Back to Journals » International Journal of Nanomedicine » Volume 13
Low-molecular-weight sulfonated chitosan as template for anticoagulant nanoparticles
Authors Heise K, Hobisch M, Sacarescu L, Maver U ![]() , Hobisch J, Reichelt T, Sega M, Fischer S, Spirk S
, Hobisch J, Reichelt T, Sega M, Fischer S, Spirk S
Received 25 April 2018
Accepted for publication 12 June 2018
Published 30 August 2018 Volume 2018:13 Pages 4881—4894
DOI https://doi.org/10.2147/IJN.S172230
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Katja Heise,1,2 Mathias Hobisch,3,4 Liviu Sacarescu,5 Uros Maver,6 Josefine Hobisch,3 Tobias Reichelt,7 Marija Sega,6 Steffen Fischer,1 Stefan Spirk3,4
Members of EPNOE and NAWI Graz
1Institute of Plant and Wood Chemistry, Technische Universität Dresden, Tharandt, Germany; 2Department of Bioproducts and Biosystems, Aalto University, Espoo, Finland; 3Institute for Chemistry and Technology of Materials, Graz University of Technology, Graz, Austria; 4Institute for Paper, Pulp and Fiber Technology, Graz University of Technology, Graz, Austria; 5“Petru Poni” Institute of Macromolecular Chemistry, Romanian Academy, Iasi, Romania; 6Faculty of Medicine, University of Maribor, Maribor, Slovenia; 7Zentrum für Bucherhaltung GmbH, Leipzig, Germany
Purpose: In this work, low-molecular-weight sulfoethyl chitosan (SECS) was used as a model template for the generation of silver core-shell nanoparticles with high potential as anticoagulants for medical applications.
Materials and methods: SECS were synthesized by two reaction pathways, namely Michael addition and a nucleophilic substitution with sodium vinylsulfonate or sodium 2-bromoethanesulfonate (NaBES). Subsequently, these derivatives were used as reducing and capping agents for silver nanoparticles in a microwave-assisted reaction. The formed silver-chitosan core-shell particles were further surveyed in terms of their anticoagulant action by different coagulation assays focusing on the inhibition of either thrombin or cofactor Xa.
Results: In-depth characterization revealed a sulfoalkylation of chitosan mainly on its sterically favored O6-position. Moreover, comparably high average degrees of substitution with sulfoethyl groups (DSSE) of up to 1.05 were realized in reactions with NaBES. The harsh reaction conditions led to significant chain degradation and consequently, SECS exhibits masses of <50 kDa. Throughout the following microwave reaction, stable nanoparticles were obtained only from highly substituted products because they provide a sufficient charge density that prevented particles from aggregation. High-resolution transmission electron microscopy images reveal that the silver core (diameter ~8 nm) is surrounded by a 1–2 nm thick SECS layer. These core-shell particles and the SECS itself exhibit an inhibiting activity, especially on cofactor Xa.
Conclusion: This interesting model system enabled the investigation of structure–property correlations in the course of nanoparticle formation and anticoagulant activity of SECS and may lead to completely new anticoagulants on the basis of chitosan-capped nanoparticles.
Keywords: chitosan ethylsulfonate, silver nanoparticles, antithrombotic activity, cofactor Xa
Introduction
Sulfated polysaccharides are a diverse class of materials, which feature a wide range of physiological properties. Heparin, a poly β-(1-4)-glycosaminoglycane, is probably the best-known congener due to its medical use as intravenous anticoagulant. Its intrinsic role throughout the physiological coagulation cascade is based on specific interactions between its sulfated pentasaccharide sequence and antithrombin III (AT III), increasing the binding affinity of AT III to thrombin (IIa) and/or cofactor Xa.1,2 As most of the pharmaceutically applied heparin, however, is currently isolated from animal tissue, implying risks of biological contaminations and a poorly predictable coagulation behavior,3,4 several approaches have been reported to (partially) replace heparin by sulfated oligo- and polysaccharides (either natural or semisynthetic).5–7 These strategies target physiological activities on the basis of well-defined, heparin-like compounds.
In this context, the amino polysaccharide chitosan features a remarkable structural similarity to heparin, and the introduction of sulfate groups into its backbone improves its hemocompatibility8 while imparting anticoagulant properties.9,10 The key role in terms of the relationship between polymer structure and bioactivity can be especially ascribed to the degree of substitution (DS) with sulfate groups. For instance, anticoagulant effects of polysaccharide sulfates were found to increase significantly with increasing DS values (>0.6).11,12 Sulfate groups, in turn, can be either introduced into the chitosan backbone as sulfate esters or in the form of sulfoalkyl ethers. The latter approach is highly interesting, as interposed alkyl spacers might affect the accessibility and, thus, activity profile of the bioactive sulfate group. Moreover, ether groups benefit from higher stabilities in aqueous solutions compared with esters.13–15 In this regard, especially chitosan ethylsulfonate has been in the focus of recent contributions due to its facile synthetic access and promising application prospects as heavy metal adsorber16–19 or potential anticoagulant.13
Two synthetic strategies have been so far used to convert chitosan into its sulfoethyl ether: reactions in gel state16–19 or heterogeneous conversions in organic solvents.13,15 In gel state, chitosan is modified regioselectively at the amine groups realizing an average DS with sulfoethyl groups (DSSE) of up to 1.0.16–19 The latter approach has been conducted either as nucleophilic substitution or Michael addition utilizing haloalkyl sulfonates or vinyl sulfonates.13,15 Both reagents unselectively lead to O6- and N-substitution of chitosan and, additionally, the required strongly alkaline medium significantly affects the molecular weight (MW) of the biopolymer.15,20 Nevertheless, in the case of cellulose sulfoalkyl ethers, comparably high DSSE of up to 2.0 were obtained using sodium vinylsulfonate (NaVS).15
In this work, we intentionally took advantage of the rather harsh conditions in the course of the heterogeneous state reactions aiming at sulfoethyl chitosans (SECSs) with a high DSSE and a low MW. The latter aspect was thereby expected to enhance the bioactivity of the derivative and increase its water solubility.21–23 As visualized in Figure 1, the obtained SECSs were further utilized as capping and reducing agents for the synthesis of silver nanoparticles (SECS@Ag). As shown in previous studies on chitosan sulfates,24,25 this specific approach serves as a convenient model system to demonstrate potential application prospects and to elucidate anticoagulant properties of SECS. As the mode of anticoagulant action strongly depends on the MW of the derivative, different anticoagulant assays (demonstrating either an inhibition of thrombin or cofactor Xa) were carried out in this work. The chosen assays thus allowed formulating a trend in terms of the anticoagulant activities of SECS and SECS@Ag.
 | Figure 1 Scheme representing the workflow in the article, starting from the synthesis of sulfoethyl chitosans (SECSs), their application as capping and reducing agents for silver nanoparticles (SECS@Ag), and the assessment of the products in anticoagulant assays. |
Materials and methods
Materials
Chitosan 95/100 [degree of deacetylation >92.6%, viscosity ≤71–150 mPas, MW 100–250 kDa (SEC)] was obtained from Heppe Medical Chitosan GmbH (Halle, Germany). NaVS [tech., 25% (aq)] was purchased from Alfa Aesar GmbH & Co. KG (Karlsruhe, Germany) and sodium 2-bromoethanesulfonate (NaBES, 98%) from Sigma-Aldrich Co. (St Louis, MO, USA). Dialysis membranes (Spectra/Por® Biotech CE, molecular weight cutoff: 100–500 Da) were received from Spectrum Laboratories, Inc. (Rancho Dominquez, CA, USA). Silver nitrate (99%; Merck, Darmstadt, Germany) was used without further purification. Human blood plasma was used for all coagulation assays and was received from a single donor to ensure comparability of consecutive tests. Citrated blood samples were obtained by clean venipuncture using 19-gauge butterfly needles and Monovette tubes (Sarstedt, Nümbrecht, Germany) containing 1/9 vol of 0.106 M sodium citrate. Plasma was prepared by double centrifugation [1,500 ×g, each for 10 minutes, at room temperature (RT)] and was stored in polypropylene tubes at −70°C until use. CaCl2 (medical grade) was received from Siemens Healthcare Diagnostics Products (Marburg, Germany). All other chemicals were utilized in analytical grade. Ultrapure water (MilliQ, Ω≤18.2 MΩ cm) was used for all applications.
Regeneration of chitosan
To activate chitosan prior to sulfoethylation, a regeneration step was carried out. In a typical procedure, 5 g of chitosan was dissolved in 500 mL of 1 wt% aqueous acetic acid at RT under continuous stirring followed by precipitation in a mixture of 500 mL methanol and 500 mL of 4 wt% aqueous NaHCO3 solution. The suspension was stirred for 2 hours at RT, and the freshly regenerated chitosan was obtained by centrifugation. After washing with an ethanol/water mixture (8/2, v/v) and isopropanol (IPA), the regenerated chitosan was suspended in 400 mL IPA in a 1 L three-neck flask, equipped with a vertical stirrer, reflux condenser, and thermometer.
Sulfoethylation with NaVS (preparation of SECSVS)
After regeneration, sulfoethylation with NaVS was carried out in two equivalent modification steps as follows: 41.25 mL NaVS solution [3 mol NaVS per mol anhydrous glucosamine units (GlcN)] was added to the chitosan suspension and the reaction mixture was stirred for 20 minutes at RT. Afterward, 52.5 mL of 3.6 N aqueous NaOH (6 mol NaOH per mol GlcN) was subjoined and the suspension was stirred for another hour at RT. The temperature was then elevated to 70°C and kept for 24 hours under continuous stirring. The reaction was stopped by cooling down the mixture to RT and removing the supernatant liquid. Subsequently, the intermediate product was suspended in 100 mL deionized water, followed by adjusting the pH to 7.5 with 50 wt% aqueous acetic acid. The product was obtained through precipitation in 500 mL of ethanol and centrifuged and further washed with an ethanol/water mixture (8/2, v/v) for removing residual salts and by-products. For the second reaction, the intermediate was again suspended in 400 mL IPA. The second sulfoethylation was performed under the conditions applied before, and the final product (SECSVS) was received as described earlier. SECSVS was washed with an ethanol/water mixture (8/2, v/v) for several times and suspended in deionized water. Soluble and insoluble fractions were separated through centrifugation, and the desired water-soluble part was dialyzed (conductivity <1 μS) and lyophilized for subsequent analysis.
DSSE by elemental analysis: DSSE 0.69 (%C 32.76, %H 5.70, %N 5.23, %S 8.22). 1H nuclear magnetic resonance spectroscopy (NMR) (600 MHz, D2O): δH 2.12 (GlcNAc-CH3), 2.72 (H2/H2SE-GlcN), 3.00 and 3.12 (NH-CH2-CH2-SO3Na and C6/3-O-CH2-CH2-SO3Na), 3.47–3.76 [H3, H5, H4, H6 and H6SE (chitosan backbone)], 3.82–3.95 (H6 and C6/3-O-CH2-CH2-SO3Na), 4.30 (C3-CH2-CH2-SO3Na), 4.45 (H1-GlcN). 13C NMR (151 MHz, D2O): δC 30.19 (GlcN-Ac-CH3), 43.68 (NH-CH2-CH2-SO3Na), 50.66 (C6/3-O-CH2-CH2-SO3Na and NH-CH2-CH2-SO3Na), 56.41 (C2), 60.12 (C6), 66.34 (C6-O-CH2-CH2-SO3Na), 67.75 (C3-O-CH2-CH2-SO3Na), 68.83 (C6SE), 73.54 (C3), 74.80 (C5), 77.59 (C4), 101.48 (C1). MW by size exclusion chromatography (SEC): Mw=162 kDa, Mn=45 kDa, Mw/Mn=3.642.
Sulfoethylation with NaBES (preparation of SECSBES)
The conversion with NaBES was performed in two equivalent steps, analogous to the reaction with NaVS. Five grams of freshly regenerated chitosan was suspended in 400 mL of IPA, 52.5 mL of 3.6 N NaOH aqueous solution was added, and the mixture was stirred at RT for 3 hours. Thereafter, 19.3 g of NaBES (3 mol per mol GlcN) was added and the suspension was stirred for another 30 minutes. Subsequently, the mixture was heated to 70°C and kept at this temperature for 24 hours under continuous stirring. After the reaction, the intermediate product was obtained and treated, as described previously. The second modification step was carried out under equivalent conditions after the addition of fresh NaOH and NaBES. The final product (SECSBES) was received, purified by dialysis, and lyophilized.
DSSE by elemental analysis: DSSE 1.05 (%C 31.80, %H 5.27, %N 4.43, %S 10.59). 1H NMR (600 MHz, D2O): δH 2.68–2.79 (H2/H2SE-GlcN), 3.00 and 3.12 (NH-CH2-CH2-SO3Na and C6-O-CH2-CH2-SO3Na), 3.47–3.74 [H3, H5, H4, H6 and H6SE (chitosan backbone)], 3.81–3.95 (H6 and C6/3-O-CH2-CH2-SO3Na), 4.29 (C3-O-CH2-CH2-SO3Na), 4.45 (H1-GlcN). 13C NMR (151 MHz, D2O, ppm): δC 43.58 (NH-CH2-CH2-SO3Na), 50.66 (C6/3-O-CH2-CH2-SO3Na and NH-CH2-CH2-SO3Na), 56.35 (C2), 60.10 (C6), 66.34 (C6-O-CH2-CH2-SO3Na), 67.65 (C3-O-CH2-CH2-SO3Na), 68.68 (C6SE), 73.52 (C3), 74.79 (C5), 77.70 (C4), 101.41 (C1). MW by SEC: Mw=25 kDa, Mn=16 kDa, Mw/Mn=1.565.
Silver nanoparticle preparation (SECS@Ag)
The preparation of silver nanoparticles (AgNPs), coated with SECS (SECS@Ag), was carried out in sealed vials in a Biotage® Initiator Robot Eight reaction microwave (Biotage, Uppsala, Sweden). Typically, 2 mL of an aqueous SECS solution (2 mg/mL) and 1 mL of aqueous silver nitrate solution (50 mM for SECSBES; 12 mM for SECSVS) were placed in a 10 mL microwave vial, rapidly heated to 120°C and kept at this temperature for 6 minutes under stirring. In the case of SECSVS, the addition of 1 mL aqueous glucose solution (1 wt%, w/w) was required for the successful reduction of silver nitrate.
Characterization of SECS and SECS@Ag
Elemental analysis was carried out by measuring contents of carbon, hydrogen, and nitrogen with an Elemental Analyzer vario El from Elementar (Hanau, Germany). Sulfur was quantified by using an Elemental Analyzer Eltra CS 500 (Eltra, Neuss, Germany). DSSE was calculated based on duplicate determinations according to the following equation: Total DSSE=(S%/32)/(N%/14). The MW of SECS was determined by SEC utilizing a ParSEC Chromatography ver 5.66 (Brookhaven Instruments Corp., Holtsville, NY, USA), equipped with a refractive index detector and a two-column set composed of ABOA CatPhil 100 and ABOA CatPhil 350 (AppliChrom, Oranienburg, Germany). The following instrument setting was applied: c(SECS)=1.5 mg/mL, T=25°C, mobile phase = 0.05 MNa2HPO4/0.1 M NaNO3, flow rate = 1 mL/min, sample injection volume = 112 μL. Pullulan standards (Mw=6.0–337 kDa; PSS, Mainz, Germany) were used for calibration. Fourier transform (FT) Raman spectra were recorded in aluminum disks using a Bruker MultiRam spectrometer (Bruker Optik GmbH, Ettlingen, Germany) equipped with a liquid nitrogen cooled Ge diode as detector and a cw-Nd:YAG-laser (exciting line of 1,064 nm) as light source. The instrument setting was as follows: spectral range = 3,500–100 cm−1, laser power output = 300 mW (defocused), operating spectral resolution = 3 cm−1, 400 scans. Average Raman spectra were formed out of four determinations with the operating software OPUS ver 6.5 from Bruker Optics. Baselines of average spectra were corrected and peak intensities were normalized (Min–Max normalization, spectral range = 1,400–1,300 cm−1). 13C DEPT-135 (distortionless enhancement by polarization transfer) equipped 1H HSQC (heteronuclear single-quantum correlation) NMR spectra were recorded on a Bruker AV-III 600 spectrometer, operating at 600 MHz for 1H and 151 MHz for 13C. SECS nanoparticles were dissolved in D2O and spectra were obtained at RT. Chemical shifts d are given in ppm relative to tetramethylsilane. Coupling constants J are given in Hertz and were determined assuming first-order spin–spin coupling. Zeta potential determinations of SECS@Ag suspensions in deionized water were carried out on a ZetaPALS Zeta Potential Analyzer (Brookhaven Instruments Corp.). Transmission electron microscopy (TEM) and energy- dispersive X-ray spectroscopy (EDX) were performed with a HITACHI HT7700 microscope equipped with a Bruker XFlash 6 EDS detector. The instrument has a dual-mode objective lens that allows high-resolution or high-contrast inspection of the samples at 120 kV accelerating voltage. The samples for TEM analysis were prepared by placing small droplets of diluted (by ~100× with toluene) dispersions (1 wt%) on copper grids (300 mesh) coated with an amorphous carbon film (Ted Pella, Redding, CA, USA) and dried at 50°C in vacuum for 24 hours prior to the measurements.
Activated partial thromboplastin time (aPTT) testing
For aPTT testing, a tilt tube technique was utilized. The aPTT reagent was prepared by suspending kaolin (10 mg/mL) together with inosithin (0.5 mg/mL) in isotonic barbiturate buffer (pH 7.35). For a regular testing procedure, 100 μL of prewarmed plasma and 100 μL of kaolin–inosithin suspension were preincubated in a glass tube at 37°C for a standard preincubation time of 1 minute. Subsequently, 100 μL 0.0025 M aqueous CaCl2 solution (prewarmed to 37°C) was added, and the clotting time was recorded in seconds. For sample testing, either 25 or 50 μL of SECS aqueous solution (0.1 mg/mL) or SECS@Ag suspension (0.1 mg/mL) was subjoined to the blood plasma.
Prothrombin time (PT) testing
The PT was assayed using a tilt tube technique. For a typical procedure, blood plasma was mixed with phospholipid-enriched tissue factor (TF) at 37°C, an excess of 0.0025 M aqueous CaCl2 solution was added to initiate coagulation, and the time for the formation of fibrin clot was recorded as PT in seconds. Clotting times after the addition of either SECS or SECS@Ag suspensions (25 or 50 μL, 0.1 mg/mL) were compared with a blank feed.
Chromogenic determination of anticoagulant activity
The chromogenic determination was performed using Berichrom→Heparin assay on a Siemens BCS XP1 system (Siemens) and based on a standard protocol for the kinetic method as prescribed by the manufacturer. Results are presented as IU/mL of LMWheparin and compared with the normal plasma. A calibration curve for the determination of respective plasma/sample mixture performance was prepared according to the manufacturer’s notes. The calibration curve is shown in Figure 2.
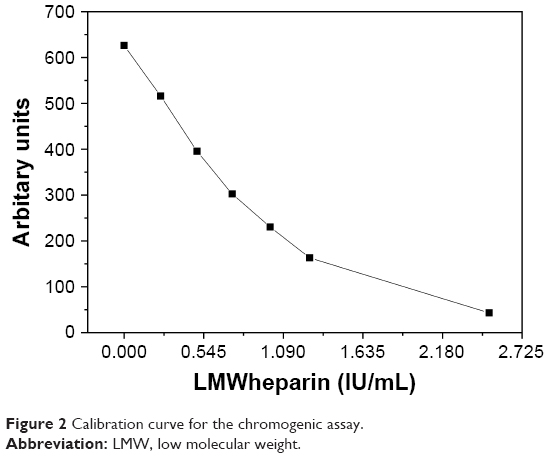 | Figure 2 Calibration curve for the chromogenic assay. |
Coagulometric determination of factor X deficiency
A plasma deficient in any of the factors comprising the extrinsic pathway will result in a prolonged prothrombin time (PT). Coagulation factor-deficient plasma can be used to confirm a factor deficiency, in general, and to identify and quantify factor deficiency in patient plasma. A mixture of the respective coagulation factor-deficient plasma and the patient plasma is tested in the PT assay, and the result is interpreted using a reference curve obtained with dilutions of Standard Human Plasma (Siemens) or a normal plasma pool mixed with the deficient plasma. A patient plasma deficient in a specific factor will not be able to compensate for the absence of the factor in the corresponding coagulation factor-deficient plasma and therefore result in a prolonged PT.
The PT was assayed as described in the previous section. Blood plasma samples were mixed with TF (containing phospholipid) at 37°C, and an excess of calcium chloride (25 mM) was added to initiate coagulation. The time taken from the addition of calcium to the formation of the fibrin clot was recorded as PT. The results are presented either as the percentage of factor Xa activity (100% means normal activity) or as PT in seconds. A calibration curve for the determination of respective plasma/sample mixture performance was prepared according to the manufacturer notes. The calibration curve is shown in Figure 3.
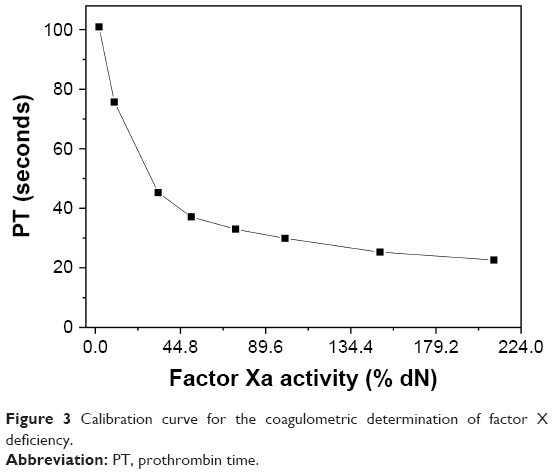 | Figure 3 Calibration curve for the coagulometric determination of factor X deficiency. |
Statistical analysis
All coagulation assays were carried out as triplicates, and measured data are given as mean±SD. Moreover, single sample t-test analysis was conducted for all sample sets using SPSS (version 25; IBM Corporation, Armonk, NY, USA). The results were compared against corresponding measurements performed using “pure blood plasma.” In the case of the Xa-specific tests, the results were compared with “pure blood plasma with added ultrapure water.” The latter was added to ensure the same dilution factor as obtained in tests adding chitosan sample suspensions. The CI used for t-test analysis was 95%. Therefore, all calculated P-values <0.05 were considered significant. Results of the t-tests are listed in the supporting information (Tables S1–S4).
Results and discussion
Preparation and characterization of SECS
Chitosan was modified in heterogeneous state in IPA via two reaction pathways using either NaVS or NaBES. Previous surveys on the reaction setting clearly demonstrated the role of the conditions (including reaction duration, temperature, and reagent quantities) on the functionalization of chitosan. For instance, a rather low DSSE of 0.19 was obtained in a single conversion with NaVS (24 hours, 70°C, molar ratio: GlcN:NaVS=1:3). By raising the temperature to 80°C, a clear increase in DSSE was achieved (from 0.19 to 0.47). However, prolonged reaction times (48 hours, 80°C) on the other hand did not lead to higher functionalization (DSSE 0.44). Furthermore, the use of an excess of sulfoalkylating agent (24 hours, 80°C, molar ratio: GlcN:NaVS=1:6) seemed to hamper the conversion of chitosan (DSSE 0.19). Therefore, to significantly increase sulfoalkyl yields, two subsequent sulfoethylation steps (2×24 hours, 70°C, GlcN:NaVS or NaBES=1:6) were carried out with an intermediate purification step to remove salts and by-products. As confirmed by elemental analysis, comparably higher DSSE of 0.69 and 1.05 can be obtained in two-step conversions with NaVS (SECSVS), as well as with NaBES (SECSBES), respectively. Moreover, these results point at a significantly higher reactivity of NaBES toward chitosan compared with the vinylsulfonate. This finding contradicts published studies on sulfoethyl cellulose15,20 and can be probably explained by the structural difference of the biopolymers.
Besides, the MW of the resulting chitosan derivative was obviously determined by the specific synthetic path (Michael addition vs nucleophilic substitution) and reagent, as shown by SEC measurements. Owing to the rather harsh conditions (pH>12), the chosen starting material (MW 100–250 kDa) strongly degraded in the course of the reaction paths. SECSBES exhibited a low MW (Mn) of ~16 kDa and a moderate MW distribution (MWD 1.6). In contrast, chain degradation was less pronounced in the reaction with NaVS (MW 45 kDa). Moreover, the heterogeneous reaction regime was more evident using this mechanism, as indicated by the comparably broad MWD of SECSVS (MWD 3.6).
In addition to elemental analysis and SEC measurements, FT Raman spectroscopy was used as a tool for gaining a qualitative insight into the structures of SECS. As shown in Figure 4, the FT Raman spectra of SECS exhibit new bands at 745 cm−1 and 1,044 cm−1 corresponding to S-C stretching vibrations and symmetric stretching vibrations of the sulfate groups, respectively.20 Compared with native chitosan, further changes [νsym/asym (CH2) at 2,940 cm−1; δ (CH2) at 1,410 cm−1; ρ (CH2) at 806 cm−1] were induced by the introduction of ethylene groups.20 Moreover, intensities of sulfoethyl group-specific bands clearly increase with increasing DSSE.
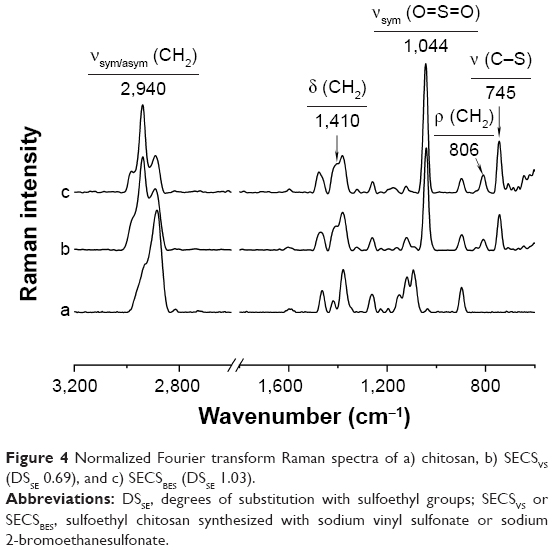 | Figure 4 Normalized Fourier transform Raman spectra of a) chitosan, b) SECSVS (DSSE 0.69), and c) SECSBES (DSSE 1.03). |
In addition to FT Raman spectroscopy, different one- (not shown here) and two-dimensional NMR techniques were applied to further elucidate substitution patterns within the individual glucosamine units. 13C-1H HSQC 2D NMR spectra of SECS were necessary to resolve the highly overlapping proton signals and complex 13C NMR spectra. 13C DEPT-135 edited HSQC spectra of SECS are illustrated in Figure 5.
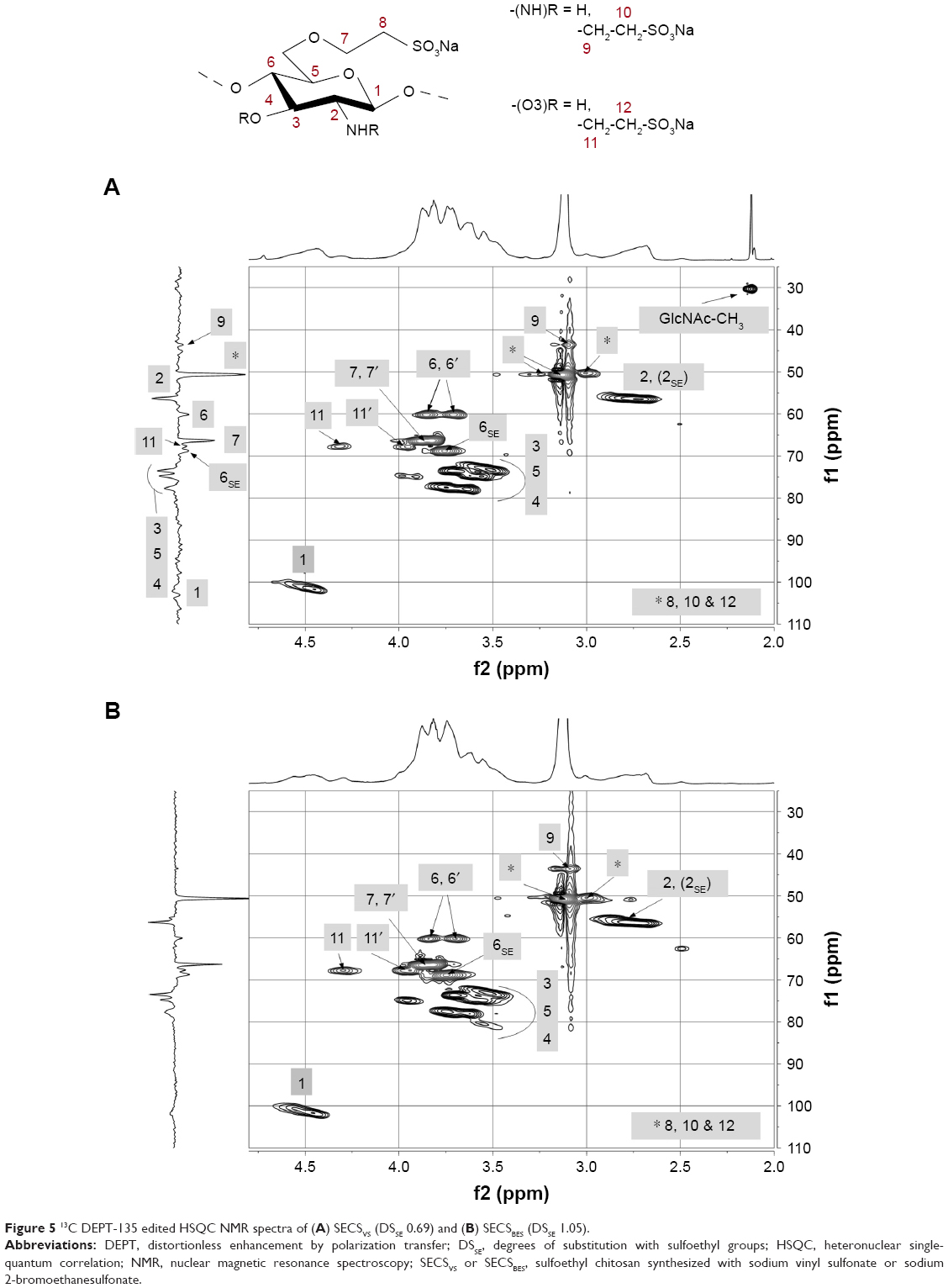 | Figure 5 13C DEPT-135 edited HSQC NMR spectra of (A) SECSVS (DSSE 0.69) and (B) SECSBES (DSSE 1.05). |
As indicated, chitosan was mainly modified in O6-position, due to the high reactivity and accessibility of primary hydroxyl groups. Peaks of unmodified C6 (60.1 ppm) and substituted C6SE (68.8 ppm) can be clearly observed in the 13C DEPT-135 NMR spectra of both chitosan derivatives. Comparing the spectra of SECSVS and SECSBES further shows that with increasing DSSE, the signal of unmodified C6 declines remarkably, which substantiates the preferred O6-substitution. The sole functionalization of primary OH-groups, however, can be most likely excluded because a considerable signal for unsubstituted C6 still appears in the spectra despite the high DSSE of the derivatives (particularly, for SECSBES–DSSE 1.05, Figure 5B). Additionally, evidence for a functionalization in N-position can be found in a slight broadening of the H2 signal (1H: 2.7 ppm), which was probably caused by overlapping protons of substituted amino groups.26
The DEPT-spectra of both samples further show strong -CH2 signals at 50.6 ppm and 66.3 ppm, which can be assigned to the newly introduced methylene carbons C8 and C7, respectively. A small peak in the 13C spectra of both derivatives at 43.6 ppm, as well as clear coupling signals in the HSQC, likely indicate that the newly introduced sulfoethylene groups are partly linked to the nitrogen in C2-position (C9/H9, 9′: NH-CH2-CH2-SO3Na). Due to the chemical similarity to C8 (O6-CH2-CH2-SO3Na), C10 (NH-CH2-CH2-SO3Na) equally appears at 50.6 ppm in the DEPT spectra. The clear preference of O6- over N-substitution is rather unexpected because primary amino groups are strong nucleophiles under alkaline conditions and were, therefore, expected to exhibit a high reactivity throughout both synthetic pathways. Considering the complex nature of the chitosan backbone and the high impact of hydrogen bonding, the accessibility to NH2 groups might be restricted resulting in a dramatically reduced reactivity. Furthermore, beside the functionalization in C6- and C2-position, a marginal substitution of available C3 OH-groups cannot be excluded. Additional 13C DEPT (C11: 67.7) and HSQC coupling signals (H11, 11′: 3.9, 4.3 ppm) rather point at a partial O3-sulfoalkylation. An interesting effect of the applied reaction mechanisms can be observed in the 1H spectrum of SECSVS. The signal at 2.1 ppm thus originates from residual acetyl-groups (GlcNAc-CH3), revealing that amino groups in SECSVS remained partially acetylated, irrespective of the high pH. Chitosan, however, was fully deacetylated in the reaction with NaBES. This effect was probably caused by the difference in water content among the synthetic approaches because an aqueous solution of NaVS was used.
Preparation and characterization of SECS@Ag
SECS-coated AgNPs were synthesized via microwave-assisted heating and silver nitrate was used as precursor. To generate stable particles, the reaction conditions (temperature, heating time, and SECS:AgNO3 ratio) were optimized. As confirmed by TEM imaging (in HR mode), stable and well-dispersed nanoparticles were obtained using SECSBES (DSSE 1.05) as reducing agent (Figure 6C). Reactions with SECSVS (DSSE 0.69) were (partially) successful after enhancing the reduction capability of the derivative by adding glucose and after reducing the AgNO3 concentrations (12 mM). However, due to the considerably lower DSSE of SECSVS compared with SECSBES, SECSVS@Ag particles were not efficiently prevented from aggregation. Probably, the broad MWD of NaVSVS negatively affected stability and uniformity of SECSVS@Ag. Therefore, several stable nanoparticles were formed, but had a high tendency to agglomerate (Figure 6A). These agglomerates obviously consist of a mixture of silver particles, as well as sulfonated chitosan (TEM/EDX, Figure 6B). Owing to the low stability, SECSVS@Ag was excluded from further characterizations.
 | Figure 6 (HR-)TEM images of (A) SECSVS@Ag (DSSE 0.69) and (C) SECSBES@Ag (DSSE 1.05); (B) TEM/EDX image of SECSVS@Ag; (D) size distribution histogram of SECSBES@Ag (based on TEM images). |
According to theory, the reduction mechanism and the particle stability require three important features of the chitosan derivative: 1) free and accessible amino groups as coordination sites, enabling polymer–metal complexation prior to the reduction; 2) aldehyde groups, for the formation of elemental silver; and 3) negatively charged functional groups to protect particles from aggregation by electrostatic repulsion.24,27 Hence, the low MW of SECSBES can be considered as a clear advantage in this regard, as higher numbers of reducing end groups were provided. Moreover, the low viscosity of the low MW chitosan derivative ensured convenient implementation without the need for organic solvents. Because chitosan was preferably modified in O6-position, a large number of free amino groups was still available. Particles with a fairly uniform size and spherical shape were realized due to the narrow MWD of SECSBES (Figure 6C). Moreover, SCESBES@Ag consisted of a highly crystalline silver core surrounded by a 1–2 nm thick layer of sulfonated chitosan. According to determinations using HR-TEM, SECSBES@Ag exhibited a mean geometric diameter of 10–12 nm in an overall size range between 6 and 16 nm (Figure 6D). Additionally, the high zeta potential of ζ = −35 mV (SECSBES@Ag in aqueous suspension, pH 8.5) on the particle surface proves the presence of negatively charged sulfonic acid groups and points at the high particle stability in water.
Anticoagulant testing
In this section, the anticoagulant properties of the SECS and AgNPs coated with SECS are investigated with respect to their action mechanism. The activity of anticoagulants strongly depends on their MW. For example, low-molecular-weight heparins (LMWHs) exhibit significantly different activity than high MW compounds.28,29 More precisely, the interaction between unfractionated heparin and thrombin (IIa) is based on a specific bridging of AT III and factor IIa, which requires an appropriate heparin MW of at least 5,400 Da.29 Thus, LMWH and other anticoagulant low-molecular-weight oligo- and polysaccharides only marginally influence IIa. Their therapeutic effect is mainly attributed to the inhibition of the plasma serine protease zymogen – cofactor Xa – which plays a central role in the coagulation cascade as activator of prothrombin (II).30–32
Clot-based assays, as PT and aPTT, are usually performed to examine bleeding abnormalities and to evaluate anticoagulant therapy. These methods use citrated plasma, and subsequently, the induced fibrin clot formation indicates the end point of coagulation.33,34 Clotting times for PT or aPTT testing typically range between 10 and 14 seconds or 22 and 40 seconds, respectively. However, a prolongation of PT and aPTT can be caused by deficiencies of coagulation factors, by coagulation antibodies, or by inhibitors of fibrinogen conversion (eg, heparin administration or other anticoagulants).33,35
As shown in Table 1, PT and aPTT of the investigated blood plasma are at the lower limit of the normal range prior to addition of our samples. To examine the anticoagulant activity of the prepared sulfonated derivatives, as well as the generated nanoparticles, different volumes (25 or 50 μL) of SECS solutions or SECSBES@Ag suspensions were added to plasma samples.
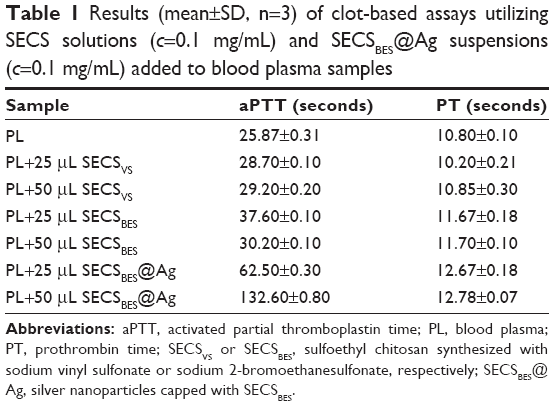 | Table 1 Results (mean±SD, n=3) of clot-based assays utilizing SECS solutions (c=0.1 mg/mL) and SECSBES@Ag suspensions (c=0.1 mg/mL) added to blood plasma samples |
Based on the comparison of the PT and aPTT values before and after sample addition, a significant increase in both values can be immediately observed in the case of plasma samples, including SECSBES@Ag suspensions. A rather small increase in PT and aPTT values is also observed after addition of SECSBES solutions (regardless of the added sample volume). Nevertheless, both measured parameters remained in the typical (expected) range for plasma samples. The addition of SECSVS, however, only resulted in a marginal increase in aPTT, whereas PT values remained almost unaffected.
At this point, it was already clear that the addition of the prepared samples (especially the SECSBES@Ag suspensions) led to an increase in the coagulation time. However, there was still not any indication about the underlying mechanism of action or any influence on a specific factor in the coagulation cascade.
Because our chitosan derivatives are of a rather low MW, it can be expected that coagulation factor Xa could be potentially influenced. The latter assumption is in line with reports that deficiency in factor Xa is a common cause for prolonged PT.35 To explore this aspect further, two additional experiments were performed in relation to the specific factor Xa activity. The first was the measurement of PT for a factor Xa-deficient plasma and the consequent comparison of the obtained values with a calibration curve (Figure 3), where PTs were measured for plasmas with known added factor Xa concentrations. The second method was conducted to evaluate the heparin amount in the plasma sample (see section on chromogenic determination of anticoagulant activity for details about the relation of heparin to the factor Xa content) based on a chromogenic assay. While clotting assays (eg, PT measurement) mostly provide a global assessment of the coagulation function, chromogenic tests are designed to more specifically determine the level or function of specific factors such as factor Xa.
The chromogenic determination of factor Xa was performed using the Berichrom® Heparin testing procedure. This test allows for indirectly assessing the similarity between the prepared samples and the native heparin, as well as the potential of the samples to mimic heparin’s activity. Heparin is known to considerably accelerate the inactivation of coagulation factor Xa. Because the prepared samples contain sulfate groups, direct interactions with factor Xa, namely via its disulfide bridges in the structure, can be expected. As mentioned earlier, the chromogenic assay indirectly evaluates the amount of free heparin. Hence, the obtained negative results (written as 0) for the prepared samples (Table 2, second column) clearly show that there is not any free heparin detectable in the respective plasma samples after addition of the SECS or SECSBES@Ag. There are two options of how our samples interact with factor Xa. First, the samples can directly interact with the cofactor hindering its transformation by heparin during the assay, or second, that our samples mimic the heparin’s activity to catalyze the transformation of factor Xa to factor Xa-AT III product.
 | Table 2 Results (mean±SD, n=3) of Xa determination assay utilizing SECS solutions (c=0.1 mg/mL) and SECSBES@Ag suspensions (c=0.1 mg/mL), added to blood plasma samples |
However, the performed chromogenic assay is not capable of distinguishing between these two options. For this purpose, another coagulation test was performed that measures the PT in relation to the activity of factor Xa. In this test, a reference curve is prepared using factor Xa-deficient plasmas of known factor Xa contents, for which respective PTs are measured. Blood plasma samples are then compared with this reference curve, allowing for the approximation of the factor Xa activity in these samples.
The last two columns in Table 2 depict the obtained results for plasmas including the prepared samples. These include either the percentage of factor Xa activity (lower values mean less factor Xa activity) or the corresponding PT of a factor Xa (a higher value therefore means less activity of Xa and a prolonged coagulation) deficient plasma as calculated using the calibration curve shown in Figure 3. As can be immediately deduced based on these results, the tested samples (SECS solutions and especially the SECSBES@Ag suspension) exhibit a decrease in the activity of factor Xa in comparison with the control sample consisting of the mixture of plasma and ultrapure water (which was the solvent for the sample suspensions). Hence, PT is increased. Based on these findings, the samples influence on the factor Xa activity is highly likely, with a direct influence on the factor Xa activity being the more likely mechanism of action. Further studies are required to explore this mechanism in more detail using complementary methods (eg, by dynamic coagulation analysis); however, this is beyond the scope of this study. It would be further of high importance to compare SECS@Ag with the effect of “neat” silver nanoparticles without chitosan coating in terms of their anticoagulant activity.
Finally, a crucial point in many studies is to translate them into real applications, which involve efficient, optimized synthetic upscaled procedures. The use of microwave-assisted heating under flow conditions is a commonly used approach allowing for continuous processing, potentially applicable also for SECS. After automatized workup, microwave-assisted heating could be further used to produce the SECS@AgNPs in a continuous, reproducible fashion. As for all new medical materials, another aspect relates to regulatory issues, which involve cytotoxicity and biocompatibility of the new materials. This step may consume extra time because nanomaterials are involved and currently many initiatives at the European level (eg, Nanosafety cluster) are addressing the challenges in judging risks and threats of new nanomaterials, with a strong focus on medical applications. In further developments, we will therefore consider risks associated with this new type of nanomaterial, both on the interaction with living organisms and release from surfaces where such materials could be potentially used such as dialysis tubes or bags for blood storage.
Conclusion
Low-molecular-weight SECSs were synthesized by applying heterogeneous reaction regimes and rather harsh conditions. The nucleophilic substitution with NaBES (DSSE 1.03) was found to be more effective than the Michael addition utilizing NaVS (DSSE 0.69). FT Raman and two-dimensional NMR spectroscopy gave detailed structural information and confirmed a preferred O6-substitution and a minor conversion of secondary hydroxyl and primary amino groups. These SECS were used as reducing and stabilizing agents for the generation of silver nanoparticles. This model system enabled the investigation of structure–property correlations in the course of nanoparticle formation and anticoagulant activity of SECS. It turned out that both the neat SECS and the nanoparticles stabilized with SECS showed anticoagulant action. However, the action of the nanoparticles was higher in all tests (PT, aPPT) than the pure SECS. Probably, the main interaction mechanism of these nanoparticles lies in the interference with factor Xa. Because factor Xa is a major target in the heparin dosage therapy, these results may lead to completely new anticoagulants on the basis of capped nanoparticles.
Acknowledgments
The authors thank Dr Margit Gruner (Technische Universität Dresden, Germany) for NMR measurements. The financial support by the Graduate Academy, Technische Universität Dresden (Excellence Initiative of the German federation and the federal states) by means of a travel grant is gratefully acknowledged. Moreover, the authors would like to acknowledge the financial support received from the Slovenian Research Agency (grant number: P3-0036). Supported by TU Graz Open Access Publishing Fund.
Author contributions
The manuscript was written through contributions of all authors. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All authors have given approval to the final version of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat Prod Rep. 2002;19(3):312–331. | ||
Weitz DS, Weitz JI. Update on heparin: what do we need to know? J Thromb Thrombolysis. 2010;29(2):199–207. | ||
Beni S, Limtiaco JF, Larive CK. Analysis and characterization of heparin impurities. Anal Bioanal Chem. 2011;399(2):527–539. | ||
Liu H, Zhang Z, Linhardt RJ. Lessons learned from the contamination of heparin. Nat Prod Rep. 2009;26(3):313. | ||
Gunay NS, Linhardt RJ. Heparinoids: structure, biological activities and therapeutic applications. Planta Med. 1999;65(4):301–306. | ||
Alban S, Franz G. Partial synthetic glucan sulfates as potential new antithrombotics: a review. Biomacromolecules. 2001;2(2):354–361. | ||
Paluck SJ, Nguyen TH, Maynard HD. Heparin-mimicking polymers: synthesis and biological applications. Biomacromolecules. 2016;17(11):3417–3440. | ||
Balan V, Verestiuc L. Strategies to improve chitosan hemocompatibility: A review. Eur Polym J. 2014;53:171–188. | ||
Muzzarelli RA, Tanfani F, Emanuelli M, Pace DP, Chiurazzi E, Piani M. Sulfated N-(carboxymethyl)chitosans: novel blood anticoagulants. Carbohydr Res. 1984;126(2):225–231. | ||
Hirano S, Tanaka Y, Hasegawa M, Tobetto K, Nishioka A. Effect of sulfated derivatives of chitosan on some blood coagulant factors. Carbohydr Res. 1985;137:205–215. | ||
Franz G, Alban S. Structure-activity relationship of antithrombotic polysaccharide derivatives. Int J Biol Macromol. 1995;17(6):311–314. | ||
Huang R, du Y, Yang J, Fan L. Influence of functional groups on the in vitro anticoagulant activity of chitosan sulfate. Carbohydr Res. 2003;338(6):483–489. | ||
Nud’ga LA, Plisko EA, Danilov SN. Synthesis and properties of sulfoethylchitosan. Zhurnal Prikl Khimi. 1974;47(4):872–876. | ||
Muzzarelli RAA. Modified chitosans carrying sulfonic acid groups. Carbohydr Polym. 1992;19(4):231–236. | ||
Nud’ga LA, Petrova VA, Ben’kovich AD, Petropavlovskii GA. Comparative study of reactivity of cellulose, chitosan, and chitin-glucan complex in sulfoethylation. Russ J Appl Chem. 2001;74(1):145–148. | ||
Pestov AV, Petrova YS, Bukharova AV, et al. Synthesis in a gel and sorption properties of N-2-sulfoethyl chitosan. Russ J Appl Chem. 2013;86(2):269–272. | ||
Petrova YS, Bukharova AV, Neudachina LK, Adamova LV, Koryakova OV, Pestov AV. Chemical properties of N-2-Sulfoethylchitosan with a medium degree of substitution. Polym Sci Ser B. 2014;56(4):487–493. | ||
Petrova YS, Neudachina LK, Mekhaev AV, Pestov AV. Simple synthesis and chelation capacity of N-(2-sulfoethyl)chitosan, a taurine derivative. Carbohydr Polym. 2014;112:462–468. | ||
Petrova YS, Pestov AV, Usoltseva MK, Neudachina LK. Selective adsorption of silver(I) ions over copper(II) ions on a sulfoethyl derivative of chitosan. J Hazard Mater. 2015;299:696–701. | ||
Zhang K, Brendler E, Gebauer K, Gruner M, Fischer S. Synthesis and characterization of low sulfoethylated cellulose. Carbohydr Polym. 2011;83(2):616–622. | ||
Feng T, du Y, Li J, Hu Y, Kennedy JF. Enhancement of antioxidant activity of chitosan by irradiation. Carbohydr Polym. 2008;73(1):126–132. | ||
Kumar BA, Varadaraj MC, Tharanathan RN. Low molecular weight chitosan – preparation with the aid of pepsin, characterization, and its bactericidal activity. Biomacromolecules. 2007;8(2):566–572. | ||
Chae SY, Jang MK, Nah JW. Influence of molecular weight on oral absorption of water soluble chitosans. J Control Release. 2005;102(2):383–394. | ||
Breitwieser D, Spirk S, Fasl H, et al. Design of simultaneous antimicrobial and anticoagulant surfaces based on nanoparticles and polysaccharides. J Mater Chem B. 2013;1(15):2022–2030. | ||
Ehmann HM, Breitwieser D, Winter S, et al. Gold nanoparticles in the engineering of antibacterial and anticoagulant surfaces. Carbohydr Polym. 2015;117:34–42. | ||
Holappa J, Nevalainen T, Savolainen J, et al. Synthesis and characterization of chitosan N-betainates having various degrees of substitution. Macromolecules. 2004;37(8):2784–2789. | ||
Travan A, Pelillo C, Donati I, et al. Non-cytotoxic silver nanoparticle-polysaccharide nanocomposites with antimicrobial activity. Biomacromolecules. 2009;10(6):1429–1435. | ||
Messmore HL, Coyne E, Wehrmacher WH, Demir AM, Fareed J. Studies comparing low molecular weight heparin with heparin for the treatment of thromboembolism: a literature review. Curr Pharm Des. 2004;10(9):1001–1010. | ||
Seo P, Locke CF. Current and potential uses of low molecular weight heparin: a review and an economic analysis. Am J Manag Care. 2000;6(4):498–506. | ||
Brooks MB. Evaluation of a chromogenic assay to measure the factor Xa inhibitory activity of unfractionated heparin in canine plasma. Vet Clin Pathol. 2004;33(4):208–214. | ||
Rosén S, Casoni MC. Heparin therapy and monitoring: a role for the chromogenic antifactor Xa assay. Am Clin Lab. 2001;20(7):35–38. | ||
Egan G, Ensom MH. Measuring anti-factor xa activity to monitor low-molecular-weight heparin in obesity: a critical review. Can J Hosp Pharm. 2015;68(1):33–47. | ||
Zia A, O’Brien SH. Global coagulation assays: a clinical perspective. J Thromb Thrombolysis. 2015;39(1):89–94. | ||
Suchman AL, Griner PF. Diagnostic uses of the activated partial thromboplastin time and prothrombin time. Ann Intern Med. 1986;104(6):810. | ||
Bates SM, Weitz JI, Assays C. Coagulation assays. Circulation. 2005;112(4):e53–e60. |
Supplementary materials
Statistical analysis coagulation assays – single sample t-test analysis
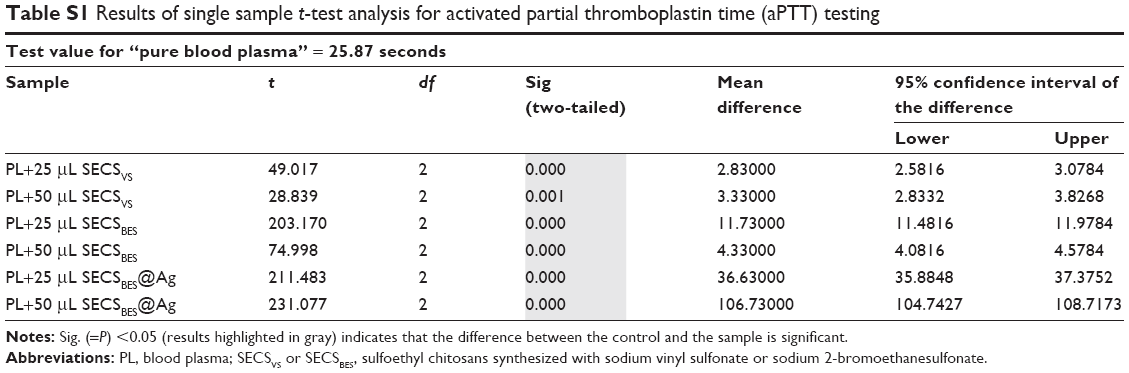 | Table S1 Results of single sample t-test analysis for activated partial thromboplastin time (aPTT) testing |
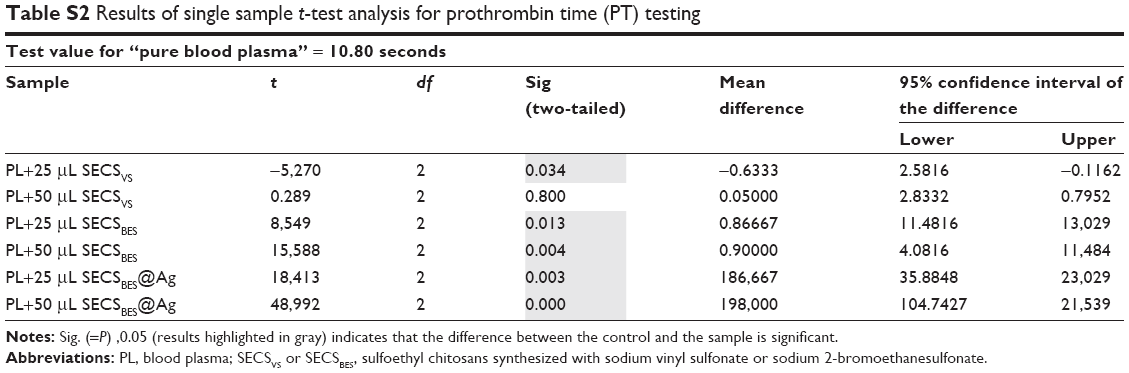 | Table S2 Results of single sample t-test analysis for prothrombin time (PT) testing |
 | Table S3 Results of single sample t-test analysis for PT-based Xa testing (% dN) |
 | Table S4 Results of single sample t-test analysis for PT-based Xa testing (seconds) |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.