Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 5
Managing congenital hyperinsulinism: improving outcomes with a multidisciplinary approach
Authors Ackermann A, Palladino A
Received 11 March 2015
Accepted for publication 21 April 2015
Published 31 July 2015 Volume 2015:5 Pages 103—117
DOI https://doi.org/10.2147/RRED.S56608
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Mingzhao Xing
Video abstract presented by Amanda M Ackermann.
Views: 2086
Amanda M Ackermann, Andrew A Palladino
Division of Endocrinology and Diabetes, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
Abstract: Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in pediatric patients and is associated with significant risk of hypoglycemic seizures and developmental delays. CHI results from mutations in at least nine genes that play a role in regulating beta-cell insulin secretion. Thus, patients with CHI have dysregulated insulin secretion that is unresponsive to blood glucose level. Each different genetic etiology of CHI is associated with particular clinical characteristics that affect management decisions. Given the broad phenotypic spectrum and relatively rare prevalence of CHI, it is important that patients with CHI be evaluated by clinicians experienced with CHI and the multiple subspecialty services that are necessary for the management of the disorder. In this review, we summarize the pathophysiology and genetic causes of CHI and then focus primarily on the most common genetic cause (mutations in the ATP-gated potassium [KATP] channel) for further discussion of diagnosis, medical and surgical management, and potential acute and chronic complications. We provide insight from relevant published studies and reports, in addition to anecdotal information from our center’s clinical experience in caring for over 400 patients with CHI. Careful assessment of each patient’s individual pathophysiology is necessary to determine the appropriate treatment regimen, and continued close follow-up and monitoring of disease- and treatment-related complications are essential. Although significant improvements have been made in the past several years with regard to diagnosis and management, given the continued high morbidity rate in patients with CHI, improved diagnostic techniques and new therapeutic options would be welcomed.
Keywords: hypoglycemia, hyperinsulinism, beta-cell, congenital, monogenic
Introduction
Congenital hyperinsulinism (CHI) is a clinically and genetically heterogeneous disorder that is the most common cause of permanent hypoglycemia in infants and children. The frequency of hyperinsulinism (HI) is estimated at one in 50,000 live births but can be as common as one in 2,500 live births in highly consanguineous communities.1 CHI can present in a variety of ways, which often leads to frequently missed or delayed diagnosis. Early detection and appropriate intervention are imperative to prevent the complications of severe and/or recurrent hypoglycemia, which include seizures, brain damage, and death.
Advances in genetic testing and imaging techniques have led to more precise diagnosis and interventional planning for patients with CHI, improving short- and long-term outcomes. In this review, we describe the multidisciplinary approach used at our institution, a large pediatric HI referral center that has cared for hundreds of patients with CHI and has performed more than 400 therapeutic partial and near-total pancreatectomies, utilizing the expertise of pediatric endocrinologists, pediatric surgeons, radiologists, pathologists, geneticists, and neonatologists.
Detection and diagnosis
Patients with HI have dysregulated insulin secretion resulting in persistent hyperinsulinemia irrespective of blood glucose level. The metabolic effects of hyperinsulinemia result in a significantly shortened fasting tolerance. Insulin secretion is normally inhibited at a blood glucose of 80–85 mg/dL (4.4–4.7 mmol/L), and glucagon, growth hormone (GH), cortisol, and epinephrine releases are stimulated at a blood glucose of 65–70 mg/dL (3.6–3.9 mmol/L).2 The result of these responses is the stimulation of hepatic gluconeogenesis and glycogenolysis, decrease in glucose uptake by peripheral tissues, and stimulation of lipolysis to produce fatty acids that are oxidized into ketones, which serve as an alternative fuel source for the heart and brain. Insulin directly counteracts these regulatory hormone changes and inhibits glucagon secretion itself, thus inhibiting gluconeogenesis, glycogenolysis, lipolysis, and fatty acid oxidation, and stimulating peripheral glucose uptake. Consequently, hyperinsulinemia results in severe hypoglycemia and the absence of alternative fuel sources, which can lead to rapid and severe consequences such as seizure and coma.
Hypoglycemia may present in neonates, infants, or young children as seizure, lethargy, or poor feeding. Primary care providers, emergency room physicians, and neonatologists should have a low threshold for checking the blood glucose level in children with any of these presenting symptoms. Ideally, patients with CHI are identified as soon as possible in order to prevent prolonged or recurrent hypoglycemia. A retrospective evaluation of 223 patients with CHI requiring partial or near-total pancreatectomy at our institution between 2004 and 2012 revealed that 50%–75% of patients were large for gestational age (LGA) and 25%–50% presented with a seizure.3 A maternal history of gestational diabetes in an infant in whom hypoglycemia does not quickly resolve may suggest a dominantly inherited form of CHI. In CHI, the focus is often on the fact that insulin does not “turn off” normally in response to a low blood glucose; however, these mutations also prevent the normal “turning on” of insulin in response to hyperglycemia, which can manifest as a failed oral glucose tolerance test in the mother.
Evaluation of hypoglycemia
Patients who meet Whipple’s criteria (Table 1) should undergo a diagnostic fasting evaluation (Table 2) to assess their fasting tolerance and to obtain plasma hormone and fuel metabolite levels when the blood glucose is <50 mg/dL (2.8 mmol/L). Close monitoring of blood glucose, beta-hydroxybutyrate (BOHB) (if possible), vital signs, and mental status during the fasting evaluation is imperative in order to prevent severe or prolonged hypoglycemia.
 | Table 1 Whipple’s triad of hypoglycemia |
 | Table 2 Diagnostic fasting evaluation of hypoglycemia |
Normal fasting stimulates lipolysis and ketogenesis, which lead to elevated ketones and free fatty acids (FFAs) at the time of hypoglycemia. Additionally, fasting stimulates glycogenolysis, depleting glycogen stores and resulting in the absence of a glycemic response to glucagon at the time of hypoglycemia. The diagnosis of HI is based on inappropriately suppressed ketones, FFAs, and insulin-like growth factor-binding protein-1 (IGFBP-1), with or without elevated insulin or C-peptide levels, at the time of hypoglycemia, as well as an inappropriate rise in blood glucose after glucagon administration (Table 3).4–7 It is important to note that low GH or cortisol levels at the time of hypoglycemia have poor sensitivity for detecting true GH or adrenal insufficiency.8 Therefore, additional tests such as insulin-like growth factor-1 (IGF-1) and insulin-like growth factor-binding protein-3 (IGFBP-3) and/or specific provocative testing are indicated if there is concern for these hormone deficiencies. The utility of the glucagon stimulation test at the end of fasting studies in evaluating GH deficiency is currently being investigated. Treatment with glucocorticoids should be restricted to those with a confirmed diagnosis of adrenal insufficiency or if clinically indicated in an emergent situation.
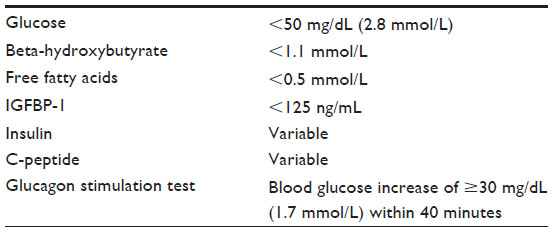 | Table 3 Laboratory findings consistent with HI |
The definition and management of neonatal hypoglycemia are the topics of debate that go beyond the scope of this review,9 and current screening guidelines are based on insufficient studies.10 However, there are data showing that term appropriate-for-gestational age neonates maintain capillary blood glucose >50 mg/dL (2.8 mmol/L) from birth to 2 days of life and >65 mg/dL (3.6 mmol/L) beyond 3 days of life11 and that impaired neurodevelopment is associated with neonatal blood glucose levels <47 mg/dL (2.6 mmol/L).12 We recommend that these thresholds serve as a guide for neonatal hypoglycemia evaluation and intervention, with the goal of preventing recurrent hypoglycemia and ensuring that the patient is safe prior to discharge. Risk factors associated with an approximate 50% risk of transient neonatal hypoglycemia (blood glucose <47 mg/dL [2.6 mmol/L]) include prematurity, small for gestational age (SGA) or LGA, and being the infant of a diabetic mother.13 Although hypoglycemia in these patients typically resolves within 48 hours,13,14 it is important to document that the hypoglycemia has resolved and that the infant is safe to skip one feeding prior to discharge. For infants with borderline blood sugars at the time of discharge, we recommend that the family be provided with a glucometer and the appropriate teaching in order to monitor pre-feeding blood glucoses at home. The endocrine service should be notified of these patients, if not already involved, so that follow-up can be arranged.
Fasting studies are used for both safety and diagnostic purposes. In neonates, safety is typically determined by skipping a single feed (6-hour fast), as term neonates over 3 days of age should be able to maintain their blood glucose >65 mg/dL (3.6 mmol/L) for the duration of the fast. Diagnostic fasting studies should be performed as described above in patients who fail the safety fast. Normal fasting tolerance, and thus the recommended maximum fasting evaluation duration, is dependent on a patient’s age (Table 4). A diagnostic fasting evaluation is ended when blood glucose is <50 mg/dL (2.8 mmol/L), when BOHB is ≥2.5 mmol/L (26 mg/dL), if the patient becomes significantly symptomatic, or if the maximum duration is achieved. Patients with shortened fasting tolerance and persistent hypoglycemia of any etiology should be referred to a pediatric endocrinologist for further evaluation and management. Patients with suspected or confirmed HI should be referred to a tertiary care center with expertise in HI.
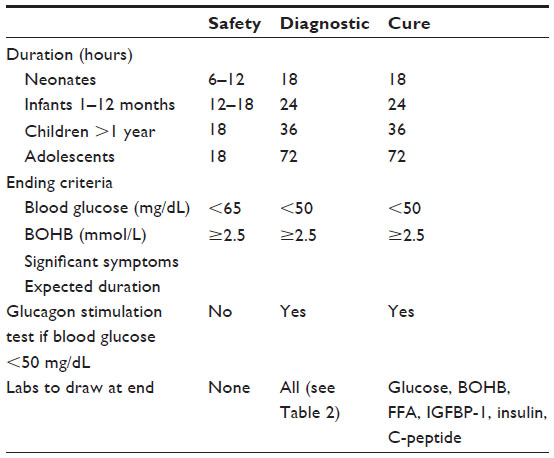 | Table 4 Types of fasting studies |
Etiology of HI
Once the diagnosis of HI is made, it can fall into one of two broad categories – suspected transient HI or suspected CHI. Transient HI (also known as perinatal stress-induced HI or prolonged neonatal HI) is often associated with perinatal risk factors such as perinatal stress, SGA, LGA, prematurity, and maternal hypertension.15 It is usually responsive to diazoxide therapy and typically resolves by 6 months of age but can last up to 1 year.15 Although most cases of transient HI do not have a known underlying genetic cause, within the past several years, genetic mutations in the transcription factors HNF4A and HNF1A have been described in infants with transient HI and macrosomia.16,17
CHI results from inherited or de novo mutations of at least nine genes involved in the regulation of insulin secretion (Table 5 and Figure 1; reviewed in Refs 18–21). Insulin secretion by the pancreatic beta-cell results from a fuel-stimulated increase in the ATP:ADP ratio, which leads to closure of the ATP-sensitive potassium (KATP) channel, depolarization of the cell membrane, influx of calcium, and release of insulin. Insulin secretion is stimulated by glucose oxidation via glucokinase and by leucine stimulation of glutamate oxidation via glutamate dehydrogenase (GDH) 1, and activating mutations of either of these enzymes result in CHI. Furthermore, inactivating mutations of uncoupling protein 2 (UCP2), which normally limits ATP production from glycolysis, or short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD), which normally inhibits GDH1, also cause CHI. Monocarboxylate transporter 1 (MCT1) is responsible for intracellular transport of pyruvate, which can enter the Krebs cycle to produce ATP; thus, activating mutations of MCT1 can also result in CHI. Because all of these pathways are upstream of the KATP channel, CHI caused by mutations in these proteins typically can be managed with diazoxide therapy. However, some patients with MCT1 mutations are not responsive to diazoxide, suggesting that there is an additional mechanism that affects insulin secretion.
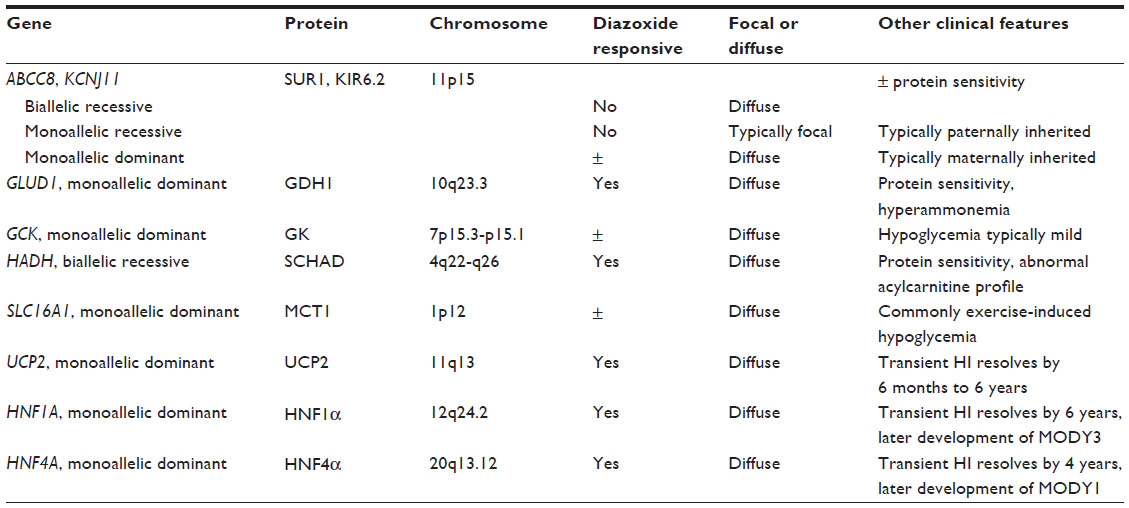 | Table 5 Genetic causes of CHI |
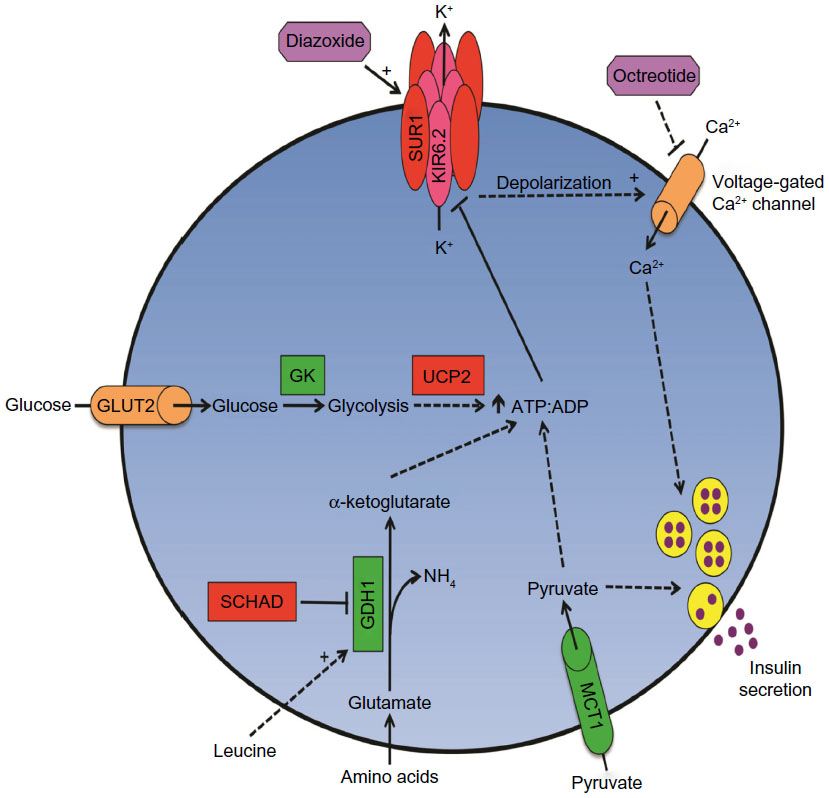 | Figure 1 Multiple genetic mutations affect beta-cell insulin secretion in CHI. |
 , and those with inactivating mutations are in
, and those with inactivating mutations are in  . Medications used to treat CHI are in
. Medications used to treat CHI are in  .
.Identification of the genetic mutation in patients with CHI is critical, as management and prognosis vary depending on which gene is affected. Mutations in ABCC8 or KCNJ11, which encode the SUR1 (sulfonylurea receptor 1) and KIR6.2 (ATP-sensitive inward rectifier potassium channel 11 isoform 2) subunits of the KATP channel, respectively, are the most common causes of CHI. Certain mutations in ABCC8 and KCNJ11 lead to defective KATP channels that are either unable to open or cannot localize to the plasma membrane. Defective channels lead to continued depolarization of the plasma membrane and subsequent insulin secretion irrespective of the plasma glucose level. Homozygous or biallelic compound heterozygous recessive mutations in either of these genes typically result in diffuse HI, characterized by insulin secretory dysfunction affecting all beta-cells.22–24 However, for patients with heterozygous ABCC8 or KCNJ11 mutations, it is imperative to perform parental genetic analysis, as maternally inherited mutations typically function differently from paternally inherited mutations. Maternally inherited heterozygous mutations classically act in a dominant fashion.25 In contrast, paternally inherited recessive ABCC8 or KCNJ11 mutations are most likely to result in focal HI, in which only a portion of the pancreas is affected due to somatic loss of heterozygosity and paternal isodisomy of the chromosome 11p15 region that contains the ABCC8 and KCNJ11 genes in addition to several imprinted genes that increase beta-cell proliferation (H19, IGF2, CDKN1c).22 This imprinted chromosomal region is also affected in Beckwith–Wiedemann Syndrome (BWS), a syndrome characterized by mosaic tissue overgrowth, increased tumor risk, and commonly hypoglycemia.
Genetic testing
With advances in gene-sequencing techniques, CHI gene panel tests are now commercially available and relatively cost- and time-efficient. We routinely perform genetic analyses on all patients with persistent or severe hypoglycemia beyond the immediate neonatal period. We initially test for gene mutations in ABCC8, KCNJ11, GCK, GLUD1, HNF1A, and HNF4A. If the initial round of testing is negative, we will then test for HADH, INSR, SLC16A1, and UCP2 mutations. For patients who are unresponsive to diazoxide, genetic analysis can be initially limited to ABCC8, KCNJ11, and GCK. We recommend sending parent blood samples with the patient’s sample for expediency, as several laboratories will perform parental genetic testing at no or reduced cost if the patient’s results reveal a mutation. Involving the genetics team or contacting an HI center once the genetic results return is important not only for discussions with the clinical team regarding patient prognosis and management but also for discussions with the parents regarding future family planning and potential testing of other family members.
Importantly, normal gene-sequencing results do not preclude the diagnosis of CHI. Typical gene sequencing, utilizing the Sanger method, sequences gene fragments and then pieces the results together and thus may not detect certain insertions or deletions. Additionally, only coding gene regions (exons) are typically sequenced, so mutations in noncoding gene regions (introns) will not be detected; although such mutations are less likely to cause disease, they can alter gene transcript splicing and have been identified in patients with CHI whose commercial genetic testing results were normal.26 Currently, such testing is limited to research laboratories but will likely be available for clinical use in the near future. Furthermore, de novo somatic mutations within pancreatic progenitor cells cannot be detected by genetic analysis of peripheral blood samples, without direct analysis of primary pancreatic tissue; although rare, we have encountered such scenarios in which the genetic mutation was only detected after therapeutic pancreatic resection (data not published).
Medical management of HI
Dextrose
Acute hypoglycemia should be treated with an enteral or intravenous (IV) dextrose bolus, followed by continuous IV dextrose infusion. Blood glucose should be measured every 10–15 minutes after treatment of the initial hypoglycemic episode until it is stably ≥70 mg/dL (3.9 mmol/L). Patients with CHI typically require a glucose infusion rate (GIR) of >10 mg/kg/min; however, a lower GIR does not exclude CHI, as the severity of CHI is highly variable among and within the different mutation groups. We recommend providing a GIR sufficient to maintain the blood glucose ≥70 mg/dL (3.9 mmol/L). While there is no theoretical upper limit for a GIR, there are practical limitations and safety considerations in terms of the dextrose concentration and fluid volume that is infused. We recommend infusing dextrose via a central venous catheter if a dextrose concentration >12.5% is needed to maintain the patient’s infusion volume within the recommended total fluid limit. We have used dextrose concentrations as high as 50% via central lines in severe cases of CHI. Central lines are also safer for patients with severe HI or tenuous peripheral access, as even brief interruptions in a glucose infusion may result in rapid and severe hypoglycemia. Additionally, many of these patients will require the long-term venous access that a central line provides (Table 6).
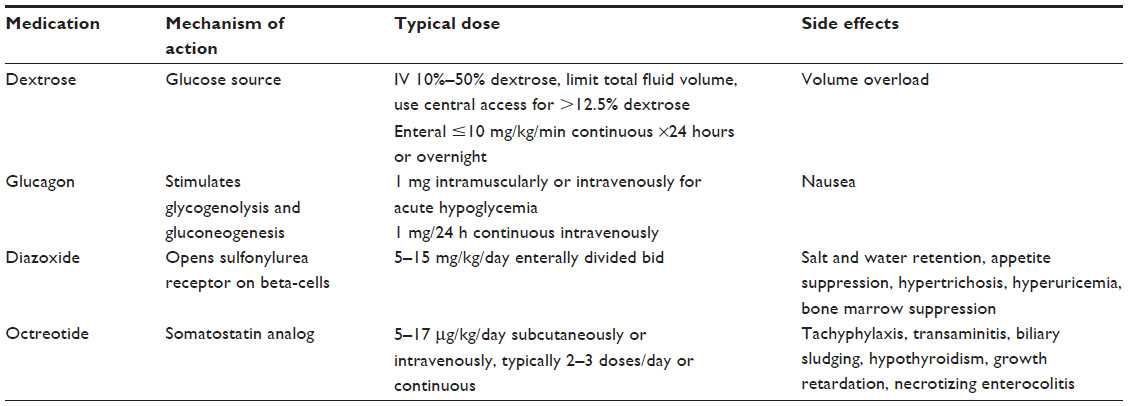 | Table 6 Medical management of HI |
Patients whose GIR is ≤10 mg/kg/min may be trialed on enteral 10% or 20% dextrose in water, either continuously for 24 hours or overnight in an attempt to determine whether they can be safely managed on such a regimen. It is important to note that enteral dextrose increases the intestinal osmotic load and can thus cause diarrhea. An enteral GIR >10 mg/kg/min is avoided due to concern for gastrointestinal intolerance and potential vascular compromise. Furthermore, in most cases of CHI, more specific medical or surgical therapy is indicated, particularly depending on the patient’s genetic cause of CHI.
Glucagon
As discussed previously, patients with HI have an inappropriate rise in blood glucose in response to glucagon which allows for glucagon (1 mg intramuscularly/intravenously) to be used as a rescue medication for acute or severe hypoglycemia.27 Additionally, glucagon can be given as a continuous IV infusion (1 mg/24 h) for patients requiring significant dextrose concentrations and fluid volumes to maintain normoglycemia.28
Diazoxide
Once the patient’s blood glucose has been stabilized by the methods described above, the first-line medical therapy for HI is diazoxide, a KATP channel agonist, which keeps the channel in an open state, resulting in decreased insulin secretion. Diazoxide is given orally and is dosed at 5–15 mg/kg/day, divided evenly into two daily doses given every 12 hours. Given diazoxide’s relatively long serum half-life of approximately 22 hours,29 it may take up to 5 days before maximal biological effect is achieved. We recommend starting diazoxide at 10–15 mg/kg/day. Importantly, diazoxide is only effective in patients with functional KATP channels that are appropriately localized on the plasma membrane. Thus, patients with heterozygous dominant ABCC8 or KCNJ11 mutations can be responsive to diazoxide, as their mutations result in dysfunctional but appropriately translated and localized KATP channels.25 In contrast, patients with homozygous recessive (diffuse HI) or heterozygous recessive (focal HI) ABCC8 or KCNJ11 mutations are unresponsive to diazoxide.22–24 We define unresponsiveness as an inability to maintain a blood glucose ≥65 mg/dL (3.6 mmol/L) while fasting without glucose support for 8–12 hours after 5 days of maximum-dose diazoxide therapy.
Sodium and fluid retention are common side effects of diazoxide that typically begin within a few days after starting the medication and are seen in at least 40% of patients.30 Therefore, careful attention should be paid to weight, edema, electrolytes, and cardiopulmonary function in all patients on diazoxide, particularly upon initiation or with a dose increase. Anecdotally, there is a higher incidence of diazoxide-related fluid overload in infants; therefore, we recommend prophylactically starting a chlorothiazide diuretic in all infants at the time of diazoxide initiation unless there are specific contraindications. Compounding this side effect is that many infants starting diazoxide are also receiving large volumes of IV fluids for glucose management. Consultation with a cardiologist should take place if there are any concerns regarding cardiopulmonary risk factors related to fluid overload, or if typical diuretic doses do not improve symptoms.
Other side effects of diazoxide include appetite suppression, hypertrichosis, elevated uric acid, and bone marrow suppression. The appetite suppressive effect of diazoxide is particularly notable in infants and young children, in whom adequate caloric intake is important not only for growth but also for blood glucose support. Hyperuricemia is due to decreased urinary excretion of uric acid. Bone marrow suppression with cytopenia is a severe side effect of diazoxide that can occur at any time.31 We recommend discontinuing diazoxide and supporting blood glucose by other means in patients with severe or worsening cytopenia until resolution. Once the cytopenia resolves, diazoxide can be trialed at a lower dose with close monitoring of blood counts and glucose levels. Hypertrichosis occurs within several months of diazoxide use and is dose dependent. It can be a source of parental distress, but as it is an esthetic effect, medical therapy is not warranted; it can be managed with shaving or with dermatological procedures in older children.
Octreotide
Octreotide is a somatostatin analog that binds to G-protein-coupled somatostatin receptors on endocrine cells, particularly on beta-cells, and indirectly inhibits voltage-gated calcium channels, resulting in reduced insulin secretion.32 It can be administered via subcutaneous injections, continuous subcutaneous infusion, or intravenously.
A side effect of octreotide is reduced splanchnic blood flow, which may explain its association with necrotizing enterocolitis (NEC).33 In our experience, treating patients with presumed diffuse disease with octreotide in hopes of avoiding a pancreatectomy was rarely successful. Given the low success rate and its association with NEC, we stopped using octreotide prior to surgery in patients in need of surgery and have since not encountered any preoperative cases of NEC. We do not recommend using octreotide in young infants or in patients with increased risk of NEC (preterm, history of cardiac anomalies, or surgery, etc). However, we have continued its use in older infants and children without incidence, save for one infant with an exceedingly unique and complicated situation involving many contributing variables. The biological half-life of octreotide is relatively short (90 minutes),34 and patients often develop tachyphylaxis resulting in diminishing effects with subsequent doses.35
The most common side effect of octreotide therapy is mild transaminitis, observed in over 40% of patients.36 This effect is typically transient but has been reported to persist and has been associated with fulminant hepatitis. Another common side effect of chronic octreotide use, seen in approximately one-third of children, is biliary sludging and cholecystitis.36 As a general inhibitor of endocrine hormone secretion, other potential side effects of octreotide include hypothyroidism and growth retardation due to direct suppression of pituitary thyroid-stimulating hormone (TSH) and GH release, respectively.36,37 Although there is some evidence that these endocrine side effects are only observed in patients on high doses of octreotide (>17 μg/kg/day), until more studies are performed, we recommend that patients treated with octreotide have evaluations of height, thyroid function tests, and growth factors (IGF-1 and IGFBP-3) performed every 6 months, along with screening of liver enzymes and yearly gallbladder ultrasounds. These side effects can typically be managed by lowering or, when possible, discontinuing the dose of octreotide. Hypothyroidism may require thyroid hormone replacement, while octreotide-related biliary pathology may require ursodiol therapy or cholecystectomy. Exogenous GH supplementation in this population has not been evaluated but would theoretically improve growth.
Goals of therapy
Given the absence of alternative fuels in hyperinsulinemic hypoglycemia and the potentially severe neurological outcomes of this dangerous combination, we recommend that the goal blood glucose in patients with CHI should be ≥70 mg/dL (3.9 mmol/L), regardless of age. Once therapy is initiated, blood glucose levels should be closely monitored to evaluate effectiveness of the intervention(s); we recommend checking the blood glucose level every 3 hours or prior to a feeding. Prior to discharging the patients, it is imperative that they undergo a fast to evaluate the efficacy and safety (“safety fast”) of the regimen that they will go home on. Although each case must be interpreted individually, we recommend that infants <1 month of age be able to fast for at least 8–12 hours, and all other patients for 12–18 hours, while maintaining their blood glucose ≥65 mg/dL (3.6 mmol/L). The longer the patients are able to fast without hypoglycemia, the more likely they are going to be able to safely tolerate situations with limited glucose intake, such as illness, vomiting, diarrhea, missed or delayed meals, or interruption of continuous dextrose or feedings.
Surgical management
Partial and near-total pancreatectomies have long been recognized as therapeutic in some cases of CHI.38,39 However, only in recent years have we had efficient genetic testing, a better understanding of the relationship between the genotype and pathophysiology, and sensitive noninvasive imaging to appropriately guide surgical management of CHI. As with any invasive procedure, it is of utmost importance to weigh the potential risks and benefits of a partial or near-total pancreatectomy on an individual basis. Surgery is recommended for patients with suspected focal disease, patients with failed medical management, and patients with a tenuous regimen or a regimen that is too complex to reasonably be sustained at home. If the patient can be safely managed at home on a tenable medical regimen, then surgery may be avoided.
Focal HI
As mentioned in previous sections, the most common gene mutations to cause CHI are in ABCC8 and KCNJ11, with most of these cases being diazoxide unresponsive.23,24 A paternally inherited heterozygous recessive mutation in either of these genes is highly associated with focal HI. In 417 patients with CHI from our center, the presence of a paternally inherited heterozygous recessive ABCC8 or KCNJ11 mutation had a 95% positive predictive value for focal disease.24 In the same study, approximately 55% of our diazoxide-unresponsive patients had focal disease.24 Focal HI is distinguished pathologically by a distinct region of endocrine cell hyperplasia with loss of p57KIP2 (CDKN1C) expression due to loss of maternal 11p15 chromosome material.40–44 Resection of the involved region cures most cases of focal HI;3,45,46 however, in rare cases, extensive focal lesions may make complete resection difficult.42,47 Out of a total of 114 patients who underwent pancreatectomy for focal HI at our center, the median extent of pancreatic resection for patients with focal HI was 27%, although the range varied from 1% to 100%, and the cure rate was 94%.3 Similar cure rates are seen at other institutions.48
Preoperative identification and localization of focal HI lesions are useful for diagnostic confirmation and surgical guidance. Pancreatic venous sampling49 and calcium arterial stimulation and venous sampling50 with insulin measurements were previously used for this purpose, but the sensitivity was <75%. More recently, noninvasive imaging with18fluoro-l-dihydroxyphenylalanine positron emission tomography and computed tomography (18F-DOPA PET/CT) has been developed and utilized to identify focal lesions based on the fact that 18F-DOPA is selectively taken up by neuroendocrine cells, and thus, dense collections of endocrine cells in focal HI can be visualized by an experienced radiologist. Our center found that18F-DOPA PET/CT has 85% sensitivity, 96% specificity, and 96% positive predictive value for diagnosis of focal HI,51 and similarly encouraging results have been seen at other institutions.52 Therefore, we recommend that all patients who are unresponsive to diazoxide with the potential for focal disease (monoallelic KATP channel mutations that are paternally inherited or of unknown origin, no identifiable mutations detected, variations of unknown significance) should have 18F-DOPA PET/CT imaging performed. 18F-DOPA PET/CT has also been used to identify ectopic islet tissue in a patient with persistent CHI after pancreatectomy.53
Intraoperative analysis of frozen sections by a pathologist with CHI experience is important for identifying the involved regions, evaluating lesion margins, and guiding the extent of pancreatic resection.43 Intraoperative pancreatic ultrasound can also be helpful in difficult cases.54
Diffuse HI
Diffuse HI is not a distinct diagnosis; essentially, it encompasses all forms of CHI that are not focal disease, as all beta-cells are affected in the other causes. However, diffuse HI is most often used to refer to cases of diazoxide-unresponsive KATP channel CHI with pathologic findings of normal islet number, size, and distribution with scattered endocrine cell nucleomegaly throughout the pancreas.42,43 Diffuse CHI is most commonly caused by homozygous or biallelic compound heterozygous recessive ABCC8 or KCNJ11 mutations, which account for approximately 43% of our patients who are diazoxide unresponsive.24
Unlike focal HI, pancreatectomy in diffuse HI is palliative, not curative, with the goal being to reduce the number of dysfunctional beta-cells and thus the amount of unregulated insulin secretion. A near-total (95%–98%) pancreatectomy is recommended for almost all patients with diffuse CHI that cannot be medically managed.55,56 However, the extent of pancreatic resection may be reduced in less severe cases in order to reduce potential long-term complications associated with near-total pancreatectomy.57 At our center, the median extent of pancreatic resection for diffuse HI is 98%, with a range of 15%–100%.3 Patients who are not medically manageable despite the initial surgery will likely require a second surgery for a more extensive pancreatectomy; this is necessary in approximately 3% of our patients with diffuse HI who initially receive a near-total pancreatectomy (data not published). In our institution, at the time of discharge, out of a total of 97 patients with diffuse HI who had undergone pancreatectomy, 23% were euglycemic without any medical support, 41% had recurrent hypoglycemia requiring medical management, and 36% had hyperglycemia requiring insulin therapy.3 Given the high incidence of recurrent hypoglycemia after pancreatectomy for diffuse HI, we recommend that a percutaneous gastrostomy tube (G-tube) be surgically placed at the time of pancreatectomy as a means to provide enteral dextrose to aid in hypoglycemia management. A smaller cohort in France, all of whom underwent near-total pancreatectomy for diffuse HI, had a 21% incidence of euglycemia, 26% incidence of recurrent hypoglycemia, 20% incidence of hyperglycemia, and 33% incidence of fasting hypoglycemia with postprandial hyperglycemia in the immediate postoperative period.48 In that same cohort, the cumulative incidence of insulin requirement was 42% at 8 years and 91% at 14 years. Another cohort who underwent near-total pancreatectomy for diffuse HI in the UK had a 60% incidence of recurrent hypoglycemia in the immediate postoperative period, 22% incidence of hyperglycemia, and 18% incidence of euglycemia.58
Beckwith–Wiedemann syndrome
As a highly heterogeneous disorder, the phenotypic spectrum of BWS is quite broad, due in part to mosaic genetic and epigenetic abnormalities that affect tissues to varying degrees.59 Hypoglycemia occurs in at least 50% of patients with BWS, but the pathophysiology is somewhat unclear, although it is likely due to alterations in gene expression from the 11p15 chromosomal region, which is also affected in focal HI.22 We recommend that geneticists be involved in the testing and management of BWS due to its complex nature. Many patients with BWS and HI are diazoxide responsive, and the HI typically spontaneously resolves within months to years.60 However, some patients with BWS and HI do not respond to diazoxide, and the HI is severe and persistent. In such cases, we have found that partial pancreatectomy (75%–95%) is therapeutic.61 Similar to focal HI, typically only a portion of the pancreas is affected in BWS, although histopathology reveals numerous large islets spread throughout the parenchyma with increased endocrine cell proliferation and nucleomegaly, as opposed to a discreet lesion as in focal HI.61 Thus, intraoperative biopsies can be extremely useful to guide extent of pancreatectomy, while taking into consideration HI severity in each patient. Of note, histologic findings from pancreatic biopsies in one of our patients who had no identified mutations led us to consider a diagnosis of BWS despite scarcely any physical stigmata of BWS. The patient was later confirmed to have BWS by genetic analysis.
Perioperative management
Preoperative considerations
CHI has been associated with neonatal hypertrophic cardiomyopathy (HCM); the incidence at our institution in infants with diffuse or focal CHI is approximately 15% and is associated with CHI severity.62 The pathophysiology of HCM in CHI is thought to be related to the growth-promoting effects of insulin excess during fetal development, although contribution of KATP chanellopathy within cardiomyocytes has not been definitively excluded. Given the relatively high incidence of HCM and the increased risks associated with fluid overload and anesthesia in this population, we recommend that all patients with severe CHI, CHI requiring surgery, or CHI and cardiorespiratory distress be evaluated by a pediatric cardiologist and have an electrocardiogram and echocardiogram.
All patients should have a central line placed prior to surgery, as they will be unable to feed enterally for at least a few days postoperatively and will therefore require IV GIR for all of their glucose requirements. Furthermore, because acute post pancreatectomy glycemia is highly variable, we recommend that patients be sent to the operating room with a titratable two-bag IV fluid system; we typically use a combination of 30% dextrose in 0.45% saline and 0.45% saline without dextrose. Octreotide should be discontinued at least 48 hours prior to pancreatectomy, and glucagon should be discontinued at least 12 hours prior to pancreatectomy due to concern for potential effects on the pancreatic endocrine tissue that could interfere with intraoperative frozen section analysis.
Acute postoperative management
During the acute postoperative period, until the patient has resumed goal enteral feeds, blood glucose levels should be monitored hourly to readily identify hypoglycemia or hyperglycemia. Hypoglycemia should be treated by increasing the patient’s IV GIR until blood glucose is stably ≥70 mg/dL (3.9 mmol/L). Hyperglycemia (blood glucose >180 mg/dL [10 mmol/L]) should be treated with an IV insulin infusion starting at a very low dose (0.005 units/kg/h) and titrated to stabilize the blood glucose between 70 mg/dL and 180 mg/dL (3.9 mmol/L and 10 mmol/L), while maintaining the IV GIR at 8 mg/kg/min to support normal infant glucose requirements.63 If the patient acutely develops hyperglycemia after having had stable blood glucose levels for at least 24 hours, infection should be highly suspected. As the patient tolerates increasing enteral feeds, the IV GIR may be decreased accordingly. Once on full enteral feeds, if hypoglycemia is persistent, then alternatives to IV GIR such as octreotide and/or enteral dextrose should be considered. If hyperglycemia persists, then the patient should be transitioned from IV to subcutaneous insulin.
Chronic management
Hypoglycemia
If postoperative hypoglycemia persists once the patient is on full enteral feeds, alternatives to IV GIR such as octreotide and/or enteral dextrose should be considered. Patients who were not responsive to diazoxide prior to surgery will not be responsive postoperatively. The required glucose support varies from patient to patient. Some patients are managed with continuous enteral dextrose via G-tube, while others are on subcutaneous octreotide every 6–8 hours, or a combination of these two options. We have had success with two daytime octreotide injections (eg, 8 am and 2 pm) with overnight (eg, 8 pm to 8 am) continuous enteral dextrose (10% or 20%) via a G-tube. An effective strategy we have employed in an attempt to overcome tachyphylaxis is to give a relatively low dose in the morning (1 μg/kg) and an increased second dose in the afternoon (3 μg/kg) with a maximum dose of 15–20 μg/kg/day. The maximum GIR we will attempt enterally is 10 mg/kg/min. Off-label use of monthly intramuscular depot octreotide (lanreotide) may be considered in patients on a stable subcutaneous octreotide regimen, although studies in patients with CHI are limited.64–66 Continuous subcutaneous infusion via a portable pump may also be used.67 Patients with the most severe postoperative hypoglycemia are often discharged home on a continuous enteral dextrose infusion plus subcutaneous octreotide injections every 6–8 hours. It is important to recognize that after a near-total pancreatectomy, the management can be complicated and there still needs to be an expertise in CHI to manage these patients.
Patients who are unmanageable with the most intensive of regimens may trial continuous subcutaneous glucagon via a portable pump.68 Unfortunately, current glucagon formulations are quite viscous and are not stable for more than 24 hours, requiring that tubing be changed multiple times per day and that pump cartridges be changed daily. However, more stable glucagon preparations are currently under development.
Hyperglycemia
Patients with preprandial hyperglycemia after pancreatectomy will require subcutaneous insulin therapy. We typically use neutral protamine hagedorn (NPH) insulin due to the frequent feeding schedule of infants but have also used glargine insulin. We recommend starting with low doses of subcutaneous insulin and titrating up slowly in order to avoid exogenous insulin-induced hypoglycemia. Patients with postoperative hyperglycemia are quite unique and should not be treated as patients with type 1 diabetes. With prolonged fasting, patients with postoperative hyperglycemia will develop hyperinsulinemic hypoglycemia in the absence of exogenous insulin. Their beta-cells remain defective and do not regulate insulin secretion in accordance with blood glucose levels. Patients with postoperative hyperglycemia need to monitor blood sugars carefully because of the risk of hypoglycemia.
Euglycemia
Patients with diffuse HI who do not require insulin or glucose support following pancreatectomy are still at risk of postprandial hyperglycemia and fasting hypoglycemia due to underlying beta-cell dysfunction. Therefore, continued blood glucose monitoring at home and regular follow-up are necessary.
Feeding
While the mainstay of CHI management acutely is glucose supplementation, chronic administration of a high carbohydrate diet will contribute to obesity and related metabolic and cardiovascular complications. We find that many families, due to an understandable fear of hypoglycemia, encourage frequent feedings and consumption of high sugar drinks and foods. However, in addition to promoting excessive weight gain, this can also complicate CHI management, due to rapid fluctuations in blood glucose levels. It is also common for patients with CHI to exhibit food avoidance behaviors, which may be due to medication side effects, oral aversion secondary to frequent forced feedings at a young age, or postoperative complications. Such food avoidance may necessitate enteral tube feeds to promote appropriate growth and glucose control. Therefore, we recommend involving a registered dietician in the care of patients with CHI to provide guidance for providers and families regarding recommended caloric intake, while taking into account the patient’s prescribed dextrose supplementation, medications, and potential post-pancreatectomy status. Furthermore, we caution caregivers from overtreating hypoglycemia or prophylactically treating perceived hypoglycemia.
Reevaluation of treatment regimen
After initial discharge, we recommend that caregivers check blood glucose levels at home before each feeding and one to two times overnight if continuous feeds or dextrose are utilized. Over time, if blood glucose proves to be stably ≥70 mg/dL (3.9 mmol/L), then blood glucose levels can be checked less frequently but at least once a day after the longest period of fasting. It is imperative that all caregivers of patients with HI have access to an emergency glucagon injection kit should the patients have a severe hypoglycemic episode or become unresponsive. Recurrent blood glucose levels <70 mg/dL (3.9 mmol/L) indicate that the patient’s regimen may need to be adjusted, which may be done at home or in the hospital, depending on each patient’s situation. Close communication and regular follow-up with a pediatric endocrinologist are necessary to determine whether feeding and/or medication regimens need to be adjusted as the child grows.
At our center, we perform annual inpatient evaluations that consist of a safety fast to ensure that the patient’s home regimen is appropriate (Table 4). If the patients require minimal support to maintain normoglycemia, they may be trialed off of their medical therapy with close supervision and then undergo a fasting evaluation (“cure fast”) to assess their fasting tolerance and ability to generate ketones (Table 4). Glucose, insulin, C-peptide, bicarbonate, BOHB, FFAs, and IGFBP-1 are checked at the end of the fast to assess if there is still evidence of hyperinsulinemic hypoglycemia.
Additional evaluations, such as an oral protein tolerance test, may also be performed, with the purpose of further characterizing the patient’s CHI and providing more specific management recommendations. Beta-cells are known to secrete insulin in response to amino acid stimulation, and several genetic causes of CHI result in exacerbated amino acid-stimulated insulin secretion that can trigger hypoglycemia (Figure 1).69,70 We recommend that patients with CHI due to KATP, GDH, or SCHAD mutations who consume a “normal” diet should have an oral protein tolerance test to assess for protein-sensitive hypoglycemia. The test is conducted by administering 1–1.5 g/kg of protein enterally (by mouth, nasogastric tube, or G-tube) and measuring blood glucose and insulin levels every 15 minutes beginning 15 minutes prior to protein administration and continuing for 3 hours after protein administration. Protein-sensitive HI is diagnosed if blood glucose decreases by at least 10 mg/dL (0.56 mmol/L) or to <70 mg/dL (3.9 mmol/L) during the test.69 Patients with such a result should be counseled to always combine protein intake with carbohydrates.
In many cases, CHI improves as the child grows, and in some cases, it spontaneously remits71,72 by mechanisms unknown despite continued insulin secretion.72,73 There is some evidence for increased beta-cell apoptosis in CHI,74 presumably due to increased beta-cell stress and damage related to persistent insulin translation and processing, making it possible that decreased beta-cell mass may play a role. It is also possible that the relative amount of insulin constitutively secreted decreases as the patient grows, diminishing the effect of HI. Along these lines, there is also evidence for decreased insulin sensitivity in older patients with CHI, possibly related to obesity and/or chronic high-carbohydrate intake.75,76 Again, the underlying mutation in these patients impairs glucose-stimulated insulin secretion, so these patients are more susceptible to diabetes because as the glucose level rises, they are unable to increase insulin secretion appropriately.
Screening for complications
As described above, patients on chronic diazoxide and/or octreotide should undergo regular screening for known side effects (Table 7). Post pancreatectomy patients should also have regular screening for related complications, including pancreatic exocrine insufficiency, glucose intolerance, and diabetes. Pancreatic exocrine insufficiency is observed in 50%–72% of patients with diffuse CHI who underwent near-total pancreatectomy.58 Hyperglycemia develops in all, and insulin-dependent diabetes eventually develops in >90% of patients by adolescence, with a sharp increase during early puberty.48,55,58 Hyperglycemia should initially be detected via routine home blood glucose monitoring. For patients with such documented hyperglycemia, we recommend that an oral glucose tolerance test and temporary continuous glucose monitor evaluations be performed. In addition to the loss of beta-cell mass due to pancreatectomy, stress-related beta-cell apoptosis and impaired glucose-stimulated insulin secretion, as discussed above, likely contribute to the rapid development of diabetes that requires insulin therapy. Initial management of glucose intolerance and diabetes in this population can be quite difficult because of their complex beta-cell dysfunction resulting in intermittent hypoglycemia and hyperglycemia. We recommend continuous glucose monitors, oral agents, and insulin pumps when feasible.
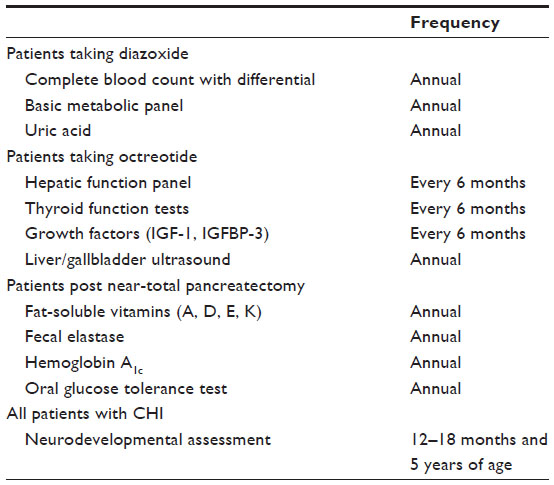 | Table 7 Recommended screening for chronic complications |
Developmental and behavioral considerations
The many complications associated with a near-total pancreatectomy for diffuse HI have led to controversy regarding how far medical therapy should be maximized or whether pancreatectomy should be recommended at all, particularly in light of the fact that CHI naturally improves over time. The concern in not performing pancreatectomies in patients who suffer from the most severe CHI is that they will have detrimental neurodevelopmental outcomes as a result of recurrent and/or severe hypoglycemia. There is strong evidence that patients with CHI have a significantly higher prevalence of developmental delays and disabilities, particularly those with severe CHI.67,77,78 While more long-term follow-up data are needed, it is clear that early severe hypoglycemic events, such as those that typically lead to the initial presentation, can have lasting neurological effects. Early screening, detection, and appropriate management of CHI are essential to prevent such outcomes. We routinely recommend an evaluation by a developmental/behavioral pediatrician at an age of 12–18 months and 5 years.
Conclusion
In summary, HI is a heterogeneous group of disorders with variable presentation, underlying etiology, and effective management strategies. As the most common cause of persistent hypoglycemia in the pediatric population, CHI represents a significant risk. Early detection and intervention are necessary to prevent both acute and chronic complications. Although our understanding of the underlying pathophysiology and genetic causes has increased in recent years, there are likely additional causes or mechanisms that have yet to be identified. Additionally, advances in genetic testing and imaging techniques have allowed for more precise diagnosis of CHI, with implications for more specific treatment. In particular, the distinction of focal vs diffuse CHI based on genetic, radiologic, and intraoperative pathologic diagnoses is very important. However, there are other types of CHI such as that due to HNF1A or HNF4A mutations or in BWS that are less well understood. Furthermore, the treatments for CHI, including diazoxide, octreotide, and partial pancreatectomy, are not without risk. There is certainly a need for further research and development in all of these areas.
We present recommendations for the diagnosis and management of CHI (summarized in Figure 2), based on the experience of a large CHI referral center. This is a complex process, and it remains to some extent an art more than a science, in terms of individualizing therapy for each patient. Successful outcomes depend on close coordination among pediatric endocrinologists, neonatologists, radiologists, pediatric surgeons, pediatric anesthesiologists, and pathologists who have significant experience with CHI. Other subspecialists, such as pediatric cardiologists and geneticists, may also need to be involved. Furthermore, as with other surgical procedures, the surgeon’s volume of experience performing partial pancreatectomies in young children with CHI likely directly correlates with improved outcomes. Therefore, we recommend that patients with suspected and/or confirmed CHI be referred to a CHI center for initial management. Following this initial management, most patients with CHI will continue to require close follow-up by a pediatric endocrinologist, both to monitor the efficacy of their therapeutic regimen and to screen for complications related to the treatments and underlying disease. As we accrue long-term outcomes data, we expect that they will further inform both acute and chronic CHI management.
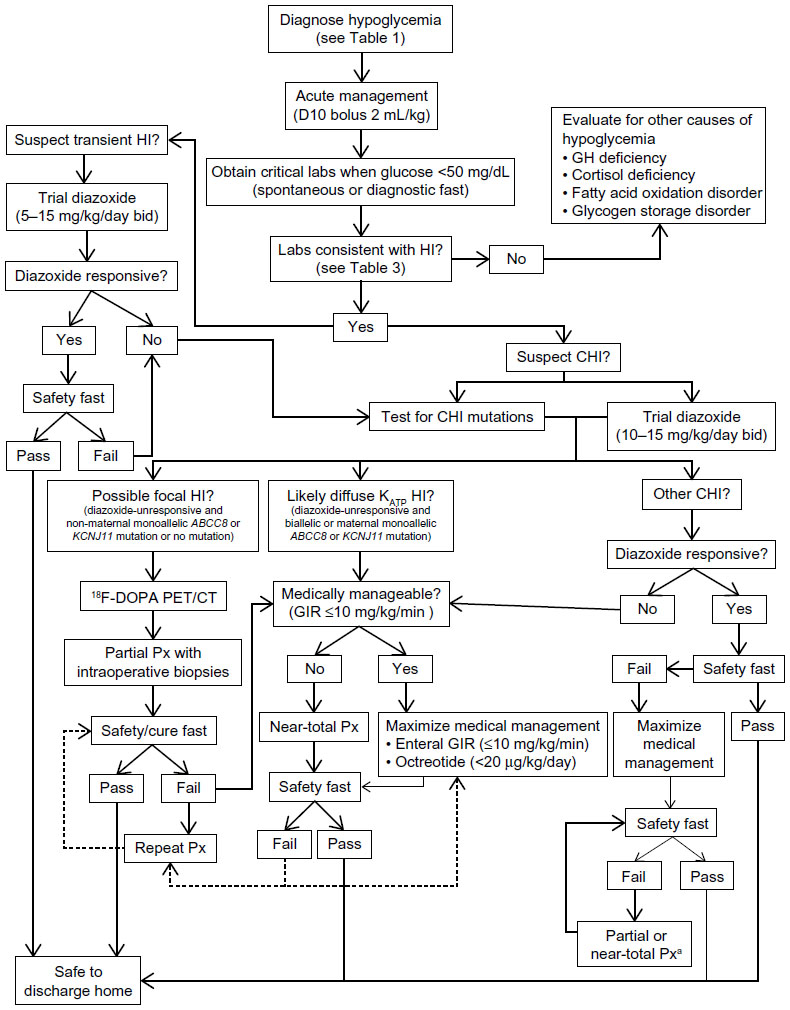 | Figure 2 Diagnostic and management algorithm for HI. |
Acknowledgments
We thank the staff of the Congenital Hyperinsulinism Center at The Children’s Hospital of Philadelphia. Specifically, we thank Dr Charles A Stanley, Dr Diva D De León, Dr Katherine Lord, and Kara E Boodhansingh for their helpful discussions. We also thank the patients and families who have received care at our center.
Disclosure
AM Ackermann is supported by a grant from the Juvenile Diabetes Research Foundation (3-PDF-2014-186-A-N). AA Palladino reports no conflicts of interest in this work.
References
Bruining GJ. Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opin Pediatr. 1990;2(4):758–765. | |
Cryer PE. Hypoglycemia, functional brain failure, and brain death. J Clin Invest. 2007;117(4):868–870. | |
Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab. 2013;98(11):E1786–E1789. | |
De León DD, Stanley CA. Determination of insulin for the diagnosis of hyperinsulinemic hypoglycemia. Best Pract Res Clin Endocrinol Metab. 2013;27(6):763–769. | |
Levitt Katz LE, Satin-Smith MS, Collett-Solberg P, et al. Insulin-like growth factor binding protein-1 levels in the diagnosis of hypoglycemia caused by hyperinsulinism. J Pediatr. 1997;131(2):193–199. | |
Stanley CA, Baker L. Hyperinsulinism in infancy: diagnosis by demonstration of abnormal response to fasting hypoglycemia. Pediatrics. 1976;57(5):702–711. | |
Finegold DN, Stanley CA, Baker L. Glycemic response to glucagon during fasting hypoglycemia: an aid in the diagnosis of hyperinsulinism. J Pediatr. 1980;96(2):257–259. | |
Kelly A, Tang R, Becker S, Stanley CA. Poor specificity of low growth hormone and cortisol levels during fasting hypoglycemia for the diagnoses of growth hormone deficiency and adrenal insufficiency. Pediatrics. 2008;122(3):e522–e528. | |
Tin W. Defining neonatal hypoglycaemia: a continuing debate. Semin Fetal Neonatal Med. 2014;19(1):27–32. | |
Adamkin DH. Newborn CoFa. Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics. 2011;127(3):575–579. | |
Cornblath M, Reisner SH. Blood glucose in the neonate and its clinical significance. N Engl J Med. 1965;273(7):378–381. | |
Lucas A, Morley R, Cole TJ. Adverse neurodevelopmental outcome of moderate neonatal hypoglycaemia. Br Med J. 1988;297(6659):1304–1308. | |
Harris DL, Weston PJ, Harding JE. Incidence of neonatal hypoglycemia in babies identified as at risk. J Pediatr. 2012;161(5):787–791. | |
Maayan-Metzger A, Lubin D, Kuint J. Hypoglycemia rates in the first days of life among term infants born to diabetic mothers. Neonatology. 2009;96(2):80–85. | |
Hoe FM, Thornton PS, Wanner LA, Steinkrauss L, Simmons RA, Stanley CA. Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J Pediatr. 2006;148(2):207–212. | |
Kapoor RR, Locke J, Colclough K, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659–1663. | |
Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97(10):E2026–E2030. | |
James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009;46(5):289–299. | |
Lord K, De Leon DD. Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and management. Int J Pediatr Endocrinol. 2013;2013(1):3. | |
Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008;54(2):256–263. | |
Marquard J, Palladino AA, Stanley CA, Mayatepek E, Meissner T. Rare forms of congenital hyperinsulinism. Semin Pediatr Surg. 2011;20(1):38–44. | |
Stanley CA, De Leon DD. Monogenic Hyperinsulinemic Hypoglycemia Disorders. 1st ed. Basel, Switzerland: Karger; 2012. | |
Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013;168(4):557–564. | |
Snider KE, Becker S, Boyajian L, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98(2):E355–E363. | |
Pinney SE, MacMullen C, Becker S, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant K(ATP)channel mutations. J Clin Invest. 2008;118(8):2877–2886. | |
Flanagan SE, Xie W, Caswell R, et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. Am J Hum Genet. 2013;92(1):131–136. | |
Cederblad F, Ewald U, Gustafsson J. Effect of glucagon on glucose production, lipolysis, and gluconeogenesis in familial hyperinsulinism. Horm Res Paediatr. 1998;50(2):94–98. | |
Miralles RE, Lodha A, Perlman M, Moore AM. Experience with intravenous glucagon infusions as a treatment for resistant neonatal hypoglycemia. Arch Pediatr Adolesc Med. 2002;156(10):999–1004. | |
Sellers EM, Koch-Weser J. Binding of diazoxide and other benzothiadiazines to human albumin. Biochem Pharmacol. 1974;23(3):553–566. | |
Hu S, Xu Z, Yan J, et al. The treatment effect of diazoxide on 44 patients with congenital hyperinsulinism. J Pediatr Endocrinol Metab. 2012; 25(11–12):1119–1122. | |
Yildizdas D, Erdem S, Kucukosmanoglu O, Yilmaz M, Yuksel B. Pulmonary hypertension, heart failure and neutropenia due to diazoxide therapy. Adv Ther. 2008;25(5):515–519. | |
Reubi JC, Schonbrunn A. Illuminating somatostatin analog action at neuroendocrine tumor receptors. Trends Pharmacol Sci. 2013;34(12):676–688. | |
Laje P, Halaby L, Adzick NS, Stanley CA. Necrotizing enterocolitis in neonates receiving octreotide for the management of congenital hyperinsulinism. Pediatr Diabetes. 2010;11(2):142–147. | |
Harris AG. Somatostatin and somatostatin analogues: pharmacokinetics and pharmacodynamic effects. Gut. 1994;35(3 Suppl):S1–S4. | |
Thorton PS, Alter CA. Short- and long-term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr. 1993;123(4):637–643. | |
Demirbilek H, Shah P, Arya VB, et al. Long-term follow-up of children with congenital hyperinsulinism on octreotide therapy. J Clin Endocrinol Metab. 2014;99(10):3660–3667. | |
Yorifuji T, Kawakita R, Hosokawa Y, et al. Efficacy and safety of long-term, continuous subcutaneous octreotide infusion for patients with different subtypes of KATP-channel hyperinsulinism. Clin Endocinol. 2013;78(6):891–897. | |
McCaughan JM. Subtotal pancreatectomy for hyperinsulinism: operative technic. Ann Surg. 1935;101(6):1336–1341. | |
Ternberg JL. Surgical treatment of idiopathic hyperinsulinemic hypoglycemia in children. First successful subtotal pancreatectomy with preservation of spleen reported by Evarts A. Graham (1934). J Thorac Cardiovasc Surg. 1984;88(5 pt 2):898–901. | |
Kassem SA, Ariel I, Thornton PS, et al. p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes. 2001;50(12):2763–2769. | |
Sempoux C, Guiot Y, Dahan K, et al. The focal form of persistent hyperinsulinemic hypoglycemia of infancy: morphological and molecular studies show structural and functional differences with insulinoma. Diabetes. 2003;52(3):784–794. | |
Rahier J, Guiot Y, Sempoux C. Morphologic analysis of focal and diffuse forms of congenital hyperinsulinism. Semin Pediatr Surg. 2011; 20(1):3–12. | |
Suchi M, Thornton PS, Adzick NS, et al. Congenital hyperinsulinism: intraoperative biopsy interpretation can direct the extent of pancreatectomy. Am J Surg Pathol. 2004;28(10):1326–1335. | |
Suchi M, MacMullen CM, Thornton PS, et al. Molecular and immunohistochemical analyses of the focal form of congenital hyperinsulinism. Mod Pathol. 2006;19(1):122–129. | |
Adzick NS, Thornton PS, Stanley CA, Kaye RD, Ruchelli EA. Multidisciplinary approach to the focal form of congenital hyperinsulinism leads to successful treatment by partial pancreatectomy. J Pediatr Surg. 2004;39(3):270–275. | |
Crétolle C, Fékété CN, Jan D, et al. Partial elective pancreatectomy is curative in focal form of permanent hyperinsulinemic hypoglycaemia in infancy: a report of 45 cases from 1983 to 2000. J Pediatr Surg. 2002;37(2):155–158. | |
Giurgea I, Sempoux C, Bellanné-Chantelot C, et al. The Knudson’s two-hit model and timing of somatic mutation may account for the phenotypic diversity of focal congenital hyperinsulinism. J Clin Endocrinol Metab. 2006;91(10):4118–4123. | |
Beltrand J, Caquard M, Arnoux JB, et al. Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care. 2012;35(2):198–203. | |
Lyonnet S, Bonnefont JP, Saudubray JM, Nihoule-Fekete C, Brunelle F. Localisation of focal lesion permitting partial pancreatectomy in infants. Lancet. 1989;334(8664):671. | |
Stanley CA, Thornton PS, Ganguly A, et al. Preoperative evaluation of infants with focal or diffuse congenital Hyperinsulinism by intravenous acute insulin response tests and selective pancreatic arterial calcium stimulation. J Clin Endocrinol Metab. 2004;89(1):288–296. | |
Laje P, States LJ, Zhuang H, et al. Accuracy of PET/CT scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg. 2013;48(2):388–393. | |
Meintjes M, Endozo R, Dickson J, et al. 18F-DOPA PET and enhanced CT imaging for congenital hyperinsulinism: initial UK experience from a technologist’s perspective. Nucl Med Commun. 2013;34(6):601–608. | |
Peranteau WH, Bathaii SM, Pawel B, et al. Multiple ectopic lesions of focal islet adenomatosis identified by positron emission tomography scan in an infant with congenital hyperinsulinism. J Pediatr Surg. 2007;42(1):188–192. | |
von Rohden L, Mohnike K, Mau H, et al. Visualization of the focus in congenital hyperinsulinism by intraoperative sonography. Semin Pediatr Surg. 2011;20(1):28–31. | |
Cherian MP, Abduljabbar MA. Persistent hyperinsulinemic hypoglycemia of infancy (PHHI): long-term outcome following 95% pancreatectomy. J Pediatr Endocrinol Metab. 2005;18(12):1441–1448. | |
Lovvorn HN 3rd, Nance ML, Ferry RJ Jr, et al. Congenital hyperinsulinism and the surgeon: lessons learned over 35 years. J Pediatr Surg. 1999;34(5):786–793. | |
Mahachoklertwattana P, Suprasongsin C, Teeraratkul S, Preeyasombat C. Persistent hyperinsulinemic hypoglycemia of infancy: long-term outcome following subtotal pancreatectomy. J Pediatr Endocrinol Metab. 2000;13(1):37–44. | |
Arya VB, Senniappan S, Demirbilek H, et al. Pancreatic endocrine and exocrine function in children following near-total pancreatectomy for diffuse congenital hyperinsulinism. PLoS One. 2014;9(5):e98054. | |
Kalish JM, Jiang C, Bartolomei MS. Epigenetics and imprinting in human disease. Int J Dev Biol. 2014;58(2–4):291–298. | |
Munns C, Batch J. Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch Dis Child Fetal Neonatal Ed. 2001;84(1):F67–F69. | |
Laje P, Palladino AA, Bhatti TR, States LJ, Stanley CA, Adzick NS. Pancreatic surgery in infants with Beckwith-Wiedemann syndrome and hyperinsulinism. J Pediatr Surg. 2013;48(12):2511–2516. | |
Huang T, Kelly A, Becker SA, Cohen MS, Stanley CA. Hypertrophic cardiomyopathy in neonates with congenital hyperinsulinism. Arch Dis Child Fetal Neonatal Ed. 2013;98(4):F351–F354. | |
Bier DM, Leake RD, Haymond MW, et al. Measurement of “true” glucose production rates in infancy and childhood with 6,6-dideuteroglucose. Diabetes. 1977;26(11):1016–1023. | |
Al-Zubeidi H, Gottschalk ME, Newfield RS. Successful use of long acting octreotide in two cases with Beckwith-Wiedemann syndrome and severe hypoglycemia. Int J Pediatr Endocrinol. 2014;2014(1):18–18. | |
Le Quan Sang KH, Arnoux JB, Mamoune A, et al. Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol. 2012;166(2):333–339. | |
Dalit M-M, Ilana K, Kineret M-A, Orit P-H, Heddy L. Treatment of congenital hyperinsulinism with lanreotide acetate (somatuline autogel). J Clin Endocrinol Metab. 2011;96(8):2312–2317. | |
Mazor-Aronovitch K, Gillis D, Lobel D, et al. Long-term neurodevelopmental outcome in conservatively treated congenital hyperinsulinism. Eur J Endocrinol. 2007;157(4):491–497. | |
Mohnike K, Blankenstein O, Pfuetzner A, et al. Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Res Paediatr. 2008;70(1):59–64. | |
Fourtner SH, Stanley CA, Kelly A. Protein-sensitive hypoglycemia without leucine sensitivity in hyperinsulinism caused by KATP channel mutations. J Pediatr. 2006;149(1):47–52. | |
Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. 2001;138(3):383–389. | |
Glaser B, Hirsch HJ, Landau H. Persistent hyperinsulinemic hypoglycemia of infancy: long-term octreotide treatment without pancreatectomy. J Pediatr. 1993;123(4):644–650. | |
Leibowitz G, Glaser B, Higazi AA, Salameh M, Cerasi E, Landau H. Hyperinsulinemic hypoglycemia of infancy (nesidioblastosis) in clinical remission: high incidence of diabetes mellitus and persistent beta-cell dysfunction at long-term follow-up. J Clin Endocrinol Metab. 1995;80(2):386–392. | |
Grimberg A, Ferry RJ, Kelly A, et al. Dysregulation of insulin secretion in children with congenital hyperinsulinism due to sulfonylurea receptor mutations. Diabetes. 2001;50(2):322–328. | |
Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49(8):1325–1333. | |
Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, et al. Novel de novo mutation in Sulfonylurea Receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes. 2008;57(7):1935–1940. | |
Al-Nassar S, Sakati N, Al-Ashwal A, Bin-Abbas B. Persistent hyperinsulinaemic hypoglycaemia of infancy in 43 children: long-term clinical and surgical follow-up. Asian J Surg. 2006;29(3):207–211. | |
Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003;149(1):43–51. | |
Rother KI, Matsumoto JM, Rasmussen NH, Schwenk WF. Subtotal pancreatectomy for hypoglycemia due to congenital hyperinsulinism: long-term follow-up of neurodevelopmental and pancreatic function. Pediatr Diabetes. 2001;2(3):115–122. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.