Back to Journals » Drug Design, Development and Therapy » Volume 9
Mechanisms underlying probucol-induced hERG-channel deficiency
Authors Shi Y, Yan C, Zhang X, Yan M, Liu L, Geng H, Lv L, Li B
Received 16 April 2015
Accepted for publication 2 June 2015
Published 20 July 2015 Volume 2015:9 Pages 3695—3704
DOI https://doi.org/10.2147/DDDT.S86724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Yuan-Qi Shi,1,* Cai-Chuan Yan,1,* Xiao Zhang,1 Meng Yan,1 Li-Rong Liu,1 Huai-Ze Geng,1 Lin Lv,1 Bao-Xin Li1,2
1Department of Pharmacology, Harbin Medical University, 2State-Province Key Laboratory of Biopharmaceutical Engineering, Harbin, Heilongjiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: The hERG gene encodes the pore-forming α-subunit of the rapidly activating delayed rectifier potassium channel (IKr), which is important for cardiac repolarization. Reduction of IhERG due to genetic mutations or drug interferences causes long QT syndrome, leading to life-threatening cardiac arrhythmias (torsades de pointes) or sudden death. Probucol is a cholesterol-lowering drug that could reduce hERG current by decreasing plasma membrane hERG protein expression and eventually cause long QT syndrome. Here, we investigated the mechanisms of probucol effects on IhERG and hERG-channel expression. Our data demonstrated that probucol reduces SGK1 expression, known as SGK isoform, in a concentration-dependent manner, resulting in downregulation of phosphorylated E3 ubiquitin ligase Nedd4-2 expression, but not the total level of Nedd4-2. As a result, the hERG protein reduces, due to the enhanced ubiquitination level. On the contrary, carbachol could enhance the phosphorylation level of Nedd4-2 as an alternative to SGK1, and thus rescue the ubiquitin-mediated degradation of hERG channels caused by probucol. These discoveries provide a novel mechanism of probucol-induced hERG-channel deficiency, and imply that carbachol or its analog may serve as potential therapeutic compounds for the handling of probucol cardiotoxicity.
Keywords: long QT, hERG potassium channels, probucol, SGK1, Nedd4-2
Introduction
The hERG gene encodes the pore-forming subunits of channels that conduct the rapid delayed rectifier potassium current.1 Mutation of the hERG gene may cause dysfunction of rapidly activating delayed rectifier potassium channel (IKr) and thus lead to cardiac arrhythmia, such as congenital long QT syndrome (LQTS).2 In addition, a wide variety of potentially dangerous proarrhythmic effects of antiarrhythmic or nonantiarrhythmic drugs have been recognized to inhibit hERG channels, which could induce acquired LQT2.3,4 Diverse therapeutic compounds, agents of distinct pharmacological classes have been removed from the market; several drugs have been limited in clinical applications, due to their risk of ventricular arrhythmias.3 Since most of these adverse reactions are preventable, increased knowledge of drug application associated with repolarization abnormalities and careful evaluation of risk factors is strongly recommended. At present, there are no effective therapeutic methods to restore or enhance hERG-channel function. Therefore, looking for compounds or therapies is extremely necessary right now.
Probucol is a cholesterol-lowering drug initially developed in the treatment of coronary artery disease,5 which reduces IhERG by decreasing membrane hERG-channel expression and thus causes LQTS.6 Although probucol was identified as the hERG inhibitor responsible for LQTS, the underlying mechanism is still not fully understood. Therefore, a better mechanistic understanding of probucol-induced hERG-channel abnormality may help us to develop therapeutic compounds that are prone to the disruption of hERG-channel processing and trafficking.
In this study, we investigated the mechanism underlying probucol-induced hERG-channel deficiency. It has been confirmed that SGK1 expression is regulated during both discrete developmental stages and pathological conditions, such as hypertension, drug interferences, diabetic neuropathy, ischemia, trauma, and neurodegenerative diseases.7 Over the past few years, there has been increasing evidence that overexpression of SGK1 is able to increase the current and expression level of the mature proteins of hERG channels localized in the membrane.8 SGK1 and its isoforms are expressed in every tissue, including the heart, and play a significant role in the regulation of hERG-channel expression. Interestingly, the downstream targets of SGK1 include the E3 ubiquitin ligase Nedd4-2, which is responsible for substrate recognition in the degradation process of proteins. Also, it has been reported that SGK inhibits Nedd4-2 activity by phosphorylating Nedd4-2 at Ser-444 residues present in the WW domains; this consequently enhances cell-surface expression of the hERG channel, which is a direct substrate of Nedd4-2.8 Also, probucol and As2O3 are the two main kinds of drugs to decrease hERG expression and subsequently cause LQTS.9,10 Therefore, we wondered whether these two drugs, which can induce hERG-channel deficiency, were involved in the ubiquitination protein-degradation pathway. Based on this, we became interested in SGK1 expression after treatment with different concentrations of probucol or As2O3 in hERG-HEK293 cells. As the research proceeded much further, our data provided evidence that the SGK1–Nedd4-2 axis represents a novel mechanism for probucol to decrease hERG-channel expression and eventually cause LQTS and torsade de pointes. In addition, we discovered that carbachol (CCh) impeded the reduction of the 155 kDa hERG expression induced by long-term treatment of probucol through enhancement of the phosphorylation level of Nedd4-2 as an alternative to SGK1.
Materials and methods
Reagents
Probucol and CCh were purchased from Sigma-Aldrich Co (St Louis, MO, USA). In cellular experiments, hERG-HEK293 cells were incubated with different concentrations of probucol for 48 hours with or without different concentrations of CCh, which were diluted in cultured medium.
Cell culture
Experiments were performed on HEK293 cells stably expressing the wild-type hERG gene. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (Hyclone, Logan, UT, USA) supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and 400 mg/mL Geneticin (G-418; Thermo Fisher Scientific) in an atmosphere of 95% humidified air and 5% CO2 at 37°C. For electrophysiological studies, the cells were harvested from the culture dishes by 0.25% trypsinase, washed twice with standard Dulbecco’s Modified Eagle’s Medium, and stored in this medium at room temperature for later research. Cells were investigated within 8 hours of harvest.11
Western blot analysis
Western blot experiments were used to detect the expression levels of interest proteins. Drugs were diluted and added to hERG-HEK293 cells for 48 hours at 37°C before analysis by Western blot. The cells were washed using ice-cold phosphate-buffered saline, and total protein was harvested with radioimmunoprecipitation-assay buffer containing 1% protease inhibitor (Sigma-Aldrich Co). Protein (100 μg) per sample was separated using 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred onto nitrocellulose membranes (Agilent Technologies, Santa Clara, CA, USA). The membrane was blocked afterward using 5% nonfat milk and 0.1% Tween 20 in Tris-buffered saline and immunoblotted overnight using appropriate primary antibodies at 4°C with gentle shaking. Goat antirabbit or goat antimouse fluorochrome-labeled Alexa Fluor 800 (dilution 1:8,000; Li-Cor Inc, Lincoln, NE, USA) was used as a secondary antibody. The Odyssey infrared fluorescent scanning system (Li-Cor Inc) was used to detect signals of membrane proteins. To quantify the Western blot data, the intensities of proteins of interest in each gel are firstly normalized to their respective actin intensities, then the normalized intensities are compared with the intensity of control group and expressed as relative values to their controls.12
Patch-clamp recording techniques
The whole-cell patch-clamp technique was used to measure hERG currents.13 Heat-polished patch pipettes had final resistances of 2–4 MW when filled with a pipette solution containing 130 mM KCl, 1 mM MgCl2·6H2O, 10 mM HEPES, 5 mM Mg–adenosine triphosphate, 5 mM ethylene glycol tetraacetic acid, and 0.1 mM guanosine triphosphate, pH 7.3 with KOH. The standard 5 mM K+ bath solution contained 136 mM NaCl, 5.4 mM KCl, 5 mM HEPES, 1 mM MgCl2·6H2O, 1 mM CaCl2, and 10 mM glucose, pH 7.4 with NaOH. For electrophysiological recordings, submaximally confluent cells were used after 1–3 days of culture at a seeding density of 1–4×105 cells per flask (Nest Biotechnology, Wuxi, People’s Republic of China). The cells were harvested from the plate by treatment with 0.25% trypsin and 0.02% ethylenediaminetetraacetic acid, and transferred to a small cell bath mounted on the stage of an inverted microscope (IX-70; Olympus Corporation, Tokyo, Japan), where they were allowed to attach to the glass bottom for about 10 minutes. The cells were then superfused continuously at a ratio of 1.5 mL/min. The experiment set a holding potential voltage from −80 mV to voltages between −70 and 70 mV in 10 mV increments. Tail currents upon repolarization to the holding potential of −80 mV after the depolarizing pulse to 50 mV were used for the current amplitudes in control and treated groups. Patch-clamp experiments were all performed at room temperature. An Axopatch 200B patch-clamp amplifier was used to record membrane current. Computer software (Clampex 9.2; Molecular Devices LLC, Sunnyvale, CA, USA) was used to generate voltage-clamp protocols and acquire data. Capacitance and series-resistance compensation were optimized. Data were recorded on a computer via Digidata 1322A, and analyzed using Clampfit 9.2 (Molecular Devices LLC) and Prism (GraphPad Software Inc, La Jolla, CA, USA) software. Graphical fits of the data were made using previously described standard equations.14,15
Statistical analyses
Data are expressed as means ± standard error of mean. A one-way analysis of variance or Student’s t-test was used to determine the significance of differences between control and test groups. A P-value of 0.05 or less was considered significant (two-tailed). Boltzmann distribution was used to fit voltage-dependent activation and inactivation curves, and a single exponential function was used to fit the curve of recovery from inactivation (reactivation). All graphs were drawn by GraphPad Prism 5.0.
Results
Probucol significantly reduced SGK1 expression
The stress-responsive SGK belongs to a family of serine/threonine kinases that is under acute transcriptional control by several stimuli.16 It has been demonstrated that overexpression of SGK1 increased the current and expression level of the membrane-localized mature proteins of hERG channels stably expressed in hERG-HEK293 cells.8 Probucol reduces hERG-channel expression in the plasma membrane in a concentration-dependent manner.6 Therefore, we used a concentration gradient of probucol to investigate the mechanisms for probucol-induced hERG-channel deficiency. Cells were cultured in either control medium or probucol (30, 50, and 100 μM) containing medium for 48 hours; hERG-expression levels and IhERG were then examined. As depicted in Figure 1A, probucol significantly reduced SGK1 expression in a concentration-dependent manner, indicating that probucol reliably downregulated SGK1 expression. In our previous research, we demonstrated that As2O3 can damage the IhERG current via disturbing its trafficking to cellular membrane.17 In order to estimate whether As2O3-induced hERG-channel inhibition was involved in the alteration of SGK1 level, we substituted the probucol with 3 μM As2O3. After incubation of 3 μM As2O3 for 24 hours, the SGK1 protein level was also detected as previously. Figure 1B shows that As2O3 had no significant effect on SGK1 expression.
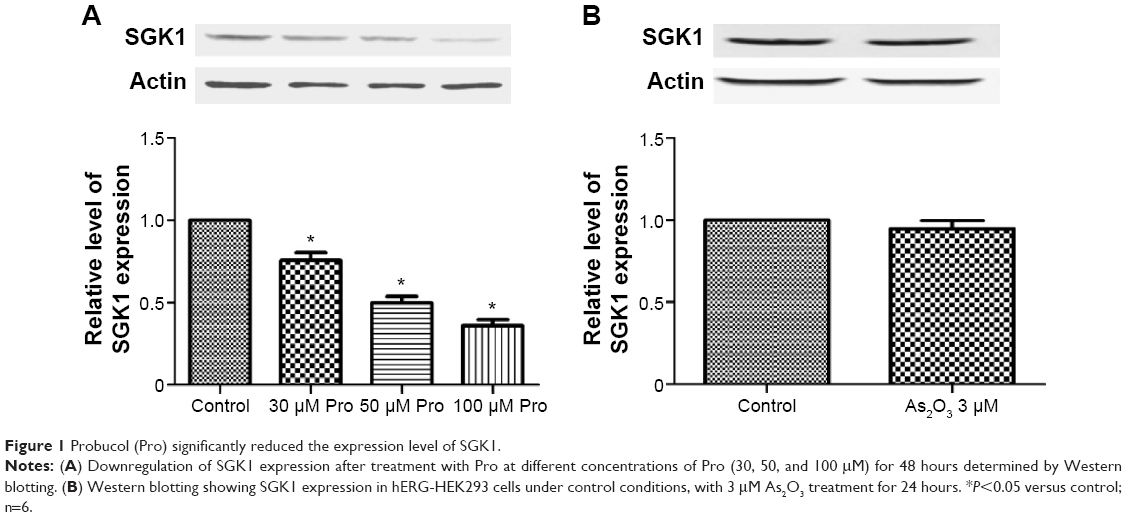 | Figure 1 Probucol (Pro) significantly reduced the expression level of SGK1. |
Phosphorylation level of Nedd4-2 decreased due to reduced expression of SGK1
Nedd4-2, also known as E3 ubiquitin ligase, is responsible for substrate recognition and ubiquitin transfer to target proteins.18 The phosphorylated form of Nedd4-2 (p-Nedd4-2) leads to the inactivation of its catalytic activity.8 It has been confirmed that Nedd4-2 can be phosphorylated by SGK1 in response to various stimuli.19 We have previously shown that probucol significantly reduces SGK1 expression. Since Nedd4-2 is the direct downstream target of SGK1,19 we suspected that reduced SGK1 levels may result in decreases of phosphorylated Nedd4-2 levels. Therefore we proposed the hypothesis that probucol treatment decreases hERG-channel expression by increasing Nedd4-2-mediated hERG ubiquitination. Interestingly, our data provide evidence that probucol treatment significantly decreased p-Nedd4-2, but not the total level of Nedd4-2 (Figure 2A and B). Meanwhile, As2O3 is also a major drug we do research on. To determine whether As2O3-induced hERG-channel abnormality results from enhanced ubiquitination, we performed our experiment by detecting the effect of As2O3 on Nedd4-2 and p-Nedd4-2 expression. As shown in Figure 2C and D, As2O3 had no significant effect on either Nedd4-2 or p-Nedd4-2. These findings indicated that As2O3-induced hERG-channel abnormality was probably due to other reasons, like trafficking deficiency.10
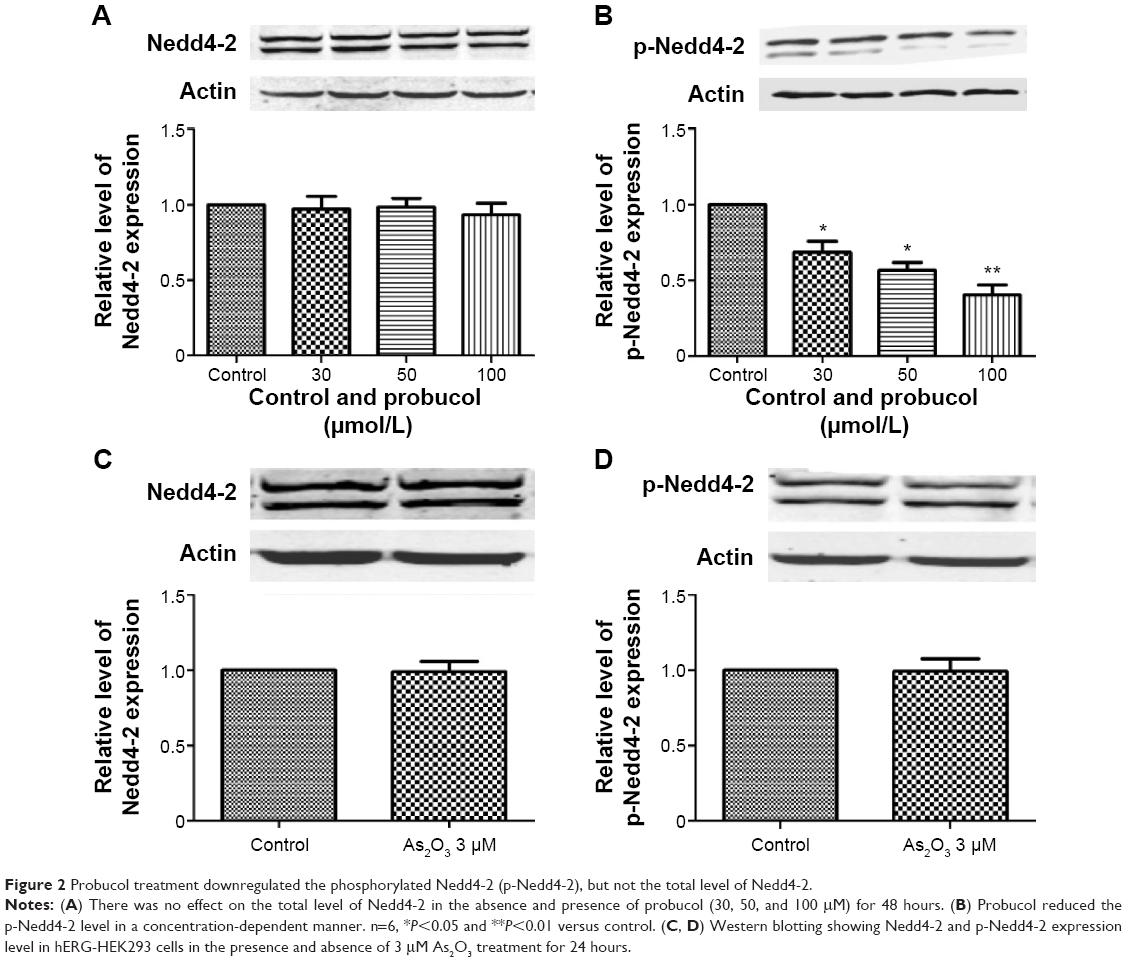 | Figure 2 Probucol treatment downregulated the phosphorylated Nedd4-2 (p-Nedd4-2), but not the total level of Nedd4-2. |
Carbachol rescued probucol-induced hERG-channel deficiency
CCh, which is a cholinomimetic drug that binds and activates the acetylcholine receptor and increases the expression and function of hERG. CCh-mediated increase in hERG expression was accomplished by enhancement of the phosphorylation level but not the total level of Nedd4-2.20 To further demonstrate probucol-induced hERG-channel deficiency caused by Nedd4-2 involved enhancement of ubiquitination, hERG-HEK293 cells were cultured with different concentrations (25 and 50 μM) of CCh and 100 μM probucol for 48 hours; hERG-expression levels and IhERG were then examined. Previous research has demonstrated that hERG-channel protein is primarily synthesized in the endoplasmic reticulum as the immature core-glycosylated form with a molecular mass of 135 kDa, and then transported to the Golgi apparatus for glycosylation to become the mature and functional form with a molecular mass of 155 kDa.21 Consistent with our previous study, the expression level of hERG, especially the mature form, declined approximately 50% compared to the control group with the incubation of probucol. However, in the probucol and CCh coincubated groups, the inhibition of hERG channels was reversed. As shown in Figure 3A, CCh (25 and 50 μM) reversed the downregulation of hERG 155 kDa channels induced by 100 μM probucol. However, CCh (50 μM) did not reverse the As2O3-induced hERG-channel deficiency (Figure 3B). Meanwhile, we also detected the hERG current after treatment with probucol and CCh. Likewise, CCh (25 and 50 μM) reversed the downregulation of IhERG induced by 100 μM probucol (Figure 3C and D).
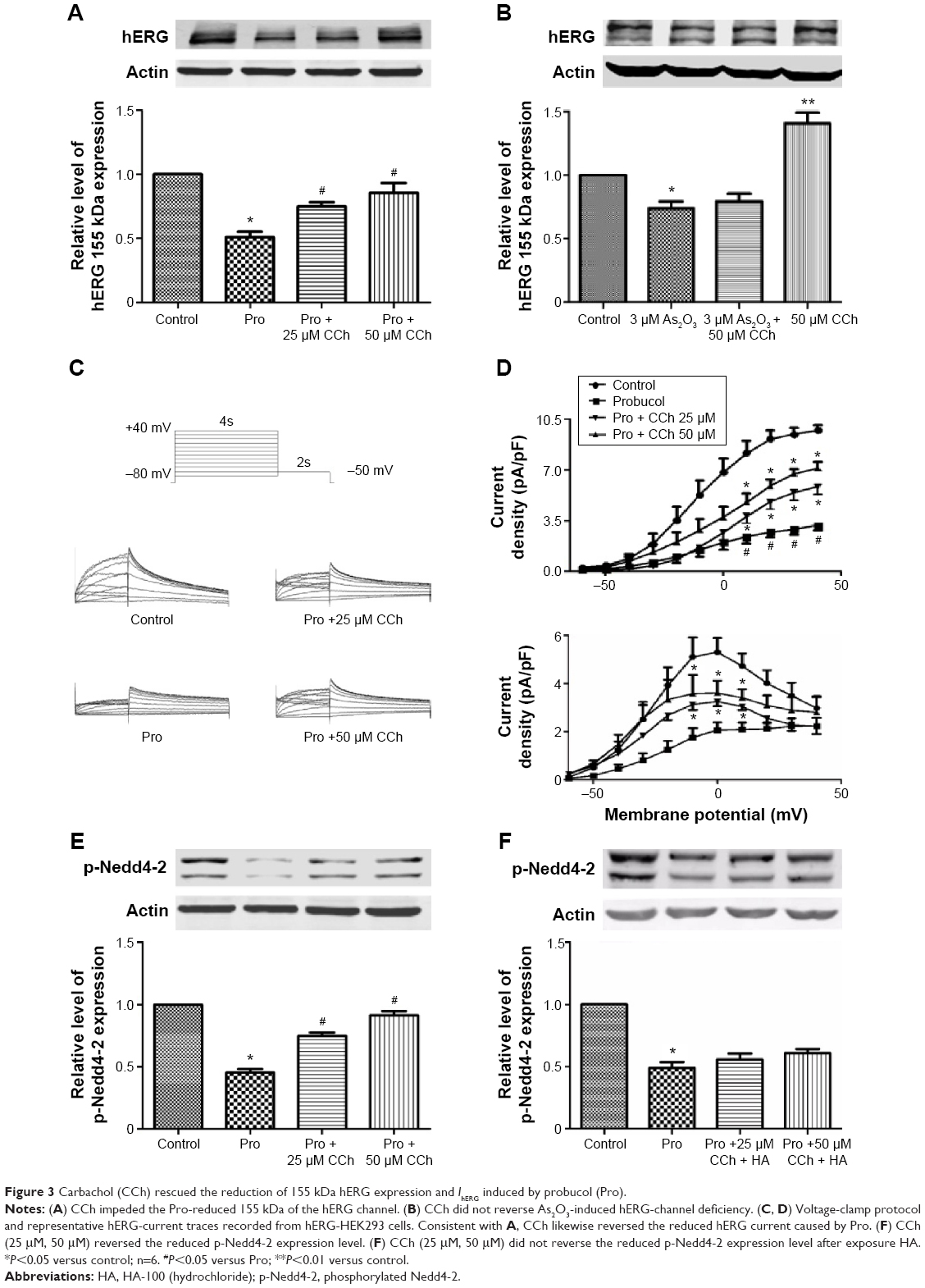 | Figure 3 Carbachol (CCh) rescued the reduction of 155 kDa hERG expression and IhERG induced by probucol (Pro). |
To demonstrate further whether CCh-impeded probucol-induced reduction of 155 kDa hERG expression was the result of alteration of p-Nedd4-2, we detected the p-Nedd4-2 expression level under equivalent conditions. Interestingly, the increased p-Nedd4-2 was accompanied by an increased expression of the mature hERG band. Figure 3E illustrates that CCh (25 and 50 μM) treatment significantly increased p-Nedd4-2 compared with the probucol-treatment group. It has been demonstrated that blockade of the PKC pathway abolishes the CCh-induced enhancement of hERG channels.20 Based on this, we used the PKC inhibitor HA to eliminate the effect of CCh on Nedd4-2, and the Western blot results showed that after treatment with HA, CCh did not reverse the reduced p-Nedd4-2 caused by probucol (Figure 3F).
Effects of CCh on APD prolonged by probucol on neonatal cardiac myocytes
To test whether our analysis in heterologous expression systems might be extended to ventricular cardiomyocytes, we studied the effects of extracellular application of probucol and CCh on action potentials (APs) evoked in neonatal cardiac myocytes. Figure 4 illustrates the representative traces showing the effects of CCh on AP duration (APD) prolonged by probucol. Changes in AP waveform were described by using 50% and 90% repolarization of APD (APD50 and APD90, respectively).
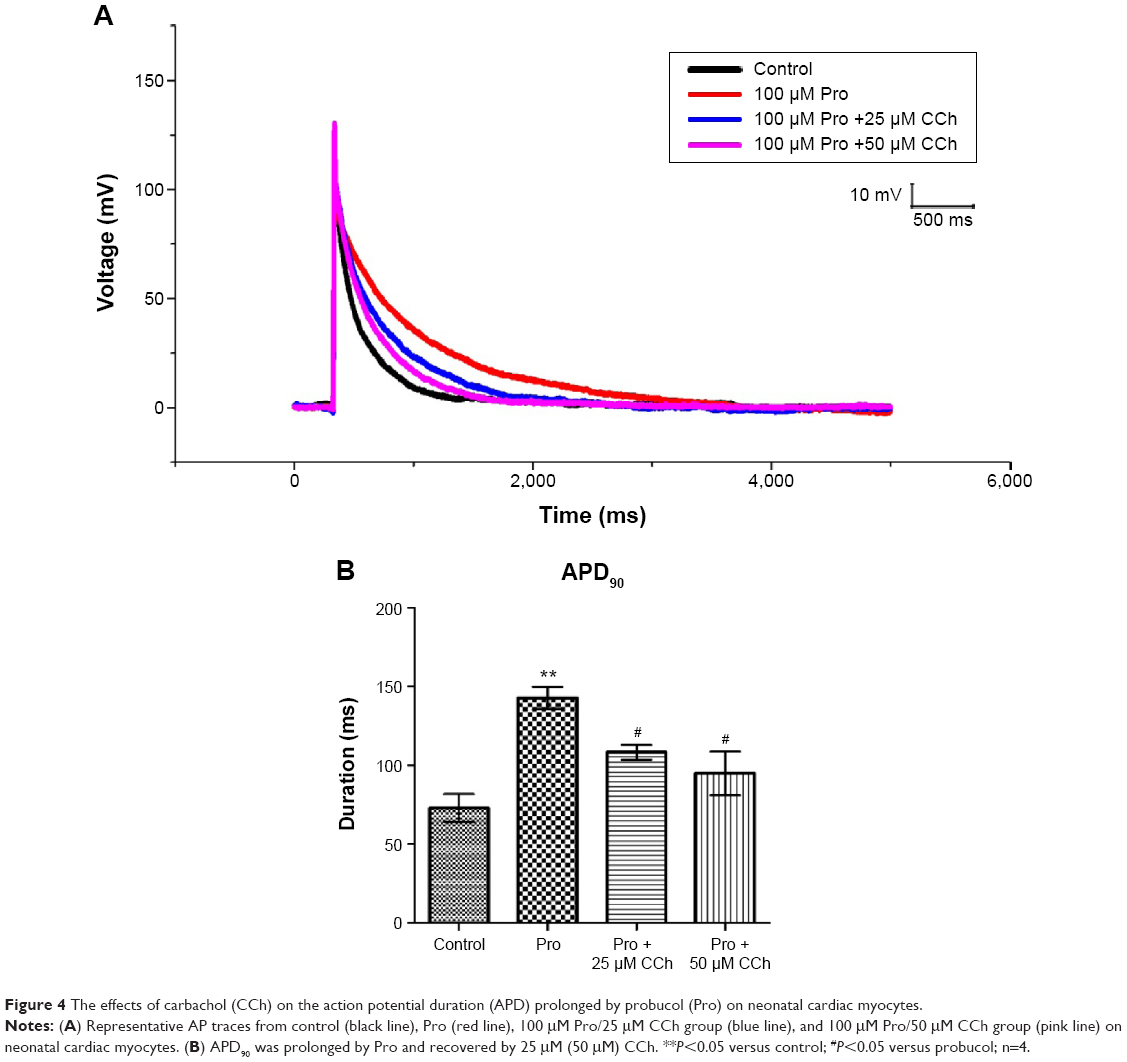 | Figure 4 The effects of carbachol (CCh) on the action potential duration (APD) prolonged by probucol (Pro) on neonatal cardiac myocytes. |
Neonatal cardiac myocytes were cultured under control conditions or in the presence of 100 μM probucol for 48 hours. We found that APD was significantly prolonged on neonatal cardiac myocytes (Figure 4). To demonstrate the effect of CCh on the prolongation of cardiac APs after probucol treatment, we added CCh to myocytes incubated with 100 μM probucol. Results showed that APD90 was prolonged to 142.6 milliseconds by probucol, 25 μM CCh shortened APD90 to 102.6 milliseconds, and 50 μM CCh shortened APD90 to 89.7 milliseconds on neonatal cardiac myocytes (Figure 4).
Probucol damaged hERG current but did not affect channel kinetics after long-term treatment
To identify the damage of hERG current induced by probucol depended on the decrease of protein level but not channel functions, patch-clamp recordings were used to detect the long-term effect of probucol on hERG-channel kinetics. Figure 5A shows the effect of probucol and coincubation of probucol with CCh on the activation curve after incubation for 48 hours. The V1/2 was −7.98±1.48 mV in the control group and −9.42±1.93 mV in the probucol group. Probucol did not shift the activation curve. Figure 5B illustrates the inhibited effect of probucol and the reversion effect of CCh (25 and 50 μM) on hERG current and its protocol, the results of which were consistent with previous studies.9,22 Figure 5C shows the effect of probucol on the time-constant curve. As depicted, probucol did not change the time constant or the onset of inactivation and recovery from inactivation. Based on these results, we came to the conclusion that long-term incubation of probucol damaged hERG current, but did not affect channel kinetics.
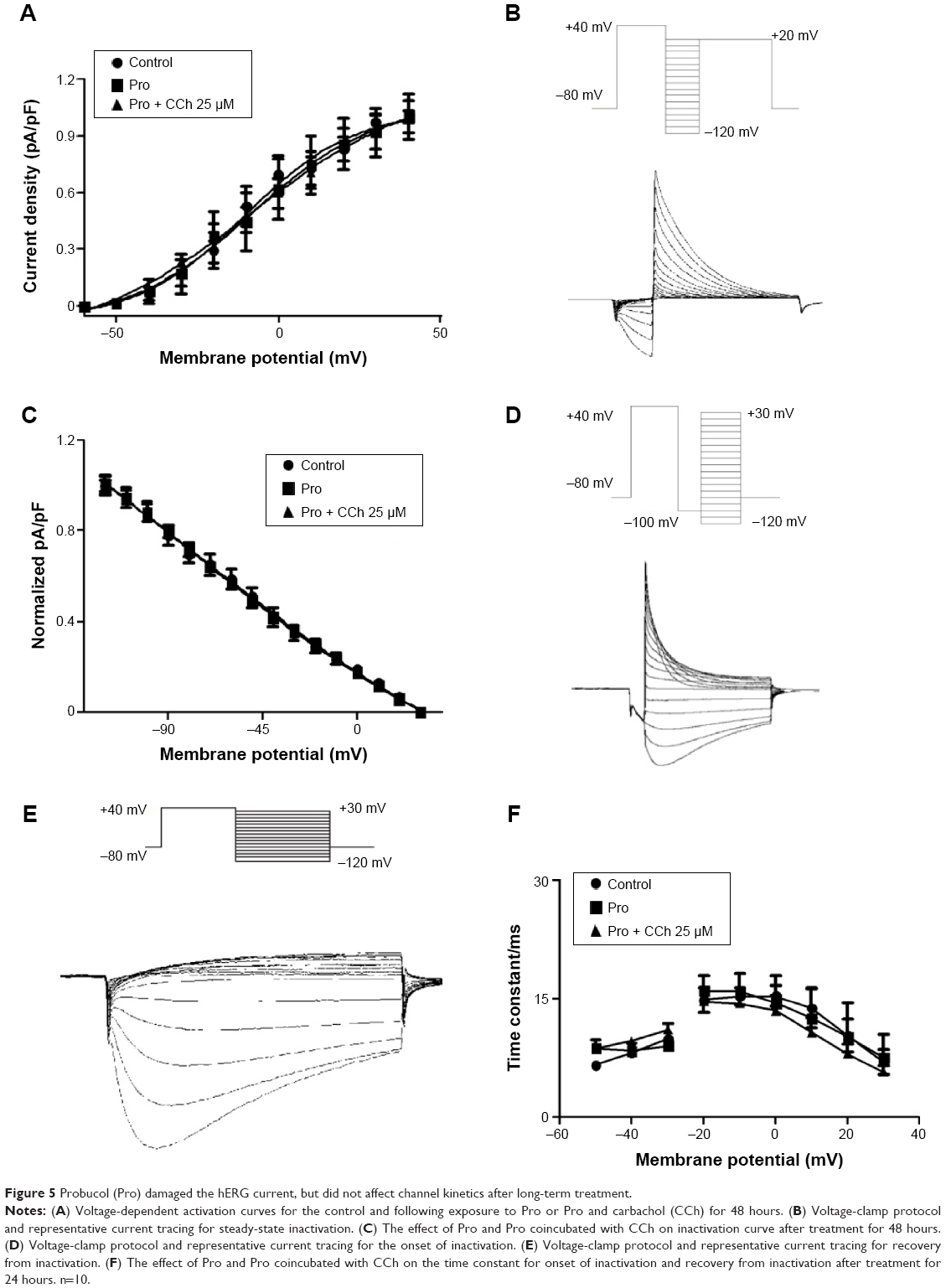 | Figure 5 Probucol (Pro) damaged the hERG current, but did not affect channel kinetics after long-term treatment. |
Discussion
A great variety of compounds interfere with hERG function and have the potential to cause LQTS by directly blocking the channel or reducing channel-membrane expression.23 Increasing awareness of drug-induced hERG channel-trafficking deficiency will help to reduce further the potentially fatal adverse cardiac events associated with acquired LQTS.
Probucol is a cholesterol-lowering drug initially developed in the treatment of coronary artery disease.5 By decreasing hERG channel-membrane density, probucol can cause LQTS and torsades de pointes arrhythmia in patients and sudden cardiac death in experimental animals.9,24 However, the mechanisms of probucol-reduced hERG-channel expression seem to be quite unspecific. Therefore, a better mechanistic understanding of probucol-induced hERG-channel deficiency may help us to develop therapeutic compounds that are prone to the disruption of hERG-channel processing and trafficking.
As we know, hERG proteins are primarily synthesized in the endoplasmic reticulum and then transported to the Golgi apparatus for glycosylation, and finally transported to the cell membrane for expression. Misfolded or misassembled proteins are retained in the endoplasmic reticulum by its quality-control system, altering forward trafficking of hERG channels to the plasma membrane.21,25 Our data show that probucol (30, 50, and 100 μM) treatment decreases the mature 155 kDa band of hERG, but does not reduce the intracellular forms of the immature hERG 135 kDa band (Figure 3A), suggesting that probucol may not block hERG forward trafficking. On the other hand, we demonstrated that probucol damaged hERG current, but did not affect channel kinetics after long-term treatment. This seems to be a result of enhanced degradation of plasma membrane hERG protein induced by probucol treatment. It has been demonstrated that Nedd4-2 plays an important role in the degradation pathway of hERG retrograde trafficking.26 The mature hERG channels are degraded by ubiquitin ligase Nedd4-2 via enhanced channel ubiquitination. In the present study, we confirmed that Nedd4-2 is involved in the process of probucol-disrupted hERG surface expression. Probucol treatment significantly reduced p-Nedd4-2 expression levels (Figure 2B). Interestingly, it has been reported that CCh can efficiently increase p-Nedd4-2, but not the total level of Nedd4-2, and eventually increase the expression level of the hERG channel.20 To address further whether CCh could rescue the probucol-disrupted IhERG and hERG-channel expression, we coincubated probucol and CCh in hERG-HEK293 cells for 48 hours. Figure 3A illustrates that CCh was able to rescue the decreased 155 kDa hERG-channel expression induced by probucol. Likewise, hERG current was also reversed by long-term coincubation with CCh (Figure 3B–D).
Cardiac toxicities caused by administration of probucol can be controlled by complex signaling pathways or molecular mechanisms. Our present research shows that the SGK1–Nedd4-2 axis may provide a plausible explanation for probucol-induced LQTS and torsade de pointes, but the results did not illustrate the involvement of any other upstream protein kinases or signaling pathways that could affect the expression and function of serine/threonine kinase SGK1. Whether there are other molecules or protein kinases that contribute to the expression and function of SGK1 needs further investigation.
As2O3 has long been of biomedical interest, since it is widely used to treat cancers like acute myeloid leukemia and other conditions.27 However, the cardiotoxicity of LQTS restricts its clinical applications. Previous studies have shown that As2O3 can decrease the expression of hERG channels by disturbing its trafficking to the cell membrane. In this research, we wanted to figure out whether As2O3 treatment could also affect the SGK1–Nedd4-2 signaling pathway. Our data indicated that in the presence or absence of 3 μM As2O3, there was no significant difference between the two groups in the activity of the SGK1–Nedd4-2 signaling pathway and CCh did not rescue the As2O3-induced hERG-channel deficiency (Figure 3B), which means CCh enhances the expression level of hERG channels mainly by the SGK1–Nedd4-2 signaling pathway.
In summary, the present study provides evidence that probucol does not block the hERG channel, but accelerates degradation of the mature hERG channel and inhibit hERG current by decreasing SGK1 expression level and eventually enhancing the ubiquitination of the hERG channel through the E3 ubiquitin ligase Nedd4-2, whereas CCh can rescue probucol-disrupted hERG surface expression by enhancing the phosphorylation level of Nedd4-2 as an alternative to SGK1. These interesting findings may help us in identifying strategies and therapeutic compound classes to prevent or treat QT-interval prolongation associated with administration of probucol.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31173050), Funds for Creative Research Groups of the National Natural Science Foundation of China (81421063), and the Research Fund for the Doctoral Program of Higher Education of China (20122307110007).
Disclosure
The authors report no conflicts of interest in this work.
References
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. | ||
Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. | ||
Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62(4):760–781. | ||
Farkas AS, Nattel S. Minimizing repolarization-related proarrhythmic risk in drug development and clinical practice. Drugs. 2010;70(5):573–603. | ||
Yamamoto A. A unique antilipidemic drug – probucol. J Atheroscler Thromb. 2008;15(6):304–305. | ||
Guo J, Massaeli H, Li W, et al. Identification of IKr and its trafficking disruption induced by probucol in cultured neonatal rat cardiomyocytes. J Pharmacol Exp Ther. 2007;321(3):911–920. | ||
Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lübbert H, Stichel CC. Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci. 2005;30(2):249–264. | ||
Lamothe SM, Zhang S. The serum- and glucocorticoid-inducible kinases SGK1 and SGK3 regulate hERG channel expression via ubiquitin ligase Nedd4-2 and GTPase Rab11. J Biol Chem. 2013;288(21):15075–15084. | ||
Cubeddu LX. Iatrogenic QT abnormalities and fatal arrhythmias: mechanisms and clinical significance. Curr Cardiol Rev. 2009;5(3):166–176. | ||
Zhao X, Zhang KP, Huang T, et al. The rescuable function and mechanism of resveratrol on As2O3-induced hERG K+ channel deficiency. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(11):1079–1089. | ||
Zhang M, Mahoney E, Zuo T, Manchanda PK, Davuluri RV, Kirschner LS. Protein kinase A activation enhances β-catenin transcriptional activity through nuclear localization to PML bodies. PLoS One. 2014;9(10):e109523. | ||
Zhang YP, Kong QH, Huang Y, Wang GL, Chang KJ. Inhibition of c-FLIP by RNAi enhances sensitivity of the human osteogenic sarcoma cell line U2OS to TRAIL-induced apoptosis. Asian Pac J Cancer Prev. 2015;16(6):2251–2256. | ||
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391(2):85–100. | ||
Gu DF, Li XL, Qi ZP, et al. Blockade of HERG K+ channel by isoquinoline alkaloid neferine in the stable transfected HEK293 cells. Naunyn Schmiedebergs Arch Pharmacol. 2009;380(2):143–151. | ||
Du X, Lu D, Daharsh ED, Yao A, Dewoody R, Yao JA. Dimethyl sulfoxide effects on hERG channels expressed in HEK293 cells. J Pharmacol Toxicol Methods. 2006;54(2):164–172. | ||
Lang F, Strutz-Seebohm N, Seebohm G, Lang UE. Significance of SGK1 in the regulation of neuronal function. J Physiol. 2010;588(Pt 18):3349–3354. | ||
Zhao X, Shi YQ, Yan CC, et al. Up-regulation of miR-21 and miR-23a contributes to As2O3-induced hERG channel deficiency. Basic Clin Pharmacol Toxicol. 2015;116(6):516–523. | ||
Flores SY, Debonneville C, Staub O. The role of Nedd4/Nedd4-like dependant ubiquitylation in epithelial transport processes. Pflugers Arch. 2003;446(3):334–338. | ||
Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004;279(44):45753–45758. | ||
Wang T, Hogan-Cann A, Kang Y, et al. Muscarinic receptor activation increases hERG channel expression through phosphorylation of ubiquitin ligase Nedd4-2. Mol Pharmacol. 2014;85(6):877–886. | ||
Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113(3):365–373. | ||
Guo J, Li X, Shallow H, et al. Involvement of caveolin in probucol-induced reduction in hERG plasma-membrane expression. Mol Pharmacol. 2011;79(5):806–813. | ||
Roy M, Dumaine R, Brown AM. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94(4):817–823. | ||
Hayashi K, Shimizu M, Ino H, et al. Probucol aggravates long QT syndrome associated with a novel missense mutation M124T in the N-terminus of HERG. Clin Sci (Lond). 2004;107(2):175–182. | ||
Dennis A, Wang L, Wan X, Ficker E. hERG channel trafficking: novel targets in drug-induced long QT syndrome. Biochem Soc Trans. 2007;35(Pt 5):1060–1063. | ||
Guo J, Wang T, Li X, et al. Cell surface expression of human ether-a-go-go-related gene (hERG) channels is regulated by caveolin-3 protein via the ubiquitin ligase Nedd4-2. J Biol Chem. 2012;287(40):33132–33141. | ||
Soignet SL, Frankel SR, Douer D, et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol. 2001;19(18):3852–3860. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.