Back to Journals » International Medical Case Reports Journal » Volume 19
Malignant Transformation and Management of Cutaneous Squamous Cell Carcinoma in a 7-Year-Old Child with Xeroderma Pigmentosum: A Case Report from Somaliland
Authors Aw Hashi MO, Ahmed SI ![]() , Abdi MA, Abdirahman RM, Abdullahi RR
, Abdi MA, Abdirahman RM, Abdullahi RR
Received 31 January 2026
Accepted for publication 25 June 2026
Published 30 June 2026 Volume 2026:19 567374
DOI https://doi.org/10.2147/IMCRJ.S567374
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Thomas E Hutson
Mohamed Osman Aw Hashi1,2, Sadam Ismail Ahmed3, Mohamed Ahmed Abdi4, Radia Mohamed Abdirahman5, Riham Rashid Abdullahi5
1Department of Medicine and Surgery, College of Medicine and Surgery, University of Hargeisa, Hargeisa, Somaliland; 2Department of Dermatology and Venereology, Hargeisa Group Hospital, Hargeisa, Somaliland; 3College of Medicine and Health Science, Department of Anesthesia, Burao Central University College, Burao, Somaliland; 4Department of Pediatric Surgery, Hargeisa Group Hospital, Hargeisa, Somaliland; 5Department of Emergency, Hargeisa Group Hospital, Hargeisa, Somaliland
Correspondence: Sadam Ismail Ahmed, Email [email protected]
Introduction: Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder characterized by defective DNA repair, leading to extreme photosensitivity Affected individuals have a profoundly elevated risk estimated to be more than 10,000-fold greater than the general population of developing cutaneous malignancies, primarily squamous cell carcinoma (SCC). Management requires rigorous photoprotection and prompt, often repeated, surgical excisions. This is critically challenging in resource-limited settings with high ultraviolet exposure and limited surgical/oncological infrastructure.
Case Presentation: We report a 7-year-old female from rural Somaliland, born to consanguineous parents, She presented with a 6-month history of an ulcerated, bleeding plaque on her right temple. Two younger siblings also exhibited milder dermatological symptoms, raising suspicion of familial XP with a 2-year history of progressive photosensitivity, xerosis, and freckling. Examination revealed characteristic XP findings including diffuse poikiloderma and ocular involvement (conjunctival growths, dry eyes). A clinical diagnosis of XP with suspected SCC was made. The lesion was initially excised with a 2mm margin; however, the SCC recurred within three months. A second, more extensive wide local excision with a 4mm margin and flap reconstruction was successful. Histopathology confirmed well-differentiated SCC invading the deep dermis.
Conclusion: This case highlights the aggressive and recurrent nature of cutaneous SCC in XP patients, It underscores the necessity of standard 4mm surgical margins even in anatomically challenging areas to prevent recurrence. Even in childhood. It underscores the immense challenges of managing this life-threatening genodermatosis in settings with limited access to specialized multidisciplinary care, advanced reconstructive surgery, and lifelong photoprotective resources. The iterative surgical approach, complicated by graft failure, illustrates the need for robust primary excision and highlights the role of pragmatic, adaptive surgical planning in low-resource contexts.
Keywords: xeroderma pigmentosum, squamous cell carcinoma, Somaliland, DNA repair disorder, pediatric oncology, reconstructive surgery
Introduction
Xeroderma pigmentosum (XP) is a rare, autosomal recessive genodermatosis resulting from mutations in genes critical for the nucleotide excision repair (NER) pathway, which is essential for correcting ultraviolet (UV) radiation-induced DNA damage.1 This molecular defect confers an extreme, lifelong photosensitivity and a profoundly elevated risk—estimated to be more than 10,000-fold greater than the general population—of developing cutaneous malignancies at a remarkably young age and while rare globally (1:1,000,000 in the US), the prevalence is significantly higher in North Africa and the Middle East (approximately 1:10,000 to 1:30,000) due to higher rates of consanguinity.2,3 The clinical spectrum is dominated by the early onset of sunburn reactions, progressive poikiloderma, and the development of actinic keratoses, basal cell carcinomas, melanomas, and notably, aggressive squamous cell carcinomas (SCCs).4 Ocular (eg., photophobia, keratitis, neoplasms) and neurological manifestations further define complex phenotypic subtypes.5
In high-resource settings, management of XP is a multidisciplinary, lifelong endeavor predicated on rigorous and absolute photoprotection, regular surveillance with prompt excision of neoplasms, and supportive neurological and ophthalmological care.6 The prognosis has improved significantly with these interventions, though it remains guarded and is heavily dependent on the severity of neurological involvement and the prevention of metastatic cancer.7
Conversely, in regions characterized by intense ambient UV radiation, subsistence economies, and constrained healthcare infrastructure—such as the Horn of Africa—the management of XP presents a formidable, often tragic, set of challenges.8 Patients frequently present late, with advanced, disfiguring, or metastatic malignancies.9 Diagnostic capabilities for both the underlying genodermatosis and its malignant sequelae are limited, while access to specialized surgeons, oncologists, and the fundamental tools of photoprotection (eg., broad-spectrum sunscreens, protective clothing) is severely restricted.10 These systemic deficiencies intersect with socioeconomic vulnerabilities, including poverty and health illiteracy, creating a context where a preventable, DNA-repair disorder translates into a near-certainty of early morbidity and mortality.11
This case report details the presentation, complex surgical management, and complications of a cutaneous SCC in a young child with XP in Hargeisa, Somaliland. It serves to illustrate the accelerated natural history of this disease in a high-UV environment and to highlight the profound diagnostic, therapeutic, and ethical challenges encountered when managing a life-threatening genodermatosis in a resource-limited setting. The case underscores the urgent need for context-appropriate guidelines, international collaboration, and advocacy for basic preventive resources to alter the devastating trajectory of XP for patients in the world’s most vulnerable regions.
Case Presentation
A 7-year-old female from a rural nomadic community near Hargeisa, Somaliland, presented to the dermatology outpatient clinic at Hargeisa Group Hospital. She had a 6-month history of a slowly enlarging, non-healing, ulcerated lesion on her right temple, associated with bleeding and hemorrhagic crusting. For two years, she had exhibited pronounced photosensitivity, dry skin and eyes, and developed numerous freckle-like macules and rough plaques on her face and forearms. She also had a history of mild deafness and delayed speech. Due to extreme sun sensitivity and speech regression, she had withdrawn from school.
Past Medical, Family, and Social History
Her parents were first cousins (consanguineous). Of her five siblings, two others (aged 3 and 5 years) exhibited milder freckling and xerosis. While their symptoms were less severe at presentation, this likely reflects a shorter duration of cumulative UV exposure rather than variable expressivity, given the recessive nature of XP. The family’s previous nomadic lifestyle involved constant sun exposure. They lived in a modest home without electricity. Care was supported by extended family.
Physical Examination
The patient was alert but visibly photophobic. Dermatologic examination revealed a single, well-demarcated, indurated, ulcerated plaque (3.5 x 2 cm) with hemorrhagic crust and tenderness on the right temple (Figures 1 and 2). No regional lymphadenopathy was palpable. There was diffuse xerosis and poikiloderma with numerous hyper- and hypopigmented macules on the face and forearms. Ophthalmic examination showed bilateral conjunctival dryness and benign growths (pterygium-like) on the right eye.
 |
Figure 1 There are multiple dyspigmentations with ulcer at right supraorbital area. |
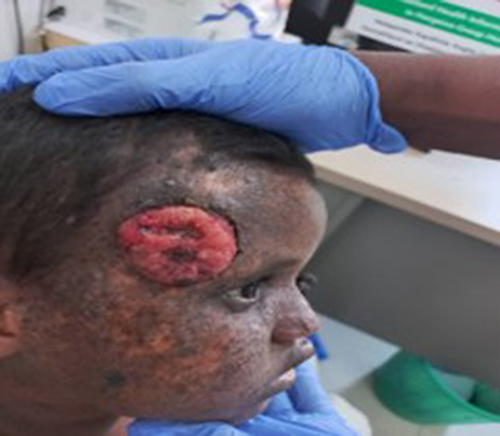 |
Figure 2 There is single 3.5x2cm well dermarcated ulcerated tender plaque with easy bleeding and crustation over left temple. |
Management and Clinical Course: A clinical diagnosis of XP with probable cutaneous SCC was made. Initially, the lesion was excised with a 2mm clinical margin. This narrow margin was chosen due to the lesion’s proximity to the orbit and a desire to preserve tissue for a full-thickness skin graft (FTSG) (Figures 3 and 4). Histopathological analysis of the excised specimen confirmed a well-differentiated squamous cell carcinoma with clear peripheral margins with invasion into the reticular dermis.
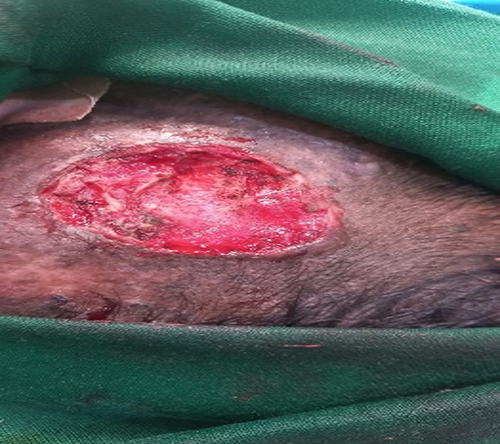 |
Figure 3 There is excision of the ulcer with 2mm margin. |
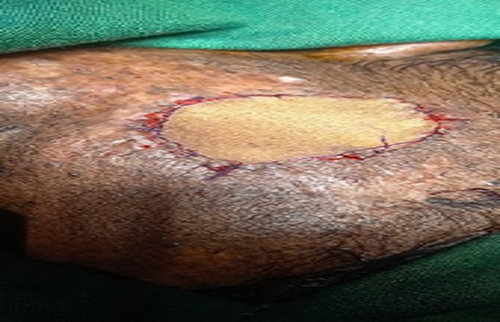 |
Figure 4 There is full thickness graft from right groin. |
The postoperative course was complicated by graft infection and failure. Three months later, the SCC recurred at the same site as a red, ulcerated Plaque (Figure 5). This rapid recurrence confirms that the initial 2mm margin was oncologically inadequate.
 |
Figure 5 There is ulcerated plaque with oozing and pus drainage with some crust. |
This was managed with daily local wound care (chlorhexidine cleansing, topical antibiotic ointment, non-adherent dressings) until secondary intention healing was achieved. Three months later, the SCC recurred at the same site, presenting as a red, ulcerated plaque with tissue overgrowth (Figure 6).
 |
Figure 6 There is red ulcer with tissue overgrowth over right supraorbital area. |
A second, definitive procedure was performed: a wide local excision with a 4mm clinical margin. Given the prior graft failure, the defect was closed using a local rotational flap (Figure 7). This procedure healed primarily without complication The patient was referred to ophthalmology for ocular lubricants and UV-protective sunglasses. The family received counseling on sun avoidance, though access to high-SPF sunscreen remains a significant financial barrier.
 |
Figure 7 There is flap performed at skin lesion site after wide local excision of 4mm margin. |
Discussion
This case illustrates the accelerated natural history of XP in high-UV environments. The most critical clinical takeaway is the management of surgical margins in pediatric SCC. While 2mm margins are occasionally used in low-risk SCC, the aggressive nature of XP-associated tumors necessitates at least 4mm margins or Mohs micrographic surgery.6 The rapid recurrence in this patient after a 2mm excision underscores that even in cosmetically sensitive areas like the temple, oncologic safety must take precedence.
Conclusion
This report serves as a reminder of the health inequities faced by patients with rare genetic disorders in resource-poor regions. While the second surgical intervention with wider margins was successful, the long-term prognosis remains guarded due to the high likelihood of future primary tumors. There is an urgent need for global health initiatives to provide affordable photoprotective resources and training in aggressive surgical management for clinicians in the Horn of Africa.
Patient Perspective
The parents expressed profound anxiety about their child’s condition and future. They were grateful for the explanation and initial care but were concerned about the long-term management and costs associated with the disease, which contributed to their decision to return to their village.
Ethical Approval
In our institute, the ethical approval is not required for publication of case reports, so our hospital is waived for case reports.
Consent for Publication
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Informed Consent
Informed consent was obtained from the patient’s parents (Primary Care giver) for the publication of this case report.
Author Contributions
All authors contributed significantly to the work reported, including conception, design, implementation, data collection, analysis, interpretation, editing, proofreading, revising, publishing, agreed on the journal to which the article was submitted, and responsibility for all aspects of the work.
Funding
No funding for this case.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132(3 Pt 2):785–7. doi:10.1038/jid.2011.426
2. Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterizes the role of DNA repair. J Med Genet. 2011;48(3):168–176. doi:10.1136/jmg.2010.083022
3. Lehmann J, Seebode C, Martens MC, Emmert S. Xeroderma pigmentosum - facts and perspectives. Anticancer Res. 2018;38(2):1159–1164. doi:10.21873/anticanres.12335
4. Lim R, Sethi M, Morley AMS. Ophthalmic manifestations of xeroderma pigmentosum: a perspective from the United Kingdom. Ophthalmology. 2021;128(8):1194–1202. doi:10.1016/j.ophtha.2020.12.025
5. Fassihi H, Sethi M, Fawcett H, et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016;113(9):E1236–E1245. doi:10.1073/pnas.1519444113
6. Tamura D, DiGiovanna JJ, Khan SG, Kraemer KH. Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol Photoimmunol Photomed. 2014;30(2–3):146–152. doi:10.1111/phpp.12108
7. Totonchy MB, Tamura D, Pantell MS, et al. Auditory analysis of xeroderma pigmentosum 1971-2012: hearing function, sun sensitivity and DNA repair predict neurological degeneration. Brain. 2018;141(1):145–161. doi:10.1093/brain/awx284
8. Seth D, Cheldize K, Brown D, Freeman EE. Global burden of skin disease: inequities and innovations. Curr Dermatol Rep. 2017;6(3):204–210. doi:10.1007/s13671-017-0192-7
9. Bouroubi A, Boudghene-Stambouli O. Xeroderma pigmentosum in Algeria: a study of 150 patients. Ann Dermatol Venereol. 2016;143(12):S44.
10. Hay RJ, Johns NE, Williams HC, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. 2014;134(6):1527–1534. doi:10.1038/jid.2013.446
11. Dolmetsch AM, Galdino da Silva D, De Andrade DA, et al. Dermatology training for primary care clinicians in rural Haiti. Int J Dermatol. 2016;55(12):1346–1351. doi:10.1111/ijd.13316
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.