Ferro M, Cogé F, et al. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J Biol Chem. 2000;275(4): 31311–31317. | |
Tan DX, Chen LD, Poeggeler B, Manchester LC, Reiter RJ. Melatonin: a potent, endogenous hydroxyl radical scavenger. Endocr J. 1993; 1(4):57–60. | |
Rodriguez C, Mayo JC, Sainz RM, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36(1):1–9. | |
Tomas-Zapico C, Coto-Montes A. A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J Pineal Res. 2005;39(2):99–104. | |
Romero MP, Garcia-Perganeda A, Guerrero JM, Osuna C. Membrane-bound calmodulin in Xenopus laevis oocytes as a novel binding site for melatonin. FASEB J. 1998;12(13):1401–1408. | |
Hotta CT, Gazarini ML, Beraldo FH, et al. Calcium-dependent modulation by melatonin of the circadian rhythm in malarial parasites. Nat Cell Biol. 2000;2(7):466–468. | |
Srinivasan V, Ahmad AH, Mohamed M, Zakaria R. Melatonin effects on Plasmodium life cycle: new avenues for therapeutic approach. Recent Pat Endocr Metab Immune Drug Discov. 2012;6(2):139–147. | |
Zhdanova IV, Wurtman RJ, Regan MM, Taylor JA, Shi JP, Leclair OU. Melatonin treatment for age-related insomnia. J Clin Endocrinol Metab. 2001;86(10):4727–4730. | |
Leger D, Laudon M, Zisapel N. Nocturnal 6-sulfatoxymelatonin excretion in insomnia and its relation to the response to melatonin replacement therapy. Am J Med. 2004;116(2):91–95. | |
Wilson SJ, Nutt DJ, Alford C, et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders. J Psychopharmacol. 2010;24(11):1577–1601. | |
Pandi-Perumal SR, Trakht I, Spence DW, Srinivasan V, Dagan Y, Cardinali DP. The roles of melatonin and light in the pathophysiology and treatment of circadian rhythm sleep disorders. Nat Clin Pract Neurol. 2008;4(8):436–447. | |
Srinivasan V, Singh J, Pandi-Perumal SR, Brown GM, Spence DW, Cardinali DP. Jet lag, circadian rhythm sleep disturbances, and depression: the role of melatonin and its analogs. Adv Ther. 2010;27(11):796–813. | |
DeMuro RL, Nafziger AN, Blask DE, Menhinick AM, Bertino JS Jr. The absolute bioavailability of oral melatonin. J Clin Pharmacol. 2000;40(7):781–784. | |
Claustrat B, Brun J, Chazot G. The basic physiology and pathophysiology of melatonin. Sleep Med Rev. 2005;9(1):11–24. | |
Arendt J. Does melatonin improve sleep? Efficacy of melatonin. BMJ. 2006;332(7540):550–553. | |
Hardeland R. New approaches in the management of insomnia: weighing the advantages of prolonged release melatonin and synthetic melatoninergic agonists. Neuropsych Dis Treat. 2009;5:341–354. | |
Miyamoto M. Pharmacology of ramelteon, a selective MT1/MT2 receptor agonist: a novel therapeutic drug for sleep disorders. CNS Neurosci Ther. 2009;15(1):32–51. | |
de Bodinat C, Guardiola-Lemaitre B, Mocaër E, Renard P, Muñoz C, Millan MJ. Agomelatine, the first melatonergic antidepressant: discovery characterization and development. Nat Rev Drug Discov. 2010;9(8):628–642. | |
Lankford DA. Tasimelteon for insomnia. Expert Opin Invest Drugs. 2011;20(7):987–993. | |
Zemlan FP, Mulchahey JJ, Scharf MB, Mayleben DW, Rosenberg R, Lankford A. The efficacy and safety of the melatonin agonist β-methyl-6-chloromelatonin in primary insomnia: a randomized, placebo-controlled, crossover clinical trial. J Clin Psychiatry. 2005;66(3): 384–390. | |
Kato K, Hirai K, Nishiyama K, et al. Neurochemical properties of ramelteon (TAK-375), a selective MT1/MT2 receptor agonist. Neuropharmacology. 2005;48(5):301–310. | |
McGechan A, Wellington K. Ramelteon. CNS Drugs. 2005;19(12): 1057–1065. | |
Miyamoto M. Pharmacology of ramelteon, a selective MT1/MT2 receptor agonist: a novel therapeutic drug for sleep disorders. CNS Neurosci Ther. 2009;15(1):32–51. | |
Stevenson S, Bryson S, Amayke D, Hibberd M. Study to investigate the absolute bioavailability of a single oral dose of ramelteon (TAK-375) in healthy male subjects. Clin Pharmacol Ther. 2004;75:22. | |
Cardinali DP, Srinivasan V, Brzezinski A, Brown GM. Melatonin and its analogs in insomnia and depression. J Pineal Res. 2012;52(4):365–375. | |
Pandi-Perumal SR, Srinivasan V, Spence DW, et al. Ramelteon: a review of its therapeutic potential in sleep disorders. Adv Ther. 2009; 26(6):613–626. | |
Pandi-Perumal SR, Srinivasan V, Poeggeler B, Hardeland R, Cardinali DP. Drug Insight: the use of melatonergic agonists for the treatment of insomnia-focus on ramelteon. Nat Clin Pract Neurol. 2007;3(4):221–228. | |
Richardson GS, Zee PC, Wang-Weigand S, Rodriguez L, Peng X. Circadian phase-shifting effects of repeated ramelteon administration in healthy adults. J Clin Sleep Med. 2008;4(5):456–461. | |
Bellapart J, Boots R. Potential use of melatonin in sleep and delirium in the critically ill. Br J Anaesth. 2012;108(4):572–580. | |
Al-Aama T, Brymer C, Gutmanis I, Woolmore-Goodwin SM, Esbaugh J, Dasgupta M. Melatonin decreases delirium in elderly patients: a randomized, placebo-controlled trial. Int J Geriatr Psych. 2011;26(7):687–694. | |
Ohta T, Murao K, Miyake K, Takemoto K. Melatonin receptor agonists for treating delirium in elderly patients with acute stroke. J Stroke Cerebrovasc Dis. 2013;22(7):1107–1110. | |
Millan MJ, Gobert A, Lejeune F, et al. The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine 2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J Pharmacol Exp Ther. 2003;306(3):954–964. | |
Racagni G, Riva MA, Molteni R, et al. Mode of action of agomelatine: synergy between melatonergic and 5-HT2C receptors. World J Biol Psychiatry. 2011;12(8):574–587. | |
Bertaina-Anglade V, La Rochelle CD, Boyer PA, Mocaer E. Antidepressant-like effects of agomelatine (S 20098) in the learned helplessness model. Behav Pharmacol. 2006;17(8):703–713. | |
Millan MJ. Multi-target strategies for the improved treatment of depressive states: Conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110(2):135–370. | |
Bogaards JJ, Hissink EM, Briggs M, et al. Prediction of interindividual variation in drug plasma levels in vivo from individual enzyme kinetic data and physiologically based pharmacokinetic modeling. Eur J Pharm Sci. 2000;12(2):117–124. | |
Dolder CR, Nelson M, Snider M. Agomelatine treatment of major depressive disorder. Ann Pharmacother. 2008;42(12):1822–1831. | |
Papp M, Litwa E, Gruca P, Mocaër E. Anxiolytic-like activity of agomelatine and melatonin in three animal models of anxiety. Behav Pharmacol. 2006;17(1):9–18. | |
Tabeeva GR, Sergeev AV, Gromova SA. [Possibilities of preventive treatment of migraine with MT1- and MT2 agonist and 5-HT2c receptor antagonist agomelatine (valdoxan)]. Zh Nevrol Psikhiatr Im S S Korsakova. 2011;111(9):32–36. Russian. | |
Rajaratnam SMW, Polymeropoulos MH, Fisher DM, et al. Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials. Lancet. 2009;373(9662):482–491. | |
Hardeland R. Tasimelteon, a melatonin agonist for the treatment of insomnia and circadian rhythm sleep disorders. Curr Opin Investig Drugs. 2009;10(7):691–701. | |
Vanda Pharma submits NDA for circadian regulator tasimelteon In totally blind. RTT News. May 31, 2011. Available from: http://archive.today/WzzaS. | |
Dhillon S, Clarke M. Tasimelteon: first global approval. Drugs. 2014;74(4):505–511. | |
Hardeland R. Investigational melatonin receptor agonists. Exp Opin Investig Drugs. 2010;19(6):747–764. | |
Hardeland R, Poeggeler B. Melatonin and synthetic melatonergic agonists: actions and metabolism in the central nervous system. Cent Nerv Syst Agents Med Chem. 2012;12(3):189–216. | |
Rivara S, Mor M, Bedini A, Spadoni G, Tarzia G. Melatonin receptor agonists: SAR and applications to the treatment of sleep-wake disorders. Curr Top Med Chem. 2008;8(11):954–968. | |
Mulchahey JJ, Goldwater DR, Zemlan FP. A single blind, placebo controlled, across groups dose escalation study of the safety, tolerability, pharmacokinetics and pharmacodynamics of the melatonin analog beta-methyl-6-chloromelatonin. Life Sci. 2004;75(15): 1843–1856. | |
Landolt HP, Wehrle R. Antagonism of serotonergic 5-HT2A/2C receptors: mutual improvement of sleep, cognition and mood? Eur J Neurosci. 2009;29(9):1795–1809. | |
Sumaya IC, Masana MI, Dubocovich ML. The antidepressant-like effect of the melatonin receptor ligand luzindole in mice during forced swimming requires expression of MT2 but not MT1 melatonin receptors. J Pineal Res. 2005;39(2):170–177. | |
Li XM, Beau J, Delagrange P, Mocaër E, Lévi F. Circadian rhythm entrainment with melatonin, melatonin receptor antagonist S22153 or their combination in mice exposed to constant light. J Pineal Res. 2004;37(3):176–184. | |
Willis JL. The role of ML-23 and other melatonin analogues in the treatment and management of Parkinson’s disease. Drug News Perspect. 2005;18(7):437–444. | |
Zlotos DP, Jockers R, Cecon E, Rivara S, Witt-Enderby PA. MT1 and MT2 melatonin receptors: ligands, models, oligomers, and therapeutic potential. J Med Chem. 2014;57(8):3161–3185. | |
Tsotinis A, Gourgourinis J, Eleutheriades A, Davidson K, Sugden D. Design and synthesis of new N-OMe fluoro-indole melatoninergics. Med Chem. 2007;3(6):561–571. | |
Tsotinis A, Afroudakis PA, Davidson K, Prashar A, Sugden D. Design, synthesis, and melatoninergic activity of new azido- and isothiocyanato-substituted indoles. J Med Chem. 2007;50(25):6436–6440. | |
El Kazzouli S, Griffon du Bellay A, Berteina-Raboin S, Delagrange P, Caignard DH, Guillaumet G. Design and synthesis of 2-phenylimidazo[1,2-a]pyridines as a novel class of melatonin receptor ligands. Eur J Med Chem. 2011;46(9):4252–4257. | |
Koike T, Hoashi Y, Takai T, et al. 1,6-dihydro-2H-indeno[5,4-b]furan derivatives: design, synthesis, and pharmacological characterization of a novel class of highly potent MT2-selective agonists. J Med Chem. 2011;54(9):3436–3444. | |
Carocci A, Catalano A, Lovece A, et al. Design, synthesis, and pharmacological effects of structurally simple ligands for MT1 and MT2 melatonin receptors. Bioorg Med Chem. 2010;18(17):6496–6511. | |
Landagaray E, Ettaoussi M, Leclerc V, et al. New melatonin (MT1/MT2) ligands: fesign and synthesis of (8,9-dihydro-7H-furo[3,2-f]chromen-1-yl) derivatives. Bioorg Med Chem. 2014;22(3): 986–996. | |
Tsotinis A, Vlachou M, Papahatjis DP, et al. Mapping the melatonin receptor. 7. Subtype selective ligands based on beta-substituted N-acyl-5-methoxytryptamines and N-acyl-5-methoxy-1-methyl-tryptamines. J Med Chem. 2006;49(12):3509–3519. | |
Dubocovich ML, Masana MI, Iacob S, Sauri DM. Melatonin receptor antagonists that differentiate between the human Mel1a and Mel1b recombinant subtypes are used to assess the pharmacological profile of the rabbit retina ML1 presynaptic heteroreceptor. Naunyn Schmiedebergs Arch Pharmacol. 1997;355(3):365–375. | |
Lucarini S, Bartolucci S, Bedini A, et al. Synthesis and configuration determination of all enantiopure stereoisomers of the melatonin receptor ligand 4-phenyl-2- propionamidotetralin using an expedient optical resolution of 4-phenyl-2-tetralone. Org Biomol Chem. 2012;10(2): 305–313. | |
Bedini A, Lucarini S, Spadoni G, et al. Toward the definition of stereochemical requirements for MT2-selective antagonists and partial agonists by studying 4-phenyl- 2-propionamidotetralin derivatives.J Med Chem. 2011;54(24):8362–8372. | |
Mésangeau C, Fraise M, Delagrange P, et al. Preparation and pharmacological evaluation of a novel series of 2-(phenylthio) benzo[b]thiophenes as selective MT2 receptor ligands. Eur J Med Chem. 2011;46(5):1835–1840. | |
Markl C, Clafshenkel WP, Attia MI, Sethi S, Witt-Enderby PA, Zlotos DP. N-acetyl-5-arylalkoxytryptamine analogs: probing the melatonin receptors for MT(1) -selectivity. Arch Pharm Chem Life Sci. 2011;334(10):666–674. | |
Spadoni G, Bedini A, Orlando P, et al. Bivalent ligand approach on N-{2-[(3-methoxyphenyl) methylamino]ethyl}-acetamide: synthesis, binding affinity and intrinsic activity for MT1 and MT2 melatonin receptors. Bioorg Med Chem. 2011;19(16):4910–4916. | |
Rivara S, Pala D, Lodola A, et al. MT1-selective melatonin receptor ligands: synthesis, pharmacological evaluation, and molecular dynamics investigation of N-{[(3-O-substituted)anilino]alkyl}amides. ChemMedChem. 2012;7(11):1954–1964. | |
Rawashdeh O, Maronde E. The hormonal Zeitgeber melatonin: role as a circadian modulator in memory processing. Front Mol Neurosci. 2012;5:27–33. | |
Bertaina-Anglade V, Drieu-La-Rochelle C, Mocaër E, Seguin L. Memory facilitating effects of agomelatine in the novel object recognition memory paradigm in the rat. Pharmacol Biochem Behav. 2011;98(4):511–517. | |
Gönenç S, Uysal N, Açikgöz O, Kayatekin BM, et al. Effects of melatonin on oxidative stress and spatial memory impairment induced by acute ethanol treatment in rats. Physiol Res. 2005;54(3):341–348. | |
Paulis L, Simko F, Laudon M. Cardiovascular effects of melatonin receptor agonists. Expert Opin Investig Drugs. 2012;21(11):1661–1678. | |
Laudon M, Urade Y, Huang Z. Neu-P11, a novel melatonin agonist: effects on sleep and EEG power spectra in rats. Sleep. 2008; 31 Suppl):A34–A35. | |
She M, Deng X, Guo Z, et al. Neu-P11, a novel melatonin agonist, inhibits weight gain and improves insulin sensitivity in high-fat/high-sucrose-fed rats. Pharmacol Res. 2009;59(4):248–253. | |
Tian SW, Laudon M. Antidepressant- and anxiolytic effects of the novel melatonin agonist Neu-P11 in rodent models. Acta Pharmacol Sin. 2010;31(7):775–783. | |
He P, Ouyang X, Zhou S, et al. A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’ disease. Horm Behav. 2013; 64(1):1–7. | |
Bustamante-García R, Lira-Rocha AS, Espejo-González O, Gómez-Martínez AE, Picazo O. Anxiolytic-like effects of a new 1-N substituted analog of melatonin in pinealectomized rats. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:133–139. | |
Mao L, Cheng Q, Guardiola-Lemaître B, et al. In vitro and in vivo antitumor activity of melatonin receptor agonists. J Pineal Res. 2010;49(3):210–221. | |
Ochoa-Sanchez R, Comai S, Lacoste B, et al. Promotion of non-rapid eye movement sleep and activation of reticular thalamic neurons by a novel MT2 melatonin receptor ligand. J Neurosci. 2011;31(50): 18439–18452. | |
Ochoa-Sanchez R, Rainer Q, Comai S, et al. Anxiolytic effects of the melatonin MT(2) receptor partial agonist UCM765: comparison with melatonin and diazepam. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):318–325. | |
Ochoa-Sanchez R, Comai S, Spadoni G, Bedini A, Tarzia G, Gobbi G. Melatonin, selective and non-selective MT1/MT2 receptors agonists: differential effects on the 24-h vigilance states. Neurosci Lett. 2014;561:156–161. | |
Schuck DC, Jordão AK, Nakabashi M, Cunha AC, Ferreira VF, Garcia CRS. Synthetic indole and melatonin derivatives exhibit antimalarial activity on the cell cycle of the human malaria parasite Plasmodium falciparum. Eur J Med Chem. 2014;78:375–382. | |
Carocci A, Catalano A, Bruno C, et al. N-(Phenoxyalkyl)amides as MT1 and MT2 ligands: Antioxidant properties and inhibition of Ca2+/CaM-dependent kinase II. Bioorg Med Chem. 2013;21(4): 847–851. | |
Peters JU. Polypharmacology – foe or friend? J Med Chem. 2013; 56(22):8955–8971. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.