Back to Journals » Hepatic Medicine: Evidence and Research » Volume 11
Lysosomal acid lipase deficiency – early diagnosis is the key
Authors Strebinger G, Müller E, Feldman A, Aigner E
Received 17 January 2019
Accepted for publication 22 February 2019
Published 23 May 2019 Volume 2019:11 Pages 79—88
DOI https://doi.org/10.2147/HMER.S201630
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Georg Strebinger, Elena Müller, Alexandra Feldman, Elmar Aigner
First Department of Medicine, Paracelsus Medical University, Salzburg, Austria
Abstract: Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease that may present from infancy to late adulthood depending on residual enzyme activity. While the severe form manifests as a rapidly progressive disease with near universal mortality within the first 6 months of life, milder forms frequently go undiagnosed for prolonged periods and typically present with progressive fatty liver disease, enlarged spleen, atherogenic dyslipidemia and premature atherosclerosis. The adult variant of LAL-D is typically diagnosed late or even overlooked due to the unspecific nature of the presenting symptoms, which are similar to common changes observed in the context of the metabolic syndrome. This review is aimed at delineating clinically useful scenarios in which pediatric or adult medicine clinicians should be aware of LAL-D as a differential diagnosis for selected patients. This is particularly relevant as a potentially life-saving enzyme replacement therapy has become available and the diagnosis can easily be ruled out or confirmed using a dried blood spot test.
Keywords: lysosomal acid lipase, microvesicular steatosis, liver cirrhosis, atherogenic dyslipidemia, low HDL
Introduction
Lysosomal acid lipase deficiency (LAL-D) is an inherited ultra-rare, autosomal-recessive lysosomal storage disease.1 The LAL serves as a non-redundant enzyme in hydrolyzing triglycerides and cholesteryl esters in lysosomes.2,3 The key mechanism of the disorder involves the progressive accumulation of cholesteryl esters and triglycerides in the lysosomes of hepatocytes and macrophages, thereby inducing organ damage over time.4 LAL-D presents with a wide clinical spectrum of severity ranging from variants with early mortality within the first months of life to mild asymptomatic forms in adulthood.5
LAL-D – Wolman disease
The most severe form of LAL-D has previously been referred to as Wolman disease (WD) and was first reported in 1956.6 WD, or early onset LAL-D, has a rapidly progressive course and is fatal within the first year of life, mostly even within the first 3–6 months at a median age of death of 3.7 months.6,7 The dismal prognosis has only recently improved with the use of enzyme replacement therapy (ERT).8 WD is characterized by absent or severely deficient (<1%) LAL enzyme activity.9,10 Infants suffering from early-onset LAL-D usually present with acute illness and failure to thrive.6,7,11,12 They have gastrointestinal symptoms including vomiting and diarrhea with steatorrhea resulting in growth failure. A further characteristic is massive hepatosplenomegaly entailing abdominal distension. The disease rapidly leads to liver failure including jaundice, portal hypertension and multi-organ failure. Additionally, in most of the patients suffering from early-onset LAL-D, calcifications of the adrenal glands are detected radiologically or in pathological examination.7,13
LAL-D – Cholesteryl ester storage disease
Cholesteryl ester storage disease (CESD) has historically been used to describe the late-onset variant of LAL-D which has a variable and milder clinical presentation.5,14,15 The classical symptom complex includes a fatty liver with mildly elevated aminotransferases, characterized by the development of fibrosis and cirrhosis with a gradual decline in liver function and an atherogenic dyslipidemia profile with low high-density lipoprotein (HDL) cholesterol, high low-density lipoprotein (LDL) cholesterol and triglycerides, triggering premature atherosclerotic vascular disease. Age at disease onset and disease severity reflect the residual LAL enzyme activity, although other modifiers of the disease course are likely to exist, which have, however, not been systematically investigated. Disease manifestation may range from early childhood to late adulthood. Liver disease is the most common cause of death, typically occurring before the age of 40 years, and hepatocellular carcinoma and liver failure may develop at young age.5,16,17 The disease spectrum of LAL-D is depicted in Figure 1.
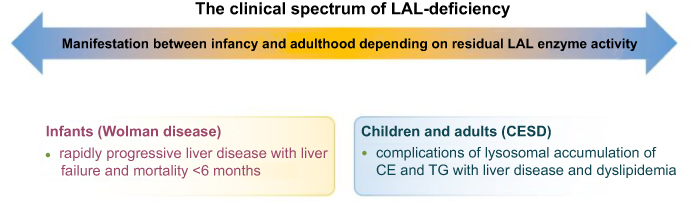 | Figure 1 The clinical spectrum of LAL-deficiency. The clinical spectrum of LAL-deficiency ranges from severe variants with early death, previously referred to as Wolman disease, to milder variants manifesting during childhood or adulthood, known as CESD. Abbreviations: CESD, cholesteryl ester storage disease; LAL, lysosomal acid lipase; CE, cholesteryl esters; TG, triglycerides. |
This review is aimed at summarizing presenting signs and symptoms of LAL-D in children and adults, only briefly touching on infantile WD when it serves the topical purpose. The diagnosis among these subjects with the milder variant of LAL-D is frequently missed and timely establishment of the diagnosis and initiation of treatment is expected to significantly improve long-term outcome.15,18
Genetics and epidemiology
Both WD and CESD are due to mutations in the LIPA gene, which is located on chromosome 10q23.2-q23.3 and contains 10 exons.19 LAL-D is inherited in an autosomal-recessive mode indicating that the presence of one intact copy is sufficient to maintain physiological functions.20 To date, a total of 120 disease-associated mutations have been reported.21 LIPA genotypes determine the level of residual enzymatic activity, being directly linked to disease progression. WD is characterized by a LAL activity between complete absence to less than 1%, while LAL activity in late-onset CESD typically ranges between <1% and 10% in peripheral leukocytes and cultured fibroblasts.5,9,10,22 The most common defect is a splice-junction mutation in exon 8 (E8SJM; rs116928232: c.894G>A, p.S275_Q298del), which is found in approximately half of all children and adults with LAL-D.1,23 Due to the rarity of the disease, with mainly case reports in the literature, there is only limited data on the prevalence of LIPA mutations. There have been studies investigating the incidence of heterozygosity for the E8SJM mutation in several populations.1,24,25 Assuming that the mutation is responsible for 50% of LAL-D causing mutations, the prevalence for homozygote or compound heterozygote patients was estimated to be 1:40,000 in Germany.1 With an allele frequency of 60% for the Hispanic and Caucasian population in the US, the prevalence was estimated to be 1:130,000.25 Further, it has been observed that the E8SJM mutation is rare in the Asian and African-American populations, illustrating that further studies are needed to assess the epidemiology.25
In a recent comprehensive genetic epidemiological analysis by Carter et al21 120 different disease-causing mutations were reported. The prevalence was calculated to 1:177.452 among a multi ancestry population. The lowest prevalence was found in people of East-Asian, South-Asian, Finnish and Ashkenazi Jewish descent. These figures represent the most reliable data currently available on the prevalence of all disease-causing mutations across various populations, hence confirming that LAL-D is an ultra-rare disease. According to these figures, one may conclude that 3–5 child or adult subjects per million Caucasians may suffer from LAL-D.
LIPA mutation heterozygosity
Muntoni et al1 hypothesized that heterozygosity for the E8SJM mutation in the LIPA gene was associated with pathological lipid profiles similar to a polygenic hypercholesterolemia phenotype. However, a main limitation of the study was the low number of only 13 E8SJM carriers. Stitziel et al24 later genotyped a larger cohort in order to investigate the lipid profile and cardiovascular risk of LIPA E8SJM heterozygotes. They found no association concerning plasma lipid profile or risk of myocardial infarction. Thus, to date no clinical phenotype for heterozygous carriers has been identified, although LAL activity may serve as a disease modifier in human non-alcoholic fatty liver disease (NAFLD).26
Mechanisms of disease
LAL is a lysosomal enzyme that hydrolyzes cholesteryl esters and triglycerides derived from LDL particles after their uptake via the LDL-receptor-endosomal pathway, mainly into hepatocytes.2 Hence, LAL activity is crucially involved in providing the cell with free cholesterol and free fatty acids. In case of a defective LAL enzyme un-hydrolyzed cholesteryl esters and, to a lower degree, also triglycerides accumulate in multiple cells throughout the body.3,4,12,27,28 Quantitatively, however, the pathway is most relevant in hepatocytes and cells of the mononuclear phagocyte system. The progressive accumulation of these lipids in lysosomes leads to the universally observed micro-vesicular hepatic steatosis.28,29 Lysosomal lipid accumulation represents an inflammatory trigger via yet unidentified mechanisms that induce fibrogenesis and subsequent development of cirrhosis over time. It can only be extrapolated from the role of lysosomal cholesterol in other diseases, such as NAFLD, that lysosomal lipid accumulation represents a potent trigger of the NLRP3-inflammosome pathway leading to liver inflammation and activation of hepatic stellate cells.30
As free cholesterol and fatty acids are not sufficiently exported from the lysosomes in LAL-D subjects, the cytoplasm features low concentrations of these compounds.2 Via activation of the key regulating transcription factors of lipogenesis, ie, sterol regulatory element binding proteins 1a, 1c and 2, the biosynthesis of cholesterol, fatty acids and consecutively apolipoprotein B-100, the major protein component of the increasingly produced very low-density lipoprotein (VLDL) particles. Further, LDL-receptor upregulation occurs in order to counteract low cytoplasmic lipid concentrations.31–34 Additionally, low HDL cholesterol concentrations arise from lack of liver X-receptor activation which is linked to low expression of ATP-binding cassette transporter A1 (ABCA1) and thus apolipoprotein A1 (ApoA1) export from liver cells.35 As ApoA1 is the key apolipoprotein of HDL particles, low concentrations of HDL cholesterol are typically found in LAL-D subjects.32 The mechanisms of disease are summarized in Figure 2.
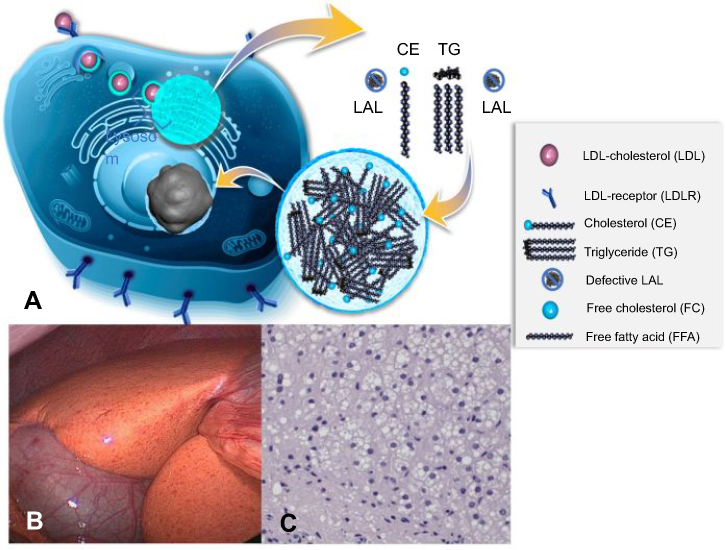 | Figure 2 Summary of the mechanisms of disease. Cholesterol is physiologically taken up as cholesteryl esters via the LDL receptor – endocytosis pathway into lysosomes and then hydrolyzed by LAL enzyme (A). In cases of defective LAL activity cholesteryl esters and triglycerides accumulate in lysosomes, leading to universal microvesicular hepatic steatosis with development of fibrosis (C). Cholesterol accumulation causes orange coloration of the fatty liver (B), which is different from the triglyceride accumulation characteristic of common-type non-alcoholic fatty liver disease. (A) Published with permission of Alexion Pharmaceuticals. (B) and (C) Reproduced from Zandanell S, Primavesi F, Aigner E. Hepatosteatosis from Lysosomal Acid Lipase Deficiency. J Gastrointest Surg. 2019;23(3):601-602 ( |
It is important to note that lysosomal degradation of LDL particles also takes place in other cells, although these are quantitatively not as important as hepatocytes and macrophages. In electron microscopy, lipid inclusions have been observed in other cell types such as biliary epithelium and endothelial cells.28 From a pathophysiological point of view, this may be relevant for the pathogenesis of atherosclerosis. Development of atherosclerotic manifestations appears out of proportion to the extent of dyslipidemia, suggesting these mechanisms via direct involvement of vessel wall macrophages and endothelial cells as potential contributors. Additionally, rapid reversal of carotid stenosis within months in response to ERT has been observed in a patient even before the correction of dyslipidemia (M Zharkova, First Moscow State Medical University, Moscow, Russia, personal communication, April 2018), which also lends support to the mechanistic relevance of these pathways in vessel wall macrophages and endothelial cells. Figure 3 summarizes the development of relevant organ involvements in LAL-D.
 | Figure 3 Development of the clinical manifestations in LAL-D. Although the mechanisms are universal as depicted in the sequence of events on the left, clinical manifestations and consequences range from liver disease to atherosclerosis and malabsorption. Abbreviations: LAL, lysosomal acid lipase; CE, cholesteryl esters; TG, triglycerides; ALT, aspartate transaminase; AST, alanine aminotransferase; LDL-c, low-density lipoprotein cholesterol; HDL-c, high-density lipoprotein cholesterol; ESLD, end-stage liver disease; MCI, myocardial infarction. |
The natural course of LAL-D and prognosis
Bernstein et al5 reviewed the clinical features of 135 CESD patients (55% females). The mean age of onset was 5 years, and younger patients were more severely affected than those with later onset of LAL-D. Only 11% had become clinically apparent after the age of 12 years. All reported subjects presented with significant liver disease with elevated serum transaminases. Hepatomegaly was present in nearly all (99.3%) and splenomegaly in 74% of the patients. In 64% of the biopsied patients liver fibrosis or cirrhosis and in 29% cirrhosis was detected in the histological work-up. Furthermore, esophageal varices were observed in 8.9% of these subjects. Eleven patients were reported dead, liver failure being the main cause of death (73%), mostly at young age; 50% of the deaths occurred before the age of 21 years and nearly universally by the age of 40 years. Cases of older subjects with liver and vascular disease up to the age of 60 have also been reported. However, long-term follow-up data were not available for most of the cases reported, possibly underestimating the extent of morbidity and mortality. On the contrary, it must be taken into account that cases with a milder course may go undiagnosed and unpublished.
Hypercholesterolemia due to an increase in LDL cholesterol was present in nearly all patients, despite many of them being treated with statins, indicating an increased cardiovascular risk.5 There are case reports of accelerated atherosclerosis in LAL-D patients including aortic plaques in a 9-year-old and cardiovascular events at young age.4,36–38 However, due to limited data with low case numbers, to our knowledge, there are currently no published studies investigating the cardiovascular outcome of CESD patients and an international registry is currently being established.
Suspecting LAL-D in the clinical context
It appears evident that there is a gap between the genetically estimated prevalence of LAL-D and the cases known in most countries worldwide.1,21,25 Due to the rarity of LAL-D, finding cases with the diagnosis is an unlikely event in any physicians’ lifetime. However, there are particular clinical scenarios in which LAL-D represents a disease to be considered on the differential diagnostic agenda.
We have aimed to illustrate the diagnosis of LAL-D using four typical clinical scenarios according to the leading presenting symptom or finding in order to augment the yield for physicians who encounter these patients.
Scenario 1 – NAFLD in lean subjects, microvesicular steatosis
As LAL-D is a disorder that primarily affects the liver, it represents a differential diagnosis in the work-up of a patient with NAFLD.39,40 NAFLD by definition is a diagnosis of exclusion, which implies that other potentially underlying liver diseases must be excluded before arriving at the diagnosis NAFLD. Both the European and the American NAFLD guidelines have included LAL-D as one of the diagnoses that need to be ruled out in the diagnostic algorithm of a potential NAFLD patient.39,40 In our experience, the practical difficulty in the care of a fatty liver patient is that the metabolic abnormalities linked to insulin resistance are so common in Western societies that these are sufficient to explain fatty liver in nearly all patients in the clinical routine. Several “red flags” are in our experience helpful to consider LAL-D as the causative disease for a patient’s fatty liver: 1) absence of obvious components and family history of the metabolic syndrome (MetS).41 Most patients with MetS have other components like hypertension or elevated blood glucose and insulin, while LAL-D subjects will present with a fatty liver and dyslipidemia that is out of proportion with the remainder of the metabolic risk profile. 2) LAL-D subjects have some degree of splenomegaly due to lipid storage in the absence of clinical evidence of advanced liver disease or portal hypertension. 3) Liver biopsy is not performed in all NAFLD patients and biopsy indications vary widely within countries and even locally. The key feature that needs to bring LAL-D to the awareness of a physician is the finding of microvesicular steatosis.28 Lysosomal storage of cholesteryl esters and triglycerides causes diffuse microvesicular steatosis which involves hepatocytes and liver macrophages. Hulkova and Elleder29 have systematically analyzed histopathological features of LAL-D in contrast to other causes of microvesicular steatosis (mainly drugs and mitochondrial disorders). In order to distinguish cytosolic from lysosomal lipid accumulation immunohistochemical staining with lysosomal markers (cathepsin D, lysosomal-associated membrane protein (LAMP) 1, LAMP 2 and lysosomal integral membrane protein (LIMP) 2) in paraffin-embedded liver specimens facilitates the diagnosis of LAL-D. Moreover, maltese cross-type birefringence exhibited by crystals of cholesteryl esters under polarized light in unfixed frozen material is pathognomonic for LAL-D.5,29 However, we consider the practical value of these recommendations to be limited, since these stainings are not performed routinely but only upon request. Thus, for the clinician the key message is to consider LAL-D in any histopathological report of microvesicular steatosis. In case of suspected LAL-D, liver biopsy does not have to be performed as the confirmation of LAL-D can be achieved by dried blood spot test.32,42 4) Only rarely will the macroscopic appearance of the liver raise the possibility of an underlying storage disease as it presents in a brilliant orange color due to the excessive storage of cholesteryl ester which is distinct from the yellow of triglyceride accumulation in common NAFLD.43,44
Scenario 2 – hepatosplenomegaly
The finding of hepatosplenomegaly needs to raise the possibility of a storage disorder once hematological causes have been ruled out.45 In LAL-D, the degree of splenomegaly appears smaller compared to the relative degree of hepatomegaly and this is different from Gaucher disease, another lipid storage disorder with dominant splenomegaly and a minor degree of hepatomegaly. Figure 4 shows MRI findings in an otherwise healthy young male with LAL-D (E Aigner, First Deparment of Medicine, Paracelsus Medical University Salzburg, Austria, unpublished own data, April, 2018).
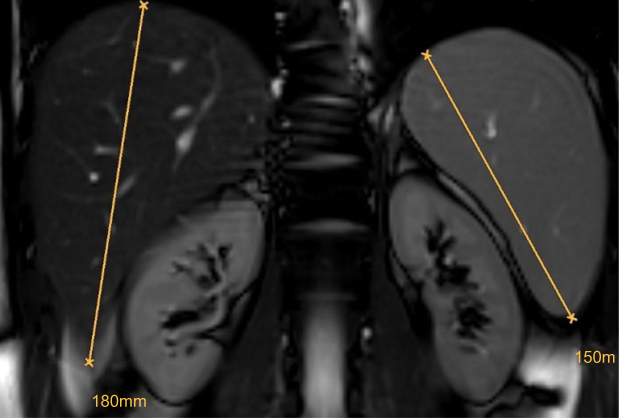 | Figure 4 Hepatosplenomegaly. In LAL-D, the relative degree of hepatomegaly is usually prevalent over the relative degree of splenomegaly. The exclusion of hematological disorders should prompt the work-up for inherited storage disorders. Abbreviation: LAL-D, lysosomal acid lipase deficiency. |
Scenario 3 – advanced liver disease and cryptogenic cirrhosis
As mentioned above, the large majority of LAL-D subjects develop advanced liver disease with end-stage liver disease, decompensation, hepatocellular carcinoma or the need for transplantation prior to the age of 40 years. Hence, the work-up for any patient with advanced liver disease at an early age should contain a LAL-D dried blood spot test since even the presence of another liver disease does not exclude concomitant LAL-D and most common liver diseases do not progress to decompensation within the fourth decade of life. Particularly when other liver disorders are not detected, LAL-D is mandatory to be tested in advanced liver disease of unknown cause.
Scenario 4 – atherogenic dyslipidemia and atherosclerotic disease
Lipid profiles are commonly determined in the hospital and the family practice routine and may thus be the first indicator for LAL-D, which, according to the underlying mechanisms, is a lipid metabolism disorder rather than a liver disease. The lipid profile of LAL-D resembles hyperlipidemia type IIb with mild to moderate elevation of triglycerides and LDL-C and a decreased concentration of HDL-C.5 In the general population, this lipid profile is quite consistently observed in subjects with type 2 diabetes or at least some degree of insulin resistance.46 From a practical point of view, when the lipid findings are the starting point and atherogenic dyslipidemia is observed in the absence of other components of the MetS, LAL-D should be considered. Ideally, the further diagnostic work-up should include at least biochemical liver tests and abdominal ultrasound in these unexplained cases of atherogenic dyslipidemia. An American and a Portuguese study investigated the prevalence of LAL-D and LIPA mutations in cohorts of familial hypercholesterolemia and from dyslipidemia clinics and found subjects with LAL-D in both settings.47,48 To the best of our knowledge, no data are available on the prevalence of LAL-D in subjects with premature atherosclerotic disease. A further diagnostic indicator can be derived from the response to medical treatment. While typically subjects with combined dyslipidemia respond well to lifestyle changes and statins or fibrates, these measures hardly affect the lipid derangements in LAL-D subjects.5 It would be particularly worthwhile to screen for LAL-D among subjects who are candidates for proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitor treatment. On the one hand, LAL-D patients are potential candidates to be selected for the treatment with PCSK9-inhibitors due to their insufficient treatment response to statins. Additionally, administration of PCSK9-inhibitors could aggravate liver disease in LAL-D due to the further up-regulation of LDL receptors on the cell surface of hepatocytes. Thus, LAL-D could, in fact, represent a medical contraindication to the administration of PCSK9-inhibitors which should be excluded prior to the administration of PCSK9 inhibitors. PCSK9 inhibitors did not show adverse effects with regard to the liver in the pivotal registration trials.49
Establishing the diagnosis LAL-D
The relatively low number of cases diagnosed with LAL-D in comparison with the genetic prevalence is likely due to a low degree of suspicion of the diagnosis and the common nature of the key clinical and biochemical abnormalities. Hence, raising awareness among clinicians is the crucial step in identifying subjects with LAL-D as establishment of the diagnosis represents the easier step once the disease is suspected. The following steps are clinically useful to establish the diagnosis in a suspected case.
Tests to confirm lysosomal storage diseases
There are two tests, that can be considered an unspecific examination for the presence or absence of a lysosomal storage disease. These are chitotriosidase levels and presence of vacuolated lymphocytes in peripheral blood smear.
Chitotriosidase is a chitinase of unknown physiological function in humans. Its production is increased in activated macrophages due to lysosomal storage.50 Excessive elevations are found in Gaucher disease but milder elevations are a common finding to all lysosomal storage disorders with macrophages involved.38 Vacuolated lymphocytes in a blood smear, which are present in LAL-D, represent an easily accessible tool to confirm the suspicion of a vacuolated lysosomal storage disorder.51,52 Figure 5 shows a vacuolated lymphocyte in a lysosomal storage disorder.
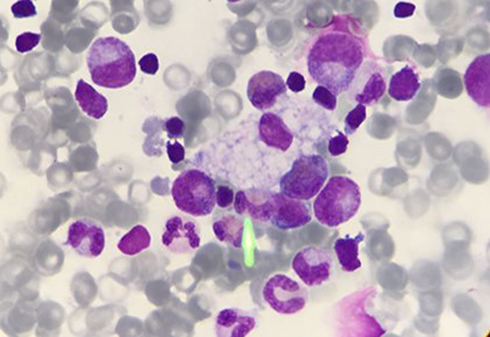 | Figure 5 Vacuolated lymphocyte. Vacuolated lymphocytes may provide a diagnostic clue in many but not all lysosomal storage disorders. Although not massive in LAL-D, the presence of vacuolated lymphocytes can provide important diagnostic hints toward an underlying lysosomal storage disorder. Abbreviation: LAL-D, lysosomal acid lipase deficiency. |
Tests to establish diagnosis LAL-D
The suspicion of LAL-D can be confirmed by measurement of enzyme activity in a dried blood spot test and sequencing of the LIPA gene.
LAL-activity – dried blood spot test
LAL enzyme activity can be determined by dried blood spot testing as published by Hamilton et al in 2012.42 In order to distinguish LAL activity from other blood lipases, Lalistat 2, a specific inhibitor of LAL, is added to the reaction. LAL activity is calculated as the difference of total lipase activity with and without the addition of Lalistat 2. This method distinguishes between healthy individuals, carriers and affected patients. The authors have further investigated long-term stability, concluding that LAL activity decreases by 6% after 4 days at room temperature, allowing sufficient diagnostic accuracy if a prompt transport of the specimens is ensured. Sample stability can be further improved at lower temperatures. Furthermore, the method is also used for LAL enzyme activity testing in fibroblasts and leukocytes.53 However, with the use of dried blood spot testing, these methods have become in fact obsolete and dried blood spot testing seems to be superior in the screening for LAL-D concerning accuracy, availability and costs to all other methods and also genetic testing.32
Genetic testing
Sequencing of the LIPA gene is required and recommended for the characterization of the underlying mutation. There are 120 known disease-causing LIPA variants, however, the 4 most common mutations account for 61% of the total allele frequency.21 We recommend the use of LIPA sequencing only in case of a positive LAL dried blood spot test and not as a screening tool in a suspected case.
Treatment of LAL-D
Statins
Taking into consideration that hyperlipidemia is one of the main features of LAL-D, lipid-lowering drugs were the mainstay in the treatment before ERT became available. A mild amelioration of the lipid abnormalities has been reported in some of the subjects treated with statins, cholestyramine and ezetimibe.5,54–58 However, no controlled clinical studies investigated the outcome of statin therapy in LAL-D patients. Despite the successful treatment of dyslipidemia with a documented decrease of hepatomegaly, histological progression of hepatic fibrosis has been reported.59 The observation that patients likely do not benefit from statin therapy concerning liver disease is further supported in the review by Bernstein et al5 demonstrating that in 12 patients treated with statins the liver histology did not improve. As the key mechanism of action of statins is the up-regulation of LDL receptors on the hepatocyte surface and thereby increasing the LDL cholesterol uptake, one might even speculate that statins can accelerate the liver disease in LAL-D.60
Stem cell transplantation
Hematopoietic stem cell transplantation has been reported to be a successful treatment in some infants suffering from WD.61–63 However, there were also reports about disease progression and fatal transplant-related complications.62,64,65
Liver transplantation
Bernstein et al66 investigated the outcome of 18 LAL-D patients that have undergone liver transplantation. In 61% of these patients, multi-systemic LAL-D progression occurred despite liver transplantation. The authors further noted the recurrence of a LAL-D typical histology 9 months after transplantation in one patient. Liver transplantation is indispensable in some patients suffering from end-stage liver failure or liver cancer; however, the deficiency in LAL enzyme activity in bone marrow-derived histiocytes and all other tissues persists. Hence, the replacement of the diseased liver fails to prevent multi-organ disease progression in most patients and hepatic recurrence of LAL-D can occur with the repopulation of the liver with recipient stem cells.66 The use of immunosuppressive agents is likely to further aggravate vascular disease.
Enzyme replacement therapy
Balwani et al67 were the first to report about the clinical use of sebelipase alfa (Kanuma©, Alexion Pharmaceuticals, Inc., Cheshire, CT, USA), a recombinant human LAL enzyme produced by applying targeted gene expression in hen oviduct cells, as an ERT in LAL-D. Sebelipase alfa is well-tolerated and leads to a reduction of transaminases, total cholesterol and triglycerides and to an improvement of HDL cholesterol.67,68 Moreover, liver volume and lipid content of the liver have been shown to decrease under ERT. The superiority of ERT to placebo in the significant reduction of serum aminotransferase levels, hepatic fat content and the amelioration of pathological lipid profiles has been confirmed in the ARISE trial.18 Even infants suffering from WD showed survival well beyond the historical life expectancy and improvement concerning gastrointestinal symptoms, hepatosplenomegaly, anemia, liver dysfunction and growth.8
Conclusion
LAL-D is a rare disease that may go unnoticed or be diagnosed late if not suspected. Although the clinical signs and symptoms are common, as they resemble components of the MetS, the disease needs to be considered in various clinical contexts as described. Establishment of the diagnosis has become easy with the use of dried blood spot tests which specifically test for LAL enzyme activity. With the availability of ERT the historically dire prognosis has dramatically improved, adding a further argument to the usefulness of timely diagnosis.
Disclosure
Elmar Aigner, MD, has received consulting and speaking honoraria from Alexion Pharmaceuticals. The other authors report no conflicts of interest in this work.
References
1. Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866–1868. doi:10.1161/ATVBAHA.107.146639
2. Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem. 1975;250(21):8487–8495.
3. Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage idisease. J Clin Invest. 1972;51(7):1923–1926. doi:10.1172/JCI106997
4. Beaudet AL, Ferry GD, Nichols BL Jr, Rosenberg HS. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr. 1977;90(6):910–914.
5. Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230–1243. doi:10.1016/j.jhep.2013.02.014
6. Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child. 1956;91(3):282–286.
7. Jones SA, Valayannopoulos V, Schneider E, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2016;18(5):452–458. doi:10.1038/gim.2015.108
8. Jones SA, Rojas-Caro S, Quinn AG, et al. Survival in infants treated with sebelipase alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study. Orphanet J Rare Dis. 2017;12(1):017–0587. doi:10.1186/s13023-017-0587-3
9. Aslanidis C, Ries S, Fehringer P, Buchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33(1):85–93. doi:10.1006/geno.1996.0162
10. Pagani F, Pariyarath R, Garcia R, et al. New lysosomal acid lipase gene mutants explain the phenotype of Wolman disease and cholesteryl ester storage disease. J Lipid Res. 1998;39(7):1382–1388.
11. Wolman M, Sterk VV, Gatt S, Frenkel M. Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics. 1961;28:742–757.
12. Crocker AC, Vawter GF, Neuhauser EB, Rosowsky A. Wolman’s disease: three new patients with a recently described lipidosis. Pediatrics. 1965;35:627–640.
13. Marshall WC, Ockenden BG, Fosbrooke AS, Cumings JN. Wolman’s disease. A rare lipidosis with adrenal calcification. Arch Dis Child. 1969;44(235):331–341.
14. Zhang B, Porto AF. Cholesteryl ester storage disease: protean presentations of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2013;56(6):682–685. doi:10.1097/MPG.0b013e31828b36ac
15. Aigner E, Feldman A, Neureiter D, Datz C, Ratziu V, Paulweber B. Benefit of treatment with sebelipase-alfa in a 63-year-old patient with advanced liver and atherosclerotic disease due to lysosomal acid lipase deficiency (LAL-D). Am J Gastroenterol. 2018;113(3):443–445. doi:10.1038/ajg.2017.486
16. Castro Narro GE, Gamboa Dominguez A, Consuelo Sanchez A, et al. Combined hepatocellular-cholangiocarcinoma in a patient with cirrhosis due to cholesteryl ester storage disease. Hepatology. 2018;29(10):30331.
17. Canbay A, Muller MN, Philippou S, Gerken G, Tromm A. Cholesteryl ester storage disease: fatal outcome without causal therapy in a female patient with the preventable sequelae of progressive liver disease after many years of mild symptoms. Am J Case Rep. 2018;19:577–581. doi:10.12659/AJCR.907755
18. Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. 2015;373(11):1010–1020. doi:10.1056/NEJMoa1501365
19. Anderson RA, Rao N, Byrum RS, et al. In situ localization of the genetic locus encoding the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in Wolman disease to chromosome 10q23.2-q23.3. Genomics. 1993;15(1):245–247. doi:10.1006/geno.1993.1052
20. Koch G, Lalley PA, McAvoy M, Shows TB. Assignment of LIPA, associated with human acid lipase deficiency, to human chromosome 10 and comparative assignment to mouse chromosome 19. Somatic Cell Genet. 1981;7(3):345–358.
21. Carter A, Brackley SM, Gao J, Mann JP. The global prevalence and genetic spectrum of lysosomal acid lipase deficiency: a rare condition that mimics NAFLD. J Hepatol. 2019;70(1):142–150. doi:10.1016/j.jhep.2018.09.028
22. Lohse P, Maas S, Elleder M, Kirk JM, Besley GT, Seidel D. Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. J Lipid Res. 2000;41(1):23–31.
23. Burke JA, Schubert WK. Deficient activity of hepatic acid lipase in cholesterol ester storage disease. Science. 1972;176(4032):309–310.
24. Stitziel NO, Fouchier SW, Sjouke B, et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2013;33(12):2909–2914. doi:10.1161/ATVBAHA.113.302426
25. Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958–965. doi:10.1002/hep.26327
26. Tovoli F, Napoli L, Negrini G. et al., A relative deficiency of lysosomal acid lypase activity characterizes non-alcoholic fatty liver disease. Int J Mol Sci. 2017;18:6.
27. Patrick AD, Lake BD. Deficiency of an acid lipase in Wolman’s disease. Nature. 1969;222(5198):1067–1068.
28. Di Bisceglie AM, Ishak KG, Rabin L, Hoeg JM. Cholesteryl ester storage disease: hepatopathology and effects of therapy with lovastatin. Hepatology. 1990;11(5):764–772.
29. Hulkova H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107–1113. doi:10.1111/j.1365-2559.2011.04164.x
30. Walenbergh SM, Shiri-Sverdlov R. Cholesterol is a significant risk factor for non-alcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2015;9(11):1343–1346. doi:10.1586/17474124.2015.1092382
31. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi:10.1172/JCI15593
32. Reiner Z, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency–an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21–30. doi:10.1016/j.atherosclerosis.2014.04.003
33. Jeon T-I, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 2012;23(2):65–72. doi:10.1016/j.tem.2011.10.004
34. Cummings MH, Watts GF, Umpleby AM, et al. Increased hepatic secretion of very-low-density lipoprotein apolipoprotein B-100 in NIDDM. Diabetologia. 1995;38(8):959–967.
35. Bowden KL, Bilbey NJ, Bilawchuk LM, et al. Lysosomal acid lipase deficiency impairs regulation of ABCA1 gene and formation of high density lipoproteins in cholesteryl ester storage disease. J Biol Chem. 2011;286(35):30624–30635. doi:10.1074/jbc.M111.274381
36. Ambler GK, Hoare M, Brais R, et al. Orthotopic liver transplantation in an adult with cholesterol ester storage disease. JIMD Rep. 2012;8:41–46. doi:10.1007/8904_2012_155
37. Elleder M, Chlumská A, Hyánek J, et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol. 2000;32(3):528–534.
38. Vom Dahl S, Harzer K, Rolfs A, et al. Hepatosplenomegalic lipidosis: what unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol. 1999;31(4):741–746.
39. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–357. doi:10.1002/hep.29367
40. Marchesini, G, Day, CP, Dufour, JF, et al.. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388–1402.
41. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415–1428. doi:10.1016/S0140-6736(05)66378-7
42. Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15–16):1207–1210. doi:10.1016/j.cca.2012.03.019
43. Schiff L, Schubert WK, McAdams AJ, Spiegel EL, O’Donnell JF. Hepatic cholesterol ester storage disease, a familial disorder. I. Clinical aspects. Am J Med. 1968;44(4):538–546.
44. Zandanell S, Primavesi F, Aigner E. Hepatosteatosis from lysosomal acid lipase deficiency. J Gastrointest Surg. 2018;6(10):018–3906.
45. Vom Dahl S, Mengel E. Lysosomal storage diseases as differential diagnosis of hepatosplenomegaly. Best Pract Res Clin Gastroenterol. 2010;24(5):619–628. doi:10.1016/j.bpg.2010.09.001
46. Goldberg IJ. Diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab. 2001;86(3):965–971. doi:10.1210/jcem.86.3.7304
47. Chora JR, Alves AC, Medeiros AM, et al. Lysosomal acid lipase deficiency: a hidden disease among cohorts of familial hypercholesterolemia? J Clin Lipidol. 2017;11(2):477–484. doi:10.1016/j.jacl.2016.11.002
48. Pullinger CR, Stock EO, Movsesyan I, et al. Identification and metabolic profiling of patients with lysosomal acid lipase deficiency. J Clin Lipidol. 2015;9(5):716–726. doi:10.1016/j.jacl.2015.07.008
49. Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–1509. doi:10.1056/NEJMoa1500858
50. Boot RG, Renkema GH, Strijland A, van Zonneveld AJ, Aerts JM. Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J Biol Chem. 1995;270(44):26252–26256.
51. Anderson G, Smith VV, Malone M, Sebire NJ. Blood film examination for vacuolated lymphocytes in the diagnosis of metabolic disorders; retrospective experience of more than 2,500 cases from a single centre. J Clin Pathol. 2005;58(12):1305–1310. doi:10.1136/jcp.2005.027045
52. Lake BD. Histochemical detection of the enzyme deficiency in blood films in Wolman’s disease. J Clin Pathol. 1971;24(7):617–620.
53. Civallero G, De Mari J, Bittar C, Burin M, Giugliani R. Extended use of a selective inhibitor of acid lipase for the diagnosis of Wolman disease and cholesteryl ester storage disease. Gene. 2014;539(1):154–156. doi:10.1016/j.gene.2014.02.003
54. Rassoul F, Richter V, Lohse P, Naumann A, Purschwitz K, Keller E. Long-term administration of the HMG-CoA reductase inhibitor lovastatin in two patients with cholesteryl ester storage disease. Int J Clin Pharmacol Ther. 2001;39(5):199–204.
55. Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis. 2005;4(26):4–26. doi:10.1186/1476-511X-4-4
56. Gasche C, Aslanidis C, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol. 1997;27(4):744–750.
57. Levy R, Ostlund RE
58. McCoy E, Yokoyama S. Treatment of cholesteryl ester storage disease with combined cholestyramine and lovastatin. Ann N Y Acad Sci. 1991;623:453–454.
59. Leone L, Ippoliti PF, Antonicelli R, Balli F, Gridelli B. Treatment and liver transplantation for cholesterol ester storage disease. J Pediatr. 1995;127(3):509–510.
60. Ma PT, Gil G, Südhof TC, Bilheimer DW, Goldstein JL, Brown MS. Mevinolin, an inhibitor of cholesterol synthesis, induces mRNA for low density lipoprotein receptor in livers of hamsters and rabbits. Proc Natl Acad Sci U S A. 1986;83(21):8370–8374.
61. Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr. 2007;166(7):663–666. doi:10.1007/s00431-006-0298-6
62. Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43(1):21–27. doi:10.1038/bmt.2008.273
63. Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant. 2000;26(5):567–570. doi:10.1038/sj.bmt.1702557
64. Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolman’s disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant. 2009;44(7):449–450. doi:10.1038/bmt.2009.57
65. Yanir A, Allatif MA, Weintraub M, Stepensky P. Unfavorable outcome of hematopoietic stem cell transplantation in two siblings with Wolman disease due to graft failure and hepatic complications. Mol Genet Metab. 2013;109(2):224–226. doi:10.1016/j.ymgme.2013.03.007
66. Bernstein DL, Lobritto S, Iuga A, et al. Lysosomal acid lipase deficiency allograft recurrence and liver failure- clinical outcomes of 18 liver transplantation patients. Mol Genet Metab. 2018;124(1):11–19. doi:10.1016/j.ymgme.2018.03.010
67. Balwani M, Breen C, Enns GM, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013;58(3):950–957. doi:10.1002/hep.26289
68. Valayannopoulos V, Malinova V, Honzik T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61(5):1135–1142. doi:10.1016/j.jhep.2014.06.022
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.