Back to Journals » Journal of Blood Medicine » Volume 10
Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: current perspectives
Authors Barcellini W, Fattizzo B, Zaninoni A
Received 10 May 2019
Accepted for publication 10 July 2019
Published 8 August 2019 Volume 2019:10 Pages 265—278
DOI https://doi.org/10.2147/JBM.S190327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Wilma Barcellini, Bruno Fattizzo, Anna Zaninoni
UOC Ematologia, Fondazione IRCCS Ca’ Grande Ospedale Maggiore Policlinico, Milano, Italy
Abstract: Autoimmune hemolytic anemia (AIHA) is increasingly observed after allogeneic hematopoietic stem cell transplantation (allo-HSCT), with a reported incidence between 4% and 6%. The disease is generally severe and refractory to standard therapy, with high mortality, and there are neither defined therapies, nor prospective clinical trials addressing the best treatment. Most of the knowledge on the therapy of AIHAs derives from primary forms, which are highly heterogeneous as well, further complicating the management of post-allo-HSCT forms. The review addresses the risk factors associated with post-allo-AIHA, including unrelated donor, the development of chronic extensive graft-versus-host disease, CMV reactivation, nonmalignant diagnosis pre-HSCT, and alemtuzumab use in conditioning regimens. Regarding therapy, we describe standard treatments, such as corticosteroids, intravenous immunoglobulin, splenectomy, rituximab, cyclophosphamide, and plasma exchange, which have lower response rates than those reported in primary forms. New therapeutic options, including sirolimus, bortezomib, abatacept, daratumumab and complement inhibitors, are promising tools for this detrimental complication occurring after allo-HSCT.
Keywords: autoimmune hemolytic anemia, allogeneic hematopoietic stem cell transplantation, rituximab, sirolimus and abatacept, bortezomib and daratumumab
Introduction
Autoimmune hemolytic anemia (AIHA) is due to increased destruction of red blood cells (RBC) mainly driven by autoantibodies and complement (C), but also to other immune players (activated macrophages, T-lymphocytes, and cytokines).1 The disease is highly heterogeneous, from mild/compensated to life-threatening forms. The major determinants of this variability are the class, thermal amplitude and ability to activate complement of autoantibodies, along with the bone marrow compensatory activity. AIHA is classified into warm (wAIHA) and cold (cAIHA) according to antibody characteristics and this distinction has important therapeutic implications. AIHA is also defined as primary (50% of cases) or secondary, depending on the presence of an underlying or associated disease (infections, autoimmune diseases, drugs, malignancies, particularly lymphoproliferative disorders).2–5
In recent years, there is accumulating evidence about AIHA arising after allogeneic hematopoietic stem cell transplantation (allo-HSCT), with a reported incidence between 4% and 6%.6–8 The diagnosis of post-allo-HSCT AIHA is challenged by several concomitant factors, including disease relapse, graft failure, drug- and treatment-related toxicity, infection, and graft-versus-host disease (GVHD). In addition, the cornerstone of AIHA, ie, the direct antiglobulin test (DAT), may be positive for alloantibodies, with associated delayed hemolytic transfusion reactions, or for isohemagglutinins produced by donor-derived immunocompetent lymphocytes in case of minor ABO-incompatibility (“the passenger lymphocyte syndrome”).9
Most of the knowledge on therapy of AIHA derives from retrospective/observational studies with few prospective trials, mostly in primary forms.10–14 There is no defined therapy for post-allo-AIHA, with the exception of clinical experience and expert recommendations, all underlining the significant morbidity and mortality of these forms and the consequent need for prompt and aggressive treatment.
Here, we will review the clinical features of post-allo-AIHA and the risk factors associated with its occurrence. In addition, we will describe standard treatments, such as corticosteroids, intravenous immunoglobulin (IVIG), splenectomy, rituximab, cyclophosphamide, and plasma exchange, along with new therapeutic options, including sirolimus, bortezomib, abatacept, daratumumab and complement inhibitors.
General concepts of AIHA
AIHA is diagnosed by the demonstration of anti-erythrocyte antibody through the DAT, and classified according to the class and thermal range of the autoantibody in wAIHA (typically DAT+ for anti-IgG, or IgG plus C), cAIHA (due to IgM with DAT+ for C3d), mixed (DAT+ for both IgG and high titer cold agglutinins) or atypical forms (DAT negative, IgA and warm IgM driven). Of note, about 5–10% of AIHAs remain DAT negative even after performing highly sensitive tests. In these cases, the diagnosis is made in exclusion after extensive diagnostic re-evaluation, possibly causing detrimental delay in therapy.1 In wAIHA, hemolysis is mainly extravascular (lymphoid organs, spleen) and mediated by the Fc fragment of IgG (antibody-dependent cellular cytotoxicity), and minimally due to complement coating. At variance, in cold forms, RBC destruction is mainly driven by complement activation and RBC coating, with erythrocyte destruction occurring directly in the circulation and/or in the liver by activated macrophages. wAIHAs are the most common forms, with percentages ranging from 60% up to 80% of all cases, cAIHA accounts for 15% of patients, and the remaining cases have mixed features. Up to 11% of patients with chronic lymphocytic leukemia and 2–3% of those with non-Hodgkin lymphoma will develop AIHA at some point in their disease course.
Considering primary AIHA, usually considered a benign disease, it should be underlined that about 20% of cases (mainly warm IgG+C, cold, mixed, and atypical) display relapsing/refractory features and increased risk of thrombotic events, representing a clinical challenge. Their reported mortality is 11%, and predictors of a fatal outcome are severe infections, particularly in splenectomized cases, acute renal failure, Evans syndrome (concomitant thrombocytopenia), and multitreatment (4 or more therapy lines).15,16 Figure 1 highlights the rate of the main complications and risks factors for fatality in primary AIHAs, and Table 1 shows the main hematologic parameters according to AIHA serologic subtypes in the largest series reported, highlighting the great heterogeneity of the disease.15,16
 |
Figure 1 Main complications and risks factors for fatality in primary AIHAs. AIHAs show great clinical heterogeneity, including symptoms related to anemia, thrombotic events, infectious complication, acute renal failure, and circulatory disabling symptoms (typical of cold AIHA). Abbreviations: DIC, disseminated intravascular coagulation; DVT, deep vein thrombosis; AIHA, autoimmune hemolytic anemia. |
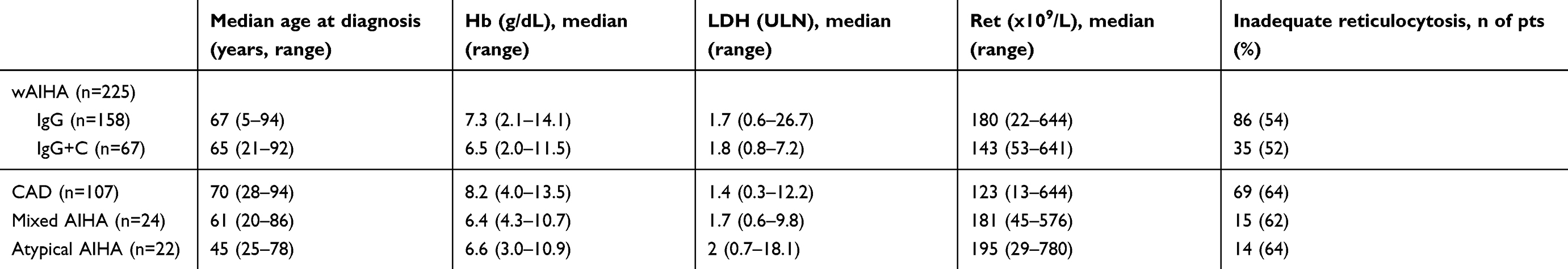 |
Table 1 Clinical and laboratory characteristics of patients at onset divided according to AIHA serological type |
Regarding therapy, steroids are the standard first-line therapy for wAIHA with a response in 75–80% of patients, but with an estimated cure rate in 20–30% only.1,2,16 At variance, steroids are much less effective in cAIHA (15–30% of cases), and unacceptably high doses are usually required. Rituximab has become the preferred second-line choice for steroid-refractory wAIHA, with an overall response rate of about 80%, a relapse-free survival of ~60% at 3 years, and a median time to response of approximately 3‒6 weeks (range 2‒16).1–3 Rituximab is now recommended as first-line in cold forms given their poor response to steroids. In fact, in cAIHA rituximab induced a response in about 50% of cases (mostly partial responses), with a median duration of <12 months. For those relapsed after a first course of rituximab, the association with bendamustine or fludarabine is a reported option.5 Splenectomy is considered the most effective second-line treatment of primary wAIHA (characterized by extravascular hemolysis in the spleen) with responses in more than 80% of cases, and the curative rate 20–50%, whereas it is contraindicated in cold agglutinin disease (where hemolysis in mainly intravascular or extravascular in the liver).1,5 Classic immunosuppressants (azathioprine, cyclophosphamide, cyclosporine) are progressively moving to the third line, due to their toxicity a limited efficacy (about 60% of cases in association with steroids).1–3,5,15,16 Table 2 summarizes available therapies for primary warm and cold AIHAs, and for the most common secondary forms.
 |
Table 2 Available therapies for primary warm and cold AIHAs, and for the most common secondary forms |
Clinical features and risk factors of post-allo-HSCT AIHA
The first studies reporting the occurrence of AIHA post-allotransplant appeared in the literature about 10–15 years ago. A study by Sanz et al17 described 12 patients with severe AIHA (eight with cold and four with warm antibodies) occurring at a median time of 147 days (range, 41–170). These cases were observed among 272 adult patients with a variety of malignant hematopoietic disorders transplanted between 1996 and 2004. Multivariate analysis showed that HSCT from unrelated donors (P=0.02) and the development of chronic extensive GVHD (P=0.018) were the only independent factors associated with AIHA. Most patients received steroids as primary treatment for AIHA with no response in the majority of cases. Two patients were treated with rituximab and achieved partial response, with an improvement in transfusion requirements. The mortality rate was elevated, with 2 patients only still alive at the time of analysis. AIHA was never the primary cause of death but increased morbidity in patients with other concomitant complications. In line with these findings, Holbro et al9 reviewed the several small series and case reports of various autoimmune complications following allo-HSCT, including 90 cases of AIHA with a reported mortality of about 60%. The authors firstly recommended the prompt administration of systemic steroids, and in case of no response, addition of rituximab with or without cyclophosphamide, along with IVIG.
In 2015, Wang et al6 reported 19 cases of AIHA (overall incidence, 3.6%) among 533 patients who received allogeneic HSCT at King’s College Hospital in London. The median time from HSCT to AIHA was 202 days, and in 3 cases, the onset of AIHA was preceded by an infective episode (adenovirus, Cytomegalovirus (CMV), and pneumonia of undetermined etiology). The analysis of risk factors showed that unrelated donor and concordant gender between the donor and the recipient were associated with the occurrence of AIHA, with a hazard risk of 5.28 (P=0.026) for the former, and of 3.52 (P=0.045) for the latter. No significant association was observed for the following variables: recipient gender, primary hematological disease, source of hematopoietic stem cells, conditioning regimen (alemtuzumab/ATG versus non-alemtuzumab/ATG, and reduced intensity versus myeloablative), HLA mismatch between donor and recipient, ABO antigen mismatch, recipient CMV status, and concurrent chronic GVHD. At the onset of AIHA, half of the patients were receiving immunosuppressive therapies, either as GVHD treatment or prophylaxis. In the majority of patients, multiple treatments were required, with only half of cases achieving complete resolution of AIHA. Patients with post-transplantation AIHA had a higher overall mortality (hazard risk 2.48; 95% CI, 1.33–4.63; P=0.004). At variance with previous finding, in this series, 36% of cases (4 of 11 cases) of deaths were directly attributable to AIHA.
In the same years, risk factors for post-allo-HSCT AIHA have been analyzed in 265 children (median age 5.1 years, range 0.5–15.4) transplanted between 1998 and 2015 in Taiwan.18 The authors identified 15 cases (incidence 6%), of whom 3 with concomitant thrombocytopenia. The median time to emergence of AIHA was 149 days (range 42–273), and patients with thalassemia had a higher rate of AIHA than patients other than thalassemia (11.5% vs 3.2%; P = 0.017). At the onset of AIHA, all patients were receiving immunosuppressive therapies, either as GVHD treatment or prophylaxis. All cases were treated with high dose methylprednisolone and IVIG, and 9 received rituximab. Resolution of AIHA was observed in 12/15 cases (80%) and death in one. No splenectomy was performed.
More recently, risk factors for post-allo-AIHA have been investigated in a pediatric population of 531 children transplanted at Leiden University Medical Center between 2000 and 2016.8 Twenty-six were complicated by autoimmune disorders (cumulative incidence 5%), of whom 12 AIHA (46%). Nonmalignant diagnosis pre-HSCT (HR, 3.5; P = 0.031), CMV reactivation (HR, 3.4; P = 0.02), and alemtuzumab use (HR, 2.5; P = 0.028) were identified as independent risk factors for autoimmune complications. Gender, donor type, stem cell source, conditioning regimen, T cell depletion, GVHD prophylaxis, ATG serotherapy pre-HSCT, ABO match, HLA mismatch, acute and chronic GVHD, EBV reactivation, adenovirus infection/reactivation, platelet recovery, absolute neutrophil count recovery, and chimerism in peripheral blood mononuclear cells were not associated with autoimmune complications. Patients were treated with corticosteroids and IVIG with responses in about one-third of cases only. Further therapies were rituximab, bortezomib, or sirolimus, with associated resolution in 40%, 57%, and 100% of cases, respectively. The same authors investigated several immunological parameters in post-transplant patients with or without autoimmune complications. The former group has a significantly lower CD3+CD8+ T cell count (P=0.002), whereas other subsets (CD3+CD4+ T cells, CD19+CD20+ B cells, CD3−CD16/56+ natural killer cells, and CD3+TCRγδ+) cells did not differ significantly between patients with or without autoimmunity. IL-4, eotaxin, platelet-derived growth factor, and RANTES were reduced in autoimmune cases, whereas IL-2, IL-6, IL-16 were increased. Finally, several Th2/Th 1 ratios (IL-5/interferon (IFN)-γ, IL-5/tumor necrosis factor (TNF)-α, IL-5/IL-12, IL-6/IFN-γ, IL-6/TNF-α, IL-13/IFN-γ, IL-13/TNF-α, IL-13/IL-12, and IL-10/TNF-α) pinpoint that autoimmune complications arise in a Th2-mediated context.
In 2018, a large Spanish collaborative study in adults and children reported 60 cases of AIHA among 4099 allo-HSCT between 2000 and 2015 (cumulative incidence 1.5%).7 AIHA occurred at a median of 6 months after the transplant, and HLA mismatch donor (P=0.005), cord blood use (P=0.005), and age <15 years (P=0.005) were associated with its development. Median lines of treatment were 3 (range 1–7), including corticosteroids (88%), immunoglobulins (63%), and rituximab (67%). Complete resolution of AIHA was achieved 55% of cases, and the cumulative incidence of AIHA-related mortality was 17%. Table 3 summarizes risk factors available in the literature.
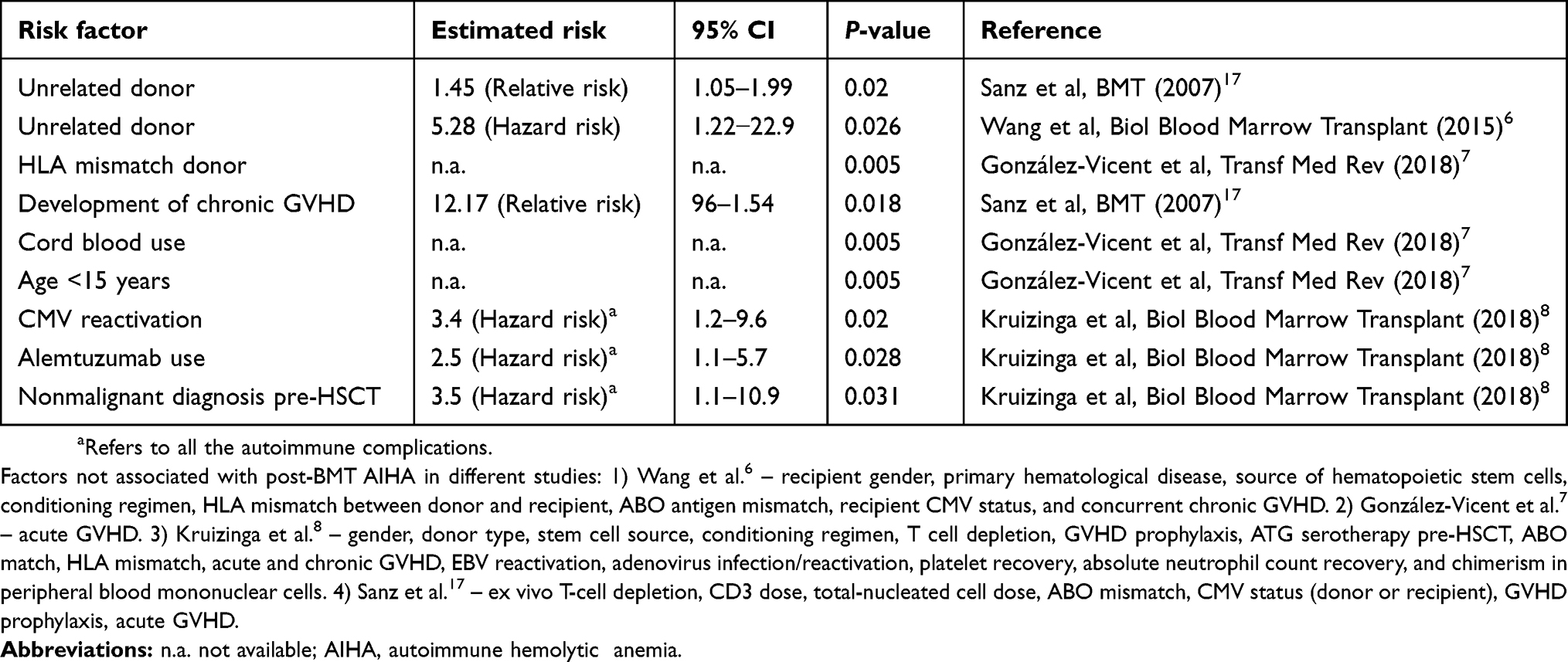 |
Table 3 Risk factors for the occurrence of post-transplant AIHA |
Finally, in a recent large pediatric study investigating 442 patients who underwent HSCT, an autoimmune complication was observed in 20 cases, including 9 AIHA, 6 multilineage cytopenias, 3 immune thrombocytopenias, and 2 autoimmune neutropenias. These patients were significantly older than the control group without autoimmune complications (8.2±7.3 years vs 4.0±4.7 years, P=0.030), and less frequently developed chronic GVHD (5% vs 26%, P=0.048). There was no significant difference in gender, source of stem cells, ABO compatibility, anti-lymphocyte serotherapy, GVHD prophylaxis, prior donor lymphocyte infusion, total body irradiation, pre-transplant autoimmunity or prevalence of acute GVHD.190
Current treatments of post-allo-HSCT AIHA
Treatment of AIHA after HSCT is challenging and mostly supported by case reports/series and some large retrospective analysis (Table 4). Although most cases usually present with severe or very severe anemia requiring transfusion and prompt intervention, a wait and see approach has also been used for selected non-life-threatening cases with spontaneous recovery in 80% of them.8 Concerning frontline therapy, the disease is often resistant to steroids (ie, methylprednisolone 1–2 mg/day IV), with response rates ranging from 10% to 55% and limited benefit of high dose IVIGs addition (2 g/kg for 2 days).6–8,17–20 These response rates are clearly lower than those observed in primary AIHA, suggesting a more profound immune dysregulation in post-HSCT AIHA. As a matter of fact, most studies did not take into account AIHA type, ie, wAIHA versus cAIHA, which respond differently to steroids. However, most post-HSCT cases are warm forms and cAIHA responds to high steroid doses. Similarly, limited efficacy has been obtained with plasma exchange, used for patients with massive hemolysis (ie, 1/10 cases); this treatment is recommended as temporizing measure in primary AIHA, particularly in cAIHA, given the predominant intravascular distribution of IgM, before the immunosuppressive therapy takes effect.21,22 Historical second-line treatments were cytotoxic immunosuppressants, such as cyclophosphamide (ie, 800 mg IV), 6-mercaptopurin (50 mg day) and mycophenolate mofetil (500 mg BID), that induced heterogeneous response rates (9–100%),7,18 with increased toxicity, similarly to what observed for primary cases. Splenectomy has also been used with variable response rates ranging from 0% to 100% (mean 39%), again without distinguishing between warm and cold AIHA type. Authors agree that splenectomy can be considered after the 2nd line in selected wAIHA patients without contraindications.6–8,17,23,24 Generally, with classical treatments, most refractory cases died to opportunistic infections, relapse of the primary disease and massive hemolysis.
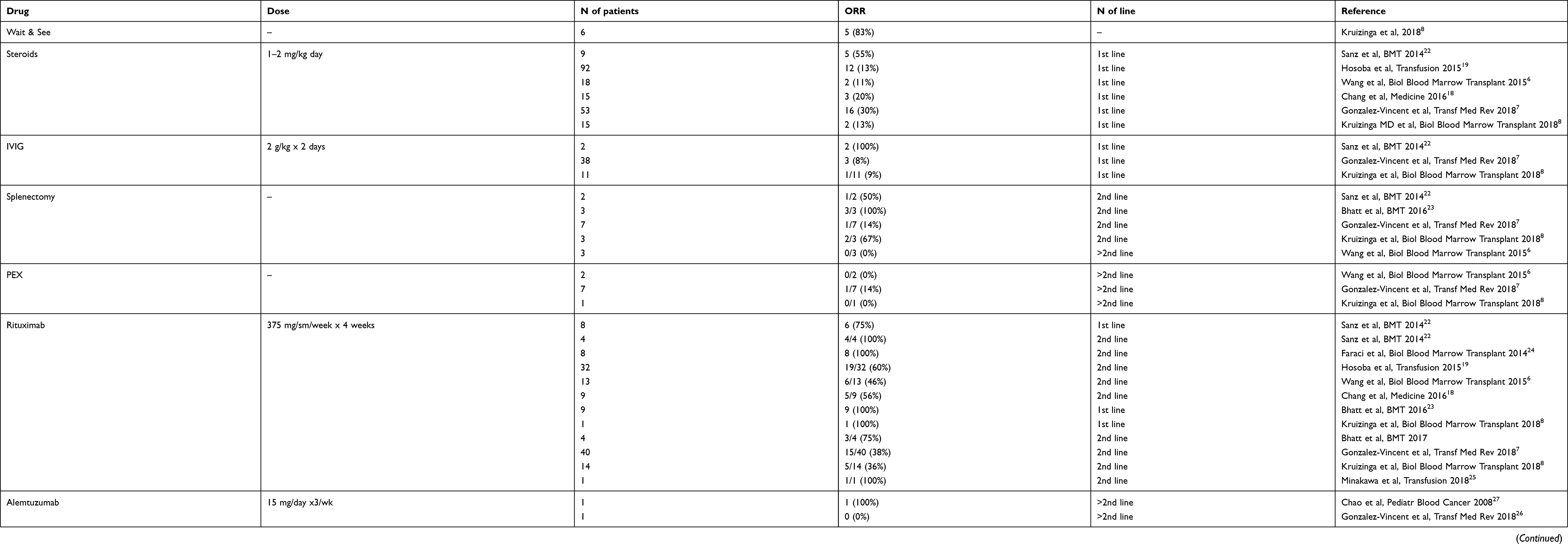 |
Table 4 Current and emerging treatments of post-allo-HSCT AIHA |
Biological treatments for post-allo-HSCT AIHA
The improving knowledge of AIHA pathogenesis has given hints for a better therapeutic approach. AIHA is a heterogeneous disease mainly due to autoantibody-mediated destruction of erythrocytes but also involves dysregulation of cellular and innate immunity with a well-established Th2 cytokine pattern. Moreover, complement activation and defective bone marrow compensatory response play an important role. Several drugs targeting these mechanisms are under development in addition to standard therapies. In particular, rituximab which acts on B-cells, bortezomib and daratumumab targeting plasma cells, or sirolimus and abatacept directed against regulatory T-lymphocytes.1
Since the advent of biological target therapies, a significant improvement of post-allo-HSCT AIHA outcome has been observed. In particular, rituximab induced a response in about 49% of cases in 2nd line (lower than in primary AIHA), and in 88% of cases as 1st line, combined to steroid treatment (comparable to primary cases), again without distinguishing for AIHA type.23–26 In particular, in a study by Sanz et al, 3/4 cases responded to rituximab frontline (2 CR and 1 PR), and 4/4 as second-line treatment (all PR).23 In another study, Bhatt et al reported 100% responses in 9 patients (mostly adult) treated with rituximab 2–18 days after diagnosis in combination with corticosteroids and none died.24 Moreover, Faraci et al observed 100% responses to rituximab (all CR) in pediatric AIHA treated in first- (1 case) or second-line (7 cases).25 At variance, in a recent large pediatric study including various autoimmune complications, the majority of patients with AIHA did not have complete response after treatment with steroids, IVIG and/or rituximab requiring further lines of therapy.19 Overall, the introduction of rituximab decreased mortality from 53% to 15%8 and response to the drug established as a new prognostic factor for post-HSCT AIHA. These data favor the use of rituximab either frontline or as a step-up early second line in patients with severe AIHA post-HSCT.
Few data are also available for the anti-CD52 MoAb alemtuzumab targeting both B and T cells and resulting in prolonged and profound lymphocytopenia. A combination of lower-dose rituximab (100 mg IV weekly for 4 weeks) and alemtuzumab (10 mg subcutaneously for 3 days) induced 100% short-term response in 8 patients with primary AIHA, but only 3 cases maintained long-term remission;27 2 patients have been treated after HSCT, one responded28 and one not.7 Beyond efficacy, the severe lymphoid suppression and increased risk of CMV reactivation question the use of this antibody following HSCT outside clinical trials.29
Another B-cell targeting agent is the proteasome inhibitor bortezomib, the backbone therapy of multiple myeloma. The drug leads to the accumulation of polyubiquitinated proteins, inducing the death of both short- and long-lived plasma cells by activation of the terminal-unfolded protein response. Bortezomib, at the dose of 1.3 mg/sm IV single course, proof safe and effective in case reports of secondary AIHA,29–31 and in a recent Phase II prospective trial in primary cAIHA, with an overall response of 31.6% (3 CR and 3 PR).32 Regarding post-HSCT AIHA even higher responses were observed, overall response of 58%, mostly as 3rd line in rituximab-refractory cases.7,8,19,20,33–36 However, data come mainly from case reports that carry the intrinsic bias of describing responding patients only. In the HSCT setting, bortezomib will be possibly used for GVHD prophylaxis in patients who received HLA mismatched allogeneic HSCT, after its inhibitory effect on alloreactive T cells and on T-helper 1 cells that produce interferon-γ and IL-2.37
Considering T-cell-directed drug used in AIHA, recent attention has raised sirolimus, an inhibitor of the serine threonine kinase mTOR (mammalian Target Of Rapamycin), part of T-cell receptor downstream pathway, that regulates protein synthesis, growth, proliferation, and activation of T cells. Sirolimus has been reported to be effective in 80% of patients with primary Evans syndrome, in 4 cases of pediatric patients with multi-drug-refractory AIHA, and in 86% of 21 cases following solid organ transplant.38–42 High efficacy has also been reported in 6 multi-refractory post-HSCT AIHA case reports, with the same caveat mentioned above. Finally, in the recent pediatric series, sirolimus has been used in 2nd or 3rd line of therapy with good outcome.19 In this highly refractory life-threatening setting, sirolimus efficacy seems linked to both the induction of abnormal T-cells apoptosis, and to the increase of IL-10 and TGF-β secreting T-regulatory cells. These additional mechanisms would target the residual “autoimmunity triggering cells” spared after the treatment with monoclonal antibodies.
Emerging treatments for post-allo-HSCT AIHA
An emerging therapeutic target in AIHA is the complement system, which plays a central role both in the intra- and in the extravascular-mediated hemolysis. Complement blockade reduces ongoing hemolysis, particularly in the acute phase. Eculizumab, a humanized monoclonal antibody that binds to the C5 fraction, has been successfully administrated to multi-refractory primary AIHA patients, particularly in cAIHA,43,44 with improvement in hemolysis (Hb increase and LDH decrease with the reduction of transfusions in 7/13 cases) and clinical symptoms. In the post-HSCT setting, responses are probably even lower, with only 1 of 3 reported cases responding to 900 mg IV infusion.7,36 As a matter of fact, the drug will only be effective on C5-mediated intravascular hemolysis, that is marginal in the pathogenesis of AIHA mainly encompassing earlier steps of the complement cascade; moreover, it carries a significant infection risk, particularly from meningococcal bacteria, that requires appropriate vaccine schedules. New complement inhibitors are under active study for primary and secondary AIHA, including the IV C1s blocker sutimlimab, that induced sustained responses in 7/10 cAIHA cases in Phase I,45–47 and the subcutaneous C3 inhibitor APL-2 whose Phase II study is currently ongoing.48
A further new drug that has recently shown efficacy in AIHA is the anti-CD38 MoAb daratumumab, approved for the treatment of multiple myeloma, but potentially targeting also CD38 expressing nonmalignant autoantibody-producing plasma cells. A case report of efficacy in primary AIHA has been recently published,49 moreover, 3 patients with life-threatening post-transplant AIHA have also been described, with complete and sustained response in 2 patients and a transient response in the other patient. Importantly, the authors suggest that after depletion of B and plasma cells by rituximab and daratumumab, immunoglobulin substitution should be administered in this setting and B-cell reconstitution after drug administration is variable.36
Finally, a recent study showed the efficacy of the T-cell inhibitor abatacept in 3 post-allo-HSCT AIHA cases.50 Abatacept is a fusion protein formed by linking extracellular domain of cytotoxic T-lymphocyte antigen 4 with the Fc region of immunoglobulin G (IgG) and inhibits T-cell activation through competitive binding of CD80 and CD86 on antigen-presenting cells, thus blocking the required CD28 costimulatory interaction. It is used for the treatment of other autoimmune diseases including rheumatoid arthritis and uveitis.51,52 The drug has also shown efficacy in acute GVHD prophylaxis and has been demonstrated to increase T-reg cells in one of the post-HSCT AIHA cases described.53
Transfusion issues
Supportive care with transfusions constitutes an important component of the management of severe AIHA in post-allo-HSCT setting. It is a common practice to give irradiated (25–30 Gy) and leuko-depleted RBC concentrates in order to reduce potentially fatal transfusion reactions. Since auto-antibodies are directed against highly prevalent antigens, cross-matching is often unable to identify compatible RBC units.9 In any case, transfusions must not be delayed in the acute setting since this option is lifesaving. In life-threatening AIHA, transfusion with ABO, Rh and K matched blood is more appropriate than waiting for full compatibility investigations that can take 4–6 hrs or more.3 In less urgent cases, extended phenotyping is advisable in order to provide the best matched RBC units. It is worth reminding that transfusion may provoke alloantibody production, an event observed in up to 30–40% of primary AIHA cases, possibly causing serious acute or delayed hemolytic transfusion reactions. Altogether, a close collaboration between clinicians, transfusion service and blood bank is advisable for the best management of transfusion support in post-allo-HSCT AIHA.
Conclusion
Post-allo-HSCT AIHA is definitely more severe and refractory than primary forms. Moreover, it arises in a complex and frail clinical context, marked by deep immunosuppression and possible multi-organ failure. All these factors lead to an increased mortality, which is hardly attributable to AIHA itself, GVHD, or infectious complications. Main risk factors for the occurrence of AIHA are unrelated donor and the development of chronic extensive GVHD, highlighting the profound dysregulation of the immunologic competence following allo-HSCT. CMV reactivation has also been associated with post-HSCT AIHA, in line with one of the mechanisms of the immunologic tolerance breakdown, ie, molecular mimicry between self-and foreign antigens. In addition, polyclonal lymphocyte activation, which plays a key role in most autoimmune conditions, certainly occurs after allo-HSCT, which is definitely a major immunological “storm.” While the degree of the immune activation is sometimes beneficial (graft-versus-leukemia), it can be greatly harmful (graft-versus-host), resembling in its chronic form a well-known immune-mediated disease, ie, progressive systemic sclerosis. Nonmalignant diagnosis pre-HSCT and alemtuzumab use have been implied too, although associated with different types of autoimmune complications and not specifically with AIHA.
Regarding therapy, steroids and rituximab, which are now consolidated therapies for primary AIHAs, show an unacceptable rate of failure in post-transplant forms, emphasizing the need for new-targeted therapies. The only satisfactory results are those observed in front-line use of the two drugs in combination, stressing the need for a prompt and aggressive treatment of post-allo-HSCT AIHA. Among new agents, either those mainly acting on plasma cells (bortezomib and daratumumab), or targeting different T-lymphocyte activation pathways (sirolimus and abatacept) or both (alemtuzumab), have shown promising results, although biased by reporting successful cases only. A new class of inhibitors would probably add powerful tools to the therapeutic arsenal of post-HSCT AIHA, ie, the several agents acting on the complement system. This would involve not only cAIHA, where complement has a recognized pivotal role, but also wAIHA, where its role is rather under-recognized but possibly detrimental. Prospective controlled trials would be obviously advisable, although difficult to conduct, given the heterogeneity and relative rarity of post-allo-HSCT AIHA. Most importantly, the understanding of the complex immunological network and positive/negative interactions occurring during immune-reconstitution will help in harnessing the most appropriate therapy/association of drugs for post-transplant AIHAs.
Acknowledgment
This work was supported by research funding from Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, grant number RC 2018-19.
Disclosure
WB received consultant honoraria from Novartis, Alexion, Bioverativ, and Apellis. BF received consultant honoraria from Apellis, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Barcellini W, Fattizzo B, Zaninoni A. Current and emerging treatment options for autoimmune hemolytic anemia. Expert Rev Clin Immunol. 2018;14(10):857–872. doi:10.1080/1744666X.2018.1521722
2. Kalfa TA. Warm antibody autoimmune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2016;1:690–697. doi:10.1182/asheducation-2016.1.690
3. Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A; British Society for Haematology. The diagnosis and management of primary autoimmune haemolytic anaemia. Br J Haematol. 2017;176(3):395–411. doi:10.1111/bjh.14478
4. Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A; British Society for Haematology Guidelines. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017;177(2):208–220. doi:10.1111/bjh.14654
5. Berentsen S. How I manage patients with cold agglutinin disease. Br J Haematol. 2018;181(3):320–330. doi:10.1111/bjh.15109
6. Wang M, Wang W, Abeywardane A, et al. Autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: analysis of 533 adult patients who underwent transplantation at King’s College Hospital. Biol Blood Marrow Transplant. 2015;21(1):60–66. doi:10.1016/j.bbmt.2014.09.009
7. González-Vicent M, Sanz J, Fuster JL, et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante de Medula Osea en Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus Med Rev. 2018;32:179–185.
8. Kruizinga MD, van Tol MJD, Bekker V, et al. Risk factors, treatment, and immune dysregulation in autoimmune cytopenia after allogeneic hematopoietic stem cell transplantation in pediatric patients. Biol Blood Marrow Transplant. 2018;24(4):772–778. doi:10.1016/j.bbmt.2017.12.782
9. Holbro A, Abinun M, Daikeler T. Management of autoimmune diseases after haematopoietic stem cell transplantation. Br J Haematol. 2012;157(3):281–290. doi:10.1111/j.1365-2141.2012.09070.x
10. Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004;103:2925–2928. doi:10.1182/blood-2003-10-3597
11. Berentsen S, Randen U, Vågan AM, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. 2010;116:3180–3184. doi:10.1182/blood-2010-06-288647
12. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biological studies. Blood. 2012;119:3691–3697. doi:10.1182/blood-2011-10-388512
13. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393–399. doi:10.1111/bjh.12541
14. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23–27. doi:10.1002/ajh.24570
15. Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124:2930–2936. doi:10.1182/blood-2014-06-583021
16. Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from 8 Italian Reference Centers. Am J Hematol. 2018;93(9):E243–E246. doi:10.1002/ajh.25212
17. Sanz J, Arriaga F, Montesinos P, et al. Autoimmune hemolytic anemia following allogeneic hematopoietic stem cell transplantation in adult patients. Bone Marrow Transplant. 2007;39(9):555–561. doi:10.1038/sj.bmt.1705641
18. Chang TY, Jaing TH, Wen YC, Huang IA, Chen SH, Tsay PK. Risk factor analysis of autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation in children. Medicine (Baltimore). 2016;95(46):e5396. doi:10.1097/MD.0000000000004864
19. Neely JA, Dvorak CC, Pantell MS, Melton A, Huang JN, Shimano KA. Autoimmune cytopenias in pediatric hematopoietic cell transplant patients. Front Pediatr. 2019;7:171. doi:10.3389/fped.2019.00171
20. Hosoba S, Jaye DL, Cohen C, Roback JD, Waller EK. Successful treatment of severe immune hemolytic anemia after allogeneic stem cell transplantation with bortezomib: report of a case and review of literature. Transfusion. 2015;55(2):259–264. doi:10.1111/trf.12815
21. Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the writing committee of the American society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145–284. doi:10.1002/jca.21276
22. Szczepiorkowski ZM, Winters JL, Bandarenko N, et al.; Apheresis Applications Committee of the American Society for Apheresis. Guidelines on the use of therapeutic apheresis in clinical practice – evidence-based approach from the apheresis applications committee of the American Society for Apheresis. J Clin Apher. 2010;25(3):83–177. doi:10.1002/jca.20240.
23. Sanz J, Arango M, Carpio N, et al. Autoimmune cytopenias after umbilical cord blood transplantation in adults with hematological malignancies: a single-center experience. Bone Marrow Transplant. 2014;49(8):1084–1088. doi:10.1038/bmt.2014.107
24. Bhatt V, Shune L, Lauer E, et al. Autoimmune hemolysis and immune thrombocytopenic purpura after cord blood transplantation may be life-threatening and warrants early therapy with rituximab. Bone Marrow Transplant. 2016;51(12):1579–1583. doi:10.1038/bmt.2016.228
25. Faraci M, Zecca M, Pillon M, et al.; Italian Association of Paediatric Haematology and Oncology. Autoimmune hematological diseases after allogeneic hematopoietic stem cell transplantation in children: an Italian multicenter experience. Biol Blood Marrow Transplant. 2014;20(2):272–278. doi:10.1016/j.bbmt.2013.11.014.
26. Minakawa K, Ohto H, Yasuda H, et al. Efficacy of D- red blood cell transfusion and rituximab therapy in autoimmune hemolytic anemia with anti-D and panreactive autoantibodies arising after hematopoietic stem cell transplant. Transfusion. 2018;58(7):1606–1610. doi:10.1111/trf.14634
27. Gomez-Almaguer D, Solano-Genesta M, Tarin-Arzaga L, et al. Low-dose rituximab and alemtuzumab combination therapy for patients with steroid-refractory autoimmune cytopenias. Blood. 2010;116(23):4783–4785. doi:10.1182/blood-2010-06-291831
28. Chao MM, Levine JE, Ferrara JL, et al. Successful treatment of refractory immune hemolysis following unrelated cord blood transplant with Campath-1H. Pediatr Blood Cancer. 2008;50(4):917–919. doi:10.1002/pbc.21187
29. Park JA, Lee HH, Kwon HS, Baik CR, Song SA, Lee JN. Sirolimus for refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: a case report and literature review of the treatment of post-transplant autoimmune hemolytic anemia. Transfus Med Rev. 2016;30(1):6–14. doi:10.1016/j.tmrv.2015.09.001
30. Carson KR, Beckwith LG, Mehta J. Successful treatment of IgM-mediated autoimmune hemolytic anemia with bortezomib. Blood. 2010;115(4):915. doi:10.1182/blood-2009-12-255992
31. Danchaivijitr P, Yared J, Rapoport AP. Successful treatment of IgG and complement-mediated autoimmune hemolytic anemia with bortezomib and low-dose cyclophosphamide. Am J Hematol. 2011;86(3):331–332. doi:10.1002/ajh.21950
32. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132(5):547–550. doi:10.1182/blood-2018-03-835413
33. Poon LM, Koh LP. Successful treatment of isohemagglutinin-mediated pure red cell aplasia after ABO-mismatched allogeneic hematopoietic cell transplant using bortezomib. Bone Marrow Transplant. 2012;47(6):870–871. doi:10.1038/bmt.2011.176
34. Rovira J, Cid J, Gutiérrez-García G, et al. Fatal immune hemolytic anemia following allogeneic stem cell transplantation: report of 2 cases and review of literature. Transfus Med Rev. 2013;27(3):166–170. doi:10.1016/j.tmrv.2013.02.004
35. Khandelwal P, Davies SM, Grimley MS, et al. Bortezomib for refractory autoimmunity in pediatrics. Biol Blood Marrow Transplant. 2014;20(10):1654–1659. doi:10.1016/j.bbmt.2014.06.032
36. Schuetz C, Hoenig M, Moshous D, et al. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood Adv. 2018;2(19):2550–2553. doi:10.1182/bloodadvances.2018020883
37. Koreth J, Kim HT, Lange PB, et al. A bortezomib-based regimen offers promising survival and graft-versus-host disease prophylaxis in myeloablative HLA-mismatched and unrelated donor transplantation: a Phase II trial. Biol Blood Marrow Transplant. 2015;21(11):1907–1913. doi:10.1016/j.bbmt.2015.05.027
38. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans Syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol. 2017;39(6):420–424. doi:10.1097/MPH.0000000000000818
39. Miano M, Calvillo M, Palmisani E, et al. Sirolimus for the treatment of multi-resistant autoimmune haemolytic anaemia in children. Br J Haematol. 2014;167(4):571–574. doi:10.1111/bjh.13010
40. Valentini RP, Imam A, Warrier I, et al. Sirolimus rescue for tacrolimus-associated post-transplant autoimmune hemolytic anemia. Pediatr Transplant. 2006;10(3):358–361. doi:10.1111/j.1399-3046.2005.00460.x
41. Acquazzino MA, Fischer RT, Langnas A, Coulter DW. Refractory autoimmune haemolytic anemia after intestinal transplant responding to conversion from a calcineurin to mTOR inhibitor. Pediatr Transplant. 2013;17(5):466–471. doi:10.1111/petr.12101
42. Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145(1):101–106. doi:10.1111/j.1365-2141.2009.07595.x
43. Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018;2(19):2543–2549. doi:10.1182/bloodadvances.2018024190
44. Lindorfer MA, Pawluczkowycz AW, Peek EM, Hickman K, Taylor RP, Parker CJ. A novel approach to preventing the hemolysis of paroxysmal nocturnal hemoglobinuria: both complement-mediated cytolysis and C3 deposition are blocked by a monoclonal antibody specific for the alternative pathway of complement. Blood. 2010;115(11):2283–2291. doi:10.1182/blood-2009-12-255992
45. Jäger U, D’Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133(9):893–901. doi:10.1182/blood-2018-06-856930
46. Clinical Trial Bioverativ Therapeutics Inc. A Study to Assess the Efficacy and Safety of BIVV009 (Sutimlimab) in Participants With Primary Cold Agglutinin Disease Without A Recent History of Blood Transfusion (Cadenza Study). Available from: https://clinicaltrials.gov/ct2/show/NCT03347422. NLM identifier: NCT03347422. Accessed November 20, 2017.
47. Clinical Trial Bioverativ Therapeutics Inc. A Study to Assess the Efficacy and Safety of BIVV009 (Sutimlimab) in Participants With Primary Cold Agglutinin Disease Who Have a Recent History of Blood Transfusion (Cardinal Study). Available from: https://clinicaltrials.gov/ct2/show/NCT03347396. NLM identifier: NCT03347396. Accessed November 20, 2017.
48. Clinical trial Apellis Pharmaceuticals, Inc.Study to Assess the Safety, Tolerability, Efficacy and PK of APL-2 in Patients With wAIHA or CAD. Available from: https://clinicaltrials.gov/ct2/show/NCT03226678. NLM identifier: NCT03226678. Accessed July 24, 2017.
49. Tolbert VP, Goldsby R, Huang J, et al. Daratumumab is effective in the treatment of refractory post-transplant autoimmune hemolytic anemia: a pediatric case report. Blood. 2016;128:4819. doi:10.1182/blood-2016-06-724161
50. Hess J, Su L, Nizzi F, et al. Successful treatment of severe refractory autoimmune hemolytic anemia after hematopoietic stem cell transplant with abatacept. Transfusion. 2018;58(9):2122–2127. doi:10.1111/trf.14907
51. Pimentel-Quiroz VR, Ugarte-Gil MF, Alarcon GS. Abatacept for the treatment of systemic lupus rythematosus. Expert Opin Investig Drugs. 2016;25(4):493–499. doi:10.1517/13543784.2016.1154943
52. Lee S, Moon JS, Lee CR, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol. 2016;137(1):327–330. doi:10.1016/j.jaci.2015.08.036
53. Koura DT, Horan JT, Langston AA, et al. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol Blood Marrow Transplant. 2013;19(11):1638–1649. doi:10.1016/j.bbmt.2013.09.003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.