Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Mechanism and therapeutic effectiveness of nerve growth factor in osteoarthritis pain
Authors Shang X, Wang Z, Tao H
Received 17 April 2017
Accepted for publication 7 June 2017
Published 1 August 2017 Volume 2017:13 Pages 951—956
DOI https://doi.org/10.2147/TCRM.S139814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor De Yun Wang
Xiushuai Shang, Zhaofei Wang, Hairong Tao
Department of Orthopedic Surgery, Shanghai Key Laboratory of Orthopedic Implant, Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, China
Abstract: Osteoarthritis (OA) is the most common form of articular joint arthritis and a cause of significant morbidity. In this review, we present the role of nerve growth factor (NGF) in pain generation, relationship between NGF and OA pain, and pathogenic factors (interleukin-1β, transforming growth factor-β1, mechanical loading, and adipokines) involved in OA development. Since NGF blocking is an efficient way to inhibit OA-associated pain, we summarize four categories of drugs that target NGF/tropomyosin receptor kinase A (TrkA) signaling. In addition, we discuss the future of NGF/TrkA antagonists and underline their potential for use in OA pain relief. A better understanding of the causes and treatment of OA will facilitate the development of more effective methods of OA pain management.
Keywords: osteoarthritis, pain, nerve growth factor, TRPV1, TrkA
Introduction
Osteoarthritis (OA) is the most common form of articular joint arthritis, the primary symptom of which is pain.1,2 It causes significant morbidity in elderly individuals, and its age of onset is gradually decreasing.3 It results in tissue disruption surrounding the affected joint, impairing its articular function and leading to disability.
Many etiologies contribute to OA development, including age, sex, hereditary factors, and trauma.2 In recent years, the risk factors, molecular basis, and medications for OA have been investigated to prevent and treat OA.
Nerve growth factor (NGF) was found to be a sort of crucial protein for neurons’ growth and survival initially. But its role in mediating pain was identified later. NGF blocking was also effective in relieving OA pain.4 In this article, we outline the molecular mechanisms underlying the role of NGF in OA pain and review evidence supporting the attenuation of OA pain by NGF blockade with the aim of promoting the discovery of new analgesics based on the antagonism of NGF signaling.
Relationship between NGF and pain
NGF was the first growth factor to be identified, and its discovery dates back to the 1950s. It belongs to a family of neurotrophic factors that include brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4. Initially, it was identified as a soluble factor produced by tumor tissue, which promotes the growth and survival of sensory neurons. Subsequent studies showed that it functions in nervous system development and tissue pain.4,5 Hefti et al6 reported that NGF levels are elevated during injury, inflammation, and chronic pain. In rats, inhibition of endogenous NGF degradation by matrix metalloproteinase (MMP)-2 and MMP-9 induces mechanical allodynia and thermal hyperalgesia.7
The molecular mechanisms underlying these actions of NGF can be explained as follows: Thermal, chemical, or mechanical stimuli activate nociceptors located in peripheral tissues, causing NGF production and release. NGF binds to NGF receptors, promoting multiple pain signaling pathways, leading to nociceptive pain.8,9 When NGF binds to tropomyosin receptor kinase (Trk) A, the complex is endocytosed and retrogradely transported to neuronal cell bodies, where it modulates the expression of bradykinin receptors, acid-sensing ion channel 2/3, voltage-gated sodium channels, and transient receptor potential vanilloid receptor 1 (TRPV1).10 These cell surface receptors involved in nociception initiate peripheral sensitization and pain hypersensitivity.11,12 The NGF–TrkA complex causes rapid posttranslational changes in the pain receptor TRPV1, evoking sustained density and increased activity in peripheral and central neurons.11 Once opened, TRPV1 facilitates Ca2+ influx and transmission of pain signals.13 Central sensitization results from transcriptional changes triggered by NGF–TrkA signaling, including increased expression of calcitonin gene-related peptide and substance P.12,14 In addition, NGF stimulates mast cells to express histamine, serotonin/5-hydroxytryptamine, and protons, as well as NGF, forming a positive-feedback loop.12,14 Thus, NGF signaling not only modulates the expression of peripheral and central pain-related substances but also sensitizes adjacent nociceptive neurons in response to inflammation (Figure 1).
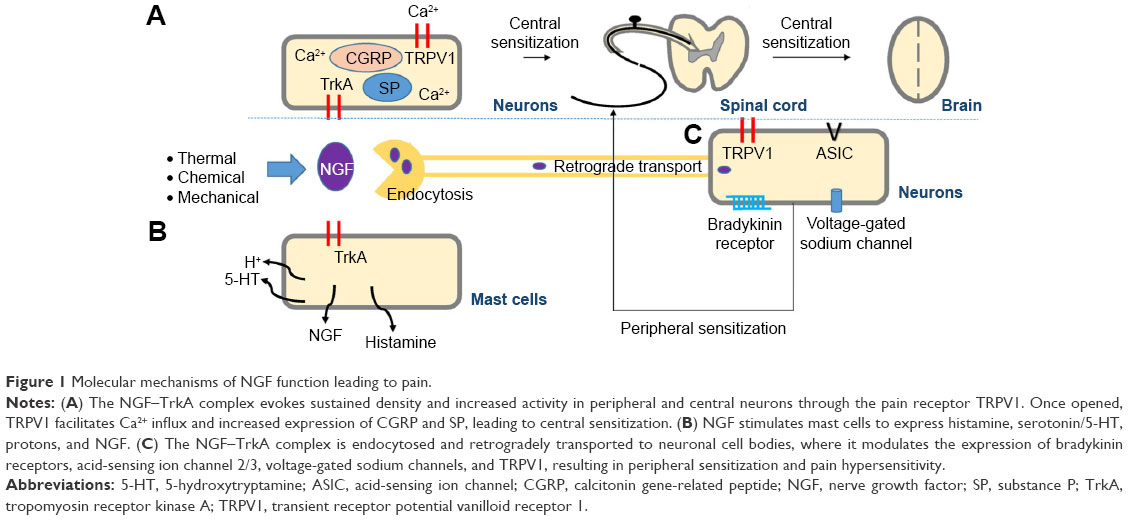 | Figure 1 Molecular mechanisms of NGF function leading to pain. |
Relationship between NGF and OA pain
Pain is the defining symptom of OA, and NGF expression is evident in joints affected by the condition. In humans, elevated NGF levels are found in the synovial fluid of patients with inflammatory or degenerative rheumatic diseases, including rheumatoid arthritis, spondyloarthritis, and OA.15 In clinical trials, anti-NGF therapy provided significant pain relief.16 In a recent article,17 NGF and bradykinin receptors were found to be significantly upregulated in the joints of mice with OA displaying pain-related behavior. In a systematic review of 13 multicenter placebo-controlled trials about hip or knee OA, Schnitzer and Marks18 identified that NGF inhibition yielded extensive pain relief compared with placebos.
The OA process involves many chemical and physical factors. Next, we present the biologic mechanisms of the risk factors for OA, including interleukin (IL)-1β, transforming growth factor-β1 (TGF-β1), and mechanical stimulation with NGF.
IL-1β elevates NGF expression in the synovium and chondrocytes
Inflammatory factors, especially IL-1β, contribute to OA progression by downregulating the expression of cartilage extracellular matrix components and stimulating the synthesis of MMPs and other cytokines.2,19 Symptoms are usually linked to inflammation.20
Interestingly, inflammation-induced NGF expression is evident in OA. In a controlled trial,21 medial tibial plateaus and synovium samples were harvested from 29 asymptomatic donors (non-OA control group, postmortem) and 29 symptomatic donors (advanced OA group, during total knee replacement). The histopathologic analysis of samples indicated that patients with advanced OA presented more severe synovitis and increased synovial NGF. NGF staining was localized to fibroblasts and some macrophages.21 Pecchi et al19 examined whether proinflammatory cytokines influence NGF synthesis by chondrocytes. Human and mouse articular chondrocytes were cultured with IL-1β. Consequently, NGF mRNA expression was increased by IL-1β in a dose-dependent manner, paralleled by NGF protein release.19 These findings are consistent with those of Blaney Davidson et al,22 who found that IL-1β elevates NGF expression through TAK1 in the human chondrocyte cell line G6, murine H4 chondrocyte cell line, and bovine primary chondrocytes. This phenomenon was also observed in two other studies investigating the effects of IL-1β exposure on cartilage stem cells and synovial tissue.23,24
TGF-β1 induces NGF expression in the synovium and chondrocytes
Although it is established that pain is linked to inflammation, some patients with OA pain lack detectable inflammation.25 Iannone et al26 reported that the interplay between NGF and pain perception or other nervous system-related functions may directly influence the growth factors and cytokines such as tumor necrosis factor (TNF)-α and TGF-β1, the roles of which have been extensively studied in the pathophysiology of cartilage.27 Using quantitative polymerase chain reaction, Blaney Davidson et al22 identified that TGF-β1 induces NGF expression via the ALK5-Smad2/3 pathway in different cells, including human, murine, and bovine chondrocytes. This induction of NGF by TGF-β1 is more potent than IL-1β stimulus.22 In a recent article17 that examined damaged joint cartilage from modified mice and pigs with OA, it was demonstrated that injury produces pro-algesic molecules, including NGF. The molecular basis of this is partly dependent on TGF-β-activated kinase 1.17 Much evidence has been published on the promotion of the OA process by TGF-β1.28–30
Mechanical loading stimulates NGF expression by chondrocytes
Mechanical overloading is considered the archetypal cause of cartilage damage and pain.31 Consequently, the influence of mechanical compression on NGF release in cartilage explants from joints with OA has become a focus of investigation. The accumulation of endogenous NGF was found to induce mechanical allodynia in rats, and similar evidence exists in humans and other models of OA.7 Recently, high-extension silicone rubber membranes were used on primary chondrocytes to examine the effect of low-frequency, high-magnitude mechanical strain.32 Mechanical strain was applied for the first 8 hours, followed by 16 hours’ rest, and then another 8 hours’ mechanical strain. Subsequently, NGF mRNA expression was significantly increased. Medium from the treated group was collected and applied to cultured PC12 cells, which are known to respond to NGF by sprouting axon-like neurites. The PC12 cells exposed to the mechanical strain-conditioned medium resembled the NGF-treated group, but differed from vehicle-treated controls or static conditioned medium-treated cells.32 Although 4 hours’ dynamic compression did not increase the NGF levels, the concentration of NGF in the medium surrounding murine costal cartilage explants after 24 hours had significantly increased (4.7-fold versus the control). These findings indicate that NGF is a mechanosensitive gene in chondrocytes.19 These findings are consistent with data obtained in neurocytes.33
Other related stimuli
Increased levels of TNF-α and adipokines are found in joints with OA and are thought to play a role in OA progression.34 In isolated synovial cells, synovial fibroblasts and macrophages form a model of OA in which the NGF gene expression increases significantly when exogenous TNF-α is added.24 However, the role of TNF-α in OA pain is controversial because anti-TNF-α therapy is effective in the early, but not the late, phases of OA.35
Anti-NGF therapies
Based on the mechanisms underlying OA, four categories of drugs have been developed and applied to relieve OA pain (Table 1). These drugs include agents that eliminate free NGF, molecules that prevent NGF from binding to the receptor, and drugs that inhibit TrkA activation. Desensitization or blocking of TRPV1 is used to prevent TrkA activation.
 | Table 1 The four categories of drugs that have been applied to relieve OA pain |
The NGF-capturing agent RN624, now known as tanezumab, is a highly selective humanized immunoglobulin G2 monoclonal antibody. It can bind NGF directly and neutralize NGF bioactivity. Tanezumab has demonstrated promising therapeutic potential for the treatment of pain, including pain related to cancer36 and OA,37 in animals38–40 and in clinical trials37 involving acute and chronic pain.39,41 In a proof-of-concept study by Lane et al,37 450 patients with or without moderate-to-severe knee OA were examined. With clinically significant reductions ranging from 45% to 62%, tanezumab improved knee pain, stiffness, and limitations of physical function.37 Other antibodies that have been evaluated in Phase III clinical trials include fasinumab (Regeneron Pharmaceuticals, Inc., Tarrytown, NY, USA) and fulranumab (Janssen [Beerse, Belgium] and Regeneron Pharmaceuticals, Inc.) and PG110 (PanGenetics BV, Utrecht, the Netherlands). In recent a systematic review, Schnitzer and Marks18 reviewed the efficacy of these antibodies. They were effective at relieving OA pain when compared with placebos, and of them, tanezumab performed the best.18,42
Molecules targeting NGF-binding sites have been developed as a result of identification of NGF–TrkA interaction domains.43 They interfere with these domains and block receptor activation. One such molecule, known as ALE0540, inhibits NGF binding to TrkA and p75NTR and is reportedly effective for the treatment of rat neuropathic and inflammatory pain.44 The identity of the molecules that bind to NGF or its receptors is an issue of debate. Recently, the development of surface plasmon resonance technology has enabled evaluation of the inhibitory potential of ALE-0540, PD90780, Ro 08-2750, and PQC083. These compounds bind NGF, rather than the TrkA and p75NTR receptors. PD90780 is the most effective, inhibiting both the NGF–TrkA and NGF–p75NTR interactions, whereas ALE-0540 only inhibits NGF–TrkA binding. Four molecules and novel bivalent naphthalimide derivatives of ALE-0540 also reportedly inhibit the binding of proNGF to the p75NTR receptor.45,46
It took a long time to develop a drug with high levels of homology for TrkA, TrkB, and TrkC. In mice, the anti-TrkA monoclonal antibody MNAC13 demonstrates a significant antiallodynic effect on neuropathic pain, promoting functional recovery.47 Intrathecal administration of antisense oligodeoxynucleotides for TrkA cause a dose-related reduction in hyperalgesia.48 In a recent study by Nwosu et al,49 AR786, a selective TrkA inhibitor, reduced pain behavior in intra-articular monosodium-iodoacetate injection-induced and meniscal transection-induced models of OA. Thus, selective inhibitors of TrkA exhibit the potential for OA pain relief in the future.
Once opened, TRPV1 promotes Ca2+ influx and calcitonin gene-related peptide and substance P expression, leading to central sensitization; therefore, agonists of TRPV1 may be effective for the treatment of OA pain. Anandamide is a TRPV1 agonist that desensitizes TRPV1, reducing Ca2+ influx.50 However, it failed to show a benefit in clinical trials.51 Recently, interfering with TRPV1 subunit association using a plasma membrane-tethered peptide was found to relieve mechanical and thermal hypersensitivity in mouse models. This may offer a new method of analgesic treatment.52 Kelly et al53 identified the clinical and mechanistic rationale for intra-articular application of TRPV1 antagonists (JNJ-17203212) to treat OA pain.
We reviewed the relationship between NGF and OA pain. It convinced us that targeting NGF/TrkA signaling is an effective method to treat OA pain. In the future, other mechanisms between NGF expression and related pro-algesic mediators will be identified, and specific NGF/TrkA antagonists will play a key role in OA pain relief.
Acknowledgments
We thank Professor Wenjie Jin for his help and guidance. This work was supported by the Scientific Research Fund of Shanghai Municipal Commission of Healthy and Family Planning (grant number 201540274).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Miller RE, Miller RJ, Malfait AM. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014;70(2):185–193. | ||
Malfait AM. Osteoarthritis year in review 2015: biology. Osteoarthritis Cartilage. 2016;24(1):21–26. | ||
Glyn-Jones S, Palmer AJ, Agricola R, et al. Osteoarthritis. Lancet. 2015;386(9991):376–387. | ||
Miller RE, Block JA, Malfait AM. Nerve growth factor blockade for the management of osteoarthritis pain: what can we learn from clinical trials and preclinical models? Curr Opin Rheumatol. 2017;29(1):110–118. | ||
Chang DS, Hsu E, Hottinger DG, Cohen SP. Anti-nerve growth factor in pain management: current evidence. J Pain Res. 2016;9:373–383. | ||
Hefti FF, Rosenthal A, Walicke PA, et al. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci. 2006;27(2):85–91. | ||
Osikowicz M, Longo G, Allard S, Cuello AC, Ribeiro-da-Silva A. Inhibition of endogenous NGF degradation induces mechanical allodynia and thermal hyperalgesia in rats. Mol Pain. 2013;9:37. | ||
Cohen SP, Mao J. Neuropathic pain: mechanisms and their clinical implications. BMJ. 2014;348:f7656. | ||
Wheeler MA, Heffner DL, Kim S, et al. TNF-alpha/TNFR1 signaling is required for the development and function of primary nociceptors. Neuron. 2014;82(3):587–602. | ||
Mizumura K, Murase S. Role of nerve growth factor in pain. Handb Exp Pharmacol. 2015;227:57–77. | ||
Eskander MA, Ruparel S, Green DP, et al. Persistent nociception triggered by nerve growth factor (NGF) is mediated by TRPV1 and oxidative mechanisms. J Neurosci. 2015;35(22):8593–8603. | ||
McKelvey L, Shorten GD, O’Keeffe GW. Nerve growth factor-mediated regulation of pain signaling and proposed new intervention strategies in clinical pain management. J Neurochem. 2013;124(3):276–289. | ||
Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Current opinion in neurobiology. 2004;14(3):362–369. | ||
Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology. 2011;115(1):189–204. | ||
Aloe L, Tuveri MA, Carcassi U, Levi-Montalcini R. Nerve growth factor in the synovial fluid of patients with chronic arthritis. Arthritis Rheum. 1992;35(3):351–355. | ||
Seidel MF, Herguijuela M, Forkert R, Otten U. Nerve growth factor in rheumatic diseases. Semin Arthritis Rheum. 2010;40(2):109–126. | ||
Driscoll C, Chanalaris A, Knights C, et al. Nociceptive sensitizers are regulated in damaged joint tissues, including articular cartilage, when osteoarthritic mice display pain behavior. Arthritis Rheumatol. 2016;68(4):857–867. | ||
Schnitzer TJ, Marks JA. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthritis Cartilage. 2015;23(Suppl 1):S8–S17. | ||
Pecchi E, Priam S, Gosset M, et al. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther. 2014;16(1):R16. | ||
Van Spil WE, Nair SC, Kinds MB, et al. Systemic biochemical markers of joint metabolism and inflammation in relation to radiographic parameters and pain of the knee: data from CHECK, a cohort of early-osteoarthritis subjects. Osteoarthritis Cartilage. 2015;23(1):48–56. | ||
Stoppiello LA, Mapp PI, Wilson D, Hill R, Scammell BE, Walsh DA. Structural associations of symptomatic knee osteoarthritis. Arthritis Rheumatol. 2014;66(11):3018–3027. | ||
Blaney Davidson EN, van Caam AP, Vitters EL, et al. TGF-β is a potent inducer of Nerve Growth Factor in articular cartilage via the ALK5-Smad2/3 pathway. Potential role in OA related pain? Osteoarthritis Cartilage. 2015;23(3):478–486. | ||
Jiang Y, Hu C, Yu S, et al. Cartilage stem/progenitor cells are activated in osteoarthritis via interleukin-1beta/nerve growth factor signaling. Arthritis Res Ther. 2015;17:327. | ||
Takano S, Uchida K, Miyagi M, et al. Nerve growth factor regulation by TNF-alpha and IL-1beta in synovial macrophages and fibroblasts in osteoarthritic mice. J Immunol Res. 2016;2016:5706359. | ||
Myers SL, Brandt KD, Ehlich JW, et al. Synovial inflammation in patients with early osteoarthritis of the knee. J Rheumatol. 1990;17(12):1662–1669. | ||
Iannone F, Perniola S, Lopalco G, Cantarini L, Lapadula G. Role of nerve growth factor and tropomyosin receptor kinase A in the pathogenesis of osteoarthritis. Might nerve growth factor be the link interwinding obesity and osteoarthritis? Ann Rheum Dis. 2015;74(12):e70. | ||
Lai Y, Bai X, Zhao Y, et al. ADAMTS-7 forms a positive feedback loop with TNF-alpha in the pathogenesis of osteoarthritis. Ann Rheum Dis. 2014;73(8):1575–1584. | ||
Zhen G, Wen C, Jia X, et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19(6):704–712. | ||
Cui Z, Crane J, Xie H, et al. Halofuginone attenuates osteoarthritis by inhibition of TGF-beta activity and H-type vessel formation in subchondral bone. Ann Rheum Dis. 2016;75(9):1714–1721. | ||
Vinatier C, Merceron C, Guicheux J. Osteoarthritis: from pathogenic mechanisms and recent clinical developments to novel prospective therapeutic options. Drug Discov Today. 2016;21(12):1932–1937. | ||
Saxby DJ, Lloyd DG. Osteoarthritis year in review 2016: mechanics. Osteoarthritis Cartilage. 2017;25(2):190–198. | ||
Rosenzweig DH, Quinn TM, Haglund L. Low-frequency high-magnitude mechanical strain of articular chondrocytes activates p38 MAPK and induces phenotypic changes associated with osteoarthritis and pain. Int J Mol Sci. 2014;15(8):14427–14441. | ||
Rana OR, Schauerte P, Hommes D, et al. Mechanical stretch induces nerve sprouting in rat sympathetic neurocytes. Auton Neurosci. 2010;155(1–2):25–32. | ||
Gomez R, Conde J, Scotece M, Gomez-Reino JJ, Lago F, Gualillo O. What’s new in our understanding of the role of adipokines in rheumatic diseases? Nat Rev Rheumatol. 2011;7(9):528–536. | ||
McNamee KE, Burleigh A, Gompels LL, et al. Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain. 2010;149(2):386–392. | ||
Sopata M, Katz N, Carey W, et al. Efficacy and safety of tanezumab in the treatment of pain from bone metastases. Pain. 2015;156(9):1703–1713. | ||
Lane NE, Schnitzer TJ, Birbara CA, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363(16):1521–1531. | ||
Shelton DL, Zeller J, Ho WH, Pons J, Rosenthal A. Nerve growth factor mediates hyperalgesia and cachexia in auto-immune arthritis. Pain. 2005;116(1–2):8–16. | ||
LaBranche TP, Bendele AM, Omura BC, et al. Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Ann Rheum Dis. 2017;76(1):295–302. | ||
Djouhri L. PG110, A humanized anti-NGF antibody, reverses established pain hypersensitivity in persistent inflammatory pain, but not peripheral neuropathic pain, rat models. Pain Med. 2016;17(11):2082–2094. | ||
Cattaneo A. Tanezumab, a recombinant humanized mAb against nerve growth factor for the treatment of acute and chronic pain. Curr Opin Mol Ther. 2010;12(1):94–106. | ||
Cohen E, Lee YC. A mechanism-based approach to the management of osteoarthritis pain. Curr Osteoporos Rep. 2015;13(6):399–406. | ||
Wiesmann C, de Vos AM. Nerve growth factor: structure and function. Cell Mol Life Sci. 2001;58(5–6):748–759. | ||
Owolabi JB, Rizkalla G, Tehim A, et al. Characterization of antiallodynic actions of ALE-0540, a novel nerve growth factor receptor antagonist, in the rat. J Pharmacol Exp Ther. 1999;289(3):1271–1276. | ||
Sheffield KS, Kennedy AE, Scott JA, Ross GM. Characterizing nerve growth factor-p75(NTR) interactions and small molecule inhibition using surface plasmon resonance spectroscopy. Anal Biochem. 2016;493:21–26. | ||
Sheffield KS, Vohra R, Scott JA, Ross GM. Using surface plasmon resonance spectroscopy to characterize the inhibition of NGF-p75(NTR) and proNGF-p75(NTR) interactions by small molecule inhibitors. Pharmacol Res. 2016;103:292–299. | ||
Ugolini G, Marinelli S, Covaceuszach S, Cattaneo A, Pavone F. The function neutralizing anti-TrkA antibody MNAC13 reduces inflammatory and neuropathic pain. Proc Natl Acad Sci U S A. 2007;104(8):2985–2990. | ||
Summer GJ, Puntillo KA, Miaskowski C, Dina OA, Green PG, Levine JD. TrkA and PKC-epsilon in thermal burn-induced mechanical hyperalgesia in the rat. J Pain. 2006;7(12):884–891. | ||
Nwosu LN, Mapp PI, Chapman V, Walsh DA. Blocking the tropomyosin receptor kinase A (TrkA) receptor inhibits pain behavior in two rat models of osteoarthritis. Ann Rheum Dis. 2016;75(6):1246–1254. | ||
Lizanecz E, Bagi Z, Pasztor ET, et al. Phosphorylation-dependent desensitization by anandamide of vanilloid receptor-1 (TRPV1) function in rat skeletal muscle arterioles and in Chinese hamster ovary cells expressing TRPV1. Mol Pharmacol. 2006;69(3):1015–1023. | ||
Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomized, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153(9):1837–1846. | ||
Flynn R, Chapman K, Iftinca M, Aboushousha R, Varela D, Altier C. Targeting the transient receptor potential vanilloid type 1 (TRPV1) assembly domain attenuates inflammation-induced hypersensitivity. J Biol Chem. 2014;289(24):16675–16687. | ||
Kelly S, Chapman RJ, Woodhams S, et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann Rheum Dis. 2015;74(1):252–259. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.