Back to Journals » Drug Design, Development and Therapy » Volume 19
Mechanisms of Remifentanil-Induced Postoperative Hyperalgesia: A Comprehensive Review
Authors Zhu K, Wen X, Mei X, Fang F, Zhang T
Received 29 June 2025
Accepted for publication 15 August 2025
Published 27 August 2025 Volume 2025:19 Pages 7445—7457
DOI https://doi.org/10.2147/DDDT.S550335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Kexin Zhu,1,2,* Xiaolin Wen,1,2,* Xuan Mei,1,2 Fang Fang,1,2 Tianyao Zhang1,2
1School of Clinical Medicine, Chengdu Medical College, Chengdu, Sichuan Province, People’s Republic of China; 2Department of Anesthesiology, The First Affiliated Hospital of Chengdu Medical College, Chengdu, Sichuan Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tianyao Zhang, School of Clinical Medicine and the First Affiliated Hospital of Chengdu Medical College, No. 278 Middle of Baoguang Avenue, Xindu District, Chengdu, Sichuan Province, 610500, People’s Republic of China, Email [email protected]
Abstract: Remifentanil, a widely used ultra-short-acting μ-opioid receptor agonist in clinical anesthesia, is strongly associated with postoperative hyperalgesia (remifentanil-induced hyperalgesia, RIH), posing significant challenges to postoperative pain management. RIH is characterized by an abnormally heightened pain perception following opioid withdrawal, and its underlying mechanisms are complex and multifactorial. Current research highlights the roles of central sensitization, peripheral sensitization, and multiple interacting molecular pathways. These include NMDA receptor activation, glial cell activation, neuroinflammation, disinhibition of inhibitory neurotransmission, and dysfunction of the descending pain modulation system. Additionally, alterations in ion channel expression, synaptic plasticity enhancement, and peripheral responses to inflammatory mediators contribute critically to RIH development. Individual factors such as age, sex, genetic polymorphisms, and surgical type significantly influence the risk of RIH. Although substantial progress has been made in elucidating the molecular mechanisms of RIH, a unified theoretical framework and effective clinical strategies remain lacking. Future studies should emphasize multi-omics approaches and clinically relevant experimental models to uncover key regulatory targets and provide a theoretical basis for individualized analgesic interventions.
Keywords: remifentanil, postoperative hyperalgesia, NMDA receptor, glial cell activation, central sensitization, NLRP3 inflammasome
Introduction
As early as the 1980s, the American Pain Society defined pain as the “fifth vital sign”, following body temperature, pulse, respiratory rate, and blood pressure.1 This initiative aimed to raise healthcare providers’ awareness of pain assessment and ensure timely and effective pain management. However, the widespread emphasis on pain has also led to concerns about opioid overuse and the associated risks of dependence. Opioids remain a cornerstone in the management of both acute and chronic pain. Despite their efficacy, they are accompanied by well-recognized adverse effects, including nausea, vomiting, constipation, respiratory depression, tolerance, addiction.2 In addition, opioids may induce a specific pathophysiological phenomenon known as opioid-induced hyperalgesia (OIH).
OIH is a paradoxical phenomenon in which patients develop increased pain sensitivity following opioid administration, especially after abrupt withdrawal. It is characterized by heightened responsiveness of both peripheral and central nervous systems, leading to exaggerated pain in response to noxious stimuli and the emergence of pain from previously non-painful stimuli.3 The occurrence of OIH is closely related to the pharmacokinetic of opioids.
Globally, remifentanil has become one of the most widely used opioids in anesthesia practice due to its ultra-short half-life, organ-independent metabolism, and precise titratability.4 In developed countries such as the United States, Germany, and Japan, remifentanil is frequently used in major surgeries and total intravenous anesthesia (TIVA).5 Despite its clinical advantages, concerns about remifentanil-induced hyperalgesia (RIH) have prompted international discussions regarding optimal dosing protocols and multimodal analgesia strategies.6 In contrast, its use in some developing countries remains limited due to economic constraints or limited awareness of perioperative pain hypersensitivity.7
Remifentanil, a short-acting μ-opioid receptor agonist, is widely used in clinical anaesthesia due to its rapid onset, short duration of action, and metabolism independent of liver and kidney function. However, studies have shown that remifentanil’s pharmacokinetics make it more likely to induce hyperalgesia (Remifentanil-Induced Hyperalgesia, RIH)—with a higher intensity compared to other opioids.8 The term remifentanil-induced hyperalgesia (RIH) refers to this specific subtype of OIH.
Clinically, RIH manifests as increased postoperative pain intensity, expansion of the pain perception area, and abnormal sensory responses such as allodynia and hyperalgesia. These symptoms typically occur within hours after remifentanil cessation and may persist for several days. Reported incidence rates vary widely, ranging from 8% to over 40%, depending on patient characteristics, remifentanil dosage and duration, and the nature of surgical procedures. Identified risk factors include high intraoperative infusion rates, prolonged exposure, advanced age, female sex, genetic polymorphisms, and surgeries involving significant tissue trauma.
Despite growing evidence for the existence of RIH, its underlying mechanisms remain incompletely understood. Current research implicates a complex interplay of central and peripheral sensitization, N-methyl-D-aspartate (NMDA) receptor activation, glial cell and neuroinflammatory responses, descending pain modulation dysfunction, and ion channel remodeling. Among these, neuroinflammation has attracted increasing attention. Studies indicate that remifentanil may activate glial cells (such as microglia and astrocytes), leading to the release of pro-inflammatory cytokines (eg, IL-1β and TNF-α), which further amplify pain transmission.9 However, a unified mechanistic framework and effective clinical strategies are still lacking.
This review aims to provide a comprehensive overview of the molecular and cellular mechanisms underlying remifentanil-induced hyperalgesia. By summarizing recent findings and identifying unresolved questions, we hope to offer theoretical insights that may guide future mechanistic studies and contribute to the development of targeted, individualized approaches to pain management.
Clinical Characteristics and Influencing Factors of RIH
There is currently no unified diagnostic standard for RIH, but its core clinical features primarily include a marked increase in postoperative pain intensity, an expanded area of pain perception, and sensory abnormalities such as hyperalgesia to noxious stimuli and allodynia in response to non-noxious stimuli.8 Patients with RIH typically experience sudden-onset, intense pain within hours of drug discontinuation, and this heightened pain sensitivity can persist far beyond the expected duration of routine postoperative pain.10 It is essential to differentiate hyperalgesia from opioid tolerance (OT) in clinical practice. Hyperalgesia is characterized by an enhanced pain response to the same stimulus (a leftward shift in the stimulus-response curve), whereas opioid tolerance is typically manifested by a need for increased opioid dosage to achieve equivalent analgesia (a rightward shift in the dose-response curve), without expansion of the pain area.6,11 Hyperalgesia is frequently conflated with opioid tolerance in clinical settings due to the overlapping nature of their symptoms.12 The distinction can be made based on the following characteristics: hyperalgesia is marked by the onset of new pain, changes in the nature of pain (such as variations in intensity or type), expansion of the pain area. Importantly, increased opioid dosage in hyperalgesia may paradoxically worsen the pain, while it is generally effective in OT13 (Figure 1).
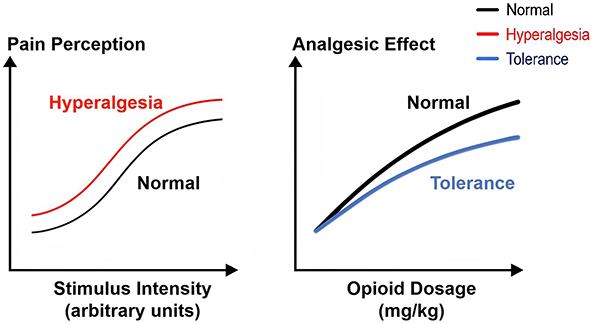 |
Figure 1 Schematic comparison between hyperalgesia and tolerance. Left: hyperalgesia manifests as increased pain response to the same stimulus. Right: tolerance manifests as reduce analgesic effect with higher opioid doses. Black curve: Normal response to stimulus or drug dose. Red curve: Hyperalgesia—same stimulus causes greater pain. Blue curve: Tolerance—higher dose needed for same analgesia. |
Although OIH and OT are different pathological concepts, studies suggest they may share common molecular mechanisms,8 such as: NMDA receptor activation,14 Glial cell activation,15 Alterations in opioid receptor signal pathways,16 Neural plasticity,17 Epigenetic regulation18 and so on.
At present, the diagnosis of RIH primarily relies on subjective pain assessment tools (eg, Visual Analog Scale VAS), combined with clinical history and exclusion of other causes. Studies have shown that high intraoperative doses of remifentanil significantly reduce the postoperative pain threshold within 24 hours,19 with RIH symptoms peaking during this period. In some patients, the hyperalgesic state can persist for up to 7 days postoperatively.10
The incidence of RIH varies widely across clinical contexts. For instance, it ranges from 8% to 30% in postoperative patients,3,6 and can exceed 40% in those with chronic pain.18 The duration of RIH also shows significant individual variability. Short-term RIH generally occurs within hours to a few days after drug withdrawal.20 However, in some patients—particularly those on high-dose or long-term opioid therapy—hyperalgesia may persist for weeks or even months.21 Research demonstrates that the duration of RIH is closely associated with cumulative opioid doses, baseline pain levels, and the presence of neuropathic pain.22
As an ultra-short-acting μ-opioid receptor agonist, remifentanil’s dosage and infusion duration are key determinants of RIH. Studies show that high-dose remifentanil infusion (>0.2 μg/kg/min) and prolonged administration (>3 hours) significantly increase the risk of RIH.8 Due to the pharmacokinetic characteristics of the rapid metabolism of remifentanil, its analgesic effect disappears swiftly postoperatively, resulting in heightened pain sensitivity and the development of primary and secondary hyperalgesic zones around surgical sites.23 Primary hyperalgesia is mainly caused by peripheral nervous system activation. Secondary hyperalgesia involves complex processing of nociceptive impulses in the spinal cord.24 Additionally, remifentanil’s analgesic effects can raise pain thresholds in primary hyperalgesia zones and reduce the extent of secondary hyperalgesia zones.25 However, the sudden withdrawal after surgery can exacerbate both types of hyperalgesia, intensifying postoperative pain. Individual differences such as age, sex, and genetic polymorphisms significantly influence the risk of developing RIH. Elderly patients may be more prone to RIH due to slower metabolism and reduced neuroplasticity.20 Gender differences suggest that female patients exhibit higher opioid sensitivity and may have a greater incidence of RIH compared to males.26 Furthermore, genetic polymorphisms (eg, OPRM1 A118G mutation) may alter opioid pharmacodynamics and pharmacokinetics, increasing RIH vulnerability.22 Surgical factors, such as procedure type and trauma severity, also critically impact RIH occurrence. Surgeries with greater trauma (eg, thoracotomy, spinal surgery) or stronger inflammatory responses (eg, laparoscopy surgery) are more likely to trigger RIH.27 A study found that a significantly higher incidence of RIH in thoracotomy patients compared to those undergoing minor procedures.3 Additionally, longer surgeries or those involving greater blood loss also carry a higher risk of RIH.6
Mechanisms of RIH
The mechanisms of Remifentanil-Induced Hyperalgesia (RIH) have not been fully elucidated. Current research primarily focuses on the multilevel interactions among central sensitization, peripheral neural mechanisms, and molecular signal pathways. However, there remains significant controversy regarding the core mechanisms of RIH. Some researchers emphasize the dominant role of central sensitization, while others highlight peripheral neural regulation and other potential factors. Additionally, there is a perspective proposing that RIH results from the synergistic effects of multiple contributing factors.
Central Sensitization
Central sensitization refers to the enhanced responsiveness of nociceptive neurons within the central nervous system (CNS) to normal or subthreshold input, leading the persistence and amplification of pain.28 Most studies indicate that RIH is primarily mediated by central sensitization, with molecular mechanisms involving multiple aspects such as NMDA receptor activation, glial cell activation, neuroinflammation, and abnormalities in neurotransmitter signal.
NMDA Receptor Activation
The NMDA receptor plays a central role in glutamatergic signal and mediates central sensitization via calcium (Ca²⁺) influx. Remifentanil may influence NMDA receptor function through the following mechanisms.
Phosphorylation of NMDA Subunits (NR1 and NR2B)
Remifentanil can significantly enhance the phosphorylation levels of these subunits in the spinal cord, promoting the transmission of pain signals. Experimental studies have confirmed that NMDA receptor antagonists can alleviate RIH.29,30 Notably, the P2Y1 receptor participates in hyperalgesia by enhancing NR1/NR2B phosphorylation, and its antagonist can reverse this effect.31 Preclinical studies have shown that the phosphorylation level of NR1/NR2B is generally upregulated in the RIH model.32 Additionally, remifentanil triggers Ca²⁺ influx by relieving Mg²⁺-mediated blockade of NMDA receptors, activating downstream protein kinase C (PKC) and calcium/calmodulin-dependent protein kinase II (CaMKII) signal pathways. Among them, excessive phosphorylation of CaMKII can enhance receptor activity and induce synaptic plasticity.17,33,34 Studies further indicate that modulating NMDA subunit phosphorylation through the PKC inhibitor (eg, Chelerythrine), CaMKII inhibitors (eg, KN93) and p38 inhibitors reduces the incidence of abnormal pain and hyperalgesia.35–37 Remifentanil enhances postsynaptic glutamate responsiveness by upregulating NMDA receptor membrane expression.38 Studies have found that CXCL13 regulates receptor membrane trafficking via the IL-17 pathway. Blocking this pathway can reduce receptor expression and alleviates hyperalgesia.39 Activation of the TRPV1 pathway can promote membrane transport of the NR1 subunit, while TRPV1 inhibitors can effectively suppress membrane localization of the NMDA receptor.40
Reactive Oxygen Species (ROS) Involvement
Continuous remifentanil infusion leads to spinal cord ROS accumulation accompanied by enhanced NR1/NR2B phosphorylation. The ROS scavenger (eg, PBN) reverses hyperalgesia, suggesting the involvement of a ROS-NMDA receptor positive feedback loop in RIH.29,41 Importantly, NMDA receptor activation promotes the generation of ROS by NADPH oxidase, a process potentially linked to neurovascular coupling and pain hypersensitivity.42 Hydrogen therapy studies further corroborate this mechanism, demonstrating significant improvement in postoperative hyperalgesia through inhibition of NR2B activation and MnSOD nitration.43
Inflammatory Cytokines
Remifentanil can also promote the release of IL-1β by activating the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, thereby enhancing the function of the NMDA receptor.44 TNF-α/IL-1β can enhance sympathetic nerve excitability through the NMDA receptor pathway and is involved in inflammatory hyperalgesia.45 Mechanism studies have shown that IL-1β can induce spinal NR1 phosphorylation, and this effect can be blocked by IL-1 receptor antagonists.46 Furthermore, IL-1β promotes NR2B tyrosine phosphorylation through Src kinase pathways and enhances the synaptic plasticity of the receptor.47
Glial Cell Activation
In recent years, Glial Cell Activation has received extensive attention as a potential mechanism of RIH. Glial cells—including astrocytes, microglia, and oligodendrocytes—play vital roles in CNS support and immune regulation. Increasing evidence links RIH to neuroinflammation mediated by glial cell activation.
Astrocyte Activation
Remifentanil induces activation of spinal astrocytes accompanied by elevated levels of pro-inflammatory cytokines (eg, TNF-α, IL-1β, IL-6). These cytokines directly participate in the pain sensitization process by enhancing the phosphorylation of NMDA receptors and neuronal excitability, and reducing the pain threshold of spinal dorsal horn neurons.48 Furthermore, studies have demonstrated that dexmedetomidine alleviates hyperalgesia through modulation of glial cell activity (eg, inhibiting glial cell proliferation).49
Microglial Activation
Notably, microglial activation plays a pivotal role in this process, as SIRT2 protein overexpression significantly reduces pro-inflammatory cytokine release and mitigates RIH by suppressing their activation.50 Additionally, related research indicates that electroacupuncture stimulation inhibits remifentanil-triggered spinal glial cell activation and reduces the expression of inflammatory factors, consequently alleviating RIH.51
NLRP3 Inflammasome Activation
NLRP3 inflammasome activation promotes IL-1β release through mechanisms involving the modulation of NMDA receptor NR1 subunit phosphorylation and astrocytic glutamate transporter GLT-1 expression. Experiments have confirmed that specific NLRP3 inhibition (eg, with MCC950) can reduce the level of IL-1β in the spinal cord and effectively alleviate the symptoms of RIH.44,52 Rodent model investigations demonstrate that continuous remifentanil infusion activates the spinal dorsal horn sphingosine kinase (SphK)/sphingosine-1-phosphate (S1P)/S1P receptor 1 (S1PR1) signal axis. S1PR1 is predominantly expressed on astrocyte membranes, and its activation stimulates mitochondrial ROS production. These ROS triggers the assembly of inflammasomes by oxidizing and modifying the NLRP3 protein, driving the release of caspase-1-dependent IL-1β and IL-18. Blocking this axis (such as SKI-II inhibitors) or using NLRP3 antagonists can simultaneously reduce the levels of NLRP3, caspase-1, IL-1β, IL-18 and ROS in the spinal cord, and significantly improve the occurrence of hyperalgesia.53 Additional studies indicate that remifentanil activates the NF-κB/NLRP3 pathway to mediate microglial activation, upregulating p21-activated kinase 4 (PAK4) expression in spinal dorsal horn cells, thereby inducing postoperative hyperalgesia. The inhibitor can block the upregulation of PAK4 expression, suggesting that the NLRP3 inflammasome and PAK4 are involved in RIH reaction after surgery.54 Remifentanil also activates Toll-like receptor 4 (TLR4) and purinergic receptors P2X4/P2X7 in microglia, shifting them toward a pro-inflammatory phenotype (M1 type).55 Among them, the activation of the P2X4 receptor can enhance synaptic plasticity through the brain-derived neurotrophic factor (BDNF)/TrkB signal pathway and indirectly intensify the inflammatory response of glial cells.56 Inhibition of the P2Y1 purinergic receptor can reduce the phosphorylation of NMDA receptors and the release of pro-inflammatory factors, suggesting that this receptor may be involved in the occurrence of RIH by regulating glial cell inflammation.31
Descending Inhibitory System (DIS)
The Descending Inhibitory System (DIS) is a critical neural network in the body for regulating pain perception. It mainly originates from the Periaqueductal Gray (PAG) of the midbrain and the rostral ventromedial medulla (RVM).57,58 The PAG receives inputs from the cerebral cortex, limbic system, and thalamus, and modulates the transmission of nociceptive signals in the spinal dorsal horn (SDH) through the RVM. The primary function of the DIS is to reduce the excitability of spinal dorsal horn neurons by releasing neurotransmitters such as serotonin 5-hydroxytryptamine (5-HT), norepinephrine (NE), and endogenous opioid peptides, thereby suppressing pain transmission.59–61
Disruption of DIS by Remifentanil
Mechanistic studies on RIH suggest that remifentanil may reduce inhibitory function and enhanced pain sensitivity by disrupting the descending inhibitory system.62,63
PAG-RVM-SDH Loop
The PAG-RVM-SDH circuit constitutes the most classical descending pain modulation system and serves as a key pathway in the endogenous analgesic system, playing a central role in the regulation of pain perception.64,65 Pain generation depends on the ascending conduction pathway transmitting pain signals to the central nervous system, and is endogenous regulated by the descending inhibitory pathway at the same time.64,66 When the function of this system is impaired, the capacity for pain inhibition diminishes, potentially leading to hyperalgesia and allodynia. This may result in pain perception from non-noxious stimuli or exaggerated pain responses to mild noxious stimuli.67–69
5-HT Imbalance
Research indicates that the PAG-RVM system regulates pain through the release of 5-HT.70 Remifentanil can imbalance the pain regulation mediated by 5-HT, thereby lowering the threshold of pain.71 Studies have shown that the increased activation of the 5-HT2A receptor leads to a decrease in descending inhibitory ability, which may further cause an increase in the excitability of sensory neurons in the spinal dorsal horn and enhance pain signal transmission, thereby resulting in the occurrence of hyperalgesia.72,73 Furthermore, studies have found that different alleles of the 5-HT transporter gene (5-HTTLPR, rs25531) affect the analgesic effect of remifentanil, and individuals with lower expression of 5-HTT have a stronger analgesic response.74
GABAergic Disinhibition
The use of remifentanil may enhance nociceptive signal transmission by reducing the activity of gamma-aminobutyric acid (GABA) ergic neurons in the RVM region, thereby weakening the inhibitory effects on descending pain-facilitating neurons. Studies have shown that remifentanil can suppress the release of GABA in the spinal cord and downregulate the expression of KCC2 (K+-Cl- cotransporter 2) and GABA receptors, jointly weakening the inhibitory regulatory ability of the spinal cord. Experimental evidence demonstrates that restoring GABAergic system function can effectively alleviate RIH.75 Local application of GABA receptor antagonists blocks the anti-pain effect produced by synaptic transmission of RVM, confirming that extracellular disinhibition is the core mechanism of pain regulation.76
Role of NMDA in Disinhibition
The disinhibition effect of RVM is also involved in the NMDA system. Acute remifentanil using triggers abnormal activation of NMDA receptors, significantly enhancing nociceptive signal transmission efficiency by promoting sensitization of SDH neurons.63
Neuron-Restrictive Silencer Factor (NRSF)
NRSF as a negative regulatory element in the PAG region, participates in the development of postoperative hyperalgesia by inhibiting the normal descending pain suppression pathway caused by the disruption of μ-opioid receptor function.77
Peripheral Sensitization
Peripheral Sensitization refers to the increased responsiveness of peripheral nociceptors (eg, C-fibers and Aδ-fibers) to painful stimuli under tissue injury or inflammation conditions, manifested as a decreased pain threshold and an amplified pain response.78–81
This change enables originally painless or mild stimuli to cause significant pain, which is considered a core mechanism of primary hyperalgesia.79 The underlying mechanisms include the activation of nociceptors by inflammatory mediators, functional changes in ion channels, and enhanced synaptic transmission efficiency, etc.
Nociceptor Activation and Inflammatory Mediators
Studies show that remifentanil induces the occurrence of RIH via the TRPV1–PKC signal pathway and spinal astrocyte activation. Remifentanil upregulates the protein expressions of TRPV1 and PKC in dorsal root ganglia (DRG), while simultaneously activating astrocytes in the spinal cord, which then release proinflammatory cytokines such as TNF-α and IL-1β. These cytokines lower the activation threshold of peripheral nociceptors, resulting in persistent nociceptive input and the development of hyperalgesia. This suggests that RIH is closely related to inflammatory peripheral sensitization.48 In addition, remifentanil also induces oxidative stress in the spinal dorsal horn, which interacts synergistically with inflammatory cytokine (such as IL-1β, TNF-α) release to amplify pain signals. Use of antioxidants (such as betulinic acid) has been shown to block this oxidative stress-inflammatory circuit and significantly alleviate RIH.82
Ion Channel Expression and Functional Changes
The onset of RIH is closely associated with functional abnormalities in various ion channels. In peripheral nervous system channels, remifentanil significantly upregulates the expression of Acid-sensing ion channels ASIC3 (acid-sensing ion channels 3) in DRG, hippocampus, and hypothalamus, and participate in hyperalgesia by enhancing nociceptive signal under tissue acidification conditions.83 In DRG neurons, remifentanil increases TRPA1 expression through TLR4, mediating mechanical hyperalgesia. Blocking the TLR4/TRPA1 pathway effectively inhibits this process.84 Remifentanil upregulates the expression of CaV3.1 in the T-type calcium channel in the ventral posterolateral nucleus (VPL) of the thalamus, enhancing bursting activity in glutamatergic neurons in the VPL (VPLGlu) and over-activating the primary somatosensory cortex (S1HLGlu), contributing to central amplification of pain signals.85 These findings highlight that ion channel dynamics form an important molecular foundation for RIH-related peripheral sensitization.
Synaptic Transmission Efficiency and Plasticity
At the synaptic level, remifentanil activates spinal δ-opioid receptors, which in turn enhance phosphorylation and membrane trafficking of AMPA receptor subunit GluR1, strengthening excitatory synaptic transmission and promoting hyperalgesia. δ-opioid receptor antagonists can alleviate this effect.86 In peripheral DRG, the downregulation of TMEM16C and Slack ion channels enhances the activity of GluA1-AMPAR, increasing excitatory synaptic signal transmission and exacerbating peripheral hyperalgesia.87
Additionally, studies have found that remifentanil can also up-regulate the reconstruction of neural excitability mediated by CaV3.1 T-type calcium channels in the VPL.85 The coordinated regulation of these receptors and ion channels at different levels of the central and peripheral nervous systems enhances synaptic transmission efficiency and plasticity, forming a multidimensional signal amplification network underlying RIH.
Other Mechanisms
The Synergistic Effect of P2Y1-NMDA Receptor in DRG
The upregulation of the P2Y1 receptor promotes the phosphorylation of the NR1 subunit of the NMDA receptor, enhances nociceptive signal, and leads to the occurrence of RIH. Targeting P2Y1R antagonists or NMDA receptor inhibitors can effectively block this process.31
GSK-3β Kinase
GSK-3β kinase exacerbates peripheral hyperalgesia by regulating the transport of the NMDA receptor NR1 subunit on the DRG cell membrane, while GSK-3β inhibitors can alleviate RIH by reversing this process.88
Iron Accumulation
Remifentanil can lead to iron ion accumulation in the spinal cord by up-regulating the expression of DMT1 (-IRE), further amplifying pain signals. This process is triggered by calcium influx caused by the activation of NMDA receptors.89 Intervention with iron chelators can significantly alleviate hyperalgesia caused by iron overload.90
Time-Dependent Regulation
Relevant studies have shown that early-phase hyperalgesia is driven by phosphorylated p38 MAPK in peripheral nerves, while in the late stage, hyperalgesia is closely related to the spinal activation of the prodynorphin–bradykinin pathway, demonstrating time-dependent regulation characteristics.91
CCL7/CCR2 Axis and IL-18/IL-18R Signal
The spinal cord CCL7/CCR2 chemokine axis enhances the activity of GluA1-AMPA receptors by up-regulating the expression of IL-18 and its receptor (IL-18R), forming a central-peripheral hyperalgesia circuit, and becomes one of the important mechanisms of RIH.92
Finally, remifentanil, as a μ-opioid receptor (MOR) agonist, its analgesic effect depends on the activation of inhibitory G protein. However, when used continuously in large doses for a long time, it will activate the excitatory G protein and induce hyperalgesia. Moreover, the mechanism of RIH also involves the dynamic imbalance of the μ-opioid receptor (MOR) and δ-opioid receptor (DOR) systems.
On the one hand, remifentanil inhibits the transcription and expression of MOR in PAG around the mesencephalic aqueduct by activating neuron-restrictive silencer factor (NRSF), resulting in the down-regulation of MOR expression, weakening the endogenous analgesic ability and enhancing pain signal transduction, thereby promoting the occurrence of RIH.77 On the other hand, the activation of DOR intensifies hyperalgesia through dual pathways-one is to upregulate the membrane expression and phosphorylation level of the GluR1 subunit of the AMPA receptor, and the other is to directly enhance the function of the NMDA receptor and improve the synaptic transmission efficiency, ultimately amplifying the pain signal.93 It is worth noting that the DOR-induced hyperalgesia effect mediated can be specifically reversed by its antagonists, such as Naltrindole.86 The imbalance of the synergistic regulation of MOR and DOR (inhibition of MOR function and excessive activation of DOR) jointly constitutes one of the key mechanisms of RIH, namely the dual drive of decreased analgesic ability and enhanced pain signals.
Conclusion
Remifentanil, as a commonly used potent and short-acting μ-opioid receptor agonist, is widely applied in clinical anesthesia and perioperative analgesia. However, its associated phenomenon of remifentanil-induced postoperative hyperalgesia (RIH) presents significant difficulties in postoperative pain management and poses new challenges to anesthetic protocols.
In recent years, research has revealed that RIH is not caused by a single abnormality but results from complex interactions among central sensitization, glial activation, inflammatory signaling, and peripheral sensitization. Central sensitization—especially NMDA receptor activation and descending inhibition failure—is widely recognized as a primary mechanism, while glial-mediated neuroinflammation and peripheral ion channel remodeling likely serve secondary, amplifying roles. In addition, the activation of glial cells and the pro-inflammatory factors they release, the participation of brain-derived neurotrophic factor (BDNF), the dysfunction of inhibitory neurotransmitters such as GABA and glycine pathways, as well as the bidirectional regulatory effect of the 5-HT pathway, are all important mechanisms for the occurrence and development of RIH (Figure 2). Additionally, emerging evidence highlights novel contributors such as the PAK4–NLRP3 signaling axis, oxidative stress, and purinergic receptor involvement. While these discoveries broaden our mechanistic understanding, a unified and dynamic model that connects these molecular events to clinical manifestations remains lacking.
 |
Figure 2 Diagram of the related mechanism pathways of remifentanil-induced hyperalgesia. Remifentanil administration may trigger central sensitization by activating N-methyl-D-aspartate (NMDA) receptors and disrupting descending inhibitory pathways, leading to enhanced excitatory synaptic transmission in the spinal dorsal horn. Additionally, remifentanil activates glial cells (microglia and astrocytes), which release pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), contributing to neuroinflammation. Brain-derived neurotrophic factor (BDNF) and serotonin (5-HT) pathways also participate in modulating pain signaling. In the periphery, changes in ion channel expression and purinergic signaling contribute to peripheral sensitization. Emerging evidence highlights the involvement of the PAK4–NLRP3 signaling axis and oxidative stress as novel mechanisms in RIH pathogenesis. Together, these factors exacerbate pain perception and contribute to postoperative hyperalgesia. Symbols and abbreviations used in the figure: “↑” Black upward arrow: Upregulation or activation of molecular expression or signaling; “↓” Black downward arrow: Downregulation or suppression of signaling pathways or neurotransmitter activity; “+” inside a circle: Activation or promotion of the downstream target or signaling pathway; “–” inside a circle: Inhibition or suppression of the downstream target or signaling pathway. Abbreviations: PAK4, p21-activated kinase 4; NLRP3, NOD-like receptor family pyrin domain-containing 3; BDNF, brain-derived neurotrophic factor; GABA, gamma-aminobutyric acid; 5-HT, 5-hydroxytryptamine (serotonin); NMDA, N-methyl-D-aspartate receptor; IL-1β, interleukin-1 beta; TNF-α, tumor necrosis factor alpha. |
It is worth noting that the occurrence of RIH may also be influenced by individual differences factors such as genetic background, age, sex, and baseline pain thresholds. These latent regulatory mechanisms that have not been fully revealed offer broader opportunities for further mechanistic studies. Although some animal models and cellular studies have preliminarily clarified the molecular signal pathways through which remifentanil induces neural hypersensitivity, the precise molecular targets and interaction networks remain undefined, and a unified theoretical framework is still lacking. Moreover, while most current studies focus on the central nervous system, the role of the peripheral nervous system in RIH also warrants further investigation.
While animal and cellular models have been instrumental in uncovering key pathways such as NMDA receptor activation, glial-mediated inflammation, and oxidative stress in RIH, important translational gaps remain. Rodents differ from humans in opioid metabolism, neural development, and pain processing, which may limit the direct applicability of findings. Moreover, experimental remifentanil protocols often involve higher doses or longer exposure durations than typical clinical practice. Thus, future studies should seek to bridge these gaps through validation in human tissues or clinically relevant patient models.
Understanding the molecular mechanisms of RIH can inform the development of precision-based perioperative strategies, such as targeting NMDA signaling, modulating glial activity, or pre-emptively adjusting opioid protocols.
Looking forward, future research should prioritize the integration of multi-omics technologies with clinically relevant models to identify key regulatory molecules, map temporal dynamics of pathway activation, and clarify the crosstalk between central and peripheral mechanisms. By deepening mechanistic insights, these efforts are expected to support the development of individualized, targeted strategies to prevent and mitigate remifentanil-induced hyperalgesia, ultimately improving postoperative pain management and patient recovery. In the era of multimodal analgesia, the role of remifentanil should be carefully reconsidered. Its use may be optimized through combination with NMDA receptor antagonists, α2-agonists, or non-opioid adjuvants to reduce the risk of hyperalgesia while maintaining effective intraoperative analgesia.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Foundation of Chengdu Medical College (24LHLNYX1-13, 23LHPDZYB27); Special Key Project of the First Affiliated Hospital of Chengdu Medical College (CYFY- 2021ZD03); Sichuan Medical health Promotion Association general project (KY2022QN0289); high-level talent introduction project of the First Affiliated Hospital of Chengdu Medical College (CYFY-GQ51).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Morone NE, Weiner DK. Pain as the fifth vital sign: exposing the vital need for pain education. Clin Ther. 2013;35(11):1728–1732. doi:10.1016/j.clinthera.2013.10.001
2. Silverman SM. Opioid induced hyperalgesia: clinical implications for the pain practitioner. Pain Physician. 2009;12(3):679–684. doi:10.36076/ppj.2009/12/679
3. Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145–161. doi:10.36076/ppj.2011/14/145
4. Stroumpos C, Manolaraki M, Paspatis GA. Remifentanil, a different opioid: potential clinical applications and safety aspects. Expert Opin Drug Safety. 2010;9(2):355–364. doi:10.1517/14740331003672579
5. Eleveld DJ, Proost JH, Vereecke H, et al. An allometric model of remifentanil pharmacokinetics and pharmacodynamics. Anesthesiology. 2017;126(6):1005–1018. doi:10.1097/ALN.0000000000001634
6. Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104(3):570–587. doi:10.1097/00000542-200603000-00025
7. Vadivelu N, Mitra S. Narayan DJTYjob, medicine. Recent Adv Postoperative Pain Manag. 2010;83(1):11–25.
8. Guignard B, Bossard AE, Coste C, et al. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology. 2000;93(2):409–417. doi:10.1097/00000542-200008000-00019
9. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–231. doi:10.1038/nri3621
10. Joly V, Richebe P, Guignard B, et al. Remifentanil-induced postoperative hyperalgesia and its prevention with small-dose ketamine. Anesthesiology. 2005;103(1):147–155. doi:10.1097/00000542-200507000-00022
11. Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24(6):479–496. doi:10.1097/AJP.0b013e31816b2f43
12. Kim SH, Stoicea N, Soghomonyan S, Bergese SD. Intraoperative use of remifentanil and opioid induced hyperalgesia/acute opioid tolerance: systematic review. Front Pharmacol. 2014;5:108. doi:10.3389/fphar.2014.00108
13. Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100(3):213–217. doi:10.1016/S0304-3959(02)00422-0
14. Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995;62(3):259–274. doi:10.1016/0304-3959(95)00073-2
15. Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “Toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30(11):581–591. doi:10.1016/j.tips.2009.08.002
16. Williams JT, Ingram SL, Henderson G, et al. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65(1):223–254. doi:10.1124/pr.112.005942
17. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. doi:10.1126/science.288.5472.1765
18. Nielsen DA, Utrankar A, Reyes JA, Simons DD, Kosten TR. Epigenetics of drug abuse: predisposition or response. Pharmacogenomics. 2012;13(10):1149–1160. doi:10.2217/pgs.12.94
19. Fletcher D, Martinez V. Opioid-induced hyperalgesia in patients after surgery: a systematic review and a meta-analysis. Br J Anaesth. 2014;112(6):991–1004. doi:10.1093/bja/aeu137
20. Colvin LA, Fallon MT. Opioid-induced hyperalgesia: a clinical challenge. Br J Anaesth. 2010;104(2):125–127. doi:10.1093/bja/aep392
21. Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. 2009;10(3):316–322. doi:10.1016/j.jpain.2008.10.003
22. Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depend. 2001;63(2):139–146. doi:10.1016/S0376-8716(00)00200-3
23. Zöllner C, Schäfer M. Remifentanil-based intraoperative anaesthesia and postoperative pain therapy. Is there an optimal treatment strategy? Der Anaesthesist. 2007;56(10):1038–1046. doi:10.1007/s00101-007-1246-1
24. Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367(9522):1618–1625. doi:10.1016/S0140-6736(06)68700-X
25. Gustorff B, Felleiter P, Nahlik G, et al. The effect of remifentanil on the heat pain threshold in volunteers. Anesthesia Analg. 2001;92(2):369–374. doi:10.1213/00000539-200102000-00017
26. Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain. 2009;10(5):447–485. doi:10.1016/j.jpain.2008.12.001
27. Koppert W, Schmelz M. The impact of opioid-induced hyperalgesia for postoperative pain. Best Pract Res Clin Anaesth. 2007;21(1):65–83. doi:10.1016/j.bpa.2006.12.004
28. Curatolo M. Central sensitization and pain: pathophysiologic and clinical insights. Curr Neuropharmacol. 2024;22(1):15–22. doi:10.2174/1570159X20666221012112725
29. Ye L, Xiao L, Bai X, et al. Spinal mitochondrial-derived ROS contributes to remifentanil-induced postoperative hyperalgesia via modulating NMDA receptor in rats. Neurosci Lett. 2016;634:79–86. doi:10.1016/j.neulet.2016.09.016
30. Roué M, Iung B, Bourgeois-Moine A, Montravers P, Kantor E. Major adverse cardiovascular events and anaesthetic management in pregnant women with cardiac disease: a retrospective, single-centre study. Br J Anaesth. 2021;127(1):e8–e10. doi:10.1016/j.bja.2021.03.032
31. Su L, Bai X, Niu T, et al. P2Y1 purinergic receptor inhibition attenuated remifentanil-induced postoperative hyperalgesia via decreasing NMDA receptor phosphorylation in dorsal root ganglion. Brain Res Bull. 2021;177:352–362. doi:10.1016/j.brainresbull.2021.10.006
32. Qi F, Liu T, Zhang X, et al. Ketamine reduces remifentanil-induced postoperative hyperalgesia mediated by CaMKII-NMDAR in the primary somatosensory cerebral cortex region in mice. Neuropharmacology. 2020;162:107783. doi:10.1016/j.neuropharm.2019.107783
33. Li S, Zeng J, Wan X, et al. Enhancement of spinal dorsal horn neuron NMDA receptor phosphorylation as the mechanism of remifentanil induced hyperalgesia: roles of PKC and CaMKII. Mol Pain. 2017;13:1744806917723789. doi:10.1177/1744806917723789
34. Zhou Z, Mao M, Cai X, Zhu W, Sun J. Store-operated calcium channels contribute to remifentanil-induced postoperative hyperalgesia via phosphorylation of CaMKIIα in rats. J Pain Res. 2021;14:3289–3299. doi:10.2147/JPR.S333297
35. Bu F, Tian H, Gong S, et al. Phosphorylation of NR2B NMDA subunits by protein kinase C in arcuate nucleus contributes to inflammatory pain in rats. Sci Rep. 2015;5(1):15945. doi:10.1038/srep15945
36. Liu Y, Liang Y, Hou B, et al. The inhibitor of calcium/calmodulin-dependent protein kinase II KN93 attenuates bone cancer pain via inhibition of KIF17/NR2B trafficking in mice. Pharmacol Biochem Behav. 2014;124:19–26. doi:10.1016/j.pbb.2014.05.003
37. Wang Z, Ma W, Chabot JG, Quirion R. Calcitonin gene-related peptide as a regulator of neuronal CaMKII-CREB, microglial p38-NFκB and astroglial ERK-Stat1/3 cascades mediating the development of tolerance to morphine-induced analgesia. Pain. 2010;151(1):194–205. doi:10.1016/j.pain.2010.07.006
38. Yuan Y, Sun Z, Chen Y, et al. Prevention of remifentanil induced postoperative hyperalgesia by dexmedetomidine via regulating the trafficking and function of spinal NMDA receptors as well as PKC and CaMKII level in vivo and in vitro. PLoS One. 2017;12(2):e0171348. doi:10.1371/journal.pone.0171348
39. Zhu M, Yuan ST, Yu WL, Jia LL, Sun Y. CXCL13 regulates the trafficking of GluN2B-containing NMDA receptor via IL-17 in the development of remifentanil-induced hyperalgesia in rats. Neurosci Lett. 2017;648:26–33. doi:10.1016/j.neulet.2017.03.044
40. Song C, Liu P, Zhao Q, Guo S, Wang G. TRPV1 channel contributes to remifentanil-induced postoperative hyperalgesia via regulation of NMDA receptor trafficking in dorsal root ganglion. J Pain Res. 2019;12:667–677. doi:10.2147/JPR.S186591
41. Gao X, Kim HK, Mo Chung J, Chung K. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain. 2007;131(3):262–271. doi:10.1016/j.pain.2007.01.011
42. Girouard H, Wang G, Gallo EF, et al. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29(8):2545–2552. doi:10.1523/JNEUROSCI.0133-09.2009
43. Zhang L, Shu R, Wang H, et al. Hydrogen-rich saline prevents remifentanil-induced hyperalgesia and inhibits MnSOD nitration via regulation of NR2B-containing NMDA receptor in rats. Neuroscience. 2014;280:171–180. doi:10.1016/j.neuroscience.2014.09.024
44. Yuan Y, Zhao Y, Shen M, et al. Spinal NLRP3 inflammasome activation mediates IL-1β release and contributes to remifentanil-induced postoperative hyperalgesia by regulating NMDA receptor NR1 subunit phosphorylation and GLT-1 expression in rats. Mol Pain. 2022;18:17448069221093016. doi:10.1177/17448069221093016
45. Gruber-Schoffnegger D, Drdla-Schutting R, Hn C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci. 2013;33(15):6540–6551. doi:10.1523/JNEUROSCI.5087-12.2013
46. Zhang RX, Li A, Liu B, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135(3):232–239. doi:10.1016/j.pain.2007.05.023
47. Guo W, Wei F, Zou S. Group I metabotropic glutamate receptor NMDA receptor coupling and signaling cascade mediate spinal dorsal horn NMDA receptor 2B tyrosine phosphorylation associated with inflammatory hyperalgesia. J Neurosci. 2004;24(41):9161–9173. doi:10.1523/JNEUROSCI.3422-04.2004
48. Hong HK, Ma Y, Xie H. TRPV1 and spinal astrocyte activation contribute to remifentanil-induced hyperalgesia in rats. Neuroreport. 2019;30(16):1095–1101. doi:10.1097/WNR.0000000000001329
49. Kimura M, Saito S, Obata H. Dexmedetomidine decreases hyperalgesia in neuropathic pain by increasing acetylcholine in the spinal cord. Neurosci Lett. 2012;529(1):70–74. doi:10.1016/j.neulet.2012.08.008
50. Zhu W, Ma Z. SIRT2 overexpression decreases remifentanil-stimulated post-surgical hyperalgesia via microglia. Cellular Mol Biol. 2023;69(12):268–274. doi:10.14715/cmb/2023.69.12.42
51. Xie Y, Ma J, Wang D, Chai X, Gao C. Electro-acupuncture stimulation prevents remifentanil-induced postoperative hyperalgesia by suppressing spinal microglia in rats. Exp Ther Med. 2018;16(1):353–359. doi:10.3892/etm.2018.6161
52. Yuan Y, Wang C, Dong B, Xie K, Yu Y. NLRP3 inflammasome activation contributes to remifentanil-induced postoperative hyperalgesia via regulation of NMDA receptor NR1 subunit phosphorylation and GLT-1. 2020.
53. Li J, Wang Q, Gao Y, et al. Blocking SphK/S1P/S1PR1 axis signaling pathway alleviates remifentanil-induced hyperalgesia in rats. Neurosci Lett. 2023;801:137131. doi:10.1016/j.neulet.2023.137131
54. Cui C, Wu X, Dong S, Chen B, Zhang T. Remifentanil-induced inflammation in microglial cells: activation of the PAK4-mediated NF-κB/NLRP3 pathway and onset of hyperalgesia. Brain Behav Immun. 2025;123:334–352. doi:10.1016/j.bbi.2024.09.018
55. Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–577. doi:10.1126/science.aaf8924
56. Fu R, Li S, Li S, et al. P2X4 receptor in the dorsal horn contributes to BDNF/TrkB and AMPA receptor activation in the pathogenesis of remifentanil-induced postoperative hyperalgesia in rats. Neurosci Lett. 2021;750:135773. doi:10.1016/j.neulet.2021.135773
57. Heinricher MM, Ingram SL. The brainstem and nociceptive modulation. Senses. 2008;5:593–626.
58. Lau BK, Vaughan CW. Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol. 2014;29:159–164. doi:10.1016/j.conb.2014.07.010
59. Serpell M. Anatomy, physiology and pharmacology of pain. Anaesth Intensive Care Med. 2005;6(1):7–10. doi:10.1383/anes.6.1.7.57133
60. Pertovaara A. Noradrenergic pain modulation. Progress Neurobiol. 2006;80(2):53–83. doi:10.1016/j.pneurobio.2006.08.001
61. Millan MJ. Descending control of pain. Progress Neurobiol. 2002;66(6):355–474. doi:10.1016/s0301-0082(02)00009-6
62. Braulio G, Passos SC, Leite F, et al. Effects of transcranial direct current stimulation block remifentanil-induced hyperalgesia: a randomized, double-blind clinical trial. Front Pharmacol. 2018;9:94. doi:10.3389/fphar.2018.00094
63. Vitin AA, Egan TD. Remifentanil-induced hyperalgesia: the current state of affairs. Curr Opin Anaesthesiol. 2024;37(4):371–378. doi:10.1097/ACO.0000000000001400
64. Tobaldini G, Sardi NF, Guilhen VA, Fischer L. Pain inhibits pain: an ascending-descending pain modulation pathway linking mesolimbic and classical descending mechanisms. Mol Neurobiol. 2019;56(2):1000–1013. doi:10.1007/s12035-018-1116-7
65. Loyd DR, Murphy AZ. The role of the periaqueductal gray in the modulation of pain in males and females: are the anatomy and physiology really that different? Neural Plast. 2009;2009:462879. doi:10.1155/2009/462879
66. Hideki O. Pain pathways and descending inhibitory pathways implicated in nociceptive modulation. J Nihon University Med Assoc. 2010;69(3):159–163. doi:10.4264/numa.69.159
67. Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Brain Res Rev. 2004;46(3):295–309. doi:10.1016/j.brainresrev.2004.07.004
68. Scholz J. Mechanisms of chronic pain. Mol Pain. 2014;10(Suppl 1):O15. doi:10.1186/1744-8069-10-S1-O15
69. Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27(8):729–737. doi:10.1016/j.neubiorev.2003.11.008
70. Chen T, Wang XL, Qu J, et al. Neurokinin-1 receptor-expressing neurons that contain serotonin and gamma-aminobutyric acid in the rat rostroventromedial medulla are involved in pain processing. J Pain. 2013;14(8):778–792. doi:10.1016/j.jpain.2013.02.002
71. Yu EH, Tran DH, Lam SW, Irwin MG. Remifentanil tolerance and hyperalgesia: short-term gain, long-term pain? Anaesthesia. 2016;71(11):1347–1362. doi:10.1111/anae.13602
72. Aira Z, Buesa I, Salgueiro M, et al. Subtype-specific changes in 5-HT receptor-mediated modulation of C fibre-evoked spinal field potentials are triggered by peripheral nerve injury. Neuroscience. 2010;168(3):831–841. doi:10.1016/j.neuroscience.2010.04.032
73. Abbott FV, Hong Y, Blier P. Activation of 5-HT2A receptors potentiates pain produced by inflammatory mediators. Neuropharmacology. 1996;35(1):99–110. doi:10.1016/0028-3908(95)00136-0
74. Kosek E, Jensen KB, Lonsdorf TB, Schalling M, Ingvar M. Genetic variation in the serotonin transporter gene (5-HTTLPR, rs25531) influences the analgesic response to the short acting opioid Remifentanil in humans. Mol Pain. 2009;5:37. doi:10.1186/1744-8069-5-37
75. Gao Y, Zhan W, Jin Y, et al. KCC2 receptor upregulation potentiates antinociceptive effect of GABAAR agonist on remifentanil-induced hyperalgesia. Mol Pain. 2022;18:17448069221082880. doi:10.1177/17448069221082880
76. Heinricher MM, Tortorici V. Interference with GABA transmission in the rostral ventromedial medulla: disinhibition of off-cells as a central mechanism in nociceptive modulation. Neuroscience. 1994;63(2):533–546. doi:10.1016/0306-4522(94)90548-7
77. Lu C, Shi L, Zhang J, et al. Neuron-restrictive silencer factor in periaqueductal gray contributes to remifentanil-induced postoperative hyperalgesia via repression of the mu-opioid receptor. J Neurol Sci. 2015;352(1–2):48–52. doi:10.1016/j.jns.2015.03.018
78. Hoegh M. Pain Science in Practice (Part 3): peripheral Sensitization. J Orthopaedic Sports Physical Ther. 2022;52(6):303–306. doi:10.2519/jospt.2022.11202
79. Vardeh D, Naranjo JF. Peripheral and central sensitization. Springer International Publishing; 2017.
80. Wang H, Ehnert C, Brenner GJ, Woolf CJ. Bradykinin and peripheral sensitization. Biol Chem. 2006;387(1):11–14. doi:10.1515/BC.2006.003
81. Rocha AP, Kraychete DC, Lemonica L, et al. Pain: current aspects on peripheral and central sensitization. Revista brasileira de anestesiologia. 2007;57(1):94–105. doi:10.1590/s0034-70942007000100011
82. Lv CC, Xia ML, Shu SJ, Chen F, Jiang LS. Attenuation of remifentanil-induced hyperalgesia by betulinic acid associates with inhibiting oxidative stress and inflammation in spinal dorsal horn. Pharmacology. 2018;102(5–6):300–306. doi:10.1159/000493144
83. Li T, Gao C, Shu S, Chai X, Xie Y. Acid-sensing ion channel 3 expression is increased in dorsal root ganglion, hippocampus and hypothalamus in remifentanil-induced hyperalgesia in rats. Neurosci Lett. 2020;721:134631. doi:10.1016/j.neulet.2019.134631
84. Liu X, Gong R, Peng L, Zhao J. Toll-like receptor 4 signaling pathway in sensory neurons mediates remifentanil-induced postoperative hyperalgesia via transient receptor potential ankyrin 1. Mol Pain. 2023;19:17448069231158290. doi:10.1177/17448069231158290
85. Todorovic SM. Opioid-induced hyperalgesia: are thalamic T-type calcium channels treatment targets? J Clin Invest. 2022;132(24):e165977. doi:10.1172/JCI165977
86. Liu A, Wang X, Wang H, Lv G, Li Y, Li H. Δ-opioid receptor inhibition prevents remifentanil-induced post-operative hyperalgesia via regulating GluR1 trafficking and AMPA receptor function. Exp Ther Med. 2018;15(2):2140–2147. doi:10.3892/etm.2017.5652
87. Li Y, Zhang L, Li J, et al. A role for transmembrane protein 16C/Slack impairment in excitatory nociceptive synaptic plasticity in the pathogenesis of remifentanil-induced hyperalgesia in rats. Neurosci Bulletin. 2021;37(5):669–683. doi:10.1007/s12264-021-00652-5
88. Zhang L, Shu R, Wang C, Wang H, Li N, Wang G. Hydrogen-rich saline controls remifentanil-induced hypernociception and NMDA receptor NR1 subunit membrane trafficking through GSK-3β in the DRG in rats. Brain Res Bull. 2014;106:47–55. doi:10.1016/j.brainresbull.2014.05.005
89. Shu R, Zhang L, Zhang H, et al. NMDA receptor modulates spinal iron accumulation via activating DMT1(-)IRE in remifentanil-induced hyperalgesia. J Pain. 2021;22(1):32–47. doi:10.1016/j.jpain.2020.03.007
90. Shu RC, Zhang LL, Wang CY, et al. Spinal peroxynitrite contributes to remifentanil-induced postoperative hyperalgesia via enhancement of divalent metal transporter 1 without iron-responsive element-mediated iron accumulation in rats. Anesthesiology. 2015;122(4):908–920. doi:10.1097/ALN.0000000000000562
91. Horii Y, Matsuda M, Takemura H, Ishikawa D, Sawa T, Amaya F. Spinal and peripheral mechanisms individually lead to the development of remifentanil-induced hyperalgesia. Neuroscience. 2020;446:28–42. doi:10.1016/j.neuroscience.2020.08.014
92. Qiang Z, Yu W. Chemokine CCL7 regulates spinal phosphorylation of GluA1-containing AMPA receptor via interleukin-18 in remifentanil-induced hyperalgesia in rats. Neurosci Lett. 2019;711:134440. doi:10.1016/j.neulet.2019.134440
93. Wang C, Li Y, Wang H, et al. Inhibition of DOR prevents remifentanil induced postoperative hyperalgesia through regulating the trafficking and function of spinal NMDA receptors in vivo and in vitro. Brain Res Bull. 2015;110:30–39. doi:10.1016/j.brainresbull.2014.12.001
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.