Back to Journals » Cancer Management and Research » Volume 11
Low-Concentration PTX And RSL3 Inhibits Tumor Cell Growth Synergistically By Inducing Ferroptosis In Mutant p53 Hypopharyngeal Squamous Carcinoma
Authors Ye J, Jiang X, Dong Z, Hu S, Xiao M ![]()
Received 31 May 2019
Accepted for publication 13 October 2019
Published 20 November 2019 Volume 2019:11 Pages 9783—9792
DOI https://doi.org/10.2147/CMAR.S217944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Jing Ye, Xiaohua Jiang, Zhihuai Dong, Sunhong Hu, Mang Xiao
Department of Otolaryngology Head and Neck Surgery, Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Mang Xiao
Department of Otolaryngology Head and Neck Surgery, Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University, East Qingchun Road Nr.3, Hangzhou, Zhejiang 310016, People’s Republic of China
Email [email protected]
Introduction: RSL3-induced ferroptosis is a cell death pathway dependent upon intracellular iron and is characterized by accumulation of lipid hydroperoxides. Glutaminolysis, a glutamine-fueled intracellular metabolic pathway, is an essential pathway of ferroptosis in cancer cells. Recent findings showed low-concentration paclitaxel (PTX) could inhibit cell death by upregulating p53 expression; downregulating glutaminolysis-related genes.
Methods: The therapeutic effect of RSL3 plus low-concentration PTX combination therapy was investigated in HPSCC cells harboring mutant p53 (mtp53). Relative cell viability, ferroptosis-specific lipid peroxidation and relevant protein expression were evaluated.
Results: We demonstrated that neither PTX nor RSL3 in low concentration caused significant cell death; however, the combination therapy is shown to induce ferroptosis and significant cell death in mtp53 HPSCC. We discovered that low-concentration PTX enhanced the RSL3-induced ferroptosis by upregulating mtp53 expression. Furthermore, mtp53-mediated transcriptional regulation of SLC7A11 could be the key determinant.
Discussion: Although gain-of-function of p53 variants remains to be characterized, our findings provide new insight into the synergistical cell death by regulating ferroptosis and p53.
Keywords: HPSCC, ferroptosis, low-concentration paclitaxel, RSL3, synthetic cell death, mtp53, SLC7A11, GOF p53 variants
Background
Head and neck cancer is the sixth most common cancer globally.1 The majority of cases arising from oral cavity, pharynx and larynx, frequently presenting as locoregional disease.2 Among them, hypopharyngeal Squamous Cell Carcinoma (HPSCC) has a poor prognosis.3 There would be an estimated 80,608 new cancer cases and 34,984 cancer deaths in 2018.1 The addition of cetuximab to combination with cisplatin and 5-fluorouracil, followed by maintenance cetuximab until disease progression (EXTREME) is the first-line treatment of patients with recurrent and/or metastatic HPSCC.4,5 Until recently, these regimens generally resulted in median progression-free survival (PFS) of 4 months, and median overall survival (OS) of 6 to 14.1 months.4,5 Paclitaxel, a mitotic poison, is widely used in the treatment of platinum-pretreated metastatic HPSCC.6 However, its acquired resistance and high toxic effect caused by high dosage represent a major barrier.7
Low-concentration paclitaxel (PTX), which ranged from 3 to 6nM, was found to be a promising scheme for some cancers.8 Rather than mitotic arrest or tumor cell apoptosis, low-concentration PTX induced the cell type-dependent p53, p21 and G1/G2 arrest, inserted strong antiangiogenic and anti-lymphangiogenic activities.8,9 What’s more, low-concentration PTX downregulated glutaminolysis-related genes and increased cellular lactate and inhibited the tumor cell growth.8 Recent studies have shown that low-concentration PTX effects glutaminolysis in colorectal carcinoma cells and redirects metabolic reprogramming from glycolysis to oxidative phosphorylation, inducing ovarian cancer stem cells suppression.8,10 Glutaminolysis, glutamine-fueled intracellular metabolic pathway, is essential pathway of ferroptosis in cancer cells.8,11 Ferroptosis is an iron-dependent, oxidative cell death characterized by iron-dependent accumulation of reactive oxygen species (ROS).12 However, few studies have investigated effects of low-concentration PTX on glutaminolysis in head and neck cancer cells; neither the effects of low-concentration PTX on ferroptosis in tumor cells were investigated.
Moreover, p53 alterations were more frequently observed in tumors of the oral cavity, oropharynx and hypopharynx.13 The p53 mutation status correlated with poor prognosis in surgically treated HPSCC patients.14 Recently it was reported that p53 inhibited cystine uptake and sensitized cells to ferroptosis by repressing expression of SLC7A11, a key component of the system Xc transporter with oxidants.11,15,16 p53 (3KR, R117, R161, and R162), acetylation-defective mutants, which abolished p53-mediated cell-cycle arrest, apoptosis and senescence, fully retains the ability to induce ferroptosis upon ROS-induced stress.15 Acetylation of K98 of p53 is required for repression of transcription of SLC7A11 and induction of ferroptosis.15 Meanwhile, p53 stabilization could delay the activation of ferroptosis in cancer cells by limiting glutathione (GSH) depletion or enhancing GSH synthesis.17
These results led us to investigate the possibility of combination therapy with ferroptosis inducer plus low-concentration PTX on mtp53 HPSCC. Here we found combination with ferroptosis inducer RSL3 and low-concentration PTX, could synergistically induce ferroptosis cell death in mtp53 HPSCC cell lines by upregulating p53 expression.
Methods
Cell Culture And Reagents
HPSCC Detroit562 (ATCC@ CCL138™) and FaDu (ATCC@ HTB-43™) cells were purchased from American type culture collection (Manassas, VA) in 2017. Detroit562 is a metastatic pharyngeal SCC cell line which was obtained from the hydrothorax. FaDu is a primary hypopharyngeal SCC cell line. They both exhibit highly invasive behavior in vivo. Immunohistochemistry assay showed p53 is expressed in more than 50% positive cells.18 Detroit562 cells harbor homozygous mutant p53. Gene sequence is c.524G>A and protein sequence is p. R175H (point mutation at codon175, Arg → His);18 FaDu cells harbor two different heterozygous mutant p53. One gene sequence is c.743G>T and protein sequence is p.R248L (point mutation at codon248, Arg → Leu),18 another is c.376-1G>A and protein sequence is unknown. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, cat.10565–018, Life Technologies), supplemented with 10% fetal bovine serum, plus antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin). When cells reached 80% confluence, they were harvested with 0.25% trypsin/0.1% EDTA (Wisent). All cells were maintained at a humidified condition at 37°C in 5% CO2 and the media were changed every 3 days. Reagents RSL3 (S7699) and Liproxstatin-1 (S7155) were purchased from Selleck Chemicals (Houston, TX, USA). Opti-MEM® I reduced serum medium (cat:31985-070) from GIBCO-Life Technologies (Thermo Fisher Scientific). Lipofectamine 3000 (cat:L3000008) from Thermo Fisher Scientific. Lipid Peroxidation Sensor BODIPY® 581/591 (Cat: D3861) from Thermo Fisher Scientific.
Transient Transfection Assays
Exponentially growing cells were seeded at 3×105 cells in 60 mm cell culture plates. The ectopic expression of wild-type p53 (p53WT) in Detroit562 and FaDu cell lines was transiently carried out by transfecting p53 Human cDNA ORF Clone p53 (NM_000546), or their negative control, pCMV6-AC Tagged Cloning Vector (PS100020). Mutant p53 protein (p53R175H and p53R248L) expression was transiently knocked down by transfected p53 siRNA (sc-29435) or its relative negative control using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instruction. p53 siRNA (sc-29435) is consisted of pools of three to five target-specific 19–25 nucleotide sequences in length.
The overexpression of GPX4 in Detroit562 and FaDu cell lines were transiently carried out by transfecting GPX4-expressing plasmid (Cat: 718676-1) or its relative negative control for 36 hrs using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instruction.
Immunofluorescence
Detroit562 cell was grown on glass slides and was transfected with pCDNA3-GFP-p53 (R175H) plasmid or relative control for 36 hrs. The cells were washed 3 times before a 24 hr treatment with paclitaxel at indicated concentration. Glass slides were then washed in cold PBS and fixed in 4% PFA (formaldehyde) containing 0.1% Triton X-100 30 min at 4°C. Then, glass slides were rinsed 5 min in cold PBS, permeabilized in PBS for 10 min and rinsed again in PBS. Slides were blocked in 5% Goat serum 30 min and incubated with Anti-TP53 (R175H) Mouse Monoclonal Antibody (Cat: 26072) at 1:50 in Immunofluorescence Staining Antibody Dilution Buffer (Solabio A1840) overnight at +4°C. Slides were washed 3 times in cold PBS, incubated for another 1 hr at 4°C with secondary antibody ab150077 Alexa Fluor® 488 goat anti-rabbit IgG (H+L) used at 2 µg/mL. DAPI was used to stain the cell nuclei. Slides were then washed twice and visualized with a Leica DM RXA fluorescence upright microscope (Leica, Wetzlar, Germany).
Cell Viability Assay
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) (LJ621, Dojindo, Japan) according to the manufacturer’s instructions. Cells were plated at a density of 3000–4000 cells/well in 96-well plates and were treated as indicated. Then, adding 10 μL CCK-8 solution to each well, cells were incubated at 37°C for additional 1–2 hrs. Absorbance was assayed at 450 nm using a microplate reader (Synergy HT, Bio-Tek, United States).
Cell Morphological Observation
Exponentially growing HPSCC cells were transferred to 6-well plates and cultured at 37C in a 5% CO2 atmosphere. Cells were treated with indicated drugs for 24 hrs. Then, images were taken using an OLYMPUS IX 71 microscope (10×10) (OLYMPUS, Tokyo, Japan).
Western Blots Analysis
Cells were washed with ice-cold PBS and whole cell extracts were prepared in SDS/β-mercaptoethanol sample buffer containing protease inhibitors. Proteins were separated by 10–15% SDS-PAGE gels and transferred to PVDF membranes (Roche, Laval, QC) and blocked in 5% skimmed milk for 1 hr at room temperature, then incubated with the primary antibodies at 4°C overnight. Antibodies against GPX4 (ab125066, dilution1:1000), p53 (ab26, dilution 1:1000), SLC7A11 (ab37185, dilution 1:1000) were purchased from Abcam (Cambridge, MA, USA). After incubation with Sheep Anti-Rabbit-IgG-HRP (ab6747) at or Sheep Anti-Mouse-IgG-HRP (ab6808) at 1:4000 dilution for 1 hr at room temperature, proteins were visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA, #32106) followed by exposure to standard X-ray films.
Lipid ROS Measurement By Flow Cytometry
Lipid Peroxidation Sensor BODIPY® 581/591 (Cat: D3861, Thermo Fisher) was used to detect reactive oxygen species (ROS) in cells and membranes according to the manufacture introduction. After HPSC cells were treated as indicated, C11-BODIPY was added (2.5 μL/mL) and incubated for 10 min. Excess dye was removed by washing the cells twice with PBS. Labeled cells were then trypsinized and resuspended in PBS and transferred to a 5 mL FACS tube. Analysis on a flow cytometer within 10 min using Cell Quest software (BD Biosciences, Franklin Lakes, NJ, USA). Oxidation of the polyunsaturated butadienyl portion of the dye results in a shift of the fluorescence emission peak from ~590 nm to ~510 nm. The signals from both non-oxidized C11 and oxidized C11 were monitored. The ratio of Mean fluorescent Intensity (MFI) of oxidized C11 to MFI of non-oxidized C11 signals was calculated for each sample. The data were normalized to control samples as shown by the relative lipid ROS.
Statistical Analysis
Paired t-test and graphical presentations were performed by GraphPad Prism 7.0. Statistically significant results were referred with **p<0.01 and *p<0.05. All data were generated with at least three independent experiments. Each experiment in the cell death analysis was carried out by 3 to 6 replicates. Values are the means of three independent experiments (mean ± SD).
Results
Low Concentration PTX Did Not Affect Cell Viability Of mtp53 HPSCC
Two HPSCC cell lines harboring p53 mutations-Detroit562-p53R175H and FaDu-p53R248L were treated with PTX for 24 h. As revealed by Cell Counting Kit-8 (CCK-8) assay, PTX induced cell death in HNSC cells in dose- and time-dependent manner (Figure 1A). The 24 hrs IC50 for Detroit562 and FaDu cells were 7.5 μM and 1.25 μM, much higher than the “low-concentration”. Previous studies demonstrated that 1 and 3nM PTX had an impact on morphology and viability of cancer cells.10,19 Therefore, we investigated whether PTX at concentration (1–3 nM) had similar effects in mtp53 HPSCC. The results showed that PTX at a concentration (1–3 nM) did not inhibit cell growth (Figure 1B). Consequently, we used 2.0 nM PTX in subsequent studies.
 |
Figure 1 Effect of PTX on mtp53 HPSCC cell viability. (A) Relative cell viability of Detroit562 and FaDu cells after 24 hrs of PTX treatment in indicated concentration. The relative cell viability was measured using the CCK8 assay. IC50 value for PTX was calculated. (B) Relative cell viability of Detroit562 and FaDu cells after 24 hrs of PTX at a low concentration (0, 1, 2, 3 nM). Data represent the means±SD, n=3 independent experiments. |
RSL3 At Concentration Of 0.2 μM Do Not Induce Intensive Cell Death In mtp53 HPSCC
Lipid peroxidation inhibitor (Liproxstatin-1, Lip-1) is a potent pharmacological inhibitor of ferroptosis. It prevents ROS accumulation and inhibits Erastin, RSL3, and BSO-induced ferroptosis in vitro.20 Detroit562 and FaDu cell lines were treated with various concentrations of RSL3 in the absence or presence of liproxstatin-1 for 24 hrs. RSL3-induced ferroptosis in dose-dependent manner and could be inhibited by Lip-1 (Figure 2A). Notably, RSL3 at a concentration of 0.2 μM did not cause intensive cell death in Detroit562 and FaDu cells (Figure 2B). Consequently, we used 0.2 μM RSL3 in the subsequent study.
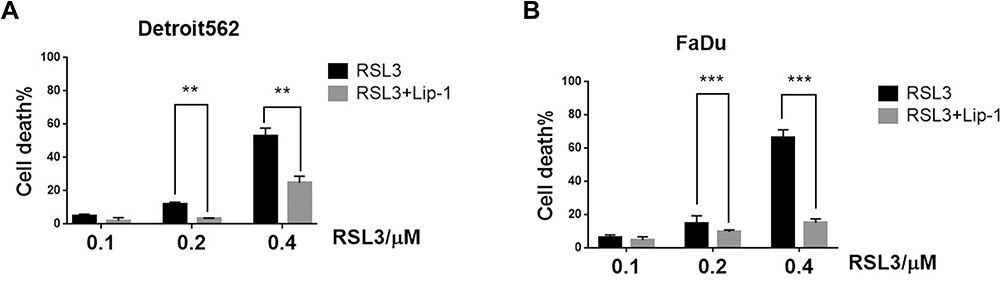 |
Figure 2 Ferroptosis contributes to RSL3-induced cell inhibition in mtp53 HPSCC. (A, B) Detroit562 and FaDu cells were treated with RSL3 (0.2 µM) for 24 h with or without the pretreatment of Lip-1 (0.02 µM). The cell death was assayed using CCK8 assay. RSL3 was dissolved in DMSO and the cell viability of DMSO was considered as 100%. Data represent the means±SD, n=3 independent experiments. **p<0.01 ***p<0.001 RSL3 vs RSL3+lip-1 treatment group, paired t-test. |
Ferroptosis Contributed To Low-Concentration PTX And RSL3-Induced Synthetic Cell Death
Low-concentration PTX has effect on the cellular metabolic process glutaminolysis, which is essential to ferroptosis.8,21,22 These results lead us to investigate the possibility of RSL3 plus low-concentration PTX. We pretreated Detroit562 and FaDu cell lines with liproxstatin-1 and determined cell death at 6 and 24 hrs. Treatment with low-concentration PTX or RSL3 alone did not inhibit tumor growth compared with control group. The combination therapy led to significant great cell inhibition in HPSCC cells, of note, the antitumor effect of the combination therapy could be abrogated by the Lip-1 pretreatment (Figure 3A). Relative cell viability increased from 17.8% to 43.5% at 24 hrs in Detroit562 cells, and from 73.4% to 93.2% at 6 hrs and from 23.8% to 58.8% in FaDu cells at 24 hrs, respectively. HPSCC cells treated with combination drug were rounded up and detached, in keeping with morphological changes of ferroptotic cells previously described (Figure 3B).23 These results indicate that RSL3 and low-concentration PTX contribute to synthetic ferroptosis.
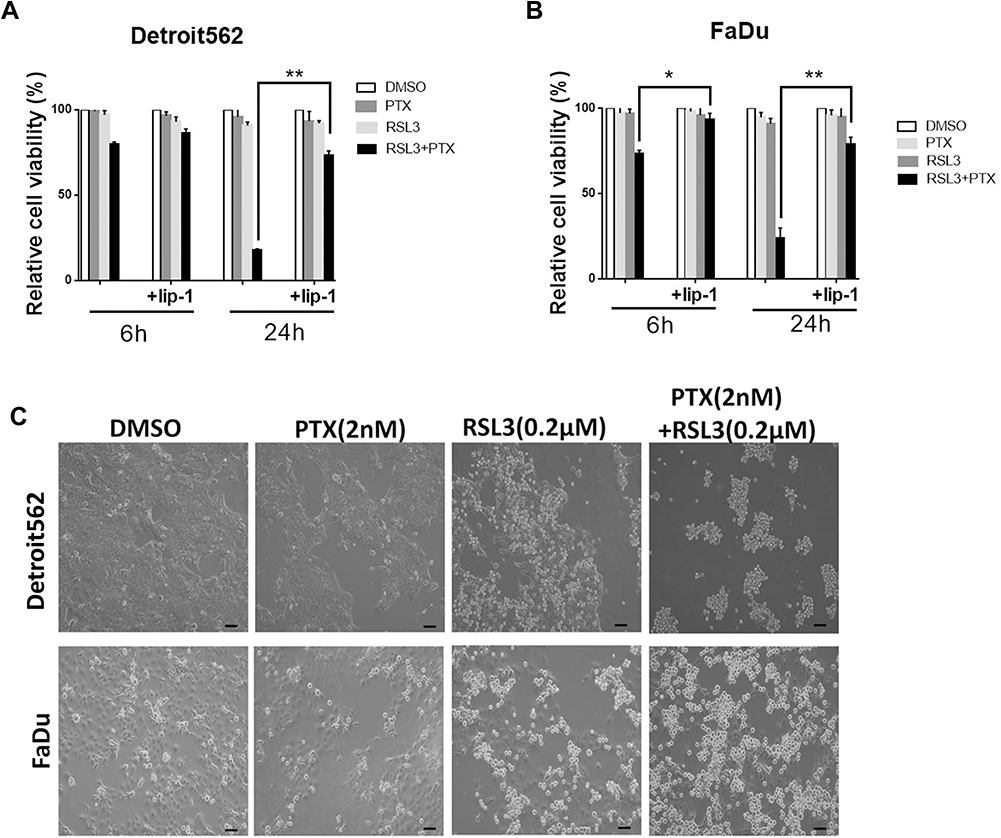 |
Figure 3 Ferroptosis contributes to low-concentration PTX and RSL3-induced synthetic cell death in HPSCC cell lines. (A) Quantification of cell viability by CCK8 assay. The effects of low-concentration PTX and RSL3 treatment with or without Lip-1 (Lip-1, 0.02 µM) pretreatment in Detroit562 and FaDu cell lines for 6 and 24 hrs, respectively. (B) Representative cell morphological changes are detected by light microscopy (Scale bar=100 µm). Exponentially growing HPSCC cells were transferred to 6-well plates and cultured at 37C in a 5% CO2 atmosphere. Cells were treated with indicated drugs for 24 hrs. Then, images were taken using an OLYMPUS IX 71 microscope (10×10) (OLYMPUS, Tokyo, Japan). (Data represent the means±SD, n=3 independent experiments. *p<0.05, **p<0.01, paired t-test). |
GPX4 Overexpression Was Failed To Suppress Low-Concentration PTX And RSL3-Induced Ferroptosis
GPX4 is a glutathione (GSH)-dependent lipid hydroperoxidase and inhibits the cysteine/glutamate amino acid transporter system that blocks GSH synthesis.8 RSL3 induced the GPX4 inactivation and triggers ferroptosis by accumulation of lipid peroxidation (lipid ROS).11,24 To investigate whether GPX4 inactivation led to ferroptotic cell death induced by drug combination, we evaluated the cell death and lipid ROS under GPX4 transient overexpression (Figure 4A). According to the Lipid ROS measurement by flow cytometry, RSL3 plus low-concentration PTX combination treatment led to upregulated lipid ROS; meanwhile, overexpression of GPX4 suppressed lipid ROS accumulation (Figure 4B and C). However, overexpression of GPX4 did not result in decreased cell death as we expected (Figure 4D). Accordingly, low-concentration PTX and RSL3 treatment alone inhibited GPX4 expression, but drug combination did not have such effect in mtp53 HPSCC cells (Figure 4E and F). These data suggest the potential rationale that synthetic ferroptosis contributed to low-concentration PTX and RSL3 but not completely through GPX4 inhibition. In other words, low-concentration PTX may exert an anticancer activity contributing to ferroptosis.
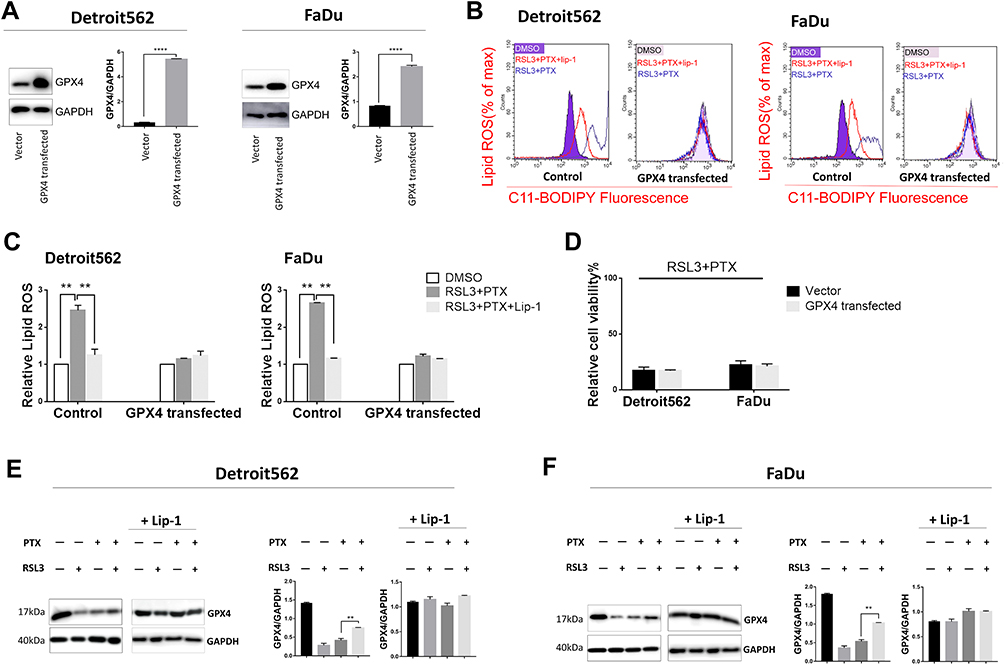 |
Figure 4 Effect of GPX4 overexpression on low-concentration PTX and RSL3-induced ferroptosis. (A) Overexpression of GPX4 as demonstrated by Western blot, respectively, in Detroit562 and FaDu cells. Cells were transfected with GPX4-expressing plasmid (Cat: 718676-1) or its relative negative control for 36 hrs. Protein bands were quantified with Image J. The quantification reflected the relative amounts as a ratio of each protein band relative to the lane’s loading control (GAPDH). (B) Detroit562 and FaDu cells were transfected with GPX4 or its negative control was treated with RSL3 (2 µM) and low-concentration PTX (2 nM) for 24 hrs with or without Lip-1 (0.02 µM) pretreatment. Lipid ROS level (Percentage of max) was determined using the C11-BODIPY, samples were examined using BD FACS Calibur. (C) Graphical representation of the histogram for relative lipid ROS . (D) Detroit562 and FaDu cells were transfected with GPX4 or its negative control was treated with combination of RSL3(2 µM) and low-concentration PTX (2nM) for 24 h. Cell viability was assayed using Cell Counting Kit-8. (E, F) Western blotting analysis of GPX4 in Detroit562 and FaDu cells treated with low-concentration PTX (2nM), RSL3 (2 µM), combination drug with or without the pretreatment of Lip-1 (0.02 µM) for 24 hrs. Data represent the means±SD, n=3 independent experiments.**p<0.01, ****p<0.0001, paired t-test |
Mtp53 Plays Crucial Role In The Low Concentration PTX And RSL3-Induced Synthetic Ferroptosis
Published reports show that PTX inhibits tumor cell growth through up-regulation of p53.8,25 Until recently, SLC7A11 was identified as a novel p53 target gene in ferroptosis,15 which allows us to investigate p53 status during treatment. Treatment of low-concentration PTX indeed resulted in a dramatic increase in p53 expression (Figure 5A). Immunofluorescence of mtp53 in Detroit562 cells was further performed. As Detroit562 cells harbor homozygous mtp53 with protein sequence p. R175H, the cells were transfected with pCDNA3-GFP-P53 (R175H) plasmid transiently. Immunofluorescence revealed that p53R175H protein was obviously increased by combination drug (Figure 5B and C). To determine the role of mtp53 in the low-concentration PTX and RSL3-induced synthetic ferroptosis, we used siRNA knockdown of p53 in HPSCC cells. Raveled by Lipid ROS measurement, drug combination failed to cause cell death and lipid ROS accumulation when p53 expression was knocked down (Figure 5D and E). These findings indicate that upregulating mtp53 by low-concentration PTX plays a pivotal role in RSL3-induced ferroptosis.
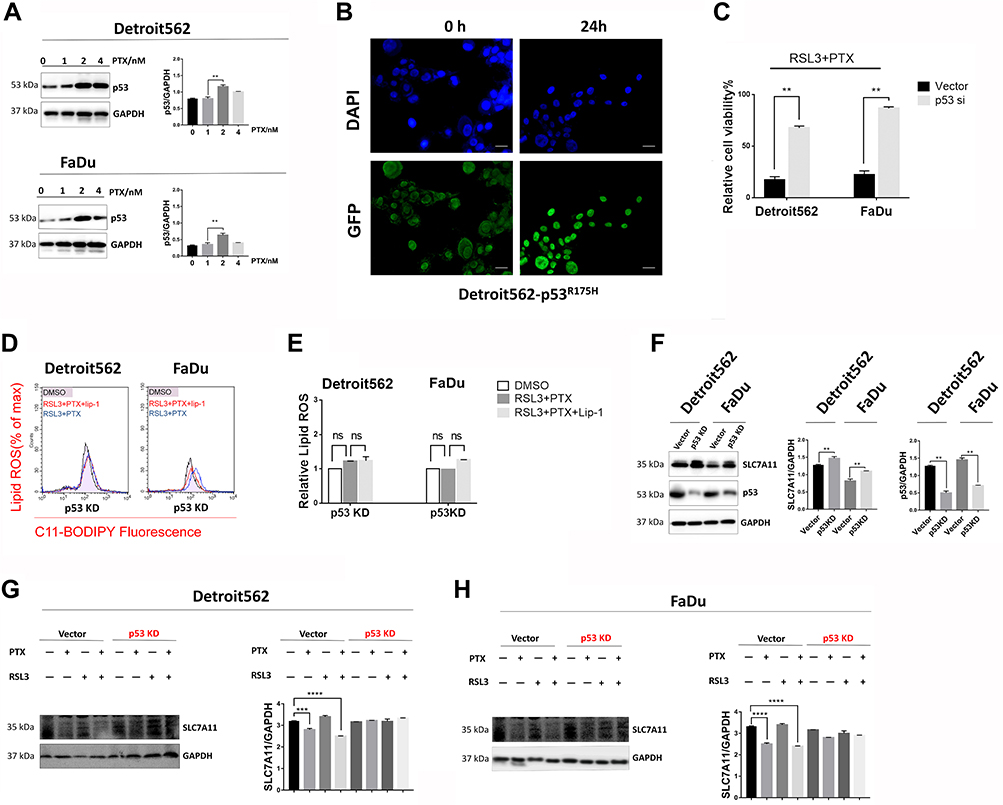 |
Figure 5 mtp53 mediated ferroptosis contributes to low-concentration PTX and RSL3-induced synthetic cell death. (A) Western blotting analysis of p53 in Detroit562 treated with PTX at indicated low concentrations for 24 hrs. Protein bands were quantified with Image J. The quantification reflected the relative amounts as a ratio of each protein band relative to the lane’s loading control (GAPDH). (B) Detroit562 cells were transfected with pCDNA3-GFP-P53 (R175H) plasmid then fixed and stained with anti-P53(R175H) monoclonal antibody (Cat. # 26072) and with secondary antibody. Fluorescent images were collected simultaneously (Scale bar=25 µm). (C) Detroit562 and FaDu cells were transfected with p53 siRNA (sc-29435) or its relative negative control was treated with combination of RSL3(2μM) and low-concentration PTX (2 nM) for 24 hrs. Cell viability was assayed using Cell Counting Kit-8. (D) Detroit562 and FaDu cells were transfected with p53 siRNA (sc-29435) or its relative negative control was treated with RSL3 (2 µM) and low-concentration PTX (2nM) for 24 hrs with or without Lip-1 (0.02 µM) pretreatment. Lipid ROS level (Percentage of max) was determined using the C11-BODIPY, samples were examined using BD FACS Calibur. (E) Graphical representation of the histogram for relative lipid ROS. (F) Western blotting analysis of p53 and SLC7A11 in Detroit562 and FaDu cells transfected with p53 siRNA (sc-29435) or its relative negative control. (G, H) Western blotting analysis of SLC7A11 in Detroit562 and FaDu cells transfected with p53 siRNA (sc-29435) or its relative negative control. Detroit562 and FaDu cells were treated with low-concentration PTX (2 nM), RSL3 (2 µM), combination drug with or without the pretreatment of Lip-1 (0.02 µM) for 24 hrs. Data represent the means±SD, n=3 independent experiments. ns p>0.05, **p<0.01, ***p<0.001, ****p<0.0001, paired t-test. |
Underlying mechanisms of mtp53 expression upregulation in ferroptosis is still largely unknown. Acetylation of K98 of p53 is required for repression of transcription of SLC7A11 and induction of ferroptosis as previously described. We attempt to evaluate SLC7A11 expression under mtp53 knock-down. Knock-down of mtp53 expression resulted in increased level of SLC7A11 (Figure 5F). Additionally, we examine whether administer of RSL3 and low-concentration PTX led to SLC7A11 suppression under mtp53 knock-down. RSL3 or low-concentration PTX alone and RSL3 plus low-concentration PTX combination therapy also failed to suppress SLC7A11 under mtp53 knock-down (Figure 5G and H). Based on the above results, mtp53 is upregulated by low-concentration PTX, and that induction of SLC7A11 inhibition would contribute to the enhancement of RSL3-induced ferroptosis in mtp53 HPSCC.
Discussion
Ferroptosis is initiated by events in a chronological signaling pathway that are yet to be well defined.11 Features of ferroptosis includes: (a) generation of ROS; (b) depletion of GPX4 in cells; (c) accumulation of lipid hydroperoxides (lipid ROS) and (d) availability of iron.26 Dependent on NADPH/H+, polyunsaturated fatty acid metabolism, and the mevalonate and glutaminolysis, metabolic pathways have been implicated in ferroptosis.27–31 Meanwhile, exploitation of ferroptosis in response to specific compounds is also attracting strategy in cancer therapy.
Our study primarily showed the GPX4 overexpression did not inhibit ferroptotic cell death induced by RSL3 and low-concentration PTX. Apart from our results, another explanation could be from the therapy-resistant status. Therapy-resistant is molecularly characterize as high-mesenchymal cell state in human cancer cell lines and organoids. It depends on a druggable lipid-peroxidase pathway that protects against ferroptosis, which induced by the build-up of toxic lipid peroxides.32 Therefore, overexpression of GPX4 could lead to PTX resistance rather than simply blocking ferroptosis. Identification of GPX4 dependency in cancer cells as the target of high-mesenchymal cell state, could be another anticancer strategy to be investigated.
It has been shown by different groups that p53 induces ferroptosis, by transcriptionally suppressing the expression on SLC7A11 (the cystine/glutamate antiporter). The 5ʹ flanking region of the human SLC7A11 gene at chromosome 4q28-31 contains one site that matches the consensus p53-binding sequence. Results observed in cancer cell lines expressing wild-type p53 tumor cells appeared to contradict the findings.17 p53WT stabilization decreases system xc – activity and simultaneously p53-p21 transcriptional axis negatively regulates ferroptosis in cancer cells.17 Until recently, underlying mechanisms of mtp53 and p53WTin ferroptosis is still largely unknown.
Several p53 missense mutations, including those two mutants in our study at codons R248 and R175, are among the most prevalent hotspot mutations in HPSCCs, occurring within the central region of the protein which serves as the p53 DNA-binding domain.33–36 They both are localized in the cytoplasm as well where it can exert its “Gain of Function (GOF)” activity. Both mutant types not only lose p53WT'stranscriptional function but also have dominant-negative activity by heterodimerization with p53WT.37 HPSCC harboring GOF mutant variant p53R175H and p53R248L were determined for the current study according to a novel computational approach termed Evolutionary Action (EAp53). It is a system to stratify patients with tumors harboring p53 mutations as high or low risk and validated this system in both in vivo and in vitro models. Patients with high-risk p53 mutations had the poorest survival outcomes and the shortest time to the development of distant metastases. Tumor cells expressing high-risk TP53 mutations were more invasive and tumorigenic.38 R248L EA score is 94.72 while R175H is 78.51, both were high-risk mutants). Our results showed both GOF p53 variants: p. R175H (point mutation at codon175, Arg → His) and p.R248L (Point mutation at codon248, Arg→Leu) here retain the regulating with SLC7A11. Unlikep53WT, which is rapidly degraded by the ubiquitin-proteasome system, the GOF mtp53 variants are highly stable and have a tendency to form higher-order aggregates.39
Our efforts identify the mechanism regulation of SLC7A11 by p53R175H and p53R248L, critical in mediating the response to ferroptosis were not successful likely because there is redundancy in ubiquitination sites on p53R175H and p53R248L. Furthermore, the diversity of the mechanisms of p53’s GOF properties strongly suggests that GOF activities may vary according to mutation type, cell type, and even stimuli, and far beyond of our investment.32 These are objective of future investigations.
Conclusion
Our data revealed that the combination of low-concentration PTX and RSL3-induced synthetic ferroptosis in mtp53 HPSCC. mtp53 is upregulated by low-concentration PTX, and that induction of SLC7A11 inhibition would contribute to the enhancement of RSL3-induced ferroptosis in mtp53 HPSCC. Until recently, underlying mechanisms of mtp53 expression upregulation in ferroptosis is still largely unknown. These findings provide new insight into the synergistical cell death by regulating ferroptosis and mtp53.
Abbreviations
Detroit562 and FaDu, Human pharyngeal Squamous Cell Carcinoma cell lines; HPSCC, Hypopharyngeal Squamous Cell Carcinomal; GPX4, Glutathione peroxidase 4; Lip-1, Liproxstatin-1; PTX, Paclitaxel; mtp53, Mutant p53; p53R175H and p53R248L, p53 variants; SLC7A11, Cationic amino-acid transporter, member 11; ROS, Reactive oxygen species; RSL3, a VDAC-independent ferroptosis activator; WT, Wild-type.
Availability Of Data And Material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
JY participated in the design of the study and performed all assays. XHJ and ZHD performed experimental studies and acquired the data. SHH did literature research and analyzed the data. MX conceived the study, participated in its design and drafted the manuscript. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
3. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
4. Yoshino T, Hasegawa Y, Takahashi S, et al. Platinum-based chemotherapy plus cetuximab for the first-line treatment of Japanese patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: results of a phase II trial. Jpn J Clin Oncol. 2013;43(5):524–531. doi:10.1093/jjco/hyt034
5. Al-Saleh K, El-Sherify M, Safwat R, et al. Phase II/III randomized controlled trial of concomitant hyperfractionated radiotherapy plus cetuximab (Anti-EGFR Antibody) or chemotherapy in locally advanced head and neck cancer. Gulf J Oncolog. 2019;1(30):6–12.
6. Ley J, Wildes TM, Daly K, Oppelt P, Adkins D. Clinical benefit of nanoparticle albumin-bound-paclitaxel in recurrent/metastatic head and neck squamous cell carcinoma resistant to cremophor-based paclitaxel or docetaxel. Med Oncol. 2017;34(2):28. doi:10.1007/s12032-017-0884-7
7. Banerji A, Lax T, Guyer A, Hurwitz S, Camargo CJ, Long AA. Management of hypersensitivity reactions to Carboplatin and Paclitaxel in an outpatient oncology infusion center: a 5-year review. J Allergy Clin Immunol Pract. 2014;2(4):428–433. doi:10.1016/j.jaip.2014.04.010
8. Lv C, Qu H, Zhu W, et al. Low-dose paclitaxel inhibits tumor cell growth by regulating glutaminolysis in colorectal carcinoma cells. Front Pharmacol. 2017;8:244. doi:10.3389/fphar.2017.00244
9. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20(29):3806–3813. doi:10.1038/sj.onc.1204487
10. Shen YA, Li WH, Chen PH, He CL, Chang YH, Chuang CM. Intraperitoneal delivery of a novel liposome-encapsulated paclitaxel redirects metabolic reprogramming and effectively inhibits cancer stem cells in Taxol((R))-resistant ovarian cancer. Am J Transl Res. 2015;7(5):841–855.
11. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170. doi:10.1007/82_2016_508
12. Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta. 2017;1861(8):1893–1900. doi:10.1016/j.bbagen.2017.05.019
13. Schneider-Stock R, Mawrin C, Motsch C, et al. Retention of the arginine allele in codon 72 of the p53 gene correlates with poor apoptosis in head and neck cancer. Am J Pathol. 2004;164(4):1233–1241. doi:10.1016/S0002-9440(10)63211-7
14. Omura G, Ando M, Ebihara Y, et al. The prognostic value of TP53 mutations in hypopharyngeal squamous cell carcinoma. BMC Cancer. 2017;17:1.
15. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62.
16. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032.
17. Tarangelo A, Magtanong L, Bieging-Rolett KT, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–575.
18. Caamano J, Zhang SY, Rosvold EA, Bauer B, Klein-Szanto AJ. p53 alterations in human squamous cell carcinomas and carcinoma cell lines. Am J Pathol. 1993;142(4):1131.
19. Stewart DA, Winnike JH, McRitchie SL, Clark RF, Pathmasiri WW, Sumner SJ. Metabolomics analysis of hormone-responsive and triple-negative breast cancer cell responses to paclitaxel identify key metabolic differences. J Proteome Res. 2016;15(9):3225–3240.
20. Friedmann AJ, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191.
21. Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–363.
22. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
23. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072.
24. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331.
25. Choi YH, Yoo YH. Taxol-induced growth arrest and apoptosis is associated with the upregulation of the Cdk inhibitor, p21WAF1/CIP1, in human breast cancer cells. Oncol Rep. 2012;28(6):2163–2169.
26. Sui X, Zhang R, Liu S, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. 2018;9.
27. Villar VH, Merhi F, Djavaheri-Mergny M, Duran RV. Glutaminolysis and autophagy in cancer. Autophagy. 2015;11(8):1198–1208..
28. Cetindis M, Biegner T, Munz A, Teriete P, Reinert S, Grimm M. Glutaminolysis and carcinogenesis of oral squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2016;273(2):495–503.
29. Lang I, Feuer L, Nekam K, Szigeti A, Gergely P, Petranyi G. Glutaurine enhances the depressed NK cell activity of tumor patients. Immunol Commun. 1983;12(5):519–527.
30. Albrecht J, Dolinska M, Hilgier W, Lipkowski AW, Nowacki J. Modulation of glutamine uptake and phosphate-activated glutaminase activity in rat brain mitochondria by amino acids and their synthetic analogues. Neurochem Int. 2000;36(4–5):341–347.
31. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2018.
32. Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017.
33. Grugan KD, Vega ME, Wong GS, et al. A common p53 mutation (R175H) activates c-Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol Ther. 2013;14(9):853–859.
34. Samassekou O, Bastien N, Lichtensztejn D, Yan J, Mai S, Drouin R. Different TP53 mutations are associated with specific chromosomal rearrangements, telomere length changes, and remodeling of the nuclear architecture of telomeres. Genes Chromosomes Cancer. 2014;53(11):934–950.
35. Rieber M, Strasberg RM. DN-R175H p53 mutation is more effective than p53 interference in inducing epithelial disorganization and activation of proliferation signals in human carcinoma cells: role of E-cadherin. Int J Cancer. 2009;125(7):1604–1612.
36. Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28(6):622–629.
37. Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Ann Oncol. 2014;25(10):1889–1900. doi:10.1093/annonc/mdu143
38. Neskey DM, Osman AA, Ow TJ, et al. Evolutionary action score ofTP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015;75(7):1527–1536.
39. Padmanabhan A, Candelaria N, Wong KK, et al. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat Commun. 2018;9(1):1270.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.