Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Management of extrapulmonary sarcoidosis: challenges and solutions
Authors Al-Kofahi K, Korsten P, Ascoli C, Virupannavar S, Mirsaeidi M ![]() , Chang I, Qaqish N, Saketkoo LA, Baughman RP, Sweiss NJ
, Chang I, Qaqish N, Saketkoo LA, Baughman RP, Sweiss NJ
Received 5 February 2016
Accepted for publication 5 April 2016
Published 7 November 2016 Volume 2016:12 Pages 1623—1634
DOI https://doi.org/10.2147/TCRM.S74476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Khalid Al-Kofahi,1,* Peter Korsten,2,* Christian Ascoli,3 Shanti Virupannavar,4 Mehdi Mirsaeidi,5 Ian Chang,6 Naim Qaqish,7 Lesley A Saketkoo,8 Robert P Baughman,9 Nadera J Sweiss3,4
1Department of Molecular Biosciences, University of Kansas, Lawrence, KS, USA; 2Department of Nephrology and Rheumatology, University Medical Center Göttingen, Göttingen, Germany; 3Division of Pulmonary, Critical Care, Sleep and Allergy, University of Illinois at Chicago, Chicago, IL, 4Division of Rheumatology, University of Illinois at Chicago, Chicago, IL, 5Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, University of Miami Miller School of Medicine, Miami, FL, 6Department of Medicine, Michigan State University College of Osteopathic Medicine, East Lansing, MI, 7Department of Gastroenterology, Hepatology and Nutrition, University of Buffalo, Buffalo, NY, 8New Orleans Scleroderma and Sarcoidosis Patient Care and Research Center, Louisiana State University Health Sciences Center, New Orleans, LA, 9Department of Medicine, University Medical Center of Cincinnati, Cincinnati, OH, USA
*These authors contributed equally to this work
Background: Sarcoidosis is a chronic multisystem disease of unknown etiology characterized by noncaseating granulomas that most often involves the lungs, but frequently has extrapulmonary manifestations, which might be difficult to treat in individual patients.
Objective: To review different disease manifestations, focusing on extrapulmonary organ systems, and to provide treatment options for refractory cases.
Materials and methods: We performed a literature search using Medline and Google Scholar for individual or combined keywords of “sarcoidosis, extrapulmonary, treatment, kidney, neurosarcoidosis, cardiovascular, gastrointestinal, transplantation, musculoskeletal, rheumatology, arthritis, and skin”. Peer-reviewed articles, including review articles, clinical trials, observational trials, and case reports that were published in English were included. References from retrieved articles were also manually searched for relevant articles.
Results and conclusion: Isolated involvement of a single organ or organ system is rare in sarcoidosis, and thus all patients must be thoroughly evaluated for additional disease manifestations. Cardiac sarcoidosis and neurosarcoidosis may be life-threatening. Clinicians need to assess patients comprehensively using clinical, laboratory, imaging, and histopathological data to recommend competently the best and least toxic treatment option for the individual patient.
Keywords: sarcoidosis, immunosuppressive agents, chronic granulomatous diseases, diagnostic tests
Introduction
“Sarcoidosis would be a benign and relatively unimportant disease but for three troublesome complications: pulmonary fibrosis, fibrotic uveitis, and nephrocalcinosis.” This notion by James et al,1 published in the Lancet in 1967, illustrates that physicians treating sarcoidosis have been aware of its frequent extrapulmonary disease manifestations for decades. Sarcoidosis is a chronic inflammatory disorder of unknown etiology, characterized by noncaseating granulomas involving the lungs in more than 90% of patients.2 Ocular, lymph-node, and cutaneous manifestations are next in frequency, but any organ system can be affected.2 Extrapulmonary disease manifestations contribute to significant morbidity, but can easily be missed. The ACCESS study revealed that 50% of the 736 sarcoidosis patients studied had pulmonary sarcoidosis and coexisting extrapulmonary sarcoidosis compared to only 2% of patients who presented with isolated extrapulmonary disease.3 Prevalence of extrapulmonary sarcoidosis was higher among African-Americans in comparison to Caucasians.3 Remission may occur in nearly two-thirds of patients after 1–2 years; still, up to a third of patients will have chronic or progressive disease.4
The diagnosis requires exclusion of other etiologies, and relies on compatible history, clinical findings, and noncaseating granulomas on histology. In patients with normal chest radiographs, evaluation and monitoring of extrapulmonary sarcoidosis with other diagnostic modalities, including ultrasound, chest computed tomography, gallium scintigraphy, magnetic resonance imaging (MRI), and positron-emission tomography (PET), can be helpful.2
Despite the lack of evidence-based studies, glucocorticosteroids (GCs), namely prednisone, are the first-line therapy of sarcoidosis. Recommended starting doses vary between 20 and 40 mg/day, and are tapered slowly to a maintenance dose of less than 10 mg/day as the disease allows. However, lack of response or drug toxicity are limiting factors, especially with long-term treatment, and can result in significant disability. GC-sparing drugs, such as disease-modifying antisarcoid drugs or biological agents, are increasingly used in these instances.5–7 Clinicians are increasingly initiating methotrexate (MTX) along with GCs in clinical profiles that portend a poor prognosis.8
Extrapulmonary disease manifestations
This review focuses on cutaneous, renal, central nervous system (CNS), cardiac, gastrointestinal, and musculoskeletal manifestations, though other organ systems not covered in this review can be involved. The following sections include the estimated frequencies, clinical presentation, relevant diagnostic tests, and management recommendations for the respective organ systems based on the current available evidence and the authors’ experience. Figure 1 gives an overview of the reported frequencies of manifestations of extrapulmonary sarcoidosis.
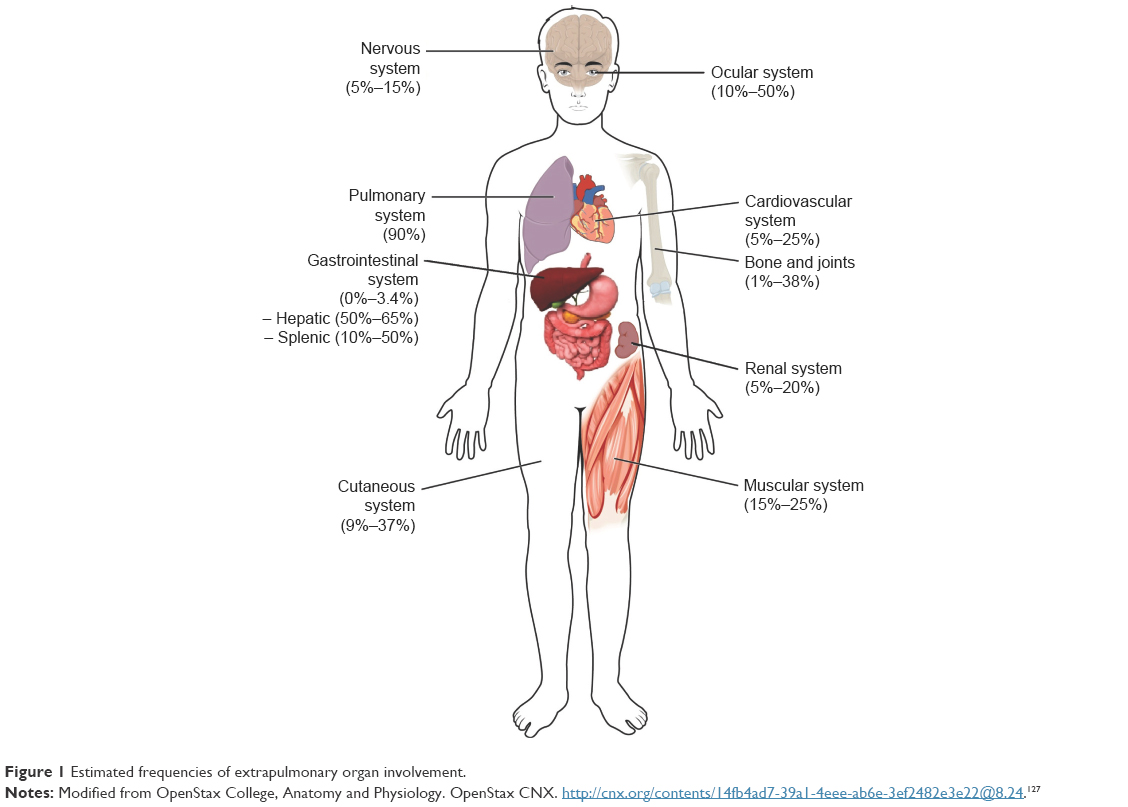 | Figure 1 Estimated frequencies of extrapulmonary organ involvement. |
Cutaneous sarcoidosis
Cutaneous lesions occur in 9%–37% of patients with extrapulmonary sarcoidosis.9 Both specific skin lesions with the presence of granulomas and nonspecific skin lesions lacking granulomatous inflammation can be histologically detected in sarcoidosis.10 One study found that 20.5% of 170 patients with sarcoidosis had erythema nodosum (EN), a nonspecific skin lesion lacking granulomas, as the most common skin manifestation.11 Also detected were skin plaques (4.3%), subcutaneous nodules (4.3%), maculopapular eruptions (3.7%), scar lesions (2.9%), psoriasiform plaques (0.9%), and lupus pernio (2.7%).11 The last, an indolent violaceous skin lesion affecting the face, had a higher association with pulmonary disease when compared to patients with EN or other lesions.11 Cutaneous sarcoidosis has also been documented to develop in scars and tattoos.12,13 The presence of EN is frequently found in Löfgren’s syndrome, the acute form of sarcoidosis with bilateral hilar lymphadenopathy and ankle (peri)arthritis, and usually portends a good prognosis.10 In addition, scarring and nonscarring alopecia,14 as well as nail changes (such as onycholysis, dystrophy, hyperkeratosis, and longitudinal ridging) may occur. Interestingly, nail changes seem to indicate bone involvement, and appropriate workup should therefore be considered. The World Association of Sarcoidosis and Other Granulomatous Diseases has proposed various end points for cutaneous sarcoidosis to be assessed in clinical trials (Physician Global Assessment, Sarcoidosis Activity and Severity Index, Lupus Pernio Activity and Severity Index, photographs, lesion counts, and skin biopsies).
Treatment of cutaneous sarcoidosis
Cutaneous lesions are treated only if they cause significant cosmetic disfigurement, psychological effects, or if the lesions are uncontrollable. Local GC therapy, with intralesional injections for thick plaque lesions, is first-line treament.15 Triamcinolone acetonide has reasonable availability, potency, and duration of action. Patients with progressive lesions will nevertheless require systemic therapy, most often GCs or hydroxychloroquine. Cases of lupus pernio achieved considerable improvement with halobetasol propionate 0.05% ointment twice daily for 10 weeks.16 Mild improvements were seen in 72% of 54 patients, and near or complete resolution was observed in 20% with systemic GCs.17 Antimalarials, antibacterials (tetracyclines, antimycobacterials), and immunosuppressive agents (MTX, thalidomide, apremilast, tumor necrosis factor alpha (TNFα) antagonists, such as infliximab and adalimumab, among others) have been investigated with variable success in refractory cases.18–26
Renal involvement
Although isolated renal involvement represents less than 5% of extrapulmonary sarcoidosis,2 the rate of renal involvement throughout the disease course is probably much higher.27 Sarcoidosis can involve the kidneys in three different ways: 1) hypercalciuria, which precedes the development of hypercalcemia, occurring in 50% and 10%, respectively, as a result of calcitriol-producing granulomatous burden, which may overwhelm the kidneys’ regulatory mechanisms and progress to renal insufficiency; 2) tubulointerstitial nephritis with or without granulomatous involvement; and 3) nephrocalcinosis and/or nephrolithiasis, which can occur as a result of hypercalcemia. These can occur independently or less frequently in combination.28 Co-occurrence with other renal disease, primarily IgA nephropathy, is not uncommon.28 Although sarcoidosis-associated end-stage renal disease is very uncommon and more likely due to other causes, a significant degree of renal insufficiency was common in one retrospective series, and hypercalcemia might be a contributing factor.29,30 Moreover, relapses may occur in up to a quarter of patients who undergo kidney transplantation because of sarcoidosis and may negatively impact graft survival.31
One cohort study revealed that up to 3.6% of sarcoidosis cases had nephrolithiasis at first visit, and another 2.7% of these patients were asymptomatic.32 Therefore, sarcoidosis should be considered if no other cause of nephrolithiasis is identified. Rare presentations include pseudotumors or mechanical complications, such as hydronephrosis, if retroperitoneal structures are affected by granulomatous inflammation.33–35
All patients diagnosed with sarcoidosis should be evaluated for the presence of renal involvement to prevent significant chronic kidney disease. Workup includes serum creatinine, blood urea nitrogen, estimated glomerular filtration rate, protein and calcium in both serum and urine, and screening of the urinary sediment for casts of red or white blood cells. 25-hydroxvitamin D3, 1,25-dihydroxyvitamin D3, and parathyroid hormone should be measured in patients with hypercalcemia or hypercalciuria to assess the degree and cause of calcium dysregulation. Renal biopsy remains the gold standard to confirm renal sarcoidosis. Nevertheless, histological findings are not specific for sarcoidosis, requiring infection and drug hypersensitivity to be excluded, as these are more frequent causes of interstitial nephritis.36
Treatment of renal sarcoidosis
GCs are the mainstay of treatment in interstitial nephritis, and are often very effective.36 They have also been used successfully in hypercalcemia and hypercalciuria, although many times discontinuation of vitamin D supplements is sufficient to normalize calcium levels.30 When GCs fail, ketoconazole or hydroxychloroquine have been used successfully in such cases.37 Additional immunosuppressive agents, such as azathioprine (AZA) or mycophenolate mofetil, have (albeit rarely) also been used in cases of granulomatous interstitial nephritis not responding to GCs.37
The long-term prognosis of renal sarcoidosis seems to correlate with response to therapy after 1 month of treatment and inversely with the initial fibrosis score on histopathological examination.29 In refractory renal sarcoidosis or failure of treatment leading to end-stage renal disease, renal transplantation is an option.38 Recurrence of sarcoidosis in transplanted kidneys has been reported and requires close surveillance, as is necessary in other diseases. Relapse after transplantation has been managed with infliximab in a steroid-resistant case.39
Neurosarcoidosis
Involvement of the CNS or peripheral nervous system is present in 5%–15% of cases, and represents one of the more serious complications of sarcoidosis.40 Postmortem studies indicate that isolated neurosarcoidosis occurs in ~17% of cases and that subclinical neurosarcoidosis may be more prevalent but underdiagnosed, with a rate of antemortem diagnosis of ~50% of cases.4,41 Symptoms relating to involvement of the cranial nerves, hypothalamus, or pituitary glands have been reported to be the initial clinical manifestation in up to 50%–70% of neurosarcoidosis cases.42 These can be present simultaneously in up to a third of patients or be isolated manifestations.42 A biopsy of the CNS is required for a definitive diagnosis, especially in isolated cases.43 However, in cases with biopsy-proven sarcoidosis of other sites, the diagnosis can be made without a CNS biopsy. The WASOG criteria for neurosarcoidosis are shown in Table 1.
 | Table 1 The WASOG organ-assessment tool for neurosarcoidosis |
The most common neurological manifestation is unilateral or bilateral cranial neuropathy of the facial or optic nerves.40,44 Cranial nerve palsy may be caused by nerve granulomas, increased intracranial pressure, or granulomatous basal meningitis. Moreover, acute or chronic aseptic meningitis may occur as well. Space-occupying cerebral lesions of sarcoidosis can involve the parenchyma and extradural or subdural spaces.45 These lesions are most commonly found in the hypothalamus and pituitary glands, and may result in endocrine manifestations, including diabetes insipidus, adrenal and pituitary failure, and amenorrhea–galactorrhea syndrome.46–48 The significance of seizures on the overall prognosis is not clear, since reports have yielded conflicting results.49,50 Psychiatric manifestations, such as psychosis, may also be present, but are quite rare, whereas cognitive failure was more prevalent in one study.51,52 Spinal cord involvement is another rare manifestation, and usually presents with leg weakness and paresthesias; most often, the thoracic segments are involved.53
Involvement of the peripheral nervous system may present with deficits of either sensory or motor function, and possibly both. Symptoms ranging from mononeuritis multiplex to Guillain–Barré-like syndromes, as well as polyradiculopathy or polyneuropathy, can occur. Electromyography often shows a multifocal axonal neuropathy pattern.54
Small-fiber neuropathy (SFN) has been frequently reported in sarcoidosis over the past few years, and can easily be missed. Symptoms include pain and dysesthesia.55 Standard nerve-conduction testing to measure large-nerve function and quantitative techniques for assessment of small-nerve fibers are not done routinely in all sarcoidosis patients. If collected carefully, skin-punch biopsy is helpful in diagnosing SFN by showing lower intraepidermal nerve-fiber density.56
A definitive diagnosis of neurosarcoidosis is challenging, due to the difficulty of obtaining biopsies without causing any damage. Therefore, brain MRI is considered the most sensitive noninvasive test for neurosarcoidosis, but lacks specificity.45 Lumbar punctures are reported to be normal in up to a third of patients, pathologic cerebrospinal fluid (CSF) findings include mild pleocytosis, increased protein levels, and mild hypoglycorrhachia, which are similar to those seen in multiple sclerosis or systemic lupus erythematosus.45 Additionally, the usefulness of CSF angiotensin-converting enzyme (ACE) levels is still a matter of debate, because it can also be found in conditions other than sarcoidosis.45,57,58
Treatment of neurosarcoidosis
Patients with confirmed systemic sarcoidosis and testing suggestive of CNS involvement should be treated immediately, due to the potentially high morbidity and mortality of neurosarcoidosis. GCs are first-line treatment, with a typical initial dose of 1 mg/kg/day. Severe cases may warrant a pulse of methylprednisolone at doses of 500–1,000 mg/day for 3 consecutive days and subsequent oral tapering. Alternative treatments, such as MTX, AZA, cyclosporine A (CsA), and cyclophosphamide (CYC), have been successfully used in refractory disease or when adverse effects to GCs are significant, but this is based on experience rather than evidence.59 The response rate to MTX in neurosarcoidosis is nearly 60%, similar to that in pulmonary sarcoidosis.60 CsA was found to reduce the GC dose to 30%–58% of the initial stabilization dose, but nonetheless deterioration occurred in four of the six reported patients.61 The use of short-course pulse CYC given every 2–4 weeks demonstrated improvement of MRI or CSF abnormalities in seven of seven patients, and symptoms improved in four of seven and nine of ten patients in the respective studies.60,62 The dose used was initially 500 mg, then 750 mg for the second dose and a maintenance dose of 1 g given approximately every 3 weeks.60,62 Mycophenolate mofetil has been reported to be effective in CNS sarcoid, but not in sarcoid myopathy,63 but this case series was small, and firm conclusions cannot be drawn.63
In corticosteroid-refractory disease or when rapid response is needed to avoid permanent disability, several case reports have indicated positive responses to infliximab.64 A review of 34 successful cases showed that 14 had improvement between the first and third infusions,64 and infliximab might be more effective than CYC.65 TNFα blockers also had positive effects on cognitive failure.52 One double-blind randomized controlled trial found a positive effect of ARA 290, an immunomodulating erythropoietin-derived compound, on symptoms of SFN in 22 patients.66 Lastly, radiation therapy with 20–25 Gy may be considered in patients who do not respond to drug therapy.67
Cardiac sarcoidosis
Cardiac sarcoidosis commonly coexists with systemic sarcoidosis, rather than as an isolated finding. The presence of ocular sarcoidosis with choroidal involvement has, for example, been suggested to reflect vascular endothelial dysfunction and coexist with cardiac disease, thus requiring evaluation for cardiac sarcoidosis.68
The ethnic background of patients is of importance, since cardiac sarcoidosis has been reported much more frequently in Japanese patients (~21%–58%) when compared to Caucasians or African-Americans (~13%–25%) based on autopsy studies.69 There is a high rate of subclinical involvement, making overt cardiac sarcoidosis a rather rare manifestation.3
Cardiac disease ranges from the incidental discovery in asymptomatic patients to life-threatening disease with sudden cardiac death. Patients commonly present with heart block, ventricular tachycardia/unsustained ventricular fibrillation, or heart failure due to cardiomyopathy.70
History-taking is an essential element at both baseline and follow-up visits to detect symptoms that warrant further investigation.71 Any structural component, as well as any layer (endocardium [heart valves], myocardium, and pericardium) of the heart can be affected. Clinical symptoms will depend on the structures involved, and may include palpitations, syncope, shortness of breath, chest pain, or peripheral edema.69,72 With the widespread use of implantable cardioverter defibrillators (ICDs), the main causes of death in patients with cardiac sarcoidosis is progressive heart failure due to granulomatous infiltration of the myocardium, the latter accounting for ~25% of deaths.73,74
The possibility of cardiac sarcoidosis should be considered in: 1) adults under 55 years of age with new-onset electrocardiogram abnormalities, decreased systolic ejection fraction, or cardiac symptoms after excluding coronary artery disease; 2) adults under 55 years of age with sustained atrioventricular block (second or third degree); 3) patients of any age with sustained ventricular tachycardia and/or cardiomyopathy; and 4) all patients with diagnosed sarcoidosis of other sites.73,75,76 The different proposed diagnostic criteria for cardiac sarcoidosis are shown in Table 2.
 | Table 2 Criteria in use for the assessment of cardiac sarcoidosis |
Workup commonly includes an electrocardiogram, 24-hour Holter monitoring and transthoracic echocardiography (TTE). The optimal diagnostic strategy still needs to be established; however, patients with cardiac sarcoidosis had significantly more abnormal Holter monitoring and TTE findings.71 The sensitivity and specificity for the detection of cardiac sarcoidosis by Holter monitoring were reported with 67% and 80%, respectively, making its usefulness controversial, but it is relatively inexpensive.77
TTE shows variable and nonspecific findings, including interventricular thinning, regional wall abnormalities, or aneurysmal distortion. These findings are similar to those found in ischemic heart disease; therefore, coronary angiography is recommended to exclude coronary artery disease.69
Over the last few years, other noninvasive imaging modalities have emerged and been tested in cardiac sarcoidosis. Cardiac MRI (CMR) and 18F-fluorodeoxyglucose PET (FDG-PET) have high sensitivity and specificity and are of prognostic relevance.78–80 Especially, patients with late gadolinium enhancement detected on CMR are at risk for adverse events, even with preserved left ventricular ejection fraction.81
Radionuclide imaging with gallium 67, thallium 201, technetium, or sestamibi are alternative tests to diagnose and monitor cardiac sarcoidosis that are useful when MRI is contraindicated or not available.82 Nevertheless, due to radiation exposure and associated imaging artifacts that may produce nonspecific results, they have been replaced with CMR and FDG-PET in most centers.82
Treatment of cardiac sarcoidosis
Cardiac sarcoidosis mandates rapid treatment, because of the risk of sudden death and progressive heart failure. GCs are the treatment of choice, and no difference in survival rates has been found between higher (>30 mg/day) and lower doses (<30 mg/day) of prednisone.83 A recent systematic review found beneficial effects in the reported literature only for atrioventricular conduction disturbances.84 The body of evidence is of very limited quality, since controlled trials for cardiac sarcoidosis have not been conducted.
Long-term improvement has been reported with the use of 6 mg MTX weekly in addition to prednisone versus prednisone alone. Ejection fraction and N-terminal prohormone brain natriuretic peptide levels were significantly better with combination therapy at 3 years, but not at 5 years.85
Other alternative treatments, including MTX, CYC and CsA, have been studied retrospectively, and resulted in improvement or recovery in ten of eleven patients with cardiac sarcoidosis.86 Infliximab has been reported as an effective therapy for cardiac sarcoidosis in case studies.87,88 TNFα blockers are, however, contraindicated in patients with advanced heart failure (New York Heart Association III or IV), as they may worsen symptoms.
Antiarrhythmic therapies have not been studied systematically, and firm recommendations cannot be made based on the available evidence. Amiodarone or sotalol are sometimes used.69 Sudden cardiac death can be prevented by ICDs, and approximately a third of patients with ICDs receive appropriate therapies for ventricular arrhythmias.89 The expert-consensus statement on management of arrhythmias associated with sarcoidosis by the Heart Rhythm Society90 recommends ICD implantation in patients with: 1) spontaneous sustained ventricular arrhythmias, including prior cardiac arrest, and/or 2) left ventricular ejection fraction <35% despite optimal medical therapy and a period of immunosuppression (if there is active inflammation).90
Ultimately, heart transplantation may be considered when all other therapies have failed, apparently without increased complications.91,92 Recurrence of sarcoidosis in the allograft has been reported, but seems to be rare.93
Gastrointestinal and hepatic sarcoidosis
Gastrointestinal sarcoidosis is very rare, accounting for only 0%–3.4% of cases, based on autopsy studies. By contrast, autopsy frequently shows hepatic involvement in as many as 80%, depending on the series.94 However, asymptomatic elevation in liver-function tests in the context of known sarcoidosis is the most common presentation in approximately a third of patients, and occurs more frequently in African-Americans than in Caucasians.95 Of note, alkaline phosphatase seems to be more consistently elevated than aminotransferases in patients with hepatic sarcoidosis.95 Clinical manifestations include hepatomegaly, fatigue, right upper-quadrant abdominal pain with pruritus (5%–15%), fever, jaundice, and weight loss (less than 5%).10 Rare complications include Budd–Chiari syndrome or hilar lymphadenopathy, cholestatic liver disease mimicking primary biliary cirrhosis, primary sclerosing cholangitis, or obstructive jaundice.96,97
Splenic involvement is most often detected by imaging rather than symptoms or laboratory abnormalities, and has been reported with similar frequency as hepatic involvement. It may present with constitutional symptoms and marked splenomegaly in up to 6% of cases.98,99
Overt gastric or intestinal sarcoidosis presents with fullness, weight loss, protein-losing enteropathy, and signs of obstruction or gastrointestinal bleeding, findings also seen in Crohn’s disease.100,101 It is thus important to rule out other common granulomatous disorders that affect the intestines, as both have been reported to coexist in rare cases.102,103
A diagnosis of gastrointestinal or hepatosplenic involvement can be made by endoscopy, computed tomography, or ultrasound with or without contrast media and subsequent biopsy.104–106 Abdominal computed tomography shows hepatomegaly or nodules, and ultrasonography can reveal hypo- or hyperechoic nodules when compared to the liver parenchyma.106 Noncaseating granulomas of the liver are present in up to 65% of patients in systemic sarcoidosis, and other causes, such as tuberculosis, fungal, or other infections, and drug toxicity, should be excluded.107
Treatment of gastrointestinal sarcoidosis
Asymptomatic hepatic and splenic sarcoidosis does not require treatment. However, in symptomatic patients, GCs can reduce liver and spleen size and number of granulomas and improve organ function to some extent. Nevertheless, the effect on disease course and development of portal hypertension or hepatic fibrosis is limited.108 Steroid-sparing agents have been used with some success, and agents such as MTX, AZA, antimalarials, and ursodeoxycholic acid all have been used.109 The latter has been used in patients presenting with signs of cholestatic jaundice.110,111 AZA is preferred over MTX by some, although both are potentially hepatotoxic.107
In end-stage liver disease, orthotopic liver transplantation has been successfully used as a treatment modality. Recurrences may occur in the allograft,112 and allograft-survival rates have been reported to be comparable with other diseases (86% after 5 years).113 Another study showed significantly lower rates compared to patients transplanted for primary sclerosing cholangitis or primary biliary cirrhosis (5-year graft-survival rates of 60% versus 75%).114
Splenectomy is indicated only in rare situations after failure of medical treatment. These include massive splenomegaly, severe hypersplenism, suspicion of lymphoma or other malignancy, and as a preventive measure against splenic rupture.115
Musculoskeletal sarcoidosis
Musculoskeletal sarcoidosis is reported in approximately 15%–25% of cases.10 From a clinical perspective, differentiation of arthritis in acute/chronic and peripheral/axial types seems most useful. Acute arthritis in the context of sarcoidosis most frequently arises in Löfgren’s syndrome (bilateral hilar adenopathy, EN, and bilateral ankle swelling). Ultrasound-based studies have revealed that true arthritis of the ankles is the exception, and swelling is predominantly due to soft-tissue swelling and tenosynovitis.116,117 This has been confirmed in a small case series using MRI.118 The presence of three of four criteria – 1) symmetrical ankle arthritis, 2) symptoms less than 2 months, 3) age below 40 years, and 4) EN reaching sensitivity and specificity of 93% and 99%, respectively – is used for diagnosing acute arthritis related to sarcoidosis.119
Chronic sarcoidosis arthritis can mimic other forms of inflammatory arthritis; dactylitis, a feature typical of psoriatic arthritis and other seronegative spondylarthropathies, may also occur in sarcoidosis.120 Conventional radiographs show a reticular, lacy pattern of the phalanges with singular or numerous cystic lesions, and in some cases, acroosteolysis may be observed and “punched-out lesions” due to granulomatous inflammations can occur.121 A comprehensive assessment to differentiate other possible causes of arthritis is necessary, since sarcoidosis and other inflammatory diseases may simultaneously be present.121
Axial sarcoidosis can involve the vertebral bodies or the sacroiliac joints. The frequency of sacroiliitis, the hallmark of seronegative spondylarthropathies, ranges from 6.6% to14.3% in the literature,122,123 and it may not always be possible to distinguish these two entities.123 A negative human leukocyte antigen B27, however, would favor a diagnosis of sarcoidosis, especially when other sarcoidosis features are present.
Imaging is an important tool for the evaluation of musculoskeletal sarcoidosis. Conventional radiographs are typically the first method. To detect inflammatory changes, ultrasound may be used, but this has mostly been studied in the context of Löfgren’s syndrome.116,117 Other, potentially useful tests include MRI and FDG-PET.124 When vertebral sarcoidosis is suspected and no other features of sarcoidosis are present, a histopathological confirmation is mandatory to exclude other possible causes, especially malignancy or tuberculosis, as these cannot be differentiated with imaging alone.
Treatment of musculoskeletal sarcoidosis
For the acute (peri)arthritis seen in Löfgren’s syndrome, nonsteroidal analgesics may be all that is necessary, but up to two-thirds of patients will nevertheless require additional treatment with GCs.125 However, the chances of complete remission after a few weeks to months are very high in Löfgren’s syndrome.10
Chronic arthritis may or may not be controlled with GCs alone. If GCs fail to control the symptoms, antimalarials and disease-modifying antisarcoid drugs have both been used with success, but controlled clinical trials for sarcoidosis arthritis have not been conducted. MTX is typically the preferred agent for these patients. In severe cases, TNFα antagonists, such as infliximab or adalimumab, may also be used. A management algorithm for patients unresponsive or intolerant to GCs has been proposed.121
Conclusion
Extrapulmonary involvement is often underdiagnosed antemortem, but can be life-threatening if CNS or cardiac sarcoidosis occur. Subclinical involvement is common, making history-taking and a standardized review of systems essential. Diagnostic tests and appropriate imaging should be guided by history and clinical findings, and biopsies performed if necessary to exclude other causes of organ dysfunction.
The majority of patients will have either spontaneous remission or resolution within 1–2 years. However, up to a third of cases relapse, remain unresponsive, or progress despite treatment with GCs. These cases are considered to have refractory or chronic sarcoidosis. Guidelines are available for many of the manifestations, but firm treatment recommendations cannot be made, due to the lack of high-quality studies. Our suggested treatment approach for various extrapulmonary manifestations of sarcoidosis is depicted in Figure 2.
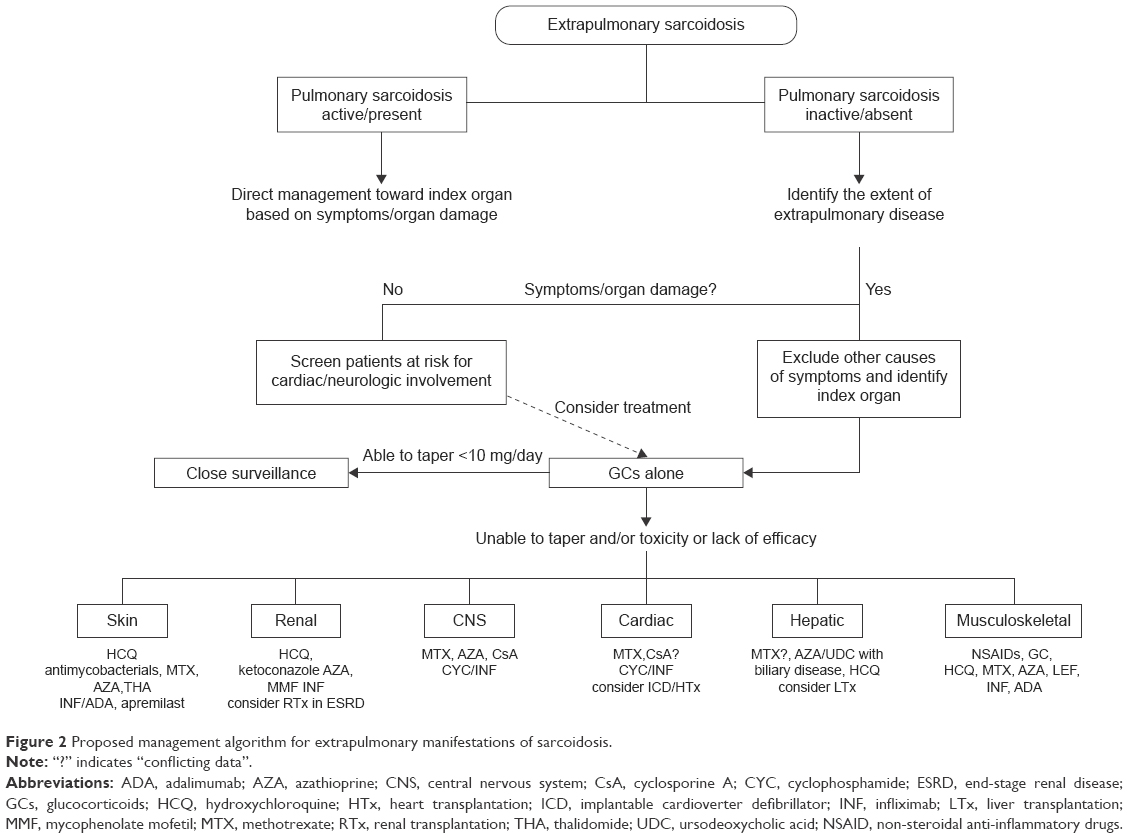 | Figure 2 Proposed management algorithm for extrapulmonary manifestations of sarcoidosis. |
Adding or initiating disease-modifying antisarcoid drugs is necessary for many manifestations, and MTX is usually the first agent used by most sarcoidosis experts. Third-line or experimental therapies may be considered if other options fail. Ultimately, organ transplantation is a viable option for cardiac, renal, and liver sarcoidosis unresponsive to medical therapy, and outcomes seem to be comparable to other allograft recipients.
Acknowledgment
The work was supported by a grant from the Bernie Mac Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
James DG, Carstairs LS, Trowell J, Sharma OP. Treatment of sarcoidosis: report of a controlled therapeutic trial. Lancet. 1967;2(7515):526–528. | ||
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155–1167. | ||
Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1885–1889. | ||
Hunninghake GW, Costabel U, Ando M, et al. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16(2):149–173. | ||
Korsten P, Mirsaeidi M, Sweiss NJ. Nonsteroidal therapy of sarcoidosis. Curr Opin Pulm Med. 2013;19(5):516–523. | ||
Korsten P, Strohmayer K, Baughman RP, Sweiss NJ. Refractory pulmonary sarcoidosis: proposal of a definition and recommendations for the diagnostic and therapeutic approach. Clin Pulm Med. 2016;23(2):67–75. | ||
Baughman RP, Grutters JC. New treatment strategies for pulmonary sarcoidosis: antimetabolites, biological drugs, and other treatment approaches. Lancet Respir Med. 2015;3(10):813–822. | ||
Wijsenbeek MS, Culver DA. Treatment of sarcoidosis. Clin Chest Med. 2015;36(4):751–767. | ||
Mangas C, Fernández-Figueras MT, Fité E, Fernández-Chico N, Sàbat M, Ferrándiz C. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33(12):772–777. | ||
Judson MA. Extrapulmonary sarcoidosis. Semin Respir Crit Care Med. 2007;28(1):83–101. | ||
Yanardağ H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97(8):978–982. | ||
Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol. 2005;141(7):869–872. | ||
Dittler EL. Boeck’s sarcoid in postoperative abdominal scar. N Y State J Med. 1954;54(19):2738. | ||
House NS, Welsh JP, English JC. Sarcoidosis-induced alopecia. Dermatol Online J. 2012;18(8):4. | ||
Callen JP. Intralesional corticosteroids. J Am Acad Dermatol. 1981;4(2):149–151. | ||
Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995;131(5):617–618. | ||
Stagaki E, Mountford WK, Lackland DT, Judson MA. The treatment of lupus pernio: results of 116 treatment courses in 54 patients. Chest. 2009;135(2):468–476. | ||
Veien NK, Brodthagen H. Cutaneous sarcoidosis treated with methotrexate. Br J Dermatol. 1977;97(2):213–216. | ||
Drake WP, Oswald-Richter K, Richmond BW, et al. Oral antimycobacterial therapy in chronic cutaneous sarcoidosis: a randomized, single-masked, placebo-controlled study. JAMA Dermatol. 2013;149(9):1040–1049. | ||
Baughman RP, Judson MA, Ingledue R, Craft NL, Lower EE. Efficacy and safety of apremilast in chronic cutaneous sarcoidosis. Arch Dermatol. 2012;148(2):262–264. | ||
Baughman R, Lo K, Guzzo C, Barnathan E. Efficacy of infliximab in improving lupus pernio, in patients receiving treatment for chronic pulmonary sarcoidosis. J Am Acad Dermatol. 2006;54(3):AB13. | ||
Pariser RJ, Paul J, Hirano S, Torosky C, Smith M. A double-blind, randomized, placebo-controlled trial of adalimumab in the treatment of cutaneous sarcoidosis. J Am Acad Dermatol. 2013;68(5):765–773. | ||
Droitcourt C, Rybojad M, Porcher R, et al. A randomized, investigator-masked, double-blind, placebo-controlled trial on thalidomide in severe cutaneous sarcoidosis. Chest. 2014;146(4):1046–1054. | ||
Steen T, English JC. Oral minocycline in treatment of cutaneous sarcoidosis. JAMA Dermatol. 2013;149(6):758–760. | ||
Jones E, Callen JP. Hydroxychloroquine is effective therapy for control of cutaneous sarcoidal granulomas. J Am Acad Dermatol. 1990;23(3 Pt 1):487–489. | ||
Baughman RP, Judson MA, Lower EE, et al. Infliximab for chronic cutaneous sarcoidosis: a subset analysis from a double-blind randomized clinical trial. Sarcoidosis Vasc Diffuse Lung Dis Off J WASOG World Assoc Sarcoidosis Granulomatous Disord. 32(4):289–295. | ||
Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20(2):126–132. | ||
Löffler C, Löffler U, Tuleweit A, Waldherr R, Uppenkamp M, Bergner R. Renal sarcoidosis: epidemiological and follow–up data in a cohort of 27 patients. Sarcoidosis Vasc Diffuse Lung Dis. 2015;31(4):306–315. | ||
Mahévas M, Lescure FX, Boffa JJ, et al. Renal sarcoidosis: clinical, laboratory, and histologic presentation and outcome in 47 patients. Medicine (Baltimore). 2009;88(2):98–106. | ||
Baughman RP, Janovcik J, Ray M, Sweiss N, Lower EE. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30(2):113–120. | ||
Aouizerate J, Matignon M, Kamar N, et al. Renal transplantation in patients with sarcoidosis: a French multicenter study. Clin J Am Soc Nephrol. 2010;5(11):2101–2108. | ||
Rizzato G, Colombo P. Nephrolithiasis as a presenting feature of chronic sarcoidosis: a prospective study. Sarcoidosis Vasc Diffuse Lung Dis. 1996;13(2):167–172. | ||
Duveau A, Sayegh J, Beloncle F, Moreau A, Subra JF, Augusto JF. Pseudotumours: an atypical presentation of renal sarcoidosis. QJM. 2013;106(10):947–949. | ||
Miyazaki E, Tsuda T, Mochizuki A, et al. Sarcoidosis presenting as bilateral hydronephrosis. Intern Med. 1996;35(7):579–582. | ||
Godin M, Fillastre JP, Ducastelle T, Hemet J, Morere P, Nouvet G. Sarcoidosis. Retroperitoneal fibrosis, renal arterial involvement, and unilateral focal glomerulosclerosis. Arch Intern Med. 1980;140(9):1240–1242. | ||
Korsten P, Müller GA. [Interstitial nephritis in rheumatic diseases]. Z Rheumatol. 2015;74(4):290–299. German. | ||
Hilderson I, Van Laecke S, Wauters A, Donck J. Treatment of renal sarcoidosis: is there a guideline? Overview of the different treatment options. Nephrol Dial Transplant. 2014;29(10):1841–1847. doi:10.1093/ndt/gft442. | ||
Göbel U, Kettritz R, Schneider W, Luft F. The protean face of renal sarcoidosis. J Am Soc Nephrol. 2001;12(3):616–623. | ||
Srivastava S, Rajakariar R, Ashman N, Raftery M, Brown H, Martin JE. Infliximab as long-term maintenance in steroid-resistant and recurrent sarcoidosis in a renal transplant with central nervous system involvement. Clin Kidney J. 2012;5(1):53–55. | ||
Stern BJ, Krumholz A, Johns C, Scott P, Nissim J. Sarcoidosis and its neurological manifestations. Arch Neurol. 1985;42(9):909–917. | ||
Iwai K, Tachibana T, Takemura T, Matsui Y, Kitaichi M, Kawabata Y. Pathological studies on sarcoidosis autopsy. I. Epidemiological features of 320 cases in Japan. Acta Pathol Jpn. 1993;43(7–8):372–376. | ||
Nozaki K, Judson MA. Neurosarcoidosis: clinical manifestations, diagnosis and treatment. Presse Méd. 2012;41(6 Pt 2):e331–e348. | ||
Zajicek JP, Scolding NJ, Foster O, et al. Central nervous system sarcoidosis – diagnosis and management. QJM. 1999;92(2):103–117. | ||
Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM. 2009;102(7):449–460. | ||
Hoitsma E, Faber CG, Drent M, Sharma OP. Neurosarcoidosis: a clinical dilemma. Lancet Neurol. 2004;3(7):397–407. | ||
Murialdo G, Tamagno G. Endocrine aspects of neurosarcoidosis. J Endocrinol Invest. 2002;25(7):650–662. | ||
Tamagno G, Murialdo G. Amenorrhea-galactorrhea syndrome as an uncommon manifestation of isolated neurosarcoidosis. Ann Ital Med Int. 2001;16(4):260–266. | ||
Bullmann C, Faust M, Hoffmann A, et al. Five cases with central diabetes insipidus and hypogonadism as first presentation of neurosarcoidosis. Eur J Endocrinol. 2000;142(4):365–372. | ||
Krumholz A, Stern BJ, Stern EG. Clinical implications of seizures in neurosarcoidosis. Arch Neurol. 1991;48(8):842–844. | ||
Joseph FG, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009;80(3):297–304. | ||
O’Brien GM, Baughman RP, Broderick JP, Arnold L, Lower EE. Paranoid psychosis due to neurosarcoidosis. Sarcoidosis. 1994;11(1):34–36. | ||
Elfferich MD, Nelemans PJ, Ponds RW, De Vries J, Wijnen PA, Drent M. Everyday cognitive failure in sarcoidosis: the prevalence and the effect of anti-TNF-α treatment. Respir Int Rev Thorac Dis. 2010;80(3):212–219. | ||
Sohn M, Culver DA, Judson MA, Scott TF, Tavee J, Nozaki K. Spinal cord neurosarcoidosis. Am J Med Sci. 2014;347(3):195–198. | ||
Scott TS, Brillman J, Gross JA. Sarcoidosis of the peripheral nervous system. Neurol Res. 1993;15(6):389–390. | ||
Hoitsma E, Marziniak M, Faber CG, et al. Small fibre neuropathy in sarcoidosis. Lancet. 2002;359(9323):2085–2086. | ||
Myers MI, Peltier AC. Uses of skin biopsy for sensory and autonomic nerve assessment. Curr Neurol Neurosci Rep. 2013;13(1):323. | ||
Tahmoush AJ, Amir MS, Connor WW, et al. CSF-ACE activity in probable CNS neurosarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19(3):191–197. | ||
Dale JC, O’Brien JF. Determination of angiotensin-converting enzyme levels in cerebrospinal fluid is not a useful test for the diagnosis of neurosarcoidosis. Mayo Clin Proc. 1999;74(5):535. | ||
Hoitsma E, Drent M, Sharma OP. A pragmatic approach to diagnosing and treating neurosarcoidosis in the 21st century. Curr Opin Pulm Med. 2010;16(5):472–479. | ||
Lower EE, Broderick JP, Brott TG, Baughman RP. Diagnosis and management of neurological sarcoidosis. Arch Intern Med. 1997;157(16):1864–1868. | ||
Stern BJ, Schonfeld SA, Sewell C, Krumholz A, Scott P, Belendiuk G. The treatment of neurosarcoidosis with cyclosporine. Arch Neurol. 1992;49(10):1065–1072. | ||
Doty JD, Mazur JE, Judson MA. Treatment of corticosteroid-resistant neurosarcoidosis with a short-course cyclophosphamide regimen. Chest. 2003;124(5):2023–2026. | ||
Androdias G, Maillet D, Marignier R, et al. Mycophenolate mofetil may be effective in CNS sarcoidosis but not in sarcoid myopathy. Neurology. 2011;76(13):1168–1172. | ||
Lorentzen AO, Sveberg L, Midtvedt Ø, Kerty E, Heuser K. Overnight response to infliximab in neurosarcoidosis: a case report and review of infliximab treatment practice. Clin Neuropharmacol. 2014;37(5):142–148. | ||
Sodhi M, Pearson K, White ES, Culver DA. Infliximab therapy rescues cyclophosphamide failure in severe central nervous system sarcoidosis. Respir Med. 2009;103(2):268–273. | ||
Heij L, Niesters M, Swartjes M, et al. Safety and efficacy of ARA 290 in sarcoidosis patients with symptoms of small fiber neuropathy: a randomized, double-blind pilot study. Mol Med. 2012;18: 1430–1436. | ||
Menninger MD, Amdur RJ, Marcus RB. Role of radiotherapy in the treatment of neurosarcoidosis. Am J Clin Oncol. 2003;26(4): e115–e118. | ||
Liu D, Birnbaum AD. Update on sarcoidosis. Curr Opin Ophthalmol. 2015;26(6):512–516. | ||
Dubrey SW, Sharma R, Underwood R, Mittal T. Cardiac sarcoidosis: diagnosis and management. Postgrad Med J. 2015;91(1077):384–394. | ||
Dubrey S, Shah S, Hardman T, Sharma R. Sarcoidosis: the links between epidemiology and aetiology. Postgrad Med J. 2014;90(1068):582–589. | ||
Mehta D, Lubitz SA, Frankel Z, et al. Cardiac involvement in patients with sarcoidosis: diagnostic and prognostic value of outpatient testing. Chest. 2008;133(6):1426–1435. | ||
Morimoto T, Azuma A, Abe S, et al. Epidemiology of sarcoidosis in Japan. Eur Respir J. 2008;31(2):372–379. | ||
Lynch JP, Hwang J, Bradfield J, Fishbein M, Shivkumar K, Tung R. Cardiac involvement in sarcoidosis: evolving concepts in diagnosis and treatment. Semin Respir Crit Care Med. 2014;35(3):372–390. | ||
Sekiguchi M, Yazaki Y, Isobe M, Hiroe M. Cardiac sarcoidosis: diagnostic, prognostic, and therapeutic considerations. Cardiovasc Drugs Ther. 1996;10(5):495–510. | ||
Hamzeh N, Steckman DA, Sauer WH, Judson MA. Pathophysiology and clinical management of cardiac sarcoidosis. Nat Rev Cardiol. 2015;12(5):278–288. | ||
Soejima K, Yada H. The work-up and management of patients with apparent or subclinical cardiac sarcoidosis: with emphasis on the associated heart rhythm abnormalities. J Cardiovasc Electrophysiol. 2009;20(5):578–583. | ||
Suzuki T, Kanda T, Kubota S, Imai S, Murata K. Holter monitoring as a noninvasive indicator of cardiac involvement in sarcoidosis. Chest. 1994;106(4):1021–1024. | ||
Blankstein R, Osborne MT, Dorbala S, Di Carli MF. Cardiac positron emission tomography as a prognostic indicator of cardiac sarcoidosis. J Am Coll Cardiol. 2014;63(23):2590. | ||
Ahmadian A, Brogan A, Berman J, et al. Quantitative interpretation of FDG PET/CT with myocardial perfusion imaging increases diagnostic information in the evaluation of cardiac sarcoidosis. J Nucl Cardiol. 2014;21(5):925–939. | ||
Ise T, Hasegawa T, Morita Y, et al. Extensive late gadolinium enhancement on cardiovascular magnetic resonance predicts adverse outcomes and lack of improvement in LV function after steroid therapy in cardiac sarcoidosis. Heart. 2014;100(15):1165–1172. | ||
Murtagh G, Laffin LJ, Beshai JF, et al. Prognosis of myocardial damage in sarcoidosis patients with preserved left ventricular ejection fraction: risk stratification using cardiovascular magnetic resonance. Circ Cardiovasc Imaging. 2016;9(1):e003738. | ||
Kouranos V, Wells AU, Sharma R, Underwood SR, Wechalekar K. Advances in radionuclide imaging of cardiac sarcoidosis. Br Med Bull. 2015;115(1):151–163. | ||
Yazaki Y, Isobe M, Hiroe M, et al. Prognostic determinants of long-term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88(9):1006–1010. | ||
Sadek MM, Yung D, Birnie DH, Beanlands RS, Nery PB. Corticosteroid therapy for cardiac sarcoidosis: a systematic review. Can J Cardiol. 2013;29(9):1034–1041. | ||
Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med. 2014;53(5):427–433. | ||
Chapelon-Abric C, de Zuttere D, Duhaut P, et al. Cardiac sarcoidosis: a retrospective study of 41 cases. Medicine (Baltimore). 2004;83(6):315–334. | ||
Chapelon-Abric C, Saadoun D, Biard L, et al. Long-term outcome of infliximab in severe chronic and refractory systemic sarcoidosis: a report of 16 cases. Clin Exp Rheumatol. 2015;33(4):509–515. | ||
Barnabe C, McMeekin J, Howarth A, Martin L. Successful treatment of cardiac sarcoidosis with infliximab. J Rheumatol. 2008;35(8):1686–1687. | ||
Schuller JL, Zipse M, Crawford T, et al. Implantable cardioverter defibrillator therapy in patients with cardiac sarcoidosis. J Cardiovasc Electrophysiol. 2012;23(9):925–929. | ||
Birnie DH, Sauer WH, Judson MA. Consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart. 2016;102(6):411–414. | ||
Valantine HA, Tazelaar HD, Macoviak J, et al. Cardiac sarcoidosis: response to steroids and transplantation. J Heart Transplant. 1987;6(4):244–250. | ||
Theofilogiannakos EK, Pettit SJ, Ghazi A, Rassl D, Lewis C, Parameshwar J. Heart transplantation for advanced heart failure due to cardiac sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(3):208–214. | ||
Oni AA, Hershberger RE, Norman DJ, et al. Recurrence of sarcoidosis in a cardiac allograft: control with augmented corticosteroids. J Heart Lung Transplant. 1992;11(2 Pt 1):367–369. | ||
Hercules HD, Bethlem NM. Value of liver biopsy in sarcoidosis. Arch Pathol Lab Med. 1984;108(10):831–834. | ||
Vatti R, Sharma OP. Course of asymptomatic liver involvement in sarcoidosis: role of therapy in selected cases. Sarcoidosis Vasc Diffuse Lung Dis. 1997;14(1):73–76. | ||
Alam I, Levenson SD, Ferrell LD, Bass NM. Diffuse intrahepatic biliary strictures in sarcoidosis resembling sclerosing cholangitis: case report and review of the literature. Dig Dis Sci. 1997;42(6):1295–1301. | ||
Delfosse V, de Leval L, De Roover A, et al. Budd-Chiari syndrome complicating hepatic sarcoidosis: definitive treatment by liver transplantation – a case report. Transplant Proc. 2009;41(8):3432–3434. | ||
Warshauer DM, Dumbleton SA, Molina PL, Yankaskas BC, Parker LA, Woosley JT. Abdominal CT findings in sarcoidosis: radiologic and clinical correlation. Radiology. 1994;192(1):93–98. | ||
Britt AR, Francis IR, Glazer GM, Ellis JH. Sarcoidosis: abdominal manifestations at CT. Radiology. 1991;178(1):91–94. | ||
Inomata M, Ikushima S, Awano N, et al. Upper gastrointestinal sarcoidosis: report of three cases. Intern Med. 2012;51(13):1689–1694. | ||
Munker M, Sharma OP. Fatal gastrointestinal haemorrhage in sarcoidosis: a previously unreported occurrence. Sarcoidosis. 1987;4(1):55–57. | ||
Sheth S, Patel R, Baddoura W, Nathan S. Metastatic sarcoidosis of terminal ileum and colon: very rare, but important to differentiate from Crohn’s disease. Poster presented at: American College of Gastroenterology Annual Scientific Meeting; October 17–22, 2014; Philadelphia, PA. | ||
Smiéjan JM, Cosnes J, Chollet-Martin S, et al. Sarcoid-like lymphocytosis of the lower respiratory tract in patients with active Crohn’s disease. Ann Intern Med. 1986;104(1):17–21. | ||
Caguiat K, Abramowitz M, Dubrovskaya V, Reka S, Garcia R, Chen NN. Gastric sarcoidosis: a case report. Poster presented at: American College of Gastroenterology Annual Scientific Meeting; October 17–22, 2014; Philadelphia, PA. | ||
Tana C, Dietrich CF, Schiavone C. Hepatosplenic sarcoidosis: contrast-enhanced ultrasound findings and implications for clinical practice. Biomed Res Int. 2014;2014:926203. | ||
Warshauer DM, Lee JK. Imaging manifestations of abdominal sarcoidosis. AJR Am J Roentgenol. 2004;182(1):15–28. | ||
Modaresi Esfeh J, Culver D, Plesec T, John B. Clinical presentation and protocol for management of hepatic sarcoidosis. Expert Rev Gastroenterol Hepatol. 2015;9(3):349–358. | ||
Ebert EC, Kierson M, Hagspiel KD. Gastrointestinal and hepatic manifestations of sarcoidosis. Am J Gastroenterol. 2008;103(12):3184–3192; quiz 3193. | ||
Ayyala US, Padilla ML. Diagnosis and treatment of hepatic sarcoidosis. Curr Treat Options Gastroenterol. 2006;9(6):475–483. | ||
Alenezi B, Lamoureux E, Alpert L, Szilagyi A. Effect of ursodeoxycholic acid on granulomatous liver disease due to sarcoidosis. Dig Dis Sci. 2005;50(1):196–200. | ||
Bécheur H, Dall’osto H, Chatellier G, et al. Effect of ursodeoxycholic acid on chronic intrahepatic cholestasis due to sarcoidosis. Dig Dis Sci. 1997;42(4):789–791. | ||
Fidler HM, Hadziyannis SJ, Dhillon AP, Sherlock S, Burroughs AK. Recurrent hepatic sarcoidosis following liver transplantation. Transplant Proc. 1997;29(5):2509–2510. | ||
Lipson EJ, Fiel MI, Florman SS, Korenblat KM. Patient and graft outcomes following liver transplantation for sarcoidosis. Clin Transplant. 2005;19(4):487–491. | ||
Vanatta JM, Modanlou KA, Dean AG, et al. Outcomes of orthotopic liver transplantation for hepatic sarcoidosis: an analysis of the United Network for Organ Sharing/Organ Procurement and Transplantation Network data files for a comparative study with cholestatic liver diseases. Liver Transpl. 2011;17(9):1027–1034. | ||
Sharma OP, Vucinic V, James DG. Splenectomy in sarcoidosis: indications, complications, and long-term follow-up. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19(1):66–70. | ||
Le Bras E, Ehrenstein B, Fleck M, Hartung W. Evaluation of ankle swelling due to Lofgren’s syndrome: a pilot study using B-mode and power Doppler ultrasonography. Arthritis Care Res. 2014;66(2):318–322. | ||
Kellner H, Späthling S, Herzer P. Ultrasound findings in Löfgren’s syndrome: is ankle swelling caused by arthritis, tenosynovitis or periarthritis? J Rheumatol. 1992;19(1):38–41. | ||
Anandacoomarasamy A, Peduto A, Howe G, Manolios N, Spencer D. Magnetic resonance imaging in Löfgren’s syndrome: demonstration of periarthritis. Clin Rheumatol. 2007;26(4):572–575. | ||
Visser H, Vos K, Zanelli E, et al. Sarcoid arthritis: clinical characteristics, diagnostic aspects, and risk factors. Ann Rheum Dis. 2002;61(6):499–504. | ||
Matuszak J, Durckel J, Sibilia J, Lipsker D, Blondet C, Imperiale A. Is sarcoid dactylitis worse than we expect? Arthritis Rheumatol. 2016;68(2):417. | ||
Sweiss NJ, Patterson K, Sawaqed R, et al. Rheumatologic manifestations of sarcoidosis. Semin Respir Crit Care Med. 2010;31(4):463–473. | ||
Erb N, Cushley MJ, Kassimos DG, Shave RM, Kitas GD. An assessment of back pain and the prevalence of sacroiliitis in sarcoidosis. Chest. 2005;127(1):192–196. | ||
Kobak S, Sever F, Ince O, Orman M. The prevalence of sacroiliitis and spondyloarthritis in patients with sarcoidosis. Int J Rheumatol. 2014;2014:289454. | ||
Mostard RL, Prompers L, Weijers RE, et al. F-18 FDG PET/CT for detecting bone and bone marrow involvement in sarcoidosis patients. Clin Nucl Med. 2012;37(1):21–25. | ||
Tejera B, Holgado S, Mateo L, Pego-Reigosa JM, Carnicero M, Olivé A. [Löfgren syndrome: a study of 80 cases]. Med Clin (Barc). 2014;143(4):166–169. Spanish. | ||
Judson MA, Costabel U, Drent M, et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(1):19–27. | ||
OpenStax College, Anatomy and Physiology. OpenStax CNX. Available from: http://cnx.org/contents/[email protected]. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.