Back to Journals » Vascular Health and Risk Management » Volume 14
Low nitric oxide level is implicated in sickle cell disease and its complications in Ghana
Authors Antwi-Boasiako C, Campbell AD
Received 20 January 2018
Accepted for publication 27 April 2018
Published 6 September 2018 Volume 2018:14 Pages 199—204
DOI https://doi.org/10.2147/VHRM.S163228
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Takashi Kajiya
Charles Antwi-Boasiako,1 Andrew D Campbell2
1Department of Physiology, School of Biomedical and Allied Health Sciences, College of Health Sciences, University of Ghana, Accra, Ghana; 2Comprehensive Sickle Cell Program Children’s National Medical Center, Division of Hematology, School of Medicine and Health Sciences George Washington University, Washington, DC, USA
Background: Nitric oxide (NO) plays a fundamental role in maintaining normal vasomotor tone. Recent clinical and experimental data suggest that NO may play a role in the pathogenesis and therapy of sickle cell disease (SCD). The aim of this study was to determine NO metabolites (NOx) in SCD patients at steady state and in vaso-occlusive crisis (VOC), as well as those with hemolytic clinical sub-phenotype that includes leg ulcers and priapism.
Methodology: This was a case–control cross-sectional study conducted on a total of 694 subjects including 148 comparison group HbAA, 208 HbSS SCD patients in steady state, 82 HbSC SCD patients in steady state, 156 HbSS SCD patients in VOC, 34 HbSC SCD patients in VOC, 34 HbSS SCD patients in post VOC, 21 HbSS SCD patients with leg ulcer and 11 HbSS SCD patients with priapism, with age ranging from 15 to 65 years. Laboratory diagnosis of SCD was done at the Sickle Cell Clinic of the Korle-Bu Teaching Hospital. Plasma nitric oxide metabolites were measured using Griess reagent system by ELISA method.
Results: Mean NOx of 59.66±0.75 µMol/L in the comparison group was significantly different from those in steady state (P=0.02). During VOC, there was a significant reduction in mean NOx levels to 6.08±0.81 µMol/L (P<0.001). Mean NOx levels were however, significantly higher (50.97±1.68 µMol/L) (P<0.001) in the immediate postcrisis period. The mean NOx levels in the leg ulcer (21.70±1.18 µMol/L) (P<0.001) and priapism (28.97±1.27 µMol/L) (P<0.001) patients were significantly low as compared to the SCD patients in the steady state and comparison group.
Conclusion: This study presents the first report on plasma NOx levels in SCD complication in Ghanaian SCD patients and confirms reduced plasma NOx levels in SCD patients in general.
Keywords: sickle cell disease, nitric oxide metabolites, vaso-occlusive crisis, priapism, leg ulcer, ELISA
Background
Sickle cell disease (SCD) has become one of the most studied inherited human diseases,1 although the condition has been described over a century ago.2 The clinical manifestations fall largely into two sub-phenotypes, defined by hyper-hemolysis and vaso-occlusion.3 The multiple pleiotropic effects of the abnormal hemoglobin S production in sickle cell disease usually range from vaso-occlusive crisis (VOC), pulmonary hypertension to osteonecrosis, stroke and leg ulcers, despite being caused by a single point mutation in the HBB gene.4–6 Among the determinants of SCD, VOC, the hallmark of SCD, is unique, with high prevalence, leading to morbidities, complications and mortalities in affected persons.7,8
The disease is usually characterized by the production of erythrocytes with increased tendency for lysis and adhesion.9 The pathophysiology of SCD is linked with adhesion of sickle erythrocytes, leukocytes and platelets to adhesive molecules on activated cells, which are found mainly in the endothelium of postcapillary venules.10 The release of free heme resulting from chronic intravascular hemolysis binds to nitric oxide (NO), causing its depletion, followed by vasoconstriction and inflammation.3 Moreover, in cases such as the priapism, plasma hemoglobin released by intravascular hemolyzed sickle red blood cells depletes NO, thereby reducing its bioavailability in the erectile tissue. Consequently, the normal balance of smooth muscle tone is skewed toward vasoconstriction.11–13 The substrate for NO production, l-arginine is destroyed by arginase (liberated by lysed erythrocytes), providing another mechanism for deficiency of endothelial NO.14
NO is a soluble gas produced by endothelial cells,15 which is a known endogenous potent vasodilator16–21 and has blood flow regulatory effect.22,23 NO excites soluble guanylyl cyclase (sGC) in the cavernosal smooth muscle, which in turn triggers increased synthesis of cyclic GMP (cGMP), thus providing the main signal for smooth muscle relaxation.24 Downstream homeostatic vascular functions of NO such as inhibition of platelet activation among others are impaired when there is a reduced endothelial NO bioavailability in sickle cell disease patients.25 NO is also known to be the primary mediator of penile smooth muscle relaxation, which leads to normal erectile function.26
The relative levels of NO, which is responsible in part for maintaining normal vasomotor tone in SCD patients, during different clinical phases and in SCD complications, seem not clear.27 A study conducted by Rees et al28 reported that NO metabolite (NOx) levels are high in patients with SCD. Our recent study conducted in Accra, Ghana, demonstrated that there is no significant difference in NO levels between the comparison group and SCD patients in the steady state as well as those in the postcrisis state.29 The current study aimed at determining the relative plasma NOx levels among patients with SCD complications including VOC, leg ulcers and priapism in the same population in the Greater Accra Region of Ghana.
Methods
Study setting and study population
This study was a case–control cross-sectional one, conducted at the Korle Bu Teaching Hospital in the Greater Accra Region of Ghana from February to July, 2014. The cases (SCD patients) and comparison group were recruited from the Center for Clinical Genetics (Sickle Cell Clinic) and Accra Area Blood Center for National blood transfusion, respectively. The study involved a total of 694 participants, including 148 comparison group HbAA, 208 HbSS SCD patients in steady state, 82 HbSC SCD patients in steady state, 156 HbSS SCD patients in VOC, 34 HbSC SCD patients in VOC, 34 HbSS SCD patients in post VOC, 21 HbSS SCD patients with leg ulcers and 11 HbSS SCD patients with priapism. The study protocol was approved by the Ethical and Protocol Review Committee of University of Ghana Medical School. Written informed consents were obtained from all participants and study subjects before samples were obtained. Consent was also sought from the parents/guardians of the children recruited into the study by signing a written informed consent agreement.
Inclusion criteria
SCD patients ranging in age from 15 and 65 years who had not used nitrite or nitrate-containing medications at least 2 weeks before the sample was taken, were included.
Exclusion criteria
Patients with renal failure (serum creatinine >2 mg/dL), pulmonary edema, cardiogenic shock, history of myocardial infarction in the last 6 months, diabetes mellitus, congestive heart failure, a cerebrovascular accident in the last 6 months, acute asthma, or angina pectoris were excluded from the study.
Patients’ selection criteria
Steady state was defined clinically as a patient who had been well and had not been in any crisis for at least 2 weeks. VOC was also clinically defined as pains in the bones, muscles and joints that were not due to any other cause and required parenteral analgesia and thus, admittance to the Sickle Cell Center for some hours. Again, leg ulcer was defined as a defect in the skin below the level of the knee and above the foot which persisted for 6 weeks or more. Priapism was also defined as a purposeless, persistent penile erection, particularly unaccompanied by any stimulation or sexual desire, which usually lasted for more than 6 hours. Post VOC was defined as 3 days after crises when patients experienced no pain in the bones and muscles. Patients with crises were followed to the post VOC crises. Patients with history of priapism were those recruited in the current study.
Blood sampling and laboratory measurements
Five milliliters of venous blood sample was collected from each of the study participants into ethylendiaminetetracetic acid (EDTA) tubes. Aliquot of 2.5 mL of the blood sample was processed into plasma and stored at –80°C. A full blood count (FBC) was done with 2.5 mL of the blood sample within 2 hours of sample collection using Labsystems Multiskan MS (Amersham Bioscience Ltd, Little Chalfont, UK). An assay which relied on a diazotization reaction originally described by Griess test was used to measure nitrite.30 The Griess Reagent System is based on the chemical reaction which uses sulfanilamide and N-l-naphthyl ethylenediamine dihydrochloride (NED) under acidic (phosphoric acid) conditions. This system is able to detect nitrite in a variety of biological and experimental liquid matrices such as plasma. The average absorbance of the triplicates of each experimental sample was recorded. The nitrite concentration (Y) of each experimental sample was determined by comparison to the Nitrite Standard reference curve. In this case the formula: Y=0.0185X+0.106, as generated from the standard curve was used, where X is the average absorbance of the experimental sample.
Data analysis
The data obtained were stored and analyzed using SPSS version-20 software (IBM Corporation, Armonk, NY, USA). Frequency table were generated for nominal and ordinal variables. The results were expressed as mean plus or minus standard deviation (mean ± SD). The Kruskal–Wallis test was used to compare differences in mean values among SCD patients in steady state and VOC as well as those with leg ulcers and priapism with the healthy comparison group. Dunn’s test was done as a post hoc analysis for multiple comparisons. Statistical significance was considered at P<0.05.
Results
Of the 546 SCD patients with and without complication recruited in the study, 430 had the HbSS genotype, while 116 had HbSC genotype. There were more male (n=349) SCD patients than females (n=345) in this study. The difference was not significant (P>0.05). The general characteristics of the SCD patients and the healthy comparison group are summarized in Table 1.
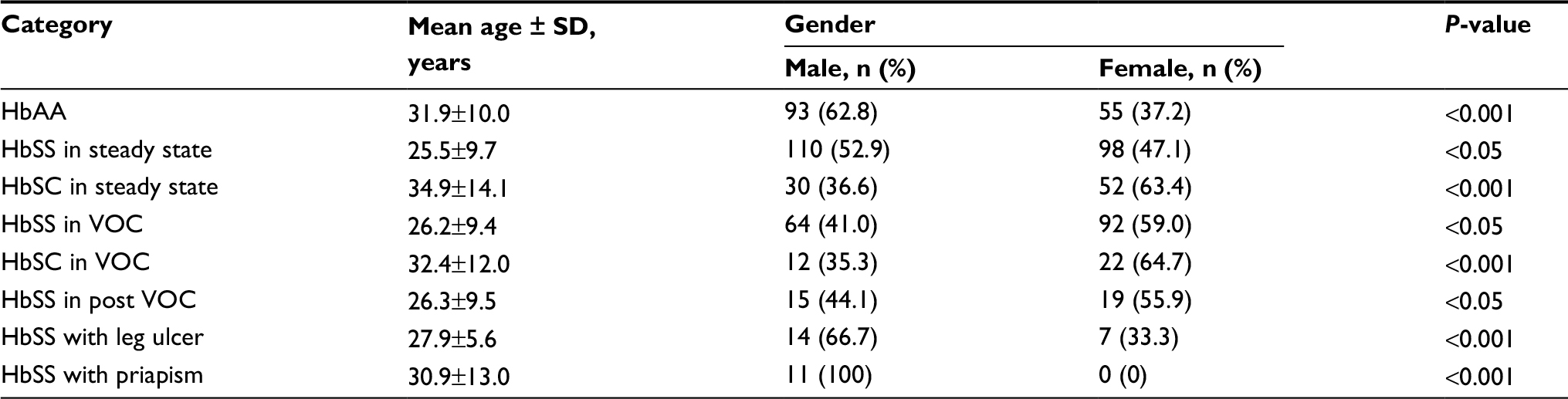 | Table 1 General characteristics of SCD patients Abbreviations: SCD, sickle cell disease; VOC, vaso-occlusive crisis. |
Nox levels in different states of SCD patients
The mean plasma NOx levels in SCD patients are illustrated in Table 2. The plasma NOx levels were lower in SCD patients with complications compared with those at the steady state, as well as SCD in the immediate postcrises state. However, the level was higher in the HbAA comparison group. The mean level of plasma NOx increased after the immediate post VOC. Among the clinical sub-phenotype studied, patients with leg ulcers had relatively lower levels compared with SCD patients with priapism.
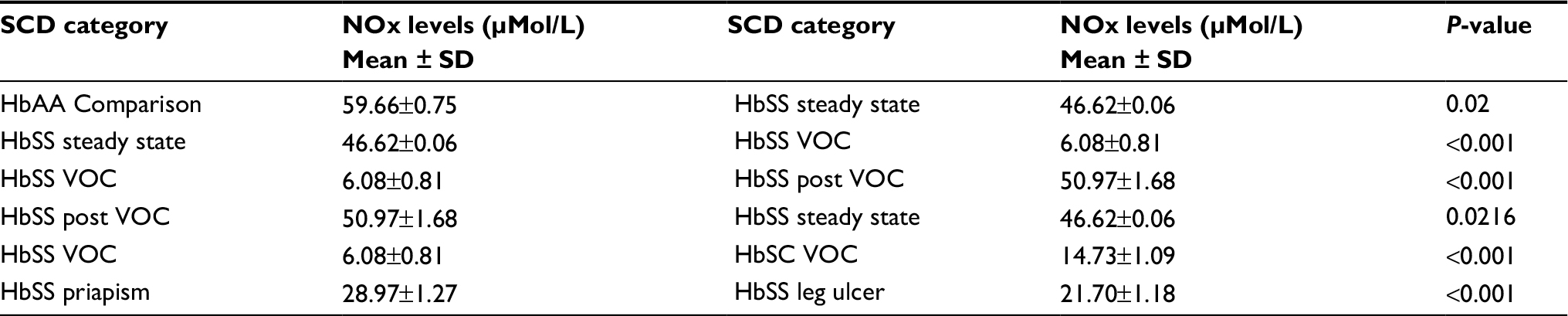 | Table 2 Plasma NOx levels in SCD sub-phenotypes and comparison group Notes: P<0.001= highly significant. P<0.05= significant. Abbreviations: NOx, nitric oxide metabolite; SCD, sickle cell disease; VOC, vaso-occlusive crisis. |
Discussion
NO is a major endothelial-derived relaxing factor in normal physiology of the vasculature and plays a central role in vascular homeostasis.31 Thus, its role in SCD and associated complications cannot be overemphasized. The significantly low plasma NOx levels recorded in the SCD patients with complications (HbSS VOC, HbSC VOC, HbSS leg ulcers and HbSS priapism) in comparison to asymptomatic SCD patients (HbSS steady state and HbSS postcrisis state) and the comparison group, could partly be the participation of NO in the compensatory response to chronic vascular injury in these SCD patients,32 especially those in VOC. This is evident in case reports from other countries where NOx levels were higher in SCD patients in steady states.20,29,33,34
Conditions including intravascular hemolysis that interfere with NO bioavailability and vasoconstriction might have partly contributed to the lower levels of plasma NOx recorded in the SCD patients with complications.25,35 The clinical presentation of SCD which is due to VOC episodes resulting from polymerization of deoxygenated Hb-S and leading to the formation of sickled red cells, has been reported in a previous study.36 The simultaneous release of erythrocyte arginase during hemolysis14 could limit the availability of arginine to NO, thus contributing to a deficiency of NO.37 The degree of hemolysis encountered in both SCD patients in the immediate postcrises state and steady state SCD patients, may not be as severe as what would have been observed in clinical complications of SCD such as VOC to limit the bioavailability of NO substrate, for NOx depletion. This partly, might have accounted for the observed similar levels of NOx in these two study groups (HbSS post VOC and HbSS steady state).
Observation from this study also suggests that severity of SCD complications may, in part, be related to the degree of NO depletion or the plasma level of NOx as a contribution to the clinical complications suffered by patients with SCD. Although HbSC disease symptoms are similar to those of homozygous (SS) sickle cell such as VOC episodes,38 it presents with milder severity and less frequency.38,39 This might explain in part the relatively lower level of NOx observed in patients with HbSS VOC as well as the higher number of HbSS VOC patients seen in this study, compared with HbSC VOC patients.
Again, findings from this study on the level of NOx among SCD patients with VOC are in line with earlier work conducted by Morris et al20 who also reported that low NOx levels consistently rose in SCD patients with VOC during hospitalization and returned to a steady state value at the time of discharge. Blood plasma levels of l-arginine are depressed in patients with SCD, particularly during VOC and acute chest syndrome, and these levels vary inversely with pain symptoms.33,36 NO-dependent blood flow is impaired in SCD patients.27,40,41 Several studies have indicated the improvement of SCD and associated complications using arginine therapy,42–44 due to the failure of NO-based therapeutic effect.45,46 Thus, arginine supplementation may be required in the SCD patients with complications studied, especially those with VOC.
Although reports are varied, some investigators have found low levels of NOx (nitrate and nitrite) in patients with SCD, and these reduced levels are consistent with impaired endothelial generation of NO, and its subsequent reactions with hemoglobin and oxygen.22 NO may have a natural effect of vasodilation on smooth muscle vasculature, anti-inflammatory effects and platelet aggregation inhibition.46 Our study had some limitations. Information on malaria in the SCD patients was not obtained, as malaria is also thought to cause hemolysis, thus, dysregulating the arginine-NO pathway. Also, we could not measure plasma arginine levels in the patients. Nonetheless, the findings add to the body of literature concerning NOx levels in SCD sub-phenotypes.
Conclusions
This study showed lower plasma NOx levels in SCD patients with complications such as VOC, priapism and leg ulcers. The levels were however, higher during the immediate post crisis period. Generally, levels of NOx in the comparison group were higher as compared to the SCD patients with or without complications.
Acknowledgments
The authors are thankful to the Office of Research, Innovation and Development (ORID), University of Ghana and University of Ghana-Carnegie Next Generation of Academics in Africa Project for funding the research. The authors are also grateful to the staff and patients of the Center for Clinical Genetics (Sickle Cell Clinic) who took part in the study. Also, the authors would like to thank the volunteers who donated blood and consented to take part in the study as comparison group at the Accra Area Blood Center for National Blood Transfusion at the Korle-Bu Teaching Hospital, Accra, Ghana. The authors are also grateful to the following persons for their contributions: Frimpong Emmanuel, Gyan Ben, Kyei-Baafour, Eric Sey Fredericka, Bartholomew Dzudzor, Mubarak Abdul-Rahman, Agbozo William, Dankwah Boatemaa Gifty, Antwi-Boasiako Kate, Ekem Ivy, and Antwi Daniel Ansong. The research was funded by the ORID, University of Ghana and University of Ghana-Carnegie Next Generation of Academics in Africa.
Disclosure
The authors report no conflicts of interest in this work.
References
Claudino MA, Fertrin KY. Sickling cells, cyclic nucleotides, and protein kinases: the pathophysiology of urogenital disorders in sickle cell anemia. Anemia. 2012;12:13. | ||
Herrick CJ. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Int Med. 1910;5:515–521. | ||
Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37–47. | ||
Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340(13):1021–1030. | ||
Kato GJ, Gladwin MT. Evolution of novel small-molecule therapeutics targeting sickle cell vasculopathy. JAMA. 2008;300(22):2638–2646. | ||
Conran N, Franco-Penteado CF, Costa FF. Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin. 2009;33(1):1–16. | ||
Ajay A, Bhatkulkar P, Khare R, Pazare K. Haematological indices and electrolyte status in sickle cell disease at rural hospital of central Maharashtra. Int J Med Sci Pub Heal. 2014:1410–1412. | ||
Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762–769. | ||
Embury S, Hebbel RP, Mohandas N, Steinberg MH. eds. Sickle Cell Disease: Basic Principles and Clinical Practice. New York: Raven Press; 1994:902. | ||
Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;24:122. | ||
Burnett AL. Pathophysiology of priapism: dysregulatory erection physiology thesis. J Urol. 2003;170(1):26–34. | ||
Ohene-Frempong K, Steinberg MH. Clinical aspects of sickle cell anemia in adults and children. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge: Cambridge University Press; 2001:611–670. | ||
Rogers ZR. Priapism in sickle cell disease. Hematol Oncol Clin North Am. 2005;19(5):917–928. | ||
Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294(1):81–90. | ||
Pearson P, Evora J, Seccombe J, Schaff H. Hypomagnesemia inhibits nitric oxide release from coronary attenuates: protective role of magnesium infusion after cardiac operations. Ann Thor Surg. 1998:967–972. | ||
Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–2012. | ||
Kam PC, Govender G. Nitric oxide: basic science and clinical applications. Anaesthesia. 1994;49(6):515–521. | ||
Kiechle FL, Malinski T. Nitric oxide. Biochemistry, pathophysiology, and detection. Am J Clin Pathol. 1993;100(5):567–575. | ||
Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 19871987;327(6122):524–526. | ||
Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22(6):515–520. | ||
Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886–895. | ||
Mack AK, Kato GJ. Sickle cell disease and nitric oxide: a paradigm shift? Int J Biochem Cell Biol. 2006;38(8):1237–1243. | ||
Thomas BN, Thakur TJ, Yi L, Guindo A, Diallo DA, Ott J. Extensive ethnogenomic diversity of endothelial nitric oxide synthase (eNOS) polymorphisms. Gene Regul Syst Bio. 2013;7:1–10. | ||
Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52(3):375–414. | ||
Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematology (Am Soc Hematol Educ Program) 2005:51–57. | ||
Burnett AL. The role of nitric oxide in erectile dysfunction: implications for medical therapy. J Clin Hypertens. 2006;8(12 Suppl. 4):53–62. | ||
Gladwin MT, Schechter AN, Ognibene FP, et al. Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation. 2003;107(2):271–278. | ||
Rees DC, Cervi P, Grimwade D, et al. The metabolites of nitric oxide in sickle-cell disease. Br J Haematol. 1995;91(4):834–837. | ||
Antwi-Boasiako C, Frimpong E, Ababio GK, et al. The role of nitric oxide in vaso-occlusive crisis in sickle cell disease patients in Ghana. Donnish J Med Medical Sci. 2015;2(4):052–055. | ||
Griess P. [Comments on the treatise of the HH. Weselsky and Benedikt “About some azo compounds”]. Ber German Chem Ges. 1879;12(1):426–428. | ||
Reiter CD, Gladwin MT. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr Opin Hematol. 2003;10(2):99–107. | ||
Enwonwu CO, Xu XX, Turner E. Nitrogen metabolism in sickle cell anemia: free amino acids in plasma and urine. Am J Med Sci. 1990;300(6):366–371. | ||
Lopez BL, Barnett J, Ballas SK, Christopher TA, Davis-Moon L, Ma X. Nitric oxide metabolite levels in acute vaso-occlusive sickle-cell crisis. Acad Emerg Med. 1996;3(12):1098–1103. | ||
Stuart MJ, Setty BN. Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood. 1999;94(5):1555–1560. | ||
Marín J, Rodríguez-Martínez MA. Role of vascular nitric oxide in physiological and pathological conditions. Pharmacol Ther. 1997;75(2):111–134. | ||
Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem. 1990;40:63–279. | ||
Morris CR. Role of arginase in sickle cell lung disease and hemolytic anemias. Open Nitric Ox J. 2010;2:41–54. | ||
Bunn HF, Nathan DG, Dover GJ, et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;11639(5):687–692. | ||
Powars DR, Hiti A, Ramicone E, Johnson C, Chan L. Outcome in hemoglobin SC disease: a four-decade observational study of clinical, hematologic, and genetic factors. Am J Hematol. 2002;70(3):206–215. | ||
Belhassen L, Pelle G, Sediame S, et al. Endothelial dysfunction in patients with sickle cell disease is related to selective impairment of shear stress-mediated vasodilation. Blood. 2001;97(6):1584–1589. | ||
Eberhardt RT, McMahon L, Duffy SJ, et al. Sickle cell anemia is associated with reduced nitric oxide bioactivity in peripheral conduit and resistance vessels. Am J Hematol. 2003;74(2):104–111. | ||
Morris CR. Alterations of the arginine metabolome in sickle cell disease: a growing rationale for arginine therapy. Hematol Oncol Clin North Am. 2014;28(2):301–321. | ||
Morris CR, Morris SM Jr, Hagar W, et al. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168(1):63–69. | ||
Nitya B, Claudia M. The role of the arginine metabolome in pain: implications for sickle cell disease. J Pain Res. 2016;9:167–175. | ||
Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: A randomized clinical trial. JAMA. 2011;305(9):893–902. | ||
Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118(4):855–864. | ||
Natarajan K, Townes TM, Kutlar A. Disorders of hemoglobin structure: sickle cell anemia and related abnormalities. In: Kaushansky K, Lichtman MA, Beutler E, editors. Williams Hematology, 8th Edn. New York: McGraw-Hill; 2010:ch. 48. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.