Back to Journals » Clinical Ophthalmology » Volume 12
Management of chronic ocular sarcoidosis: challenges and solutions
Authors Matsou A, Tsaousis KT
Received 20 October 2017
Accepted for publication 20 January 2018
Published 19 March 2018 Volume 2018:12 Pages 519—532
DOI https://doi.org/10.2147/OPTH.S128949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Artemis Matsou,1 Konstantinos T Tsaousis2
1Second Department of Ophthalmology, Papageorgiou General Hospital, Aristotle University of Thessaloniki, Greece; 2Ophthalmology Department, Leicester Royal Infirmary, University Hospitals of Leicester, Leicester, UK
Background: Sarcoidosis constitutes one of the leading causes of ocular inflammation. Chronic ocular sarcoidosis can affect any segment of the eye and its adnexa, producing a wide range of clinical manifestations and severity. If left untreated, permanent visual impairment or even blindness may ensue. Treatment approaches vary from topical therapy to systemic agents that induce immunosuppression to different levels according to disease severity.
Objective: To review the published literature on the management options for chronic ocular sarcoidosis and provide a comprehensive list of available treatment strategies, including the newer biologics.
Summary: Ocular disease remains a challenging aspect of sarcoidosis and may even be the presenting sign of the disease. Prompt and effective therapy may reverse visual damage and prevent permanent loss of vision. Because of the complexity of the disease, a multidisciplinary approach is often required, with a view to addressing both the ocular and other systemic manifestations of sarcoidosis. Recent data suggest that achieving overall optimal systemic control is of paramount importance in controlling eye inflammation as well. Cytotoxic immunosuppressive agents for refractory chronic ocular disease, as well as biologic anti-TNFα therapies, have advanced the management of chronic disease and should be considered corticosteroid-sparing strategies before the onset of significant steroid-induced morbidity.
Keywords: ocular sarcoidosis, uveitis, immunosuppression, biologic agents, anti-TNFα
Introduction
Sarcoidosis is a chronic idiopathic granulomatous inflammatory disease that was first described by Sir Jonathan Hutchinson in 1878 as a dermatological disorder.1 It was later in 1909 that Heerfordt, a Danish ophthalmologist, reported for the first time the uveoparotid-fever syndrome (“Heerfordt syndrome”), thus introducing ocular involvement as a clinical manifestation of sarcoidosis.2 Known to be a systemic disorder, sarcoidosis affects multiple major organ systems, primarily the lungs in more than 90% of cases, which tend to be in the spotlight of clinical attention. Extrapulmonary disease frequently involves the lymph nodes, skin, eye, cardiovascular, musculoskeletal, gastrointestinal, renal, and central nervous systems.3 Despite it being known for more than 100 years, sarcoidosis remains an enigmatic disease, the etiology of which has still to be resolved, demonstrating a heterogeneous clinical course that often poses a diagnostic and treatment challenge for the treating physician.
Prevalence of this entity varies around the world, and large regional diversities exist. Some of the variability in prevalence and phenotype might relate to differences in surveillance procedures, diagnostic criteria employed by physicians, and genetic factors and exposure to environmental agents.4 Within Europe, it has been noted that Western Europeans are more commonly affected than Eastern Europeans, with Scandinavians having an incidence of as high as 19 per 100,000/year.5 UK estimates report an annual incidence of five per 100,000 of the population with an Irish preponderance. Recent data reveal that sarcoidosis is the prevailing cause of uveitis in Japan, where the highest rates of prevalence of ocular involvement have been reported (up to 79%) compared to any other racial or ethnic population.6,7 In the US, African Americans are reportedly affected at rates of 40 per 100,000/year, also being twice as likely to suffer ocular disease compared to Caucasian patients.5 Disease severity also appears to be greater among those of African origin in the US, while females are consistently affected to a greater extent across all racial and ethnic groups.4,5,8 Although it may occur at all ages, there is generally a predilection for young adults <40 years old, peaking in those aged 20–29 years, with a second peak observed in female patients of Japanese or Scandinavian origin at the age of 50 years.5,9 Sarcoidosis in children is relatively rare.
Due to the remarkable heterogeneity in presentation, clinical findings, and natural history, along with the absence of a unique identifying feature and biomarker, diagnosing sarcoidosis remains a challenge. It is usually based on compatible history and a constellation of clinical findings, combined with histological confirmation of nonnecrotizing granulomas in affected tissue and the exclusion of other granulomatous disease, such as tuberculosis.10 In the case of ocular sarcoidosis, biopsies of intraocular tissue in order to obtain a definitive diagnosis are not performed, due to the high risk of visual loss. In 2009, the International Workshop on Ocular Sarcoidosis, published criteria for diagnosing ocular sarcoidosis, identifying seven clinical signs suggestive of ocular sarcoidosis, five laboratory investigations in suspected ocular sarcoidosis, and four levels of certainty (Table 1) of a sarcoid diagnosis.11 Ocular adnexal involvement is not included in these criteria, as it can theoretically be confirmed by histopathological analysis of the accessible affected lesions. Despite efforts to determine universal diagnostic criteria for correctly recognizing ocular sarcoidosis, diagnosis still poses great difficulties, making sarcoidosis an underrecognized or misclassified disorder.
 | Table 1 IWOS criteria for diagnosing ocular sarcoidosis: introducing seven clinical signs suggestive of ocular sarcoidosis, five laboratory investigations in suspected ocular sarcoidosis, and four levels of certainty |
Pathogenesis and genetics
Histologically, the hallmark of sarcoidosis is a noncaseating granuloma, consisting of epithelioid cells, histiocytes, multinucleated giant cells, CD4+ lymphocytes and macrophages/monocytes that are surrounded by CD8+ lymphocytes, plasma cells, fibroblasts, and collagen.12 Granulomas possess significant biosynthetic and secretory properties. Among the enzymes and other chemicals secreted by granulomas are angiotensin-converting enzyme, lysozyme, glucuronidase, collagenase, and calcitriol. Granulomatous inflammation in sarcoidosis may persist, causing tissue damage during the active phase, completely resolve, or lead to obliterative fibrosis during the healing process.
Although the etiology of sarcoidosis remains highly elusive, it is conventionally believed to derive from an interaction between environmental antigens and genetic factors (the genetic–environmental theory).13 ACCESS, a US-based multicenter epidemiologic study of 706 clinically and histologically confirmed subjects with sarcoidosis, failed to identify a single predominant environmental, infective, or occupational “cause” of sarcoidosis.14 This large case–control study suggests that multiple environmental sources of exposure may trigger an aberrant immunomediated granulomatous response in genetically susceptible individuals. Proposed environmental “antigens” encompass infective (viral/bacterial/fungal) agents and their components, chemical exposure, and inorganic particulate matter.5,14
Presumably, in response to antigen exposure, an exaggerated cellular immunoresponse ensues in target organs, promoting the development and accumulation of granulomas. Granuloma formation is a distinctive tissue reaction to a persistent (although unspecified in this case) inciting agent, in order to wall off the pathogen and protect surrounding tissue by restricting inflammation.15,16 Antigen-presenting cells interact with CD4+ lymphocytes, which subsequently differentiate into T-helper (TH)-1 effector cells.14 The resultant highly polarized TH1-cell immunoresponse promotes the secretion of a cascade of cytokines, predominantly IFNγ, IL2, as well as TNFα, with parallel activation of macrophages and other cytotoxic T cells. This process drives the development and maintenance of granulomas in an effort to defend the host from intracellular pathogens, indicating an immune mechanism in the pathogenesis of sarcoidosis.17
In recent years, genome-wide association studies have revolutionized genetic research, facilitating substantially the study of the genetic background of sarcoidosis and subsequently providing considerable evidence supporting a genetic predisposition to sarcoidosis. Variation in incidence rates based on geographic location and racial features, the observation of familial clustering in approximately 5%–16% of patients, and a reported 80-fold risk of sarcoidosis development in monozygotic twins compared to dizygotic twins suggest a genetic element to the occurrence and clinical course of the disease.18,19 There appears to be a strong association between certain human leukocyte antigen (HLA) alleles and sarcoidosis susceptibility and disease behaviour.12,13 Both HLA class I and II human leukocyte antigens have been implicated, with HLA class II genes reported to be more strongly and consistently associated. Certain alleles, such as HLA-DRB1 (HLA-DRB1*11, *12, *14, *15) are principally correlated with susceptibility, disease phenotype, and prognosis. One of the most frequently described in literature gene–environment associations is between HLA-DRB1*1101 and occupational insecticide exposure. HLA-DRB1*03 has been closely linked with Löfgren’s syndrome, a distinct sarcoidosis phenotype.18 Some HLA genetic markers have been linked with specific organ involvement, such as HLA-DRB1*0401, DRB1*0401-DQB1*0301, and DRB1*03-DQB1*02, that have been implicated in eye involvement.19
Continued research into the genetic and autoimmune mechanisms of sarcoidosis is essential not only for their theoretical value but also for their tremendous clinical implications and potential for breakthrough health care delivery. Better knowledge of the role of HLA molecules in the pathogenesis of the disease may aid in identifying possible triggers of the granulomatous response that characterizes sarcoidosis, in addition to offering a predictive ability of disease development, phenotype, and course.
Ocular manifestations
Ocular sarcoidosis constitutes one of the leading causes of inflammatory eye disease, exhibiting a wide range of clinical manifestations (Table 2) and potentially affecting any structure of the visual system and the orbit.20 An estimated 20%–30% of patients present with de novo ophthalmic findings compatible with sarcoidosis but lack of evident extraocular disease.20–22 Up to 80% (25%–80%) of sarcoidosis patients will develop ocular involvement at some point during the course of the disease. This estimate is subject to considerable variation, based on the population studied and the duration and frequency of ophthalmic follow-up.22–24 Ocular involvement has been reported in 64%–89% of patients with sarcoidosis in Japanese studies.23 Although ocular sarcoidosis has been described to occur in the absence of any apparent systemic involvement, the frequency of this has yet to be investigated.
 | Table 2 Spectrum of ocular manifestations of sarcoidosis |
Intraocular involvement
Anterior segment
The uveal tract is arguably the most common site of ocular involvement, making uveitis the most typical ocular manifestation, reported in 30%–70% of cases.21 Due to the diversity in anatomical location and severity of inflammation, sarcoidosis should be considered in the differential of almost any uveitis case. There is potential for severe visual impairment and morbidity if effective treatment is not initiated in a timely fashion. Sarcoidosis-related uveitis can affect any part of the uvea, leading to anterior, intermediate, and posterior uveitis or panuveitis, with anterior uveitis being the most frequent.22 The most common form of uveitis encountered in sarcoidosis is acute, bilateral anterior uveitis without posterior-segment involvement. The severity of acute inflammation ranges from mild to severe, with affected patients experiencing varying degrees of redness, ocular pain, photophobia, lacrimation, and decreased vision. Besides the classic picture of acute iridocyclitis, chronic anterior-chamber inflammation may also occur, with a more insidious course of granulomatous inflammation, accompanied by keratic precipitates (“mutton-fat” type), iris nodules, posterior synechiae, peripheral-angle synechiae, and trabecular meshwork obstruction. Complications may develop, such as ocular hypertension, secondary glaucoma, corneal band keratopathy, cataract formation, epiretinal membrane, and cystoid macular edema (CME).22,23 The severity and chronicity of anterior-segment inflammation in conjunction with the need for long-term topical corticosteroids are both to account for these complications.
Posterior segment
Posterior-segment involvement encompasses intermediate and posterior uveitis and panuveitis (Figure 1). A minority of patients (6%–10%) diagnosed with sarcoidosis develop intermediate uveitis compared to other forms of uveitis, and present with vitreous opacities, “snowball” infiltrates, snowbanking, string-of-pearls appearance, and peripheral vasculitis.7,23,24 Posterior uveitis, presenting in just over 20% of ocular sarcoidosis cases with symptoms of painless decrease in vision and floaters, encompasses a wide range of presentations. Nonobstructive segmental retinal periphlebitis in the mid-periphery with perivenous exudative infiltrates referred to as “candle-wax drippings” or “tâches de bougies” is the classic type of vascular involvement seen in sarcoidosis patients.24,25 Capillary obstruction, ischemia, and neovascularization associated with vitreous hemorrhage may ensue. Occlusive retinal vascular disease has also been reported, although more rarely. Peripheral or even central multifocal chorioretinitis with multiple round punched-out lesions is another common presentation in the spectrum of posterior-segment involvement, and significant visual loss in these cases usually results from CME or pigment epithelial atrophy upon resolution of centrally occurring lesions.26 Choroidal granulomas, multiple or solitary, can be observed anywhere in the fundus, and normally do not cause any symptoms unless located in the optic disk or macula (Figure 2). Resolution of the granulomatous lesions leaves areas of hypopigmentation and scarring.27 Panuveitis, where inflammation of all three segments occurs, affects approximately 10%–30% of sarcoidosis cases and is usually bilateral.28,29
 | Figure 1 Sarcoid-related panuveitis with vitreous haze and right-eye optic disk swelling. |
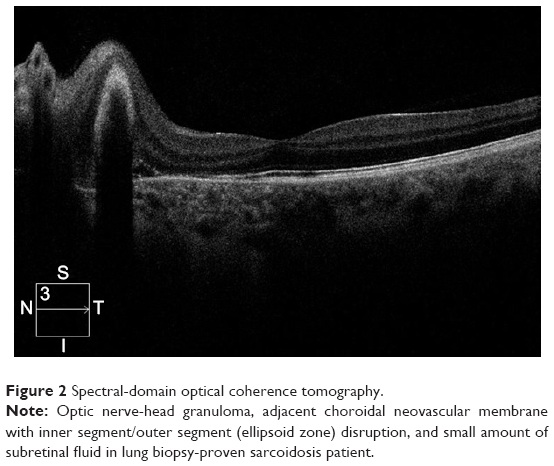 | Figure 2 Spectral-domain optical coherence tomography. |
Neuro-ophthalmic and optic nerve involvement
Neurosarcoidosis, also known as “the great imitator” because of the variety of symptoms and lack of disease-specific clinical signs, occurs in approximately 5%–15% of cases and can manifest with optic neuropathy, cranial neuropathy, papilledema, nystagmus, pupillary abnormalities, visual field defects, raised intracranial pressure, abnormal eye movement, visual hallucinations, encephalopathy, hydrocephalus, seizures, aseptic meningitis, and psychiatric symptoms.21,22,29,30 Cranial neuropathy is arguably the most common neuro-ophthalmic presentation of sarcoidosis, with the optic nerve and facial nerve more commonly affected.31
Ocular surface disease
Conjunctival involvement is frequent but often overlooked, as it tends to cause fewer if any symptoms. Follicular conjunctivitis, conjunctival nodules, and granulomas in the palpebral conjunctiva can be observed and respond well to topical steroid treatment. The cornea is rarely affected directly by the disease, but patients can ultimately develop corneal complications, such as keratoconjunctivitis sicca, superficial punctate keratitis, keratic precipitates, and band keratopathy, due to calcium deposition in Bowman’s subepithelial layer.32–35
Ocular adnexa and orbit
The lacrimal gland and lacrimal drainage system are also a common site of ocular involvement, with a reported frequency of 10%–69%.21 Although initially asymptomatic, extensive inflammation/infiltration of the lacrimal gland leads to decreased reflex aqueous tear production and subsequently to severe keratoconjunctivitis sicca and dry-eye symptoms. Significant enlargement of the main lacrimal gland due to infiltration can present as a palpable orbital mass, with resultant mass-effect symptoms, such as diplopia, ptosis, and proptosis. Nasolacrimal duct obstruction as a consequence of granulomatous inflammation causes excessive tearing, dacryocystitis, and nasal stuffiness.21
Orbital sarcoidosis can also involve other structures besides the nasolacrimal system, such as the orbital adipose tissue, extraocular muscles and optic nerve sheath, resembling to a great extent other inflammatory disorders affecting the orbit, such as Graves’s ophthalmopathy and idiopathic orbital inflammatory syndromes.33,34 Inflammation-related and treatment-related complications may lead to severe vision loss in sarcoidosis patients, and various treatment strategies are employed to restore vision by controlling inflammation and prevent permanent ocular structural changes, such as CME, macular scarring, cataract, glaucoma, and corneal opacification.
Because of the high prevalence of ophthalmic involvement in systemic sarcoidosis and the risk of significant visual impairment if left untreated, it is highly recommended that all patients with systemic disease be screened for ocular signs, albeit being asymptomatic.24,34 The lack of a robust system and accepted guidance on the optimal timing and frequency of ocular examination in asymptomatic patients partly explains why ocular sarcoidosis remains underrecognized and underreported. The detection of ocular signs requires meticulous eye examination, which should include a baseline ophthalmic examination of visual acuity (VA), pupillary reflexes, and slit-lamp examination of the anterior and posterior structures. Appropriate attention should be paid to anterior-chamber angle for tent-shaped peripheral anterior synechiae, as they could result from subclinical anterior uveitis and progressively induce an increase in intraocular pressure (IOP) and secondary glaucoma. In contrast to asymptomatic systemic disease that does not always necessitate treatment due to side effects that outweigh the benefits, asymptomatic ocular disease requires treatment when detected, because it may lead to permanent visual impairment.
Treatment of chronic ocular sarcoidosis
The management of ocular sarcoidosis, especially intraocular inflammation in the form of uveitis, which is by far the most common and serious presentation, has traditionally been a challenge for the treating ophthalmologist, due to the complexity of the disease, difficulty in reaching the diagnosis, and lack of a standardized algorithm. A stepwise approach is usually recommended, with sight-threatening situations requiring a more aggressive scheme of therapy. Corticosteroids, either topical (eyedrops, periocular, intraocular injections) or systemic, have been the pillar of ocular sarcoidosis management for many decades and continue to be. However, in recent years, the introduction of immunomodulators and the more contemporary biologic agents have tremendously altered the landscape in managing ocular sarcoidosis, establishing a new era in the successful treatment of many inflammatory ocular diseases (Table 3).
 | Table 3 Treatment options in sarcoidosis induced uveitis according to location of inflammation and severity |
We conducted a review of the recent literature of the last 5 years (September 2012–September 2017) regarding treatment modalities in chronic ocular sarcoidosis, including available topical and systemic therapies. We searched PubMed for all published literature on the management of chronic sarcoid-related eye disease, focusing mainly on the principal manifestation of intraocular inflammation (uveitis). All clinical studies reporting the employment of topical, periocular, and intraocular systemic corticosteroids, systemic immunomodulatory therapy (alkylating agents, antimetabolites), and biologic agents (anti-TNFα monoclonal antibodies) in adult patients with uninfectious uveitis inclusive of sarcoidosis are included in this review. Studies with no data on uveitis underlying etiology or not inclusive of sarcoidosis of uveitis cases were excluded. We focused our review on newer trends in intravitreally used agents and biologics.
Corticosteroids
Topical ophthalmic solutions
Topical corticosteroid eyedrops are generally used for newly presented cases of acute anterior uveitis, with frequency of administration ranging from hourly to once a day according to severity of inflammation. They are commonly used along with cycloplegics to prevent posterior synechiae formation and to ease ciliary muscle spasm. A list of different preparations is currently available, with prednisolone acetate 1% being the most popular and widely prescribed steroid for uveitis therapy.36 Sheppard et al demonstrated that difluprednate 0.05% – a newer ophthalmic steroid solution considered more potent than the traditionally used prednisolone – dosed four times daily was well tolerated and noninferior to prednisolone acetate 1% dosed eight times daily in improving the signs of acute endogenous acute uveitis.37 Rimexolone 1% ophthalmic suspension has been confirmed to have less propensity than prednisolone acetate 1% ophthalmic suspension in causing IOP rise, and thus stands as a safer option in steroid responders. Nevertheless, rimexolone is a weaker agent, and so should be preferred in milder degrees of anterior inflammation.38–40 Loteprednol etabonate 0.5% ophthalmic suspension is another alternative for steroid responders, as it has been found to impact less on IOP elevation; however, it was observed to be less effective than prednisolone acetate 1% in inflammation control.41,42
Although generally the preferred strategy to control anterior-chamber inflammation, prolonged application of corticosteroid eyedrops may lead to local adverse events, such as raised IOP and induction of glaucoma, early-onset cataract, delayed wound healing, and impaired resistance to infection in a dose- and time-dependent manner. Besides anterior uveitis, topical corticosteroids can also be used for sarcoid-related ocular surface inflammation, such as episcleritis, scleritis, conjunctivitis, and conjunctival granulomas.
Periocular corticosteroids
Periocular corticosteroid injections are usually employed when topical administration proves inadequate to achieve effective control of intraocular inflammation, especially in sarcoid cases with posterior-segment involvement or uveitic CME, and are particularly useful in unilateral cases.43 They can be administered by different routes: into the subconjunctival space, sub-Tenon’s capsule, orbital floor, and retrobulbarly. Sub-Tenon’s and orbital floor injections are the most popular techniques. Triamcinolone acetonide (TA; 20–40 mg) is currently the most frequently used formulation for periocular injection.44 The anti-inflammatory effect is transient, lasting for an estimated 2–4 months, so repeated injections or additional systemic treatment are often required to maintain a therapeutic result over an extended period. Raised IOP, cataract progression, and injection-related side effects (ptosis, extraocular muscle injury, globe penetration, skin depigmentation) are the most frequently reported complications.
Intraocular corticosteroids
Intravitreal administration of corticosteroids is a well-established way to deliver the drug more directly to the target tissue in a predictable intraocular concentration over a prolonged period of time. Intravitreal TA (IVTA) injections – Triesence (Alcon, Fort Worth, TX, USA) and Trivaris (Allergan, Riverside, CA, USA), which are US Food and Drug Administration (FDA)-approved for intraocular use, and off-label Kenalog 40 (Bristol-Myers Squibb, Princeton, NJ, USA) and the sustained-release intraocular implants Ozurdex (Allergan, Irvine, CA, USA), Iluvien, and Retisert – are intravitreal steroid therapeutic options in uveitis of various etiologies, among them sarcoidosis.
IVTA 1–4 mg has been used in ophthalmology for decades for a number of medical retinal conditions, including uveitic CME. Patients receiving such treatment need to be monitored for raised IOP. The anti-inflammatory effect usually lasts approximately 3–6 months. There is a paucity of new published clinical studies over recent years on the use of IVTA in uveitides, because of the advent of and preference toward intraocular corticosteroid implants. Shin and Yu showed that IVTA injection achieves earlier reduction of leakage and retinal edema compared to systemic anti-inflammatory treatment alone in patients with uveitic CME.45 In a pilot study, IVTA was found to be superior to intravitreal diclofenac in the treatment of a sarcoid-associated refractory uveitic CME.46 A preservative-free triamcinolone preparation (Triesence) has recently emerged and been used in sarcoid CME (17.5% of the uveitic population studied), with favorable central retinal thickness and visual outcomes and similar risks to other intravitreal steroid formulations, including raised IOP necessitating medical therapy and cataract.47 Polymeric triamcinolone acetonide nanoparticles are an emerging biodegradable formulation of IVTA, but only in vitro and animal model studies exist so far.48
Dexamethasone (Dex) intravitreal implant (0.7 mg Ozurdex) is a sustained-release, biodegradable, injectable dexamethasone-containing implant that has been FDA-approved for intermediate and posterior uninfectious uveitis. It is dispensed through the pars plana into the vitreous cavity with a 22-gauge single-use applicator and is designed to release Dex for up to 6 months. The use of the implant in uninfectious uveitis was first evaluated in the HURON trial, which demonstrated that a single injection led to adequate control of intraocular inflammation and good visual outcomes for up to 6 months in uninfectious intermediate and posterior uveitis.49 Since then, more studies have come to light. A multicenter study by Zarranz-Ventura et al in 82 eyes with uninfectious uveitis (six of 82, 7.3% sarcoidosis), with the main indication for the implant being CME, showed that the Dexamethasone implant can achieve adequate control of vitreous haze and ME with significant improvement in vision in eyes with uninfectious uveitis. In eyes that completed 12-month follow-up (54 of 82), 40.7% underwent two injections and 11.2% required three or more injections, while the main adverse event was IOP increase >21 mmHg in 40.2% of the eyes, requiring ocular hypotensive treatment in 39% and glaucoma surgery in 2.4%.50 Khurana and Porco reported that a single Dex implant in 18 eyes with uninfectious uveitis, including sarcoidosis-related, produced sustained improvements in both VA and retinal thickness in the majority of eyes with persistent uveitic CME (no CME in 89% and 72% in 1 month and 3 months, respectively); however, CME did gradually recur in most eyes, so close monitoring was advised.51 In a retrospective study by Miserocchi et al in 2012, the implant showed promising results in terms of reduction of uveitic activity, best-corrected VA (BCVA), and mean retinal thickness improvement (two of 12 patients with sarcoidosis).52 Ryder et al, Pleyer et al, and Cao et al53–55 have also studied the efficacy of Ozurdex implant in both active posterior inflammation and inactive inflammation with CME, demonstrating satisfactory results for inflammatory control and macular thickness improvement.
Overall, the safety and efficacy of the Dex implant has been illustrated in a number of large studies with favorable outcomes in terms of BCVA and central macular thickness improvement in uninfectious uveitis patients;56 however, recurrence of inflammation and CME posttreatment are the main disadvantages, while raised IOP is usually medically managed. Repeated implants seem to work with the same efficacy, with minimal additional side effects. However, among the limitations of the existing literature are that most studies are retrospective in nature with relatively small samples, few studies have looked into the long-term effects of the implant, and no studies have investigated the response to treatment according to underlying etiology of uveitis and concomitant therapy in subgroup analysis. This is particularly important in sarcoidosis cases, where patients usually receive additional systemic therapy.57
A sustained-release fluocinolone acetonide (FA) implant in two dosage formulations (0.59 mg and 0.19 mg) has been approved for use in uninfectious posterior uveitis the US. The MUST trial is the largest randomized comparative trial (RCT) to date that has investigated the efficacy, safety, and impact on quality of life of the FA implant in comparison with systemic immunosuppression. The implant achieved better outcomes in terms of inflammation control and CME resolution in the short term, but had an increased rate of IOP rise requiring treatment.58
A recent large RCT attempted to compare the association between intravitreous FA implant vs systemic therapy and long-term visual and other outcomes in patients with severe intermediate uveitis, posterior uveitis, or panuveitis. Seven-year data were obtained for 161 uveitic eyes (70% of 90 patients assigned to implant) and 167 uveitic eyes (71% of 90 patients assigned to systemic therapy). Analysis of the data revealed that those randomized to receive systemic therapy had better VA than those randomized to receive intravitreous FA implants.59 However, this study had significant limitations, due to loss at follow-up of 30% at 7 years, a 20% crossover treatment between the groups, and incomplete masking. Jaffe et al compared outcomes in patients with recurrent uninfectious intermediate uveitis, posterior uveitis, or panuveitis receiving either a low-dose (0.19 mg) or high-dose (0.59 mg) FA implant. No significant differences were observed in any of the assessed efficacy or safety parameters between eyes that received these two doses, both implant types effectively controlled intraocular inflammation in all eyes in the study, and at the last follow-up, all implanted eyes demonstrated an improvement in VA, and thus the authors concluded that it is a promising approach for patients with uninfectious intermediate uveitis, posterior uveitis, or panuveitis who do not respond to or are intolerant to conventional therapy.60
A number of reports from MUST, a landmark trial in uveitis comparing the FA implant to systemic immunosuppression, disclosed among other results that: FA-implant therapy is associated with a clinically important increased risk of glaucoma and cataract, suggesting careful monitoring and early intervention to prevent glaucoma;61 based on cost-effectiveness and side-effect considerations, systemic therapy may be indicated as the initial treatment for many bilateral uveitis cases, while FA implant is a reasonable alternative for unilateral cases and intolerable or failed systemic therapy;62 and immediate improvement of vision-related quality of life in patients with uninfectious uveitis in the implant group compared to patients receiving systemic therapy.63
Sustained-release corticosteroid implants are a promising strategy in managing chronic, refractory cases of sarcoidosis-related uveitis. Different devices with variable duration and side effects are available. More data with well conducted RCTs are available for the Dex implant in achieving satisfactory control of inflammation with fewer side effects than the FA implants; however, the duration of effect is shorter. Further studies are warranted to determine long-term efficacy, as well as comparative studies between the Dex and FA implants.64 While steroid implants are a new area in ocular inflammation, clearly the application in sarcoidosis has been limited. The main cause is that sarcoidosis is a systemic disease, and aggressive topical therapy is often replaced by systemic treatment early on during the course of the disease. In published literature, there have been a limited number of sarcoidosis patients studied, because most will require local and systemic therapy.
Systemic corticosteroids
Systemic (oral or intravenous) corticosteroids are the mainstay of therapy for chronic, bilateral, vision-threatening ocular sarcoidosis, and are effective for both intraocular and orbital/adnexal disease, especially when systemic inflammation coexists. Usual initial doses of oral prednisone can be as high as 1–1.5 mg/kg/day and should be tapered gradually to the lowest effective dose to avoid a flare-up. However, although short-term therapy, even at high doses, is usually well tolerated with reversible adverse effects, long-term systemic administration is associated with substantial systemic morbidity. Among the most common serious side effects of oral steroids are hyperglycemia/diabetes mellitus, hypertension, myopathy, susceptibility to infections, impaired wound healing, adrenal suppression, hyperlipidemia, osteoporosis, Cushing’s syndrome, and mental-status changes, even psychosis. Therefore, in refractory cases or when more prolonged or high-dose steroid treatment is required to maintain control of inflammation, corticosteroid-sparing agents are employed.
Immunosuppressives
Systemic immunosuppressive agents are used as a corticosteroid-sparing strategy when prolonged steroid therapy is required, in cases of failure of treatment, or relapse. The corticosteroid-sparing approach is generally employed earlier in ocular disease than in other forms of sarcoidosis, because of the significant risk of ocular complications caused by both the disease itself and the steroid use.
Preferred medications in patients with chronic ocular or systemic sarcoidosis are antimetabolites. Methotrexate (Mtx) and mycophenolate mofetil (Mmf), a selective suppressor of T- and B-lymphocyte proliferation, are favored as first-line steroid-sparing agents. Mtx is currently the most studied cytotoxic agent in chronic ocular disease and sarcoidosis in general. Several retrospective studies have shown satisfactory outcomes in terms of inflammatory control with such agents, but only a small number of prospective comparative studies evaluating their safety and efficacy exist. A recent study by Browne et al inquiring uveitis experts on their preference on antimetabolite use in uninfectious posterior uveitis revealed that experts believed Mmf 2 g/day was more effective than Mtx 25 mg/week in controlling inflammation.65 Despite that, an RCT by Rathinam et al comparing the relative effectiveness of Mtx and Mmf for uninfectious uveitis showed no statistically significant difference in corticosteroid-sparing control of inflammation between the two groups, but Mtx was favored by a 22% difference in treatment success.66 In 2012, Baughman et al conducted a retrospective review of 465 patients with ocular sarcoidosis, including 365 treated with Mtx and a small number that received anti-TNFα antibodies (19 patients with infliximab, six with adalimumab). Immunosuppressive therapy – either Mtx monotherapy or combination of immunosuppressive agents – achieved adequate control of most cases of chronic ocular sarcoidosis, while adverse events, recurrent infections, or financial constraints were the main reasons for failure in the small cohort of patients who received anti-TNFα.67 In a comparative study by Bitoun et al, Mtx was found to be superior to Mmf in reducing the risk of relapses in patients with neurosarcoidosis.68 The majority of recently reported outcomes in the literature on the use of Mtx or Mmf in uveitis concern Vogt–Koyanagi–Harada disease or birdshot chorioretinopathy.
Calcineurin inhibitors (cyclosporine A 2.5–10 mg/kg/day twice daily and more rarely tacrolimus 0.15–0.3 mg/kg/day and sirolimus) have also been occasionally used for sarcoid ocular inflammation.69 Mtx, Mmf, azathioprine, and cyclosporine are the most popular immunosuppressive options for chronic ocular sarcoidosis.70 Intravitreal Mtx, although a known treatment modality for intraocular lymphoma, has also been studied off-label in uninfectious uveitis by Taylor et al, where a large proportion (73%) of patients entered an extended period of remission of up to 18 months.71
Combination immunosuppressive therapy also exists as a treatment approach, with the most commonly recommended being Mtx with azathioprine and Mtx with leflunomide, although this concept is more popular when managing systemic rather than ocular disease. Overall, no standardized algorithm currently exists for the use of systemic immunomodulators for ocular sarcoidosis or other types of intraocular inflammation. Experienced practitioners should closely monitor patients receiving such treatment, due to the risk of significant systemic side effects and cumulative toxicity.
Biologic agents
When corticosteroids and cytotoxic immunosuppressive therapy fail to produce complete and sustainable remission or in cases of intolerance, the relatively novel option of biologic-response modifiers is often considered. These are recombinant proteins or antibodies regarded as targeting specific molecules or cytokines involved in the inflammatory cascade of the disease pathway.72 A fundamental feature of these agents is specifically targeted suppression of the immunoeffector responses that are accountable for damaging tissue. The advent of biologic agents and the recent FDA approval of adalimumab for uninfectious intermediate uveitis, posterior uveitis, and panuveitis has been a game changer in the field of uveitis management. While antimetabolites have been established as steroid-sparing agents without necessarily being more potent than them, biologic therapy offers the option of an equally or more efficacious choice for chronic disease, capable of inducing complete and prolonged quiescence. In ocular sarcoidosis in particular, where systemic therapy holds an important place in achieving control of inflammation, biologic agents seem to stand as an increasingly popular choice. Although growing evidence from randomized controlled trials regarding the use of biologic agents are gradually coming to light with promising results, on the grounds of limited long-term safety and efficacy data, these agents widely remain a second-line treatment choice among uveitis experts and reserved for use if and when antimetabolites fail to induce and sustain remission.
TNFα inhibitors
TNFα is a powerful, pleiotropic, multifunctional, proinflammatory cytokine that plays a principal role in a wide range of autoinflammatory conditions. High levels of TNFα are secreted by alveolar macrophages, thereby contributing to the formation and preservation of sarcoidal granulomas in patients with active disease,32 indicating that selective blockage of this cytokine could accomplish satisfactory control and remission of inflammation. In fact, it has been advocated that in a considerable number of refractory sarcoidosis cases, failure of corticosteroid or antimetabolite therapy might be due to the significantly increased TNFα release.73 As such, TNFα inhibition could conceivably be a favorable option in sarcoidosis-related intraocular inflammation compared to other types of uveitis, such as nongranulomatous inflammation. These agents have demonstrated a relatively safe profile with acceptable tolerability, although a small number of case reports describing autoimmune adverse events, paradoxically listing uveitis as one of them, have emerged.74
Anti-TNFα agents, including infliximab, adalimumab, certolizumab, and golimumab, are monoclonal antibodies that bind the TNFα molecule, which plays a crucial role in the granulomatous inflammation of sarcoidosis.12 These biologic agents have been most studied in the treatment of sarcoid-related uveitis. By blocking the TNFα receptors and competitively binding circulating TNFα, these molecules facilitate the dissolution of granulomas in sarcoidosis.
Adalimumab (Humira; AbbVie, Chicago, IL, USA) is a fully humanized monoclonal antibody against TNFα. It has been in the spotlight recently, as it is the first biological agent ever approved by the FDA for the treatment of uninfectious intermediate uveitis, posterior uveitis, and panuveitis in adults, being as such the only steroid-sparing option having a clear indication for uveitis, while all other steroid-sparing treatments continue to be used off-label. With FDA approval, the drug can now be prescribed, hence addressing the issue of financial restrictions which has been a significant hindrance in utilizing it up until recently. Two double-blind, placebo-controlled, multinational, Phase III studies evaluated the therapeutic efficacy of subcutaneous adalimumab in adults with active (VISUAL I) or inactive (VISUAL II) uninfectious intermediate uveitis, posterior uveitis, or panuveitis. In these two studies, adalimumab significantly reduced the risk of treatment failure relative to placebo following the tapered withdrawal of corticosteroid therapy.75,76 VISUAL I enrolled 217 patients, 18 of whom had sarcoid uveitis, and demonstrated that patients receiving adalimumab were 50% less likely to experience treatment failure than the placebo group, also exhibiting better results in the secondary end points of anterior-chamber inflammation, vitreous haze, and BCVA, significantly lowering the risk of uveitic flare and visual impairment. Likewise, VISUAL II assessed the safety and efficacy of adalimumab in 229 patients with inactive disease (32 patients with sarcoid uveitis as the underlying etiology) controlled by systemic corticosteroids against placebo. Time to treatment failure was significantly longer for patients receiving adalimumab compared to placebo, reducing by half the risk of uveitic flare and loss of VA. The most common adverse events of the drug in these studies were injection-site reactions and allergic reactions, while serious adverse effects include an increased risk of infection, a lupus-like syndrome, and demyelinating disorders.75,76 Nevertheless, the safety profile of adalimumab overall seems comparable to that in other approved indications, with acceptable tolerability. Future studies exploring long-term safety and efficacy of adalimumab in uninfectious uveitis are on the way. Before VISUAL I and II, Erckens et al studied the use of adalimumab in 26 sarcoidosis patients with refractory posterior uveitis unresponsive to corticosteroids or Mtx: 85% of patients showed improvement of intraocular inflammatory signs, as well as other indicators of disease activity, while 15% of them had stable outcomes. 77–80 The approval of adalimumab for uveitis is a huge step forward in the management of refractory sarcoid uveitis. Table 4 contains a summary of the latest studies on the use of adalimumab in sarcoid-related uveitis.75–80
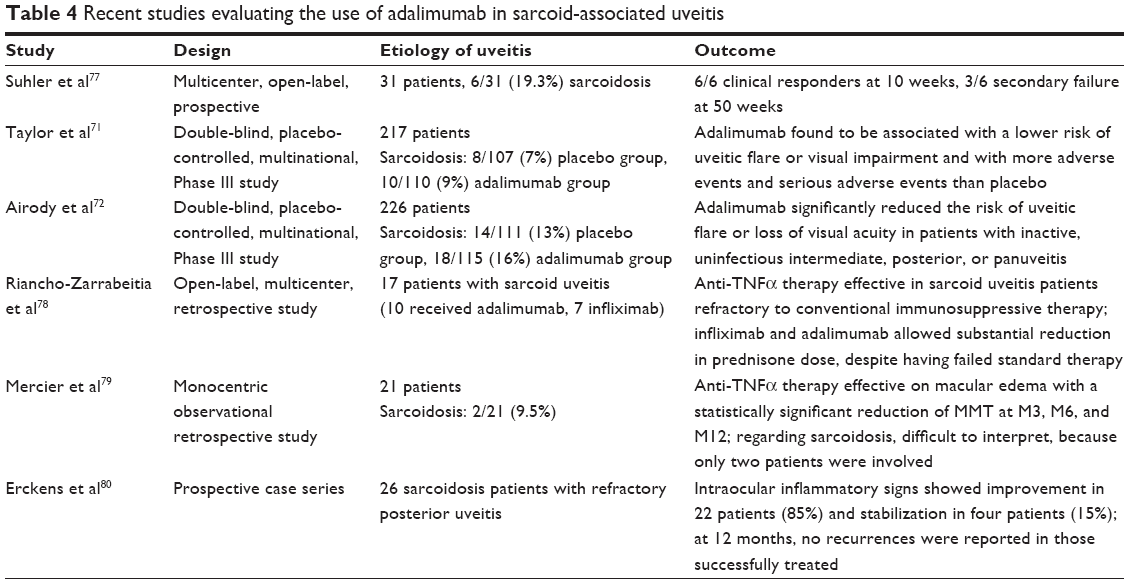 | Table 4 Recent studies evaluating the use of adalimumab in sarcoid-associated uveitis |
Infliximab (Remicade; Janssen Biotech, Horsham, PA, USA), a chimeric IgG1 monoclonal antibody against TNFα, binds to TNFα with high affinity, thereby blocking the binding to its receptor.81 For ocular use, it is normally given intravenously at a dose range of 5–10 mg/kg every 4 weeks. Infliximab has mainly been studied in vision-threatening Behçet’s disease and juvenile idiopathic arthritis-associated uveitis and spondyloarthritides, while medium-level evidence exists for its use in sarcoidosis-related uveitis. In a multicenter study from the French Uveitis Network analyzing a large cohort of patients treated with anti-TNF for refractory uveitis, infliximab and adalimumab were found to have equivalent efficacy, with an overall response of 97% among patients who received infliximab and 95% in the adalimumab group. Both drugs were similar in terms of safety, although there was a nonsignificant trend toward a higher incidence of serious side effects with infliximab.82 Kruh et al published the largest single-center retrospective, interventional, noncomparative cohort study evaluating the safety and efficacy of infliximab in various types of uveitis that failed to respond to other treatments. In this 6.5-year study of a cohort of 88 patients with recalcitrant uveitis, infliximab induced clinical remission in 81.8% of cases (including six sarcoid patients), and most patients tolerated the treatment well. The most common adverse effects were skin rash (9.1%) and fatigue (8%).83 Notwithstanding the lack of prospective comparative studies between infliximab and adalimumab, adalimumab arguably seems to have a number of advantages over infliximab, including route of administration, lower rate of adverse reactions, and proven efficacy against placebo.
Etanercept, a fusion protein of a human Fc molecule and two p75 TNF receptors that binds free TNFα, not only proved to be less effective than other agents and placebo in the treatment of uveitis in general and sarcoidosis in particular, but could even precipitate uveitis and sarcoid-like reactions; therefore, its use in uveitis management and sarcoidosis specifically is not recommended.84–87 Golimumab (Simponi; Janssen Biotech), a fully human anti-TNFα monoclonal antibody, gained FDA approval in 2009 for the treatment for rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and ulcerative colitis. A nonrandomized retrospective interventional case series by Cordero-Coma et al published in 2014 revealed that golimumab was well tolerated and associated with control of inflammation in 92.3% of a heterogeneous group of immunomediated uveitis patients (two of 13 sarcoidosis, intermediate uveitis, and panuveitis) resistant to traditional therapy and other biologic agents.88 Based on a few retrospective published case series of its use after either inadequate response to or intolerance of other biologics, golimumab seems to be a promising additional option in the treatment of immunomediated uveitis; however, evidence is scarce and more studies are required.89,90
Certolizumab pegol (Cimzia; UCB, Smyrna, GA, USA) is a PEGylated humanized antibody Fab fragment of a monoclonal antibody against TNFα, which has a longer half-life thanks to the delayed elimination provided by the PEGylation of the antibody. It is administered subcutaneously every other week.91 There is currently no published report on the use of certolizumab pegol in sarcoid uveitis; however, this agent has potential advantages over other anti-TNFα drugs in terms of increased half-life and less risk of efficacy loss.92
Other biologic disease-modifying antirheumatic drugs
Daclizumab (Zenapax; Genentech, San Francisco, CA, USA) is a humanized monoclonal antibody that binds to the CD25 portion of the IL2 receptor on activated T and B lymphocytes and natural killer cells. The most recent study on the use of daclizumab in sarcoid-related uveitis amongst other etiologies comes from Wroblewski et al, who demonstrated efficacy in the reduction of concomitant immunosuppressive medication, stabilization of VA, and the prevention of uveitic flares in most of the 78 cases; however, solid-tumor malignancies were observed in four patients during the 11-year follow up.93
Rituximab (Rituxan; Genentech) is a CD20-directed cytolytic antibody that has proven efficacious in ocular cicatricial pemphigoid peripheral ulcerative keratitis and scleritis associated with antineutrophil cytoplasmic antibody-positive vasculitis and sarcoidosis, as well as sarcoid-related uveitis. Few data on its use in sarcoid uveitis and systemic sarcoidosis exist in the literature, mainly in the form of case reports or case series and only one Phase II trial on the safety of the drug in pulmonary sarcoidosis.94,95
Conclusion
Ocular disease is an important manifestation of sarcoidosis, and can lead to significant visual disability or blindness if not adequately or promptly treated. A wide variation in clinical presentation renders this entity one of the most challenging to diagnose and manage, usually requiring a multidisciplinary approach. With the ever-growing spectrum of available and emerging therapies targeting different aspects of the granuloma-formation pathway, successfully managing ocular sarcoidosis is becoming more and more achievable. Although corticosteroids have traditionally been the cornerstone of treatment, and appropriate escalation to steroid-sparing medications is crucial to avert significant side effects from prolonged or high-dose intake. Corticosteroid-sparing options have expanded over recent years to include intravitreal therapy and biologic modifiers. Admittedly, systemic immunomodulators are currently the second-line treatment when a steroid-sparing option is required. TNFα inhibitors are gaining ground rapidly, with randomized controlled studies generating more robust evidence on safety and efficacy in disease remission. adalimumab is the first anti-TNFα agent with FDA approval for use in active and inactive refractory cases of uninfectious, intermediate uveitis, posterior uveitis, or panuveitis through two large successful Phase III studies, introducing a new era in intraocular inflammation management. Along with better understanding of the mechanisms of the granulomatous inflammation in sarcoidosis comes the need for a targeted treatment approach with maximum efficacy and the fewest possible side effects. Further evolution seems to be on the way.
Disclosure
The authors report no conflicts of interest in this work.
References
Young RC Jr, Rachal RE, Cowan CL Jr. Sarcoidosis – the beginning: historical highlights of personalities and their accomplishments during the early years. J Natl Med Assoc. 1984;76:887–896. | ||
Heerfordt CF. On febris uveo parotidea subchronica localized in the parotid gland and uvea of the eye, frequently complicated by paralysis of the cerebrospinal nerves. Ugeskr Laeger. 1909;71:417–421. | ||
Al-Kofahi K, Korsten P, Ascoli C, et al. Management of extrapulmonary sarcoidosis: challenges and solutions. Ther Clin Risk Manag. 2016;12:1623–1634. | ||
Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153–2165. | ||
Dubrey S, Shah S, Hardman T, Sharma R. Sarcoidosis: the links between epidemiology and aetiology. Postgrad Med J. 2014;90:582–589. | ||
Goto H, Mochizuki M, Yamaki K, Kotake S, Usui M, Ohno S. Epidemiological survey of intraocular inflammation in Japan. Jpn J Ophthalmol. 2007;51(1):41–44. | ||
Groen F, Rothova A. Ocular involvement in sarcoidosis. Semin Respir Crit Care Med. 2017;38:514–522. | ||
Chesnutt AN. Enigmas in sarcoidosis. West J Med. 1995;162:519–526. | ||
Costabel U, Hunninghake GW. ATS/ERS/WASOG statement on sarcoidosis. Eur Respir J. 1999;14:735–737. | ||
Acharya NR, Browne EN, Rao N, Mochizuki M. Distinguishing features of ocular sarcoidosis in an international cohort of uveitis patients. Ophthalmology. 2018;125:119–126. | ||
Herbort CP, Rao NA, Mochizuki M. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop On Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009;17:160–169. | ||
Saketkoo LA, Baughman RP. Biologic therapies in the treatment of sarcoidosis. Expert Rev Clin Immunol. 2016;12:817–825. | ||
Kiszałkiewicz J, Piotrowski WJ, Brzeziańska-Lasota E. Selected molecular events in the pathogenesis of sarcoidosis: recent advances. Pneumonol Alergol Pol. 2015;83:462–475. | ||
Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324–1330. | ||
Fischer A, Rybicki BA. Granuloma genes in sarcoidosis: what is new? Curr Opin Pulm Med. 2015;21:510–516. | ||
Williams GT, Williams WJ. Granulomatous inflammation: a review. J Clin Pathol. 1983;36:723–733. | ||
Rivera NV, Hagemann-Jensen M, Ferreira MA, et al. Common variants of T-cells contribute differently to phenotypic variation in sarcoidosis. Sci Rep. 2017;7:5623. | ||
Spagnolo P, Grunewald J. Recent advances in the genetics of sarcoidosis. J Med Genet. 2013;50:290–297. | ||
Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569–584. | ||
Baughman RP, Lower EE, Kaufman AH. Ocular sarcoidosis. Semin Respir Crit Care Med. 2010;31:452–462. | ||
Pasadhika S, Rosenbaum JT. Ocular sarcoidosis. Clin Chest Med. 2015;36(4):669–683. | ||
Yang SJ, Salek S, Rosenbaum JT. Ocular sarcoidosis: new diagnostic modalities and treatment. Curr Opin Pulm Med. 2017;23:458–467. | ||
Jamilloux Y, Kodjikian L, Broussolle C, Sève P. Sarcoidosis and uveitis. Autoimmun Rev. 2014;13:840–849. | ||
Rothova A. Ocular involvement in sarcoidosis. Br J Ophthalmol. 2000;84:110–116. | ||
El-Asrar AM, Herbort CP, Tabbara KF. Differential diagnosis of retinal vasculitis. Middle East Afr J Ophthalmol. 2009;16:202–218. | ||
Koop A, Ossewaarde A, Rothova A. Peripheral multifocal chorioretinitis: complications, prognosis and relation with sarcoidosis. Acta Ophthalmol. 2013;91:492–497. | ||
Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina. 2001;21:40–47. | ||
Crick RP. Ocular sarcoidosis. Trans Ophthalmol Soc U K. 1955;75:189–206. | ||
Uyama M. Uveitis in sarcoidosis. Int Ophthalmol Clin. 2002;42:143–150. | ||
Delaney P. Neurologic manifestations in sarcoidosis: review of the literature, with a report of 23 cases. Ann Intern Med. 1977;87:336–345. | ||
Phillips YL, Eggenberger ER. Neuro-ophthalmic sarcoidosis. Curr Opin Ophthalmol. 2010;21:423–429. | ||
Kefella H, Luther D, Hainline C. Ophthalmic and neuro-ophthalmic manifestations of sarcoidosis. Curr Opin Ophthalmol. 2017;28:587–594. | ||
Umur KA, Tayfun B, Oguzhan O. Different ophthalmologic manifestations of sarcoidosis. Curr Opin Ophthalmol. 2012;23:477–484. | ||
Birnbaum AD, Oh FS, Chakrabarti A, Tessler HH, Goldstein DA. Clinical features and diagnostic evaluation of biopsy-proven ocular sarcoidosis. Arch Ophthalmol. 2011;129:409–413. | ||
Heiligenhaus A, Wefelmeyer D, Wefelmeyer E, Rösel M, Schrenk M. The eye as a common site for the early clinical manifestation of sarcoidosis. Ophthalmic Res. 2011;46:9–12. | ||
Tempest-Roe S, Joshi L, Dick AD, Taylor SR. Local therapies for inflammatory eye disease in translation: past, present and future. BMC Ophthalmol. 2013;13:39. | ||
Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS. Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study. Invest Ophthalmol Vis Sci. 2014;55:2993–3002. | ||
Biswas J, Ganeshbabu TM, Raghavendran SR, Raizada S, Mondkar SV, Madhavan HN. Efficacy and safety of 1% rimexolone versus 1% prednisolone acetate in the treatment of anterior uveitis: a randomized triple masked study. Int Ophthalmol. 2004;25:147–153. | ||
Foster CS, Alter G, DeBarge LR, et al. Efficacy and safety of rimexolone 1% ophthalmic suspension vs 1% prednisolone acetate in the treatment of uveitis. Am J Ophthalmol. 199;122:171–182. | ||
Agrawal RV, Murthy S, Sangwan V, Biswas J. Current approach in diagnosis and management of anterior uveitis. Indian J Ophthalmol. 2010;58:11–19. | ||
Pavesio CE, Decory HH. Treatment of ocular inflammatory conditions with loteprednol etabonate. Br J Ophthalmol. 2008;92:455–459. | ||
[No authors listed]. Controlled evaluation of loteprendnol etabonate and prednisolone acetate in the treatment of acute anterior uveitis. Am J Ophthalmol. 1999;127:537–544. | ||
Sen HN, Vitale S, Gangaputra SS, et al. Periocular corticosteroid injections in uveitis: effects and complications. Ophthalmology. 2014;121:2275–2286. | ||
Salek SS, Leder HA, Butler NJ, Gan TJ, Dunn JP, Thorne JE. Periocular triamcinolone acetonide injections for control of intraocular inflammation associated with uveitis. Ocul Immunol Inflamm. 2013;21:257–263. | ||
Shin JY, Yu HG. Intravitreal triamcinolone injection for uveitic macular edema: a randomized clinical study. Ocul Immunol Inflamm. 2015;23:430–436. | ||
Soheilian M, Eskandari A, Ramezani A, Rabbanikhah Z, Soheilian R. A pilot study of intravitreal diclofenac versus intravitreal triamcinolone for uveitic cystoid macular edema. Ocul Immunol Inflamm. 2013;21:124–129. | ||
Steeples LR, Anand N, Moraji J, Jones NP. Clinical outcomes of intravitreal preservative-free triamcinolone preparation (Triesence) for cystoid macular oedema and inflammation in patients with uveitis. Ocul Immunol Inflamm. 2017;20:1–8. | ||
Sabzevari A, Adibkia K, Hashemi H, et al. Polymeric triamcinolone acetonide nanoparticles as a new alternative in the treatment of uveitis: in vitro and in vivo studies. Eur J Pharm Biopharm. 2013;84:63–71. | ||
Lowder C, Belfort R Jr, Lightman S, et al. Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol. 2011;129:545–553. | ||
Zarranz-Ventura J, Carreño E, Johnston RL, et al. Multicenter study of intravitreal dexamethasone implant in noninfectious uveitis: indications, outcomes, and reinjection frequency. Am J Ophthalmol. 2014;158:1136.e5–1145.e5 | ||
Khurana RN, Porco TC. Efficacy and safety of dexamethasone intravitreal implant for persistent uveitic cystoids macular edema. Retina. 2015;35:1640–1646. | ||
Miserocchi E, Modorati G, Pastore MR, Bandello F. Dexamethasone intravitreal implant: an effective adjunctive treatment for recalcitrant noninfectious uveitis. Ophthalmologica. 2012;228:229–233. | ||
Ryder SJ, Iannetta D, Bhaleeya SD, Kiss S. Efficacy and tolerability of bilateral sustained-release dexamethasone intravitreal implants for the treatment of noninfectious posterior uveitis and macular edema secondary to retinal vein occlusion. Clin Ophthalmol. 2015;9:1109–1116. | ||
Pleyer U, Klamann M, Laurent TJ, et al. Fast and successful management of intraocular inflammation with a single intravitreal dexamethasone implant. Ophthalmologica. Epub 2014 Nov 21. | ||
Cao JH, Mulvahill M, Zhang L, Joondeph BC, Dacey MS. Dexamethasone intravitreal implant in the treatment of persistent uveitic macular edema in the absence of active inflammation. Ophthalmology. 2014;121:1871–1876. | ||
Adán A, Pelegrín L, Rey A, et al. Dexamethasone intravitreal implant for treatment of uveitic persistent cystoid macular edema in vitrectomized patients. Retina. 2013;33:1435–1440. | ||
Tan HY, Agarwal A, Lee CS, et al. Management of noninfectious posterior uveitis with intravitreal drug therapy. Clin Ophthalmol. 2016;10:1983–2020. | ||
Kempen JH, Altaweel MM, Holbrook JT, et al. Randomized comparison of systemic anti-inflammatory therapy versus fluocinolone acetonide implant for intermediate, posterior, and panuveitis: the multicenter uveitis steroid treatment trial. Ophthalmology. 2011;118:1916–1926. | ||
Kempen JH, Altaweel MM, Holbrook JT, Sugar EA, Thorne JE, Jabs DA. Association between long-lasting intravitreous fluocinolone acetonide implant vs systemic anti-inflammatory therapy and visual acuity at 7 years among patients with intermediate, posterior, or panuveitis. JAMA. 2017;317:1993–2005. | ||
Jaffe GJ, Lin P, Keenan RT, Ashton P, Skalak C, Stinnett SS. Injectable fluocinolone acetonide long-acting implant for noninfectious intermediate uveitis, posterior uveitis, and panuveitis: two-year results. Ophthalmology. 2016;123:1940–1948. | ||
[No authors listed]. Quality of life and risks associated with systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, or panuveitis: fifty-four-month results of the multicenter uveitis steroid treatment trial and follow-up study. Ophthalmology. 2015;122:1976–1986. | ||
Kempen JH, Altaweel MM, Drye LT, et al. Benefits of systemic anti-inflammatory therapy versus fluocinolone acetonide intraocular implant for intermediate uveitis, posterior uveitis, and panuveitis: fifty-four-month results of the multicenter uveitis steroid treatment (MUST) trial and follow-up study. Ophthalmology. 2015;122:1967–1975. | ||
Sugar EA, Venugopal V, Thorne JE, et al. Longitudinal vision-related quality of life for patients with noninfectious uveitis treated with fluocinolone acetonide implant or systemic corticosteroid therapy. Ophthalmology. 2017;124:1662–1669. | ||
Cabrera M, Yeh S, Albini TA. Sustained-release corticosteroid options. J Ophthalmol. 2014;2014:164692. | ||
Browne EN, Rathinam SR, Kanakath A, et al. A Bayesian analysis of a randomized clinical trial comparing antimetabolite therapies for non-infectious uveitis. Ophthalmic Epidemiol. 2017;24(1):63–70. | ||
Rathinam SR, Babu M, Thundikandy R, et al. A randomized clinical trial comparing methotrexate and mycophenolate mofetil for noninfectious uveitis. Ophthalmology. 2014;121:1863–1870. | ||
Baughman RP, Lower EE, Ingledue R, Kaufman AH. Management of ocular sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2012;29:26–33. | ||
Bitoun S, Bouvry D, Borie R, et al. Treatment of neurosarcoidosis: a comparative study of methotrexate and mycophenolate mofetil. Neurology. 2016;87:2517–2521. | ||
Kaçmaz RO, Kempen JH, Newcomb C, et al. Cyclosporine for ocular inflammatory diseases. Ophthalmology. 2010;117:576–584. | ||
Pasadhika S, Kempen JH, Newcomb CW, et al. Azathioprine for ocular inflammatory diseases. Am J Ophthalmol. 2009;148:500.e2–509.e2. | ||
Taylor SR, Banker A, Schlaen A, et al. Intraocular methotrexate can induce extended remission in some patients in noninfectious uveitis. Retina. 2013;33:2149–2154. | ||
Airody A, Heath G, Lightman S, Gale R. Non-infectious uveitis: optimising the therapeutic response. Drugs. 2016;76:27–39. | ||
Ziegenhagen MW, Rothe ME, Zissel G, et al. Exaggerated TNFα release of alveolar macrophages in corticosteroid resistant sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:185–190. | ||
Lim LL, Fraunfelder FW, Rosenbaum JT. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum. 2007;56:3248–3252. | ||
Jaffe GJ, Dick AD, Brezin AP, et al. Adalimumab in patients with active noninfectious uveitis. N Engl J Med. 2016;375:932–943. | ||
Nguyen QD, Merrill PT, Jaffe GJ, et al. Adalimumab for prevention of uveitic flare in patients with inactive noninfectious uveitis controlled by corticosteroids (VISUAL II): a multicentre, double-masked, randomised, placebo-controlled phase 3 trial. Lancet. 2016;388:1183–1192. | ||
Suhler EB, Lowder CY, Goldstein DA, et al. Adalimumab therapy for refractory uveitis: results of a multicentre, open-label, prospective trial. Br J Ophthalmol. 2013;97:481–486. | ||
Riancho-Zarrabeitia L, Calvo-Río V, Blanco R, et al. Anti-TNF-α therapy in refractory uveitis associated with sarcoidosis: multicenter study of 17 patients. Semin Arthritis Rheum. 2015;45:361–368. | ||
Mercier AE, Ribeiro E, Korobelnik JF, Delyfer MN, Rougier MB. Efficacy of anti-TNF-α therapy for the treatment of non-infectious uveitis: a retrospective study of 21 patients. Ocul Immunol Inflamm. Epub 2016 Oct 24. | ||
Erckens RJ, Mostard RL, Wijnen PA, Schouten JS, Drent M. Adalimumab successful in sarcoidosis patients with refractory chronic non-infectious uveitis. Graefes Arch Clin Exp Ophthalmol. 2012;250:713–720. | ||
Agrawal R, Iyer J, Connolly J, Iwata D, Teoh S. Cytokines and biologics in non-infectious autoimmune uveitis: bench to bedside. Indian J Ophthalmol. 2014;62:74–81. | ||
Vallet H, Seve P, Biard L, et al. Infliximab versus adalimumab in the treatment of refractory inflammatory uveitis: a multicenter study from the French Uveitis Network. Arthritis Rheumatol. 2016;68(6):1522–1530. | ||
Kruh JN, Yang P, Suelves AM, Foster CS. Infliximab for the treatment of refractory noninfectious Uveitis: a study of 88 patients with long-term follow-up. Ophthalmology. 2014;121:358–364. | ||
Brito-Zerón P, Perez-Alvarez R, Ramos-Casals M. Etanercept and uveitis: friends or foes? Curr Med Res Opin. 2015;31:251–252. | ||
Daïen CI, Monnier A, Claudepierre P, et al. Sarcoid-like granulomatosis in patients treated with tumor necrosis factor blockers: 10 cases. Rheumatology (Oxford). 2009;48:883–886. | ||
Hashkes PJ, Shajrawi I. Sarcoid-related uveitis occurring during etanercept therapy. Clin Exp Rheumatol. 2003;21:645–646. | ||
Smith JA, Thompson DJ, Whitcup SM, et al. A randomized, placebo-controlled, double-masked clinical trial of etanercept for the treatment of uveitis associated with juvenile idiopathic arthritis. Arthritis Rheum. 2005;53:18–23. | ||
Cordero-Coma M, Calvo-Río V, Adán A, et al. Golimumab as rescue therapy for refractory immune-mediated uveitis: a three-center experience. Mediators Inflamm. 2014;2014:717598. | ||
Pasadhika S, Rosenbaum JT. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;15:67–81. | ||
Cordero-Coma M, Yilmaz T, Onal S. Systematic review of anti-tumor necrosis factor-α therapy for treatment of immune-mediated uveitis. Ocul Immunol Inflamm. 2013;21:19–27. | ||
Borrás-Blasco J, Casterá DE, Cortes X, Abad FJ, Rosique-Robles JD, Mallench LG. Effectiveness of infliximab, adalimumab and golimumab for non-infectious refractory uveitis in adults. Int J Clin Pharmacol Ther. 2015;53:377–390. | ||
Llorenç V, Mesquida M, de la Maza MS, et al. Certolizumab pegol, a new anti-TNF-α in the armamentarium against ocular inflammation. Ocul Immunol Inflamm. 2016;24:167–172. | ||
Wroblewski K, Sen HN, Yeh S, et al. Long-term daclizumab therapy for the treatment of noninfectious ocular inflammatory disease. Can J Ophthalmol. 2011;46:322–328. | ||
Lower EE, Baughman RP, Kaufman AH. Rituximab for refractory granulomatous eye disease. Clin Ophthalmol. 2012;6:1613–1618. | ||
Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J. 2014;43:1525–1528. | ||
Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130:492–513. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.