Back to Archived Journals » Research and Reports in Neonatology » Volume 4
Management of refractory neonatal seizures
Authors Neubauer D, Soltirovska-Salamon A, Osredkar D, Paro-Panjan D
Received 27 October 2013
Accepted for publication 19 November 2013
Published 26 February 2014 Volume 2014:4 Pages 17—29
DOI https://doi.org/10.2147/RRN.S39714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
David Neubauer,1 Aneta Soltirovska-Salamon,2 Damjan Osredkar,3 Darja Paro-Panjan2
1Department of Child, Adolescent and Developmental Neurology, 2Department of Neonatology, University Children's Hospital Ljubljana, Ljubljana, Slovenia; 3University of Oslo, Oslo, Norway
Abstract: The risk of seizures is highest during the neonatal period. Currently used therapies have limited efficacy and the treatment of neonatal seizures has not significantly changed in recent decades. Controversies still exist as to whether neonatal seizures can cause damage to the developing brain per se, or if the associated neurodevelopmental problems are caused by the underlying condition(s). However, during recent years, there has been substantial progress in the diagnostic possibilities for uncovering the etiologic cause of seizures. This article addresses the most common and important (treatable) etiologic causes of neonatal seizures, and discusses some of the diagnostic tools available. Current therapeutic approaches and their efficacy are discussed, with the aim to present the current knowledge on the topic.
Keywords: neonatal seizures, electroencephalography, etiologic considerations, treatment options
Introduction
Seizures are the most common clinical manifestation of neurologic dysfunction in a newborn.1 Seizures are usually the first (and sometimes only) sign of a central nervous system disorder in a newborn infant.2 Seizures are more common in the neonatal period than at any other age during infancy and childhood. Newborn infants with seizures are at risk for neonatal death and survivors are at risk for neurologic impairment, developmental delay, and post-neonatal epilepsy.
The estimated incidence of seizures is 1–3.5 per 1,000 births in term newborns and even higher in preterm infants. Ronen et al found an overall incidence of 2.6 per 1,000 in a population-based study in Newfoundland, with a much higher incidence in preterm infants compared with their term counterparts (11.1 per 1,000 versus 2 per 1,000), which was even higher in those with birth weight below 2,500 g.3 Similar figures were found in another regional study by Saliba et al, who reported the incidence of neonatal seizures in infants born between1992 and 1994 in Harris County, TX, USA, to be 1.8 per 1,000 live births and 19 per 1,000 for those weighing less than 1,500 g.4 Although the population was quite ethnically diverse in this study, no significant difference was found in incidence according to ethnicity. Lanska and Lanska analyzed National Hospital Discharge Survey data for the period 1980–1991 to derive a nationally representative estimate of neonatal seizures for the US,5 and reported an overall risk for neonatal seizures of 2.84 per 1,000 and a relative risk for low birth weight infants (<2,500 g) of 3.9. The authors compared their results with those of similar studies in developed countries, and reported seizure risks in the range of 1.5–14.2 per 1,000 live births. Sheth et al studied the influence of gestational age on seizures using a cohort of 4,165 neonates admitted to a neonatal intensive care unit.6 They discovered that the seizure rate had a parabolic relationship with gestational age, with the seizure rate being lowest at 30–36 weeks’ gestation (4.8%) and highest at both ends, ie, 11.9% in infants below 30 weeks’ gestation and 14.1% in those above 36 weeks’ gestation. The authors commented also that, not surprisingly, intracranial hemorrhage and hypoxic-ischemic encephalopathy occurred most frequently at the extremes of gestational age.6,7
Neonatal seizures are clinically significant because they are rarely idiopathic. Prompt diagnosis of the underlying condition is important because many of the etiologies have specific treatments which, when applied early, may improve the outcome.8 Neonatal seizures will most probably cause further neuronal compromise, but it is not clear whether this leads to further clinically significant neuronal injury in all cases. Many of the clinical studies examining the relationship between seizure activity and outcome have been biased because the outcome is often associated with the underlying etiology. It is also unclear if an adverse neurodevelopmental outcome, occurring as a consequence of seizures, can be prevented by currently available treatment. Many clinicians are therefore uncertain when to treat seizures and how to assess the adequacy of treatment.9 However, many experimental studies in animals seem to indicate that prolonged (and perhaps also resistant) seizures in the developing brain might have long-lasting sequelae, and many studies have shown the adverse effects on the developing brain.10–13 This is also the reason why some authors consider neonatal seizures to be an epiphenomenon rather than a cause of underlying disease.14 Further, Pisani et al confirmed that age at appearance of prolonged seizures (status epilepticus) is the determining factor for severe neurologic disability and post-neonatal epilepsy only in full-term newborns.15
Seizures in newborns thus frequently signal significant brain pathology, such as hypoxic-ischemic injury, stroke, intracranial infection, hypoglycemia, inborn error of metabolism, or brain malformation. Global cerebral hypoxia-ischemia, focal cerebral hypoxia-ischemia, and intracranial hemorrhage were shown to be the most common causes of seizures in one series, causing nearly two thirds of all cases.16 The same study also showed that, among the infants with a favorable outcome, 41 (64%) were considered to be normal in all areas evaluated, whereas 23 (36%) had neurologic abnormalities without apparent functional impact on examination, while among the infants with overall poor neurologic outcome (28%), 20 had severe and five had moderate neurologic impairment, with a mortality rate of 7%.16 In the cohort reported by Pisani et al, hypoxia-ischemia was present in around 50% of 106 newborns with confirmed neonatal status epilepticus, defined as continuous seizure activity lasting 30 minutes or more or recurrent seizures lasting more than 30 minutes without definite return to the baseline neurologic condition between seizures. In this group, 20 died (19%) and 50 (47%) had an adverse outcome, described as severe neurologic disability and post-neonatal epilepsy, while the rest (36 infants, 34%) had a normal outcome.15 Of 26 newborns who presented with status epilepticus, only one had a normal outcome, while 12 of 22 with post-neonatal epilepsy had status epilepticus at birth. Another study reported a significantly higher incidence of status epilepticus in full-term babies in comparison with preterm ones.17 In one of our unpublished studies, we identified 42 newborns with neonatal seizures of differing etiology during a 3-year period (2010–2012), ten (24%) of whom have had status epilepticus or recurrent seizures. Two children died (5%), while the remaining ten have had post-neonatal epilepsy (compared with only 21% in the rest of the group with neonatal seizures) and severe developmental delay. The etiologies in these ten patients were as follows: hypoxia-ischemia in five (50%), cortical maldevelopment in three (30%), epileptic encephalopathy in one (10%), and confirmed neonatal tuberous sclerosis in one (10%).
In the series reported by Ronen et al, 5% of newborns had prolonged seizures (for more than 30 minutes) and the mortality rate was 9%.3 It seems that mortality and morbidity increase significantly when recurrent neonatal seizures or neonatal status epilepticus are present, and that full-term infants are particularly at risk. Tekgul et al studied only full-term infants and found post-neonatal epilepsy in 35.7% of infants with seizures and global developmental delay in 50.8%.16 In our own cohort from the Department of Neonatology in Ljubljana, Slovenia, we found that 36 of 204 full-term newborns hospitalized in the neonatal department due to neonatal seizures developed post-neonatal epilepsy (18.6%), and 22 of those 36 (61%) have had a poor neurologic outcome (mental developmental index below 60 or cerebral palsy). We also found that early appearance of seizures and/or drug resistance were poor prognostic signs.18,19
The long-term neurodevelopmental prognosis is not only poor for those with electroencephalographic (EEG) and clinical seizures, but also for those who are merely at risk.15,16,20,21 While the mortality due to neonatal seizures has dropped significantly, the long-term neurodevelopmental sequelae remain unchanged.16,22–24 It seems that the most reliable early predictors of a subsequent adverse neurologic outcome are severe encephalopathy (due to hypoxia-ischemia or cerebral dysgenesis),25 need for multiple antiepileptic drugs (AEDs),22,25 presence of moderately to severely abnormal background EEG patterns,26 amount of EEG seizures,27 and being full-term.15Although there are AEDs available to treat neonatal seizures, they are still quite ineffective. The goal of future bench and clinical research should be development of more effective therapies for neonatal seizures, regardless of etiology.28
Clinical versus EEG seizures
A standard operating definition for clinical neonatal seizures is lacking, as pointed out by Shewmon as far back as 1990.29 Despite the proliferation of the literature on neonatal seizures since his statement, this comment is still valid today.30 Further, there is no consensus regarding the management of neonatal seizures.31 Some authors use the following description: “an epileptic seizure in a neonate refers to an abnormal, stereotypical clinical event, that results from an abnormal ‘hypersynchronous’ discharge of neurons, located in the cerebral cortex or thalami, which is most often sudden in onset and paroxysmal or self-limiting”.30 However, clinical judgment is essential, and Mizrahi and Kellaway have tried to characterize and classify neonatal seizures by incorporating EEG information using a portable, crib-side video EEG polygraphic system.32 They found that eleven of 71 newborns with electrical seizure activity had no clinical correlate (subclinical seizures). However, we should also be aware that episodes with and without an EEG correlate can occur in the same neonate, and many newborns may have more than just one type of clinical seizure. There are certain clinical characteristics that strongly suggest an epileptic seizure, while on the other hand, there are many nonepileptic motor events that should not be mistaken for neonatal seizures.33,34
Among the most common nonepileptic motor events is jitteriness, the so-called recurrent tremor,35 which is characterized by tremulousness of all limbs and chin or may involve only a single limb. It may have a pathologic basis and can be a fairly prominent sign in neonates with hypoxia, hypoglycemia, hypocalcemia, or drug withdrawal. Jitteriness can be differentiated from seizures by the fact that it can be stopped with gentle restraint and also by an absence of the features commonly associated with seizures, such as tachycardia and/or apnea.36
The next entity is hyperekplexia, which is an exaggerated startle response to stimuli in the awake state. It is an autosomal dominant condition that can present in a mild or more severe form, where startle is accompanied with generalized stiffness, with no clear-cut precipitating stimulus, and can cause life-threatening apnea during periods of stiffness.30,36,37 The genetic defect is linked to chromosome 5q.
Benign sleep myoclonus refers to often multifocal, migrating limb movements during drowsy and sleep states only, which stops on arousal with painful stimuli and may worsen on light touch, sound, or even AEDs, and can frequently be observed beyond the neonatal period.30,33,34 Benign sleep myoclonus is observed most commonly in the arms, and the movements can occur in rapid succession, causing concern to caregivers.38 Tremor may be fine (high frequency of more than 6 Hz and low amplitude of less than 3 cm) or coarse (lower frequency and higher amplitude); if it is repetitive and recurrent, it is referred to as jitteriness.39
Benign myoclonus of infancy usually starts beyond the neonatal period, but can be found also in neonates. It is characterized by recurrent episodes of nonepileptic myoclonus while awake (and usually not provoked by stimuli) and the neonate is otherwise normal. It has been proposed that this condition also be called “benign nonepileptic infantile spasms” or Fejerman syndrome.34,37,40–42 Recently, it has been suggested that the so-called secondary Fejerman syndrome could be found in children with neurodevelopmental impairment.40,43 It is recommended to use polymyography for determining the duration of these abnormal movements, given that, by definition, cortical myoclonus lasts less than 0.1 seconds, while EEG activity lasting longer than 0.1 seconds and less than 2 seconds is defined as a jerk, and if lasting more than 2 seconds is defined as a sustained contraction. It has been argued that a subcortical myoclonus would then be called a jerk, while a spasm (which is defined as an initial phasic contraction lasting less than 2 seconds followed by a less intense tonic contraction that is 2–10 seconds in duration) might be classified either as a jerk or as a sustained contraction.43,44
Identification of neonatal seizures is critical in the management of newborns at risk. It has been shown that only one third of neonatal EEG seizures are accompanied by clinical signs on simultaneous video recordings, and even these clinical manifestations are often (in two thirds of cases) unrecognized or misinterpreted by experienced neonatal staff.45 We should be aware that so-called electroclinical dissociation or decoupling/uncoupling is an outstanding feature of neonatal seizures, which means that EEG seizures are frequently not accompanied by any identifiable clinical symptom. Moreover, motor phenomena, which have been considered to be seizures, are often not associated with an ictal EEG correlate.45–47 Uncoupling is more frequently found in infants who are treated by phenobarbital or phenytoin.46
Until recently, EEG seizures per se were not considered harmful to the developing brain, and if clinical manifestations accompanying a seizure discharge disappeared after anticonvulsant therapy, treatment was considered successful. However, Boylan et al have clearly shown that EEG seizures often continue after treatment when clinical signs have already subsided, and that babies with severe seizures and a significantly abnormal EEG background often do not respond even to high doses of phenobarbitone (40 mg/kg) and tend to have an adverse outcome.48 They also recommended that babies who do not respond to phenobarbitone within 2 hours should be treated with a second-line anticonvulsant, preferably not phenytoin, which has been proven to be as equally inefficient as phenobarbitone.49 With regard to electroclinical dissociation, the same study also shows the importance of the EEG for monitoring the efficacy of AEDs in neonates, given that we cannot rely on clinical assessment only.48
Detection of neonatal seizures is thus most reliable with continuous EEG monitoring, which requires special expertise but is not always available, or a simplified method of EEG recording, ie, amplitude-integrated EEG (aEEG). aEEG monitors usually consist of two compressed aEEG channels and a raw EEG channel, and have become an accepted way to monitor brain function in neonatal intensive care units.50–52 Using a standard EEG channel together with aEEG channels increased the identification of nonstatus neonatal seizures to 76%.53 Monitoring of the following groups of newborns for possible EEG seizures is recommended: babies with suspected clinical seizures, those exhibiting abnormal movements, those with a known hypoxic-ischemic insult, those with moderate to severe encephalopathy, those undergoing therapeutic hypothermia or any other neuroprotective intervention, and also very preterm babies during their first days of life and those with abnormal cranial ultrasound imaging.45–47,52
The use of continuous aEEG is increasing worldwide.54 Boylan et al showed that 90% of neonatal intensive care units are using either standard EEG or aEEG and that more than 50% are using both. However, the standard EEG is still usually interpreted by a neurophysiologist whereas 80% of aEEGs are interpreted by neonatologists.55 Time to seizure recognition can be significantly reduced if we use the aEEG and the rate of seizure diagnosis of those without EEG confirmation can also be significantly reduced.56
As far back as 20 years ago, Mizrahi and Kellaway32 showed that around two thirds of suspected clinical seizure events were not accompanied by a seizure pattern on EEG, and we are now quite aware of the fact that only the EEG can confirm the epileptic nature of suspicious movements and is thus considered essential in evaluation and confirmation of neonatal seizures.57 It has also been confirmed that neonatal EEG patterns are different from those in older children and adults. These patterns will usually start focally (regardless of whether the brain injury is diffuse or more localized) and gradually evolve in amplitude, duration, and waveform morphology. There is no clear relationship between etiology and location of onset or morphology of the discharges.58 We also know that 78% of all seizures are detected on the C3–C4 channel.59 The usual diagnostic description of the pattern in the neonatal EEG is: “a sudden, repetitive, evolving and stereotyped ictal pattern with a clear beginning, middle and ending and a minimum duration of 5–10 seconds”.57
Finally, we should be aware that even with the more sophisticated tools available nowadays for recognition and confirmation of neonatal seizures, the claim by Mizrahi and Kellaway from more than 20 years ago that clinical judgment is essential still holds.32 Because of the unique nature of neonatal anatomic brain connections and considerable developmental changes in the nature of neurotransmitters, seizures in newborns are quite different from those in older children. Volpe has proposed a clinical classification that is still widely accepted.1 Neonatal seizures are usually focal (but display multifocal characteristics as the seizure develops), brief, and subtle, and because the newborn brain is largely unmyelinated and synapses are still immature, well organized tonic-clonic seizures are extremely rare. While clonic seizures are readily recognized by nursing and/or medical staff clinically as well as electrographically, myoclonic, tonic, and subtle seizures may need repeated EEG tracing to confirm their epileptogenic nature.60 Neonatal seizures can be classified as clonic, myoclonic, tonic, or subtle.36,52,60
Clonic seizures
Clonic seizures are rhythmic movements involving the face, arms, legs, or trunk, either on one side of the body (focal) or multifocal, and shifting rapidly from one side to the other. They typically present with rhythmic jerking at 1–4 Hz and are not stopped by putting the examiner’s hand against the limb. They are most easily identified by clinical observation, and represent 25%–30% of all neonatal seizures. Ictal EEG correlation is a prominent feature, and repetitive spikes are the most common graphoelement. The etiology is various, but these seizures are frequently associated with neonatal stroke, and are frequent in preterm babies with major cerebral pathology.
Myoclonic seizures
Myoclonic seizures resemble clonic movements but are “quicker”, and hence affected newborns appear more “jerky”. The predilection area is the flexor muscles. These seizures can be erratic, fragmentary, or generalized, and represent around 15%–20% of all neonatal seizures. Ictal EEG correlation can be a prominent feature or may be completely absent. The etiology is various, but metabolic causes are found quite frequently.
Tonic seizures
Tonic seizures resemble decerebrate rigidity (generalized, involving both upper and lower extremities in extension) or decorticate posturing (tonic flexion of the arms with extension of the legs). Sometimes just deviation of the head or eyes can be present. Only around 5% of neonatal seizures appear like tonic ones. As with myoclonic seizures, ictal EEG correlation can be a prominent feature or can be completely absent, and sometimes rhythmic delta activity will be present. Etiologically, quite often structural brain abnormalities can be present.
Subtle seizures
Subtle seizures may take the form of horizontal eye deviation or nystagmoid movements, sustained eye opening, blinking, chewing movements of the mouth, repetitive sucking, limb posturing or pedaling movements, vasomotor change, and repetitive apneic spells. The latter can sometimes be difficult (especially in preterm babies where this type is the most common) to distinguish from idiopathic apnea of prematurity; however, such apneas are usually accompanied by bradycardia, whereas apneas presenting as the seizures are not. They are quite common and represent 50%–70% of all neonatal seizures. Ictal EEG correlation may be absent at first, and more than one EEG will be required to show epileptic discharges, and flattening of background activity can sometimes be associated with subtle seizures. The etiology is various, but hypoxic-ischemic encephalopathy is frequently encountered (see Table 1).
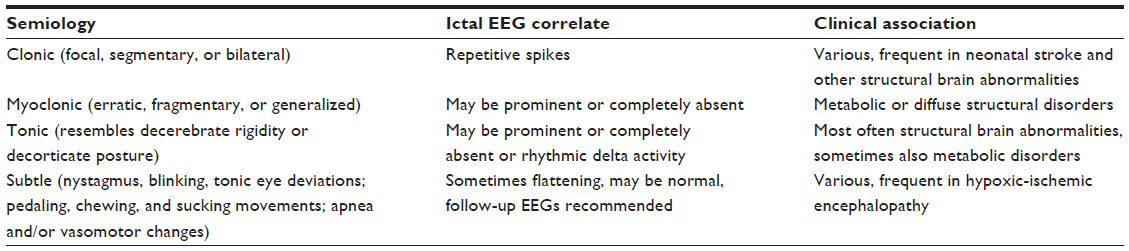 | Table 1 Different seizure types, ictal electroencephalographic patterns, and clinical associations |
Etiologic considerations
Identification of the underlying etiology is also an important issue when considering appropriate management of neonatal seizures. It is presumed that 75%–90% of neonates with seizures will have an underlying etiology.61 Treatment of the underlying etiology is more important than treatment of the seizures themselves.47 The majority of neonatal seizures are attributed to hypoxic-ischemic encephalopathy, which accounts for 35%–70% of all cases. In our study of a small cohort of infants, seizures occurred in only 15% of babies with mild or moderate birth hypoxia, but the percentage was much higher in a study by Tekgul et al.16 The incidence is even more variable, likely as a result of the inconsistent diagnostic criteria used. Seizures appear between 4 and 24 hours after birth.61 During this time, acute cell death and damage by secondary cytotoxins will have maximum impact. Seizures are often accompanied by other signs, such as apathy, hypotonia or hypertonia, absence of the Moro reflex, and bulbar signs, often presenting as difficulties in sucking and swallowing.60
Although hypoxic-ischemic brain injury is often the etiologic cause of seizures, special care should be taken not to overlook some potentially treatable conditions. Among other causes of seizures are infections, intracranial hemorrhage and stroke, congenital malformations and abnormalities of brain development, inborn errors of metabolism (including potentially treatable ones such as pyridoxine-dependent epilepsy, pyridoxal phosphate-dependent epilepsy, folinic acid-responsive seizures, and biotinidase deficiency, see Table 2), mitochondrial encephalomyopathies, transient metabolic disturbances, maternal drug abuse, exposure to toxins (especially lead and mercury), and neurocutaneous syndromes (like tuberous sclerosis, Sturge–Weber syndrome, and incontinentia pigmenti), all of which can present with seizures during neonatal life.
 | Table 2 Treatment options for neonatal seizures* |
We should also include rare neonatal epilepsy syndromes with a presumed underlying etiology and familial neonatal epilepsy syndromes. Among the former are Ohtahara syndrome (early infantile epileptic encephalopathy), which was first described in 1976 by Ohtahara et al,62 and early myoclonic epileptic encephalopathy, first described by Aicardi and Goutieres in 1978.63 These epileptic syndromes have a characteristic clinical presentation (multiple tonic seizures over the course of the day in the former and fragmentary myoclonus and partial seizures in the latter) and EEG findings, burst suppression being common.30,36 Often an underlying brain malformation is noted on imaging and/or manifestations of underlying inborn error of metabolism.63,64 On the other hand, we can identify chromosomal and/or molecular genetic abnormalities in an increasing number of familial (benign) neonatal epilepsy syndromes.65–67 Often we cannot find any clinical abnormalities in these newborns, but not all have a benign prognosis.68,69 Recently, a multicenter study confirmed that SCN2A mutations are an important genetic cause of Ohtahara syndrome.70 The authors concluded that given the wide clinical spectrum associated with SCN2A mutations, genetic testing for SCN2A should be considered for children with different epileptic conditions. Inheritances are usually autosomal dominant, although de novo mutations may occur and mutations may differ between families.30
Neonatal seizures due to a symptomatic cause can be associated with other features, which can give us clues to the etiology, like fever or low body temperature (infection), different metabolic and/or hepatic derangements (inborn error of metabolism), dysmorphic features (abnormal brain development), skin lesions (neurocutaneous syndromes), or clear focal signs (stroke, focal encephalitis [herpes]).30 It should also be mentioned that acute-stage EEG abnormalities, characterized by suppression of background, are often observed in infants with neonatal seizures of acute symptomatic etiology.47,71 In contrast, chronic-stage EEG abnormalities, characterized by abnormal morphology of background activity, are frequently seen in infants with a remote symptomatic origin of their seizures, while a normal EEG is frequently encountered in newborns with benign familial or nonfamilial seizures.72
Treatment options
When considering the treatment of neonatal seizures, we must always consider two aspects, ie, treatment of the seizures themselves and treatment of a possible underlying etiology.47,60 Neonatal seizures are an emergency because their adverse effects may affect the long-term outcome.15,27 Before starting antiepileptic (anticonvulsive) treatment, one should consider seizures that could be associated with an acute metabolic disorder, central nervous system or systemic infection, and above all, treatable inborn errors of metabolism, given that inappropriate treatment of the underlying cause will result in failure of treatment with AEDs. The so-called group of vitamin-responsive seizures constitute a subgroup peculiar to the neonatal period. In therapy-resistant neonatal seizures or status epilepticus, 100 mg of pyridoxine or, if ineffective, 500 mg, should be given intravenously during EEG monitoring, and should be continued intravenously or orally with 30 mg/kg/day divided into two or three single doses over 3 consecutive days.36,60
Plecko advocates so-called second-line investigations to rule out possible rare etiologies of vitamin-responsive neonatal seizures in newborns with seizures that are repeated or refractory to the first-line AEDs in addition to normal first-line investigations (including blood sugar, electrolytes, blood gases, lactate, liver enzymes, creatine kinase, ammonia, procalcitonin and C-reactive protein, lumbar puncture, and cranial ultrasound).60 Second-line investigations include more sophisticated neuroimaging techniques (different modes of magnetic resonance), metabolic investigations (plasma, urine, and cerebrospinal fluid to be stored at −20°C for later work-up), and cofactor trials. The first in the line is the above-mentioned pyridoxine, followed by trials of pyridoxal-5′-phosphate (PLP) 30–50 mg/kg/day in 3–4 single doses over 3 days and then folinic acid 5 mg/kg in 2–3 single doses over 3 days.60,73 One should also be aware that about 30% of patients with pyridoxine-dependent epilepsy have a history of complicated delivery, and in addition to resistance to therapy, signs like erratic myoclonus (similar to drug withdrawal syndrome), irritability, restlessness, and sleepiness should alert the neonatologist to such a possibility.74 In PLP-dependent epilepsy, preterm birth and fetal distress are common as well as therapy-resistant myoclonic seizures with a suppression-burst EEG pattern; however, even early treatment seems to be associated with a poor outcome, in contrast with pyridoxine-dependent epilepsy where the outcome (apart from the need for long-life treatment) is rather good. Very low PLP concentrations in cerebrospinal fluid are the only reliable biomarker, and would need collection and preservation by deep-freezing before treatment. Today we know that pyridoxine-dependent seizures are caused by an autosomal recessive defect of the antiquitin gene, and pipecolic acid and alpha-aminoadipic semialdehyde in plasma, urine, and cerebrospinal fluid are reliable diagnostic biomarkers.74,75
In biotinidase deficiency, seizures can quite often be the presenting symptom (being myoclonic and therapy-resistant), while other symptoms include hypotonia and breathing problems. Measurement of biotinidase activity in serum will establish the diagnosis, and treatment with biotin (vitamin H) 5–10 mg/day leads to complete resolution of symptoms. There are some other rare metabolic conditions which should be thought of in therapy-resistant cases, like molybdenum cofactor deficiency and sulfite oxidase deficiency. Rare cases of neonatal GLUT1 deficiency can sometimes be found; seizures, often multiple types, start during the neonatal period, but diagnosis is delayed and the biomarker is a low cerebrospinal fluid/blood glucose ratio (below 0.5). The majority have mutations in the SLC2A1 gene and respond to the ketogenic diet.76
We should bear in mind that neonatal seizures can be refractory to anticonvulsive drugs, especially when the seizures are symptomatic (as in cerebral dysgenesis or intraventricular hemorrhage), and that hypoxic/ischemic encephalopathy is the most common cause of refractory neonatal seizures.16 After isolating potentially treatable metabolic and infectious causes, we should concentrate on refractory seizures due to asphyxia, where early treatment (due to the short course of seizures and poor prognosis) is essential.77 While neonatal mortality has lowered (being less than 20%), lifelong consequences after prolonged neonatal seizures are still seen in one third to nearly one half of survivors.16,25 The most troublesome are cognitive deficits, which range from learning problems to developmental delay and intellectual disability as well as post-neonatal epilepsy.25 Current practice in such cases includes early treatment with phenobarbitone up to 40 mg/kg, phenytoin or fosphenytoin up to 20 mg/kg, benzodiazepines (for example, lorazepam), and newer anticonvulsants, such as lidocaine, topiramate, and levetiracetam (see Table 2).77 However, it is not known how long to continue treatment or how the length of treatment impacts the outcome. In addition to pharmacologic treatment, newborns with hypoxic/ischemic encephalopathy have been increasingly treated with therapeutic hypothermia (for whole-body hypothermia, the current practice is to decrease core body temperature to 33.5°C for 72 hours).78 Recent clinical studies have shown that early whole-body or limited cranial hypothermia improves the neurologic outcome in treated newborns; however, the effect of hypothermia on the incidence and severity of neonatal seizures is yet to be determined.77–81
First-line and second-line AEDs
Phenobarbitone is still the AED of first choice in over 80% of neonatal units worldwide, despite concerns about its adverse effects on the developing brain.54,58,82,83 In a recent study, a high level of agreement concerning initiation of treatment of neonatal seizures as well as first-choice of treatment (phenobarbital) was found among neonatologists and child neurologists in Sweden.84 Most centers use a loading dose of 20 mg/kg, and if seizures persist, additional doses are given (maximum total dose 40 mg/kg), but significant side effects are more likely to be experienced at higher doses and higher phenobarbital levels in blood. Nevertheless, there are limited studies demonstrating good efficacy of phenobarbital. The response rate is usually below 50% in situations where more than the loading dose of 20 mg/kg is needed.85 Other treatment options within the first-line AEDs, depending on the clinical situation, include the following: phenytoin (phosphenytoin is better in neonates because it is usually associated with fewer side effects), benzodiazepines (midazolam, lorazepam, and clonazepam, not necessarily in this order), and lidocaine (recently having the “honor” to be placed within this category, despite some pros and cons).86–90 Painter et al reported the effect of phenobarbital compared with phenytoin in a randomized crossover study.49 Seizures were controlled in 43% of newborns in the phenobarbital-treated group compared with 45% in the phenytoin-treated group; addition of the other drug when seizures were not controlled with the first drug did not significantly improve seizure control. Despite this fact, Co et al advocate phenytoin (especially in developing countries) as the second choice and lorazepam as an additional option.91 Because of its unique pharmacokinetics, phenytoin (phosphenytoin) is recommended only for acute treatment and generally not for long-term maintenance. The recommended dosage of phenytoin is 15–20 mg/kg (or the equivalent active dose of phosphenytoin).92 Benzodiazepines are also used in cases refractory to phenobarbital (and/or phenytoin), as we tend to term seizures refractory or intractable when they fail to respond to adequate doses (and levels) of two AEDs. The efficacy of benzodiazepines is reported to be between 0% and 100%; however, the most often cited response rates are 67% and 80%, respectively.92–94 Cost, availability, and individual clinical situations determine the choice. Lorazepam has a longer half-life than diazepam or midazolam, so administration is less frequent and dosing is intermittent (cumulative exposure to toxic excipients is thereby minimized). This can be the reason why lorazepam might be the preferred benzodiazepine in most cases. Midazolam is short-acting and has a more rapid onset of action, the disadvantage being its metabolic pathway, via which pharmacologically active metabolites are formed. Clonazepam has also been effective in newborns unresponsive to phenobarbitone and phenytoin. In one study, two patients who failed phenobarbital (more than 30 mg/kg) and phenytoin (15–20 mg/kg) had seizure cessation within 120 minutes of administration of clonazepam.95 The benzodiazepines doses are as follows:
- lorazepam, loading dose 0.05–0.15 mg/kg in 5 minutes, without maintenance doses (but loading dose can be repeated)57,96,97
- midazolam, loading dose 0.05–0.2 mg/kg in 10 minutes, followed by 0.1–0.5 mg/kg/hour (maximum 1.0 mg/kg/hour) in increasing steps,92,98 while side effects (especially with maximum doses) are decreased consciousness and hypotension with multiorgan failure92,97
- clonazepam, loading dose by 0.1 mg/kg bolus, followed by 0.01 mg/kg in repeated doses up to five times within 24 hours.54,57,99
Although not widely used, several studies indicate that lidocaine is an effective drug for refractory seizures, and a recent study showed moderate efficacy as second-line therapy following benzodiazepines.57,90 For term neonates undergoing hypothermia treatment, the initial bolus loading dose of 2.0 mg/kg is given followed by continuous infusions of 7 mg/kg/hour for 3.5 hours, 3.5 mg/kg/hours for 12 hours, and 1.75 mg/kg/hour for 12 hours, then stopping.87 During normothermia, the regimen is slightly changed as follows: 2.0 mg/kg in 10 minutes, followed by infusions of 7.0 mg/kg/hour over 4 hours, and 3.5 mg/kg/hour for the next 12 hours, and 1.75 mg/kg/hour for the last 12 hours (completed within 28 hours).54
Third-line and second-generation AEDs
Levetiracetam seems to be an effective and well tolerated adjunctive AED for seizure control in newborns.100 In one study, it was observed that seizure control was achieved in one hour in 86% of neonates treated with levetiracetam intravenously.101 Levetiracetam can be given intravenously as a loading dose of 20–50 mg/kg, followed by an initial maintenance dose of 5 mg/kg, which may be increased if required.57 However, in different studies, the doses have ranged from 10 mg to 115 mg/kg daily, so many authors believe that more systematic studies of levetiracetam the same holds true for topiramate are needed.30
Topiramate is an AED with multiple mechanisms of action, including glutamate receptor inhibition and sodium blockade.102 In one trial, topiramate was given to 13 term newborns via orogastric tube at a dose of 5 mg/kg once per day for the first 3 days of life.103 A randomized controlled trial in infants beyond one month of age suggested that oral topiramate in doses up to 25 mg/kg/day may not be effective as add-on AED therapy.104 A recent study in adults with epilepsy or migraine has yielded promising results with the use of intravenous topiramate, and controlled clinical trials should be designed to determine the efficacy and safety of intravenous topiramate also for neonatal seizures.105 Valproic acid is an effective AED for recurrent seizures and status epilepticus.30 However, it should be used with extreme caution because of a relatively high risk of fatal hepatotoxicity in neonates, especially with polytherapy and/or if an underlying inborn error of metabolism is suspected. Limited pharmacokinetic data are currently available to guide appropriate intravenous dosages; initial loading doses studied have been in the range of 20–25 mg/kg (orally), 20–30 mg/kg (rectally), and 10–25 mg/kg (intravenously), while maintenance dosages have been in the range of 5–10 mg/kg per dose (orally).106 A single study reported the pharmacokinetics of vigabatrin in neonates with uncontrolled seizures.107 Peripheral vision loss is an adverse effect that has been reported in around 15% in children,108 and because the risk of vision loss cannot be tested in newborns, vigabatrin should not be used as an option to treat neonatal convulsions.57 Lamotrigine is well tolerated in children, but evidence of its clinical application and efficacy in neonates is very limited. In one published case report, the drug was used at 4.4 mg/kg as a single daily dose as add-on therapy and was followed by rapid and sustained control of seizures in a newborn.109
Drugs rarely used for neonatal seizures
Carbamazepine prevents repetitive firing of action potentials in depolarized neurons by blocking frequency, use, and voltage-dependent sodium channels.102 In neonates, there are limitations to the use of carbamazepine due to a lack of parenteral formulations as well as low activity of the cytochrome P450 3A4 isoenzyme at birth, but an “ideal” carbamazepine dose for neonates would be a loading dose of 5–20 mg/kg and maintenance doses of 5–8 mg/kg per dose, administered every 8–12 hours.110
Paraldehyde has been used historically for control of refractory seizures, but its exact mechanism of action is unclear. In one trial of intravenous paraldehyde in the treatment of neonatal seizures, it was given as an infusion at a rate of 150 mg/kg/hour for 3 hours.111 Pharmacokinetic data for intravenous paraldehyde are limited, and there are no data available regarding its rectal use (as it is used in older children) to guide dosing.112 Bumetanide is still an experimental drug, but has been found to augment the effectiveness of phenobarbital (decreased seizure frequency and duration) in rat hippocampal slices.113 Bumetanide dosages for neonatal seizure control are currently under investigation in NEMO1 (NEonatal seizure using Medication Off-patent), a multicenter pan-European clinical trial with the aim of developing new strategies for the treatment of neonatal seizures using this loop diuretic. There is evidence that bumetanide improves the GABAergic function of phenobarbitone, the current standard drug. Bumetanide has been used as a diuretic in term and preterm babies for around 30 years. This trial should confirm if bumetanide in addition to standard treatment results in better seizure control.114 Felbamate inhibits the N-methyl-D-aspartate receptor and potentiates GABAergic action by interacting with the strychnine-insensitive glycine recognition site of the N-methyl-D-aspartate receptor.115 It is only available as an oral preparation and has slow absorption, so oral felbamate is probably unsuitable for reaching adequate neuroprotective levels in a timely fashion in newborns with hypoxic-ischemic encephalopathy.116 Dexamethasone is rarely mentioned as an add-on treatment for neonatal seizures; however, some authors do recommend it in their algorithms.117,118 The dosage is 0.6–2.8 mg/kg intravenously and is usually divided into four daily doses given for 3–10 consecutive days118 (see Table 2).
Concluding remarks
Once the diagnosis of refractory neonatal seizures and/or HIE is made and treatment has been more or less successful, the major focus should be on the parents’ concerns regarding the expected shorter-term and long-term outcome, which will definitely influence the child’s and family’s future life and challenge the family’s ability to cope with the child’s problems, as well as show the adequacy of society to meet his/hers special needs.119 The prognosis is highly unpredictable and seizures may recur at any time. Therefore, the parents should be well prepared and instructed not only regarding their child’s seizure medication, but also to recognize any unusual symptoms in their child which could possibly reflect seizure relapse. They should be very well instructed on how to reach the nearest emergency department, as well as whom to contact in case of an emergency. Regular clinical follow-up over time will enable the prognosis to be estimated appropriately and any retarded/disrupted development trends to be recognized as soon as possible and managed accordingly. The parents should also be told that any estimate of long-term prognosis made before one year of an infant’s corrected age has a wide margin of error.119 Attention must also be paid to less severe forms of disability, such as minor motor impairment, attention deficit-hyperactivity disorder, perceptual-motor and/or cognitive difficulties, as well as the major ones, such as autism.119–121
Finally, we must be aware of the fact that for more than 50 years phenobarbitone (which was introduced in 1914) and phenytoin (introduced in 1938) were the only AEDs used during the neonatal period. We cannot attribute this endurance to their efficacy but rather to their availability in parenteral formulations.122 During the past decade, a substantial number of newer AEDs have become available to treat neonatal seizures, and progress has been made in the diagnosis of neonatal seizures. However, the AEDs currently used to treat neonatal seizures are relatively ineffective, but their effectiveness appears to improve under hypothermia therapy.57 Therefore, there is an urgent need for prospective, randomized, controlled trials of some newer adult and pediatric AEDs. It seems that development of safe AMPA antagonists (such as topiramate) that can also be used for intravenous application will be a priority in clinical trials in the future. However as de Vries and Toet, so nicely put it in one of their recent articles: “Once effective AEDs are available, it will become feasible to answer the last important question: whether treatment of electrographic seizures has a beneficial effect on postneonatal epilepsy and neurocognitive outcome.”22
Disclosure
The authors report no conflict of interest in this work.
References
Volpe JJ. Neonatal seizures. In: Volpe JJ, editor. Neurology of the Newborn. Philadelphia, PA, USA: WB Saunders; 2001. | |
Glass HC, Glidden D, Jeremy RJ, Barkovich AJ, Ferriero DM, Miller SP. Clinical neonatal seizures are independently associated with outcome in infants at risk for hypoxic ischemic brain injury. J Pediatr. 2009;155: 318–323. | |
Ronen GM, Penney S, Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr. 1999;134:71–75. | |
Saliba RM, Annegers JF, Waller DK, Tyson JE, Mizrahi EM. Incidence of neonatal seizures in Harris County, Texas, 1992–1994. Am J Epidemiol. 1999;150:763–769. | |
Lanska MJ, Lanska DJ. Neonatal seizures in the United States: results of the National Hospital Discharge Survey, 1980–1991. Neuroepidemiology. 1996;15:117–125. | |
Sheth RD, Hobbs GR, Mullett M. Neonatal seizures: incidence, onset and etiology by gestational age. J Perinatol. 1999;19:40–43. | |
Camfield PR, Camfield CS. Neonatal seizures: a commentary on selected aspects. J Child Neurol. 1987;2:244–251. | |
Evans D, Levene M. Neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 1998;78:70–75. | |
Massingdale TW, Buttross S. Survey of treatment practices for neonatal seizures. J Perinatol. 1993;13:107–210. | |
De Lorenzo RJ, Pellock JM, Towne AR, Boggs JG. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316–325. | |
Holmes GL, Ben-Ari Y. Effects of seizures on developing processes in the immature brain. Lancet Neurol. 2006;5:1055–1063. | |
Jensen FE. Acute and chronic effects of seizures in the developing brain: experimental models. Epilepsia. 1999;40:S51–S58. | |
Holmes GL. Effects of seizures on brain development: lessons from the laboratory. Pediatr Neurol. 2005;33:1–11. | |
Scher MS. Neonatal seizures and brain damage. Pediatr Neurol. 2003;29:381–390. | |
Pisani F, Fusco C, Sisti L. Neonatal status epilepticus vs recurrent neonatal seizures. Neurology. 2007;69:2177–2185. | |
Tekgul H, Gauvreau K, Soul J, et al. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics. 2006;117:1270–1280. | |
Scher MS, Hamid MY, Steppe DA, Beggarly ME, Painter MJ. Ictal and interictal electrographic seizure durations in preterm and term neonates. Epilepsia. 1993;34:284–288. | |
ParoPanjan D, Neubauer D. Electroclinical characteristics and late outcome of neonatal seizures. In: Velic?kovic´Perat M, editor. New Developments in Child Neurology. Presented at the VIII International Child Neurology Congress, Ljubljana, Slovenia, September 13–17, 1998. | |
Danojevic S, Salamon Soltirovska A, Neubauer D, Paro-Panjan D. Risk factors for postneonatal epilepsy in a group of children with neonatal seizures. Eur J Pediatr. 2009;13 Suppl 1:S29–S30. | |
Bye AM, Cunningham CA, Chee KY, Flanagan D. Outcome of neonates with electrographically identified seizures, or at risk of seizures. Pediatr Neurol. 1997;16:225–231. | |
Legido A, Clancy RR, Berman PH. Neurologic outcome after electroencephalographically proven neonatal seizures. Pediatrics. 1991;88:583–396. | |
Toet MC, De Vries LS. Seizures in the term newborn infant. In: Shevell M, Miller SP, editors. Acquired Brain Injury in the Fetus and Newborn. London, UK: Mac Keith Press; 2012. | |
Bergman I, Painter MJ, Hirsch RP, Crumrine PK, David R. Outcome in neonates with convulsions treated in an intensive care unit. Ann Neurol. 1983;14:642–647. | |
Lombroso CT. Prognosis in neonatal seizures. Adv Neurol. 1983;34: 101–113. | |
Ronen GM, Buckley D, Penney S, Streiner DL. Long-term prognosis in children with neonatal seizures. Neurology. 2007;69:1816–1822. | |
Laroia N, Guillet R, Burchfiel J, McBride MC. EEG Background as predictor of electrographic seizures in high-risk neonates. Epilepsia. 1998;39:545–551. | |
McBride MC, Laroia N, Guillet R. Electrographic seizures in neonates correlate with poor neurodevelopmental outcome? Neurology. 2000;55:506–513. | |
Zupanc ML. Neonatal seizures. Pediatr Clin North Am. 2004;51: 961–978. | |
Shewmon DA. What is a neonatal seizure? Problems in definition and quantification for investigative and clinical purposes. J Clin Neurophysiol. 1990;7:315–368. | |
Seshia SS, Huntsman RJ, Lowry NJ, Seshia M, Yager JY, Sankaran K. Neonatal seizures: diagnosis and management. Zhongguo Dang Dai Er Ke Za Zhi. 2011;13:81–100. | |
Expert Committee on Pediatric Epilepsy, Indian Academy of Pediatrics. Guidelines for diagnosis and management of childhood epilepsy. Indian Pediatr. 2009;46:681–698. | |
Mizrahi EM, Kellaway P. Characterization and classification of neonatal seizures. Neurology. 1987;37:1837–1844. | |
Hrastovec A, Hostnik T, Neubauer D. Benign convulsion in newborns and infants: occurrence, clinical course and prognosis. Eur J Paediatr Neurol. 2012;16:64–73. | |
Paro-Panjan D, Neubauer D. Benign neonatal sleep myoclonus: experience from the study of 38 infants. Eur J Paediatr Neurol. 2008;12:14–18. | |
Parker S, Zuckerman B, Bauchner H, Frank D, Vinci R, Cabral H. Jitteriness in full term neonates: prevalence and correlates. Pediatrics. 1990;85:17–23. | |
Sivaswamy L. Approach to neonatal seizures. Clin Pediatr. 2012;51: 415–425. | |
King MD, Stephenson JBP. Epileptic and non-epileptic disorders together. In: King MD, Stephenson JBP, editors. A Handbook of Neurological Investigations in Children. London, UK: Mac Keith Press; 2009. | |
Maurer VO, Ritzi M, Bianchetti MG, Ramelli GP. Benign neonatal sleep myoclonus: a review of the literature. Pediatrics. 2010;125: e919–e924. | |
Huntsman RJ, Lowry NJ, Sankaran K. Nonepileptic motor phenomena in the neonate. Paediatr Child Health. 2008;13:680–684. | |
Canavese C, Canafoglia L, Costa C, et al. Paroxysmal non-epileptic motor events in childhood: a clinical and video-EEG-polymyographic study. Dev Med Child Neurol. 2012;54:334–338. | |
Lombroso CT, Fejerman N. Benign myoclonus of early infancy. Ann Neurol. 1977;1:138–143. | |
Dalla Bernardina B. Benign myoclonus of early infancy or Fejerman syndrome. Epilepsia. 2009;50:1176–1183. | |
Stephenson JBP. Paroxysmal non-epileptic motor events in childhood. Dev Med Child Neurol. 2012;54:299–300. | |
Kellaway P, Hrachovy RA, Frost JD Jr, Zion T. Precise characterization and quantification of infantile spasms. Ann Neurol. 1979;6:214–218. | |
Murray DM, Boylan GB, Ali I, Ryan CA, Murphy BP, Connolly S. Defining the gap between electrographic seizure burden, clinical expression and staff recognition of neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 2008;93:F187–F191. | |
Scher MS, Alvin J, Gaus L, Minnigh B, Painter MJ. Uncoupling of EEG-clinical neonatal seizures after antiepileptic drug use. Pediatr Neurol. 2003;28:277–280. | |
Okumura A. The diagnosis and treatment of neonatal seizures. Chang Gung Med J. 2012;35:365–372. | |
Boylan GB, Rennie JM, Pressler RM, Wilson G, Morton M, Binnie CD. Phenobarbitone, neonatal seizures, and video-EEG. Arch Dis Child Fetal Neonatal Ed. 2002;86:F165–F170. | |
Painter MJ, Scher MS, Stein AD, et al. Phenobarbital compared with phenytoin for treatment of neonatal seizures. N Engl J Med. 1999;341: 485–489. | |
Neubauer D, Osredkar D, Paro-Panjan D, Skofljanec A, Derganc M. Recording conventional and amplitude-integrated EEG in neonatal intensive care unit. Eur J Paediatr Neurol. 2011;15:405–416. | |
Boylan GB, Stevenson NJ, Vanhatalo S. Monitoring neonatal seizures. Semin Fetal Neonatal Med. 2013;18:202–208. | |
Shah DK, Boylan GB, Rennie JM. Monitoring seizures in the newborn. Arch Dis Child Fetal Neonatal Ed. 2012;97:F65–F69. | |
Shah DK, Mackay MT, Lavery S, et al. Accuracy of bedside electroencephalographic monitoring in comparison with simultaneous continuous conventional electroencephalography for seizure detection in term infants. Pediatrics. 2008;121:1146–1154. | |
van Rooij LG, Hellström-Westas L, de Vries LS. Treatment of neonatal seizures. Semin Fetal Neonatal Med. 2013;4:1–7. | |
Boylan G, Burgoyne L, Moore C, O’Flaherty B, Rennie J. An international survey of EEG use in the neonatal intensive care unit. Acta Paediatr. 2010;99:1150–1155. | |
Shellhaas RA, Barks AK. Impact of amplitude-integrated electroencephalograms on clinical care for neonates with seizures. Pediatr Neurol. 2012;46:32–35. | |
van Rooij LG, van den Broek MP, Rademaker CM, de Vries LS. Clinical management of seizures in newborns: diagnosis and treatment. Pediatr Drugs. 2013;15:9–18. | |
Clancy RR, Legido A. The exact ictal and interictal duration of electroencephalographic neonatal seizures. Epilepsia. 1987;28: 537–541. | |
Shellhaas RA, Clancy RR. Characterization of neonatal seizures by conventional EEG and single-channel EEG. Clin Neurophysiol. 2007;118: 2156–2161. | |
Plecko B. Neonatal seizures, including metabolic epileptic encephalopathies. In: Kennedy C, editor. Principles and Practice of Child Neurology in Infancy. London, UK: Mac Keith Press; 2012. | |
Perlman JM. Brain injury in the term infant. Semin Perinatol. 2004;28: 415–424. | |
Ohtahara S, Ishida T, Oka E, et al. [On the specific age-dependent epilepsy syndromes: the early infantile epileptic encephalopathy with suppression burst]. No To Hattatsu. 1976;8:270–280. Japanese. | |
Aicardi J, Goutieres F. [Neonatal myoclonic encephalopathy]. Rev Electroencephalogr Neurophysiol Clin. 1978;8:99–101. French. | |
Prasad AN, Hoffmann GF. Early onset epilepsy and inherited metabolic disorders: diagnosis and management. Can J Neurol Sci. 2010;37: 350–358. | |
Heron SE, Cox K, Grinton BE, et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet. 2007;44:791–796. | |
Herlenius E, Heron SE, Grinton BE, et al. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia. 2007;48:1138–1142. | |
Fister P, Soltirovska-Salamon A, Debeljak M, Paro-Panjan D. Benign familial neonatal convulsions caused by mutation in KCNQ3, exon 6: a European case. Eur J Paediatr Neurol. 2013;17:308–310. | |
Heron SE, Hernandez M, Edwards C, et al. Neonatal seizures and long QT syndrome: a cardiocerebral channelopathy? Epilepsia. 2010;51: 293–296. | |
Heron SE, Scheffer IE, Grinton BE, et al. Familial neonatal seizures with intellectual disability caused by a microduplication of chromosome 2q24.3 Epilepsia. 2010;51:1865–1869. | |
Nakamura K, Kato M, Osaka H, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;8:992–998. | |
Hayakawa F, Okumura A, Kato T, Kuno K, Watanabe K. Determination of timing of brain injury in preterm infants with periventricular leucomalacia with serial neonatal electroencephalography. Pediatrics. 1999;104:1077–1081. | |
Watanabe K, Hayakawa F, Okumura A. Neonatal EG: a powerful tool in assessment of brain damage in preterm infants. Brain Dev. 1999;21: 361–372. | |
Gospe SM. Neonatal vitamin-responsive epileptic encephalopathies. Chang Guang Med J. 2010;33:1–12. | |
Plecko B, Stockler S. Vitamin B6 dependent seizures. Can J Neurol Sci. 2009;36 Suppl 2:S73–S77. | |
Stockler-Ipsiroglu S, Plecko B. Metabolic epilepsies: approaches to a diagnostic challenge. Can J Neurol Sci. 2009;36 Suppl 2:S67–S72. | |
Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010;133:655–670. | |
Jensen FE. Neonatal seizures: an update on mechanisms and management. Clin Perinatol. 2009;36:881–898. | |
Shankaran S, Laptoook AR, Ehrenkranz RA, et al; National Institute of Child Health and Human Development Neonatal Research Network. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574–1584. | |
Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663–670. | |
Jacobs S, Hunt R, Tarnow-Mordi W, et al. Cooling for newborns with hypoxic-ischaemic encephalopathy. Cochrane Database Syst Rev. 2007;4:CD003311. | |
Azzopardi D, Brocklehurst P, Edwards D, et al. The TOBY study. Whole body hypothermia for the treatment of perinatal asphyxia encephalopathy: a randomized controlled trial. BMC Pediatr. 2008;8: 17–21. | |
Clancy RR. Summary proceedings from the neurology group on neonatal seizures. Pediatrics. 2006;117 Suppl 2:S23–S27. | |
Vento M, de Vries LS, Alberola A, et al. Approach to seizures in the neonatal period: a European perspective. Acta Paediatr. 2010;99: 497–501. | |
Wickström R, Hallberg B, Bartocci M. Differing attitudes toward phenobarbital use in the neonatal period among neonatologists and child neurologists in Sweden. Eur J Paediatr Neurol. 2013;17:55–63. | |
Gilman JT, Gal P, Duchowny MS, Weaver PL, Ransom JL. Rapid sequential phenobarbital treatment of neonatal seizures. Pediatrics. 1989;83:674–678. | |
Painter MJ, Pippenger C, MacDonald H, Pitlick W. Phenobarbital and diphenylhydantoin levels in neonates with seizures. J Pediatr. 1987;92: 315–319. | |
van den Broek MP, Rademaker CM, van Straaten HL, et al. Anticonvulsant treatment of asphyxiated newborns under hypothermia with lidocaine: efficacy, safety and dosing. Arch Dis Child Fetal Neonatal Ed. 2013;98:F341–F345. | |
Lundqvist M, Agren J, Hellstrom-Westas L, Flink R, Wickstrom R. Efficacy and safety of lidocaine for treatment of neonatal seizures. Acta Paediatr. 2013;102:863–867. | |
Hakan N, Aydin M, Dilli D, Zenciroglu A, Okumus N. Lidocaine should only be used in neonatal seizures that do not respond to first-generation AEDs. Acta Paediatr. July 25, 2013. [Epub ahead of print.] | |
Lundqvist M, Agren J, Hellström-Westas L, Flink R, Wickström R. Adverse effects following lidocaine treatment are limited with current dosing regimens. Acta Paediatr. August 12, 2013. [Epub ahead of print.] | |
Co JP, Elia M, Engel J Jr, et al. Proposal of an algorithm for diagnosis and treatment of neonatal seizures in developing countries. Epilepsia. 2007;48:1158–1164. | |
Castro Conde JR, Hernández Borges AA, Doménech Martínez E, González Campo C, Perera Soler R. Midazolam in neonatal seizures with no response to phenobarbital. Neurology. 2005;64:876–879. | |
Shany E, Benzaqen O, Watemberg N. Comparison of continuous drip of midazolam or lidocaine in the treatment of intractable neonatal seizures. J Child Neurol. 2007;22:255–259. | |
Yamamoto H, Aihara M, Niijima S, et al. Treatments with midazolam and lidocaine for status epilepticus in neonates. Brain Dev. 2007;29: 559–564. | |
Bye A, Flanagan D. Electroencephalograms, clinical observations and the monitoring of neonatal seizures. J Paediatr Child Health. 1995;31: 503–507. | |
Glass HC, Sullivan JE. Neonatal seizures. Curr Treat Options Neurol. 2009;11:405–413. | |
Slaughter LA, Anup DP, Slaughter JL. Pharmacological treatment of neonatal seizures: a systematic review. J Child Neurol. 2013;28: 351–364. | |
Van Leuven K, Groenendaal F, Toet MC, et al. Midazolam and amplitude-integrated EEG in asphyxiated full-term neonates. Acta Paediatr. 2004;93:1221–1227. | |
Andre M, Boutroy MJ, Dubruc C, et al. Clonazepam pharmacokinetics and therapeutic efficacy in neonatal seizures. Eur J Clin Pharmacol. 1986;30:585–589. | |
Krief P, Li K, Maytal J. Efficacy of levetiracetam in children with epilepsy younger than 2 years of age. J Child Neurol. 2008;23:582–584. | |
Khan O, Chang E, Cipriani C, et al. Use of intravenous levetiracetam for management of acute seizures in neonates. Pediatr Neurol. 2011;44:265–269. | |
Tulloch J, Carr RR, Ensom MH. A systematic review of the pharmacokinetics of antiepileptic drugs in neonates with refractory seizures. J Pediatr Pharmacol Ther. 2012;17:31–44. | |
Fillipi L, La Marca G, Fiorini P, et al. Topiramate concentrations in neonates treated with prolonged whole body hypothermia for hypoxic ischemic encephalopathy. Epilepsia. 2009;50:2355–2361. | |
Glass HC, Poulin C, Shevell MI. Topiramate for the treatment of neonatal seizures. Pediatr Neurol. 2011;44:439–442. | |
Clark AM, Kriel RL, Leppik IE, et al. Intravenous topiramate: safety and pharmacokinetics following a single dose in patients with epilepsy or migraines taking oral topiramate. Epilepsia. 2013;54:1106–1111. | |
Gal P, Oles KS, Gilamn JT, Weaver R. Valproic acid efficacy, toxicity, and pharmacokinetics in neonates with intractable seizures. Neurology. 1988;38:467–471. | |
Vauzelle-Kervroëdan F, Rey E, Pons G, et al. Phamacokinetics of the individual enantiomers of vigabatrin in neonates with uncontrolled seizures. Br J Clin Phamacol. 1996;42:779–781. | |
Wild JM, Chiron C, Ahn H, et al. Visual field loss in patients with refractory partial epilepsy treated with vigabatrin: final results from an open-label, observational, multicenter study. CNS Drugs. 2009;23: 965–982. | |
Barr PA, Buettiker VE, Antony JH. Efficacy of lamotrigine in refractory neonatal seizures. Pedaitr Neurol. 1999;20:161–163. | |
Singh B, Singh P, al Hifzi I, Khan M, Majeed-Saidan M. Treatment of neonatal seizures with carbamazepine. J Child Neurol. 1996;11: 378–382. | |
Giacoia GP, Gessner PK, Zaleska MM, Boutwell WC. Phamacokinetics of paraldehyde disposition in neonate. J Pediatr. 1984;104: 291–296. | |
Appleton RE. Giving drugs per rectum for systemic effect. Arch Dis Child. 1987;62:771–772. | |
Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63: 222–235. | |
ClinicalTrials.gov. A service of the US. Available from: http://clinicaltrials.gov/show/NCT01434225. Accessed October 5, 2013. | |
Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and gamma-aminobutyric acid A receptors. Ann Neurol. 1994;35: 229–234. | |
Laroia N, Guillet R, McBride M. Felbamate in term infants with hypoxic ischemic encephalopathy. J Pediatr Neurol. 2007;5:301–304. | |
Rennie JM. Neonatal seizures. Eur J Pediatr. 1997;54:83–87. | |
Boor R, Schmitt-Mechelke T, Stopfkuchen H, Reitter B. Dexamethasone in refractory seizures of premature infants. Acta Paediatr. 1993;82: 100–101. | |
Chiaffoni GP, Trevisanuto D. Neonatal encephalopathy. In: Kennedy C, editor. Principles and Practice of Child Neurology in Infancy. London, UK: Mac Keith Press; 2012. | |
Simms V, Cragg L, Gilmore C, Marlow N, Johnson S. Mathematics difficulties in children born very preterm: current research and future directions. Arch Dis Child Fetal Neonatal Ed. 2013;98:F457–F463. | |
Moore T, Hennessy EM, Myles J, et al. Neurological and developmental outcome in extremely preterm children born in England in 1995 and 2006: the EPICure studies. BMJ. 2012;345:e7961. | |
Sankar R, Painter MJ. Neonatal seizures. After all these years we still love what doesn’t work. Neurology. 2005;64:776–777. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.