Back to Journals » Clinical Ophthalmology » Volume 13
Loteprednol etabonate (submicron) ophthalmic gel 0.38% dosed three times daily following cataract surgery: integrated analysis of two Phase III clinical studies
Authors Fong R, Cavet ME, DeCory HH ![]() , Vittitow JL
, Vittitow JL
Received 30 March 2019
Accepted for publication 2 July 2019
Published 1 August 2019 Volume 2019:13 Pages 1427—1438
DOI https://doi.org/10.2147/OPTH.S210597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Raymond Fong,1 Megan E Cavet,2 Heleen H DeCory,2 Jason L Vittitow3
1Manhattan Eye, Ear and Throat Hospital and Lenox Hill Hospital, New York, NY, USA; 2Medical Affairs, Bausch + Lomb, Rochester, NY, USA; 3Clinical Affairs, Bausch + Lomb, Bridgewater, NJ, USA
Purpose: To evaluate the efficacy and safety of a submicron formulation of loteprednol etabonate (LE) gel 0.38% instilled three times daily (TID) compared with vehicle for the treatment of inflammation and pain following cataract surgery with intraocular lens implantation, integrated across two multicenter, double-masked, randomized, parallel-group, Phase III studies.
Patients and methods: Subjects ≥18 years of age with anterior chamber (AC) cells ≥grade 2 (6–15 cells) on day 1 after cataract surgery were randomized to receive 1 drop of LE gel 0.38% TID, twice daily (not reported/analyzed herein), or vehicle instilled in the study eye for 14 days. Primary endpoints were the proportion of subjects with resolution of AC cells and grade 0 (no) pain at postoperative day 8. Safety outcomes included adverse events (AEs), ocular signs, fundoscopy results, visual acuity, intraocular pressure (IOP), and tolerability (drop comfort and ocular symptoms).
Results: The integrated intent-to-treat population included 742 subjects (LE gel 0.38% TID, n=371; vehicle, n=371). Significantly more subjects in the LE gel 0.38% TID group compared with the vehicle group had complete resolution of AC cells (29.6% vs 15.1%) and grade 0 pain (74.4% vs 48.8%) at day 8 (P<0.0001 for both). LE gel 0.38% TID was safe and well tolerated, with only 1 LE-treated subject experiencing an IOP elevation ≥10 mm Hg. Most treatment-related AEs were mild and occurred less frequently with LE gel 0.38% than with vehicle. The majority (>75%) of subjects in each treatment group reported no drop discomfort. There were no reports of blurred vision with LE gel.
Conclusion: The results of this integrated analysis indicate that LE (submicron) gel 0.38% administered TID is safe and effective for the treatment of ocular inflammation and pain following cataract surgery, with minimal risk of IOP elevation.
Keywords: cataract surgery, postoperative pain, postoperative inflammation, loteprednol etabonate, submicron, integrated analysis
Introduction
The use of topical corticosteroids following cataract surgery is routine for the management of postoperative inflammation and pain.1 Potential side effects of concern with the postsurgical use of ocular corticosteroids include increased intraocular pressure (IOP), susceptibility to infection, and delayed wound healing.1–3 Loteprednol etabonate (LE) is a retrometabolically designed topical corticosteroid that was engineered via modification of prednisolone in order to achieve the desired anti-inflammatory effect followed by rapid metabolism to inactive metabolites.4,5 This approach aims to preserve the beneficial corticosteroid effects while reducing the potential for adverse reactions. LE has been shown to pose a low risk of clinically significant IOP elevations with both short-term and long-term use and has demonstrated lower rates of clinically significant IOP elevation in comparison to prednisolone acetate or in comparison to dexamethasone/tobramycin when used in combination with tobramycin.6 The anti-inflammatory effects of LE across a range of clinical ocular conditions and its reduced potential to increase IOP have been reviewed in detail elsewhere.5
The first ophthalmic formulation of LE, a 0.5% suspension, was approved in 1998 for the treatment of postoperative inflammation and pain following ocular surgery as well as for steroid-responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea, and anterior segment of the eye (eg, allergic conjunctivitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis).5,7 LE ophthalmic suspension 0.5% administered four times daily (QID) demonstrated safety and efficacy for the management of postoperative inflammation and exhibited similar efficacy with less fluctuation in IOP assessments compared with prednisolone for reducing postsurgical ocular inflammation.5,8 Subsequently, a nonsettling ophthalmic gel formulation of LE 0.5% containing micron-sized drug particles was approved by the United States Food and Drug Administration (FDA) in September 2012.5,9–11 This gel formulation provides consistent, uniform dosing and eliminates the requirement to shake to resuspend the drug prior to instillation.11 The safety and efficacy of LE ophthalmic gel 0.5% QID in reducing inflammation and pain following cataract surgery have been demonstrated in a number of studies both in comparison to vehicle and to difluprednate 0.05%.12–15
A lower-dose (0.38%) ophthalmic gel formulation of LE was recently developed using SM TechnologyTM in which the drug particle size was reduced from a median diameter of ~3–5 µm (in the 0.5% suspension and gel formulations) to ~0.4–0.6 µm,16,17 representing a ~5- to 10-fold reduction in diameter or ~125- to as much as 2000-fold reduction in volume. This new submicron drug particle formulation has the same rheological properties as LE gel 0.5%;11 upon ocular instillation, the gel transitions from a semisolid to a mucoadhesive liquid on dilution with tears but has sufficient viscosity to facilitate extended retention time on the ocular surface.16 Both gel formulations have a pH of 6.5 which is similar to that of human tears and have a low concentration of the preservative benzalkonium chloride (0.003%), features which may improve the comfort of the instilled drop.9,11,18 In preclinical studies, LE (submicron) gel 0.38% demonstrated faster dissolution compared with the larger micronized LE particles in the 0.5% gel formulation16 as well as comparable or superior penetration of LE into clinically relevant anterior segment tissues following a single topical instillation despite the 24% lower dose of LE in the new gel formulation.16 Based on these findings, it was hypothesized that LE gel 0.38% may be clinically effective at a lower dosing frequency than LE gel 0.5%,16 which has a recommended QID dosing frequency.9 Less frequent dosing may improve patient convenience and potentially increase dosing compliance.19
The clinical development program designed to evaluate the safety and efficacy of LE (submicron) gel 0.38% included two similarly designed, Phase III, randomized, vehicle-controlled trials20,21 of LE gel 0.38% or vehicle instilled either BID or TID, for the treatment of postoperative inflammation and pain following cataract surgery with intraocular lens implantation. In both studies, LE gel 0.38% administered TID was safe and effective in the treatment of ocular inflammation and pain following cataract surgery, leading to the United States FDA approval of this new LE (submicron) gel formulation with TID dosing in February 2019 with the brand name of Lotemax® SM.18 Herein, we report on integrated data for LE (submicron) gel 0.38% administered TID compared with vehicle from these two trials to provide a robust analysis of the efficacy, safety, and tolerability of LE gel 0.38% at this dosing regimen in the postoperative setting.
Methods
Study design
The two studies included in this integrated analysis (Study 1/NCT01996839,20 Study 2/NCT0278690121) were randomized, multicenter, double-masked, parallel-group, vehicle-controlled trials conducted in the United States. Study 1 was conducted at 45 sites and study 2 at 43 sites.20 Both studies were performed in accordance with the International Conference on Harmonisation, Good Clinical Practices as required by the Declaration of Helsinki, the US Code of Federal Regulations, and applicable local regulations. For both studies, the protocol was approved by a central Institutional Review Board (Schulman Associates; Cincinnati, OH) prior to screening of subjects; all study subjects provided written informed consent.
Study subjects
Both studies enrolled subjects ≥18 years of age who had routine uncomplicated cataract surgery by phacoemulsification with posterior chamber intraocular lens implantation in one eye and not combined with any other surgery. Subjects were required to have potential postoperative corrected distance pinhole Snellen visual acuity (VA) of at least 20/200 in the study (surgical) eye at screening and ≥grade 2 anterior chamber (AC) cells (6–15 cells) in the study eye on postoperative day 1. Female subjects of childbearing potential were required to have a negative urine pregnancy test result at screening and on postoperative day 1 in order to participate.
Patients were excluded from study participation if they had a severe or serious ocular condition; had a history or presence of chronic generalized systemic disease; had ocular surgery in the study eye within 3 months or fellow eye within 2 weeks of screening; had a current diagnosis of cystoid macular edema; had ocular hypertension (IOP ≥21 mm Hg) at screening or baseline (postoperative day 1), glaucoma, or any glaucoma-related incisional or laser surgery in the study eye; were monocular; or had a known hypersensitivity or contraindication to study drug(s) or their components. Both studies additionally excluded patients who used ocular therapy with NSAIDs, mast cell stabilizers, antihistamines, or decongestants within 7 days prior to surgery or who were expected to require any of these treatments (with the exception of ≤81 mg/day of acetylsalicylic acid) during the 18 days following cataract surgery. Patients were also excluded if they had used systemic or ocular corticosteroids within 14 days prior to cataract surgery; if they had used ocular immunosuppressants within 30 days before surgery; or if they were expected to require systemic or ocular use of corticosteroids or glucocorticoids or concurrent systemic or ocular therapy with immunosuppressants during the 18 days following cataract surgery.
Study treatments and assessments
The investigational product in both studies was LE gel containing submicron LE at a concentration of 0.38%, the preservative benzalkonium chloride 0.003%, and the excipients disodium ethylenediaminetetraacetic acid, sodium chloride, polycarbophil, hypromellose, poloxamer 407, glycerin, propylene glycol, and sodium hydroxide and/or hydrochloric acid to maintain the pH at 6.5 (manufactured by Bausch & Lomb Incorporated, Tample, FL, USA). The vehicle did not contain LE but was otherwise identical to the investigational product. To ensure masking, study treatments were packaged in identical polyethylene bottles containing equal volumes and were supplied in identical subject kit boxes, and vehicle was matched in dosing to the LE gel groups. Study drug treatments were assigned using numbered kits containing bottles of study treatment. Kits were packaged along with instructions appropriate for the assigned dosing frequency, and an assigned designee at each site who was not involved in assessing safety or efficacy dispensed treatments and instructed subjects on dosing in order to maintain investigator masking to treatment regimen (ie, BID versus TID dosing).
Studies were conducted over approximately 4 weeks during which eligible subjects completed 7 study visits beginning with a screening visit (visit 1) within 14 days prior to surgery. Visit 2 occurred on the day of surgery. On postoperative day 1 (baseline; visit 3), subjects with AC cell grade ≥2 were randomized by computer in a 2:2:1:1 ratio to LE gel 0.38% BID, LE gel 0.38% TID, vehicle BID, or vehicle TID. Following randomization, subjects completed postoperative study visits 4, 5, 6, and 7 on postoperative days 3, 8, 15, and 18, respectively. Subjects instilled 1 drop of their assigned treatment into the study eye at approximately 12-hr (BID) or 8-hr (TID) intervals for 14 days, with the first dose administered in the clinic on postoperative day 1 and the final dose administered on the evening before postoperative day 15. Subjects documented compliance with dosing instructions by recording the date and time of each study treatment instillation into diaries.
At any time during the study, subjects could be placed on anti-inflammatory rescue medication at the investigator’s discretion. Subjects who required anti-inflammatory rescue medication discontinued study treatment and were considered treatment failures but were followed until the end of the study. Intracameral injection of antibiotic at the end of cataract surgery and/or the use of perioperative topical antibiotics (not combined with a steroid) was permitted at the discretion of the investigator. Subjects using topical antibiotics were instructed to instill the topical antibiotic at least 15 mins before administration of study treatment.
Efficacy assessments were performed on postoperative days 3 (visit 4), 8 (visit 5), 15 (visit 6), and day 18 (visit 7). At each site, AC cells and AC flare were evaluated by the same investigator, whenever possible, using a 1.0 mm × 1.0 mm high-power-field slit beam and graded on a 5-point scale (cells: 0=no cells, 1=1–5 cells, 2=6–15 cells, 3=16–30 cells, 4=>30 cells; flare: 0=none, 1=mild, 2=moderate, 3=severe, 4=very severe), with complete resolution of AC cells defined as a grade of 0 (ie, no cells observed). Subjects assessed and graded ocular pain (defined as foreign body sensation, stabbing, throbbing, or aching) on a 6-point scale (0=none, 1=minimal, 2=mild, 3=moderate, 4=moderately severe, 5=severe).
Adverse events (AEs) and concomitant medications were recorded at each study visit. Treatment-emergent AEs (hereafter “AEs”) were defined as those that began or worsened after the first administration of study drug. Pinhole Snellen VA, IOP (Goldman applanation tonometry), and ocular signs (ciliary flush, conjunctival chemosis, eyelid erythema, conjunctival injection, corneal staining, corneal edema, hyphema, posterior synechiae, anterior vitreous haze, precipitates, and hypopyon; evaluated using slit-lamp biomicroscopy) were assessed at screening and all postoperative visits. A dilated fundus examination was performed at screening and on postoperative day 15.
Subjects evaluated discomfort with study treatment at visit 5 (postoperative day 8) by grading their overall impression of drop sensation experienced within 1 min after drop instillation on a scale from 0 (none, or no discomfort from study treatment) to 3 (severe discomfort from study treatment). Ocular symptoms (photophobia, itching, tearing, and discharge) were assessed by subjects at baseline (screening) and at each postoperative visit and graded on a scale from 0 to 3 (absent, mild, moderate, and severe). At each postbaseline visit, subjects were classified as either “improved/no change” or “worsened” relative to visit 3 (the day following surgery) for each ocular symptom.
Outcome measures
Primary efficacy endpoints were the proportion of subjects with complete resolution of AC cells (cell score=0) at postoperative day 8 (visit 5) and the proportion of subjects with grade 0 (no) pain at postoperative day 8 (visit 5). Secondary efficacy endpoints included the proportion of subjects at each on-treatment visit with complete resolution of AC cells, grade 0 pain, complete resolution of AC flare, complete resolution of both AC cells and AC flare in the study eye and the change from baseline at each on-treatment visit in AC cells, AC flare, and in the AC cell and flare composite score (defined as sum of scores for AC cells and AC flare).
Safety and tolerability endpoints included the incidence of ocular and nonocular AEs; worsening from baseline in ocular signs (biomicroscopy), in dilated fundus examination results, or in VA; the change from baseline in IOP; ocular symptoms other than pain (ie, photophobia, itching, tearing, and discharge); and study drug sensation.
Statistical analyses
Approximately 161 participants were planned to be enrolled per treatment group in study 1, while 196 were planned to be enrolled per treatment group in study 2. These sample sizes were estimated to yield 90% and 87% of power, respectively, to detect a difference in the rate of complete resolution of AC cells, on postoperative day 8 between LE gel 0.38% and vehicle based on proportions reported for studies completed at the time each of these studies was initiated—for study 1, this included studies of LE gel 0.5%13,15; for study 2, this included studies of LE gel 0.5%,13,15 a study evaluating LE gel 0.38% BID dosing only (NCT02208297), and study 1.20
Efficacy and safety data from study 1 and study 2 were pooled for this integrated analysis, and endpoints were analyzed for the pooled LE gel 0.38% TID group and pooled combined vehicle groups (vehicle BID and TID). Efficacy analyses were conducted in the intent-to-treat (ITT) population, which included all randomized subjects, according to assigned treatment. The analysis of the primary efficacy endpoints tested the difference in the proportion of subjects with complete resolution of AC cells and the difference in rates of grade 0 pain between LE gel 0.38% TID and combined vehicle groups on postoperative day 8 using the asymptotic Pearson Chi-squared (χ2) statistic at a 2-sided α=0.05 level. In these analyses, missing data and subjects placed on rescue medication prior to day 8 were imputed as treatment failures. If the difference in the proportion of subjects with complete resolution of AC cells was statistically significant in favor of LE gel 0.38% TID, then success was claimed for resolution of inflammation. If the difference in the proportion of subjects with grade 0 pain was statistically significant in favor of LE gel 0.38% TID, then success was claimed for treatment of pain. The superiority of LE gel 0.38% TID over vehicle for each outcome was claimed if the difference in proportions was greater than 0.
For secondary endpoints, differences between treatment groups in proportions of subjects with complete resolution of AC cells, AC flare, combined AC cells and flare, and the difference in the proportions of subjects with grade 0 pain at each study visit were likewise analyzed using Pearson χ2 tests. Missing data and subjects placed on rescue medication prior to the visit being summarized were imputed as treatment failures. Changes from baseline (visit 3) in AC cells, AC flare, and the composite score of AC cells and flare were analyzed by treatment and visit using both continuous and discrete statistical methods. Differences between treatment groups in change from baseline in AC cells, AC flare, and the composite score were analyzed at each study visit with a Cochran-Mantel-Haenszel mean score test.
Safety and tolerability analyses were conducted in the safety population, which included all subjects who received at least 1 dose of study drug; subjects were analyzed according to the treatment received. Safety endpoints were summarized by visit and treatment group, with data summarized for each visit as categorical or continuous variables. AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA®) and presented using the preferred term by body system. Biomicroscopy and fundoscopy results were summarized using discrete summary statistics, and changes from baseline (% with worsening) were analyzed using Pearson χ2 tests. VA was summarized for each visit as a categorical variable and as a line change from baseline (visit 3). Subjects with worsening of at least 3 lines from baseline in the LE gel 0.38% TID arm were compared to the combined vehicle using a Pearson χ2 test. IOP was summarized at each visit for each subject using continuous variable summaries, including change from baseline. The proportion of subjects with change from screening in IOP at any postbaseline visit ≥10 mm Hg as well as the proportion of subjects with IOP ≥30 mm Hg at any visit was also determined. Ocular symptoms excluding pain were summarized at each visit by category and by treatment group using observed data. For each postbaseline visit, the proportion of subjects classified as “worsened” for each symptom was compared between the LE gel TID group and the combined vehicle group using a Pearson χ2 test. Study drug sensation was assessed on postoperative day 8 and summarized by treatment group.
AEs are reported regardless of rescue medication use. Other safety and tolerability findings are presented for the safety population for study visits prior to rescue medication use.
Unless otherwise indicated, all statistical testing was 2-sided and performed at the α=0.05 significance level. All analyses were conducted using SAS® Version 9.4 or higher.
Results
Subjects
A total of 742 participants were included in the pooled ITT population comprising 371 subjects, each assigned to either LE gel 0.38% TID or vehicle (combined BID or TID groups) (Figure 1). One subject who was randomized to the vehicle group and included in the ITT population did not receive study treatment and was therefore excluded from the safety population. Two subjects randomized to LE gel 0.38% TID and included in the ITT population were excluded from the safety population; one received an LE gel BID kit in error and the other did not receive the study drug. Overall in the ITT population, 298 subjects (80.3%) in the LE gel group and 202 in the vehicle group (54.4%) completed the study (Figure 1). There were 73 and 169 discontinuations in the LE gel and vehicle groups, respectively. In both treatment arms, the primary reason for discontinuation was the use of rescue medication (Figure 1).
 |
Figure 1 Participant flow. Notes: aBased on the intent-to-treat population. Reasons for discontinuations are primary reasons for withdrawal from the intent-to-treat population.Abbreviation: LE, loteprednol etabonate; TID, three times daily. |
Demographics were well-balanced across treatment groups (Table 1). The majority of subjects were white (77.6%) and female (58.6%), with a mean age of 68.9 years (range 36–92 years). Ocular and nonocular medical histories were comparable across treatment groups and consistent with the age of the study population. At baseline, the mean (SD) AC cell and flare scores were comparable between the LE gel 0.38% TID group and vehicle group (AC cells: 2.4 [0.54] and 2.4 [0.59]; flare: 0.8 [0.65] and 0.7 [0.64]). Pain was present in 50.1% and 49.9% of subjects in the LE gel 0.38% and vehicle groups, respectively, which was mostly minimal (23.2% and 23.7%, respectively) or mild (18.6% and 16.4%, respectively).
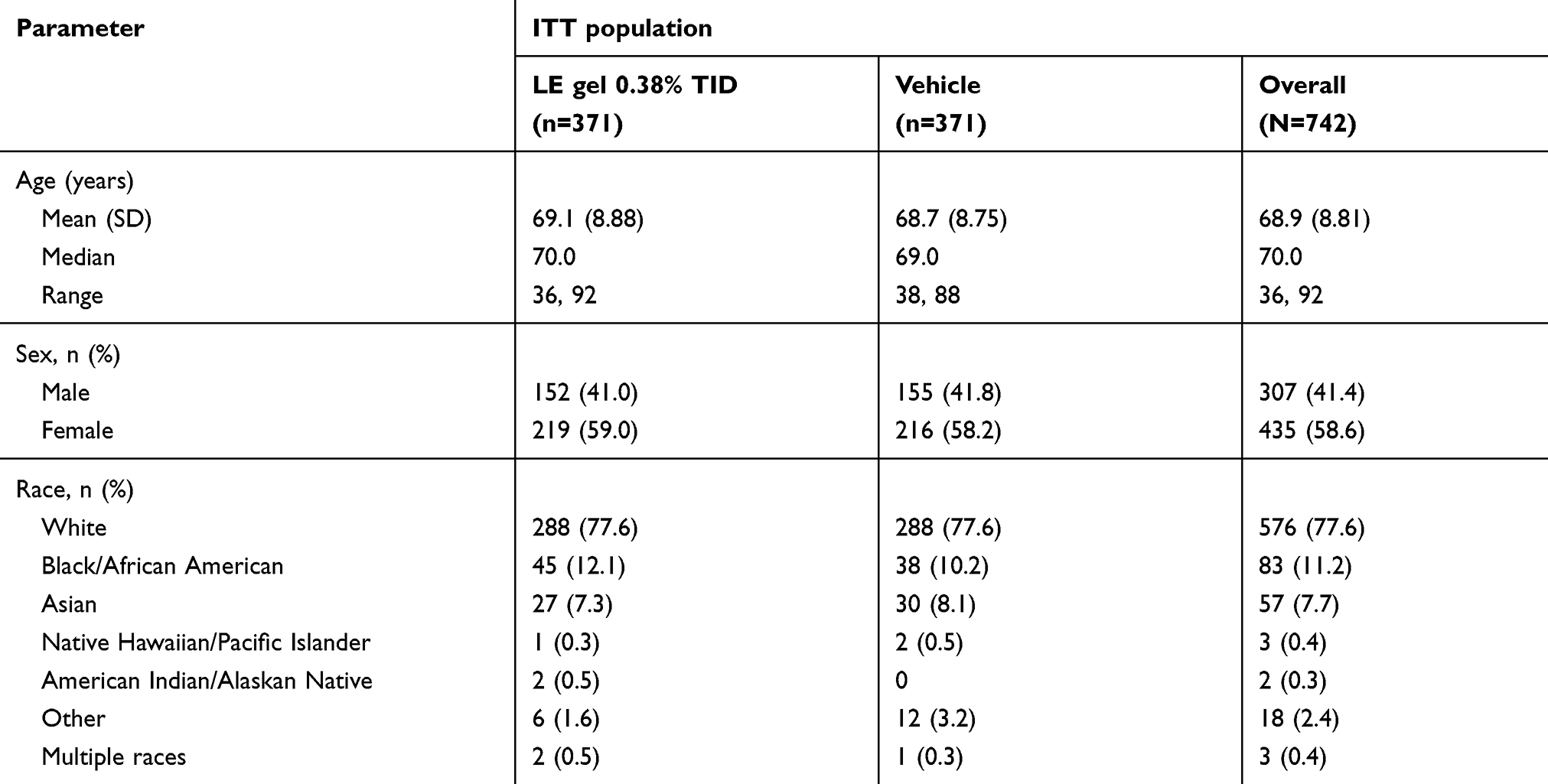 |
Table 1 Subject demographics for the integrated dataset |
Rescue medication
Over the course of the study, rescue medication was required by approximately twice as many subjects in the vehicle group (163 [44.1%]) compared with the LE gel 0.38% TID (68/369 [18.4%]) group. Similarly, less subjects receiving LE gel 0.38% required rescue medication as compared to those receiving vehicle prior to postoperative day 8 (39 [10.5%] vs 134 [36.1%]; P<0.0001). The most common classes of rescue medication used were topical corticosteroids (most frequently, difluprednate or prednisolone) and NSAIDs (most frequently, nepafenac or bromfenac).
Efficacy
Primary efficacy endpoints
Figure 2 presents the percentage of subjects with complete resolution of AC cells and grade 0 pain at day 8. A significantly greater proportion of subjects achieved complete resolution of AC cells at day 8 (visit 5) in the LE gel 0.38% TID group compared with the vehicle group (29.6% vs 15.1%; P<0.0001). The proportion of subjects with complete resolution of ocular pain at day 8 (visit 5) was also significantly higher in the LE gel 0.38% TID group than in the vehicle group (difference, 25.6%; P<0.0001).
 |
Figure 2 Percentage of subjects with complete resolution of AC cells and grade 0 pain at day 8 (visit 5) in the ITT population. Note: aPearson Chi-squared test P<0.0001 vs vehicle.Abbreviations: AC, anterior chamber; ITT, intent-to-treat; LE, loteprednol etabonate; TID, three times daily. |
Secondary efficacy endpoints
The proportion of subjects with complete resolution of AC cells, flare, AC cells and flare combined, and ocular pain at each postoperative visit are summarized in Table 2. For all of these measures, the proportions were significantly greater with LE gel 0.38% TID versus vehicle, with the exception of AC cells and AC cells and flare combined at day 3.
 |
Table 2 Proportion of subjects with complete resolution of AC cells, flare, AC cells and flare combined, and grade 0 (no) pain 3–18 days postoperatively (ITT population, missing values, and post-rescue values imputed as treatment failures) |
Mean reductions from baseline in AC cell and flare scores, individually or combined, were significantly greater in the LE gel 0.38% TID group compared with the vehicle group from day 3 onward (Figure 3).
 |
Figure 3 (A) Mean (SD) change from baseline in anterior chamber cells (ITT population). (B) Mean (SD) change from baseline in anterior chamber flare (ITT population). (C) Mean (SD) change from baseline in anterior chamber cells and flare combined (ITT population). Notes: aP<0.01 vs vehicle; bP<0.0001 vs vehicle. Negative values denote improvement. Missing values and post-rescue values were imputed using LOCF, and data were analyzed using a Cochran-Mantel-Haenszel mean score test.Abbreviations: LE, loteprednol etabonate; ITT, intent-to-treat; LOCF, last observation carried forward; TID, three times daily. |
Safety
The mean (SD) duration of exposure was 13.1 (3.05) days in the LE gel TID group and 10.7 (4.62) days in the vehicle group. At least 1 AE was reported for 30/369 (8.1%) subjects in the LE gel 0.38% TID group and 43/370 (11.6%) of those in the vehicle group. Ocular AEs in the study eye were reported for 9/369 (2.4%) subjects in the LE gel 0.38% TID group and 23/370 (6.2%) subjects in the vehicle group. The most commonly reported AEs in the study eye included eye pain (LE gel, 0.3%; vehicle, 2.2%), photophobia (LE gel, 0.8%; vehicle, 1.4%), and corneal edema (LE gel, 0%; vehicle, 1.4%). An AE of increased IOP in the study eye was reported by 2 subjects (0.5%) in the LE gel TID group (1 mild, 1 moderate; neither considered treatment related). Table 3 presents the ocular AEs in the study eye considered treatment related. At least 1 treatment-related AE in the study eye was experienced by 3/369 (0.8%) of subjects in the LE gel TID group and by 10/370 (2.7%) in the vehicle group.
 |
Table 3 Treatment-related ocular AEs (safety population) |
Nonocular AEs occurred in 9/369 (2.4%) of subjects in the LE gel 0.38% TID group and 5/370 (1.4%) in the vehicle group. None were considered related to study medication. The most common nonocular AE was headache (LE gel TID, 3/369 [0.8%]; vehicle, 1/370 [0.3%]). All nonocular AEs were reported in <1% of subjects in each treatment group. With the exception of headache (incidence reported above) and bronchitis (reported in 2 subjects [0.5%] in the LE gel 0.38% TID group and 0 [0%] in the vehicle group), all other nonocular AEs were reported in single subjects in only 1 of the treatment groups.
The majority of ocular AEs in the study eye were mild in severity, as were the majority of nonocular AEs. No severe ocular AEs were reported. A severe nonocular non-treatment related AE was experienced by 1 subject each in the LE gel TID (nasopharyngitis) and vehicle (migraine with aura) groups. Two subjects in the vehicle group experienced serious AEs, including 1 subject with moderate endophthalmitis and 1 subject with moderate hypokalemia; both serious AEs were considered not related to study treatment.
Overall, 5 subjects receiving LE gel 0.38% TID (1.4%) and 8 subjects in the vehicle group (2.2%) discontinued the study drug due to an AE. AEs leading to study drug discontinuation were considered treatment related for 2 subjects in the vehicle group. Withdrawal from the study due primarily to an AE was reported in 2/369 (0.5%) subjects in the LE gel 0.38% TID group (IOP increased; acute bronchitis) and 3/370 (0.8%) in the vehicle group (cataract operation complication [not serious, moderate]; endophthalmitis [serious, moderate, not resolved at study exit]; plastic iritis [moderate severity]). None of these events were considered related to treatment.
Fewer subjects in the LE gel 0.38% TID treatment group compared with the vehicle group experienced worsening of various ocular signs on biomicroscopy. Relative to subjects in the vehicle group, fewer subjects in the LE gel TID group had worsening of bulbar conjunctival injection, corneal edema, ciliary flush, and conjunctival chemosis on days 3, 8, and 15; AC cells, AC flare, and palpebral conjunctival injection on days 3 and 8 (all P≤0.04); and corneal staining on day 8 (P<0.02). More subjects in the LE gel TID group had worsening of AC cells and of bulbar conjunctival injection on day 18 (both P<0.05) compared with vehicle. Biomicroscopy revealed no significant differences between LE gel TID and vehicle on external adnexa lids, external adnexa lashes, hyphema, posterior synechiae, precipitates, hypopyon, or anterior vitreous haze.
Fundoscopy findings were comparable between the LE gel 0.38% TID and vehicle groups at screening and day 15. No abnormalities were reported in the majority of study eyes.
Compared with the vehicle group, a significantly lower proportion of subjects in the LE gel 0.38% TID group had a decrease of 3 lines or more in VA from baseline to day 8 (LE gel TID, 4/346 [1.2%]; vehicle, 17/289 [5.9%]; P<0.001). There were no significant differences between treatment groups at other time points.
The mean (SD) IOP in the study eye at screening was similar in the LE gel 0.38% TID group (15.5 [2.67] mm Hg) and the vehicle group (15.2 [2.47] mm Hg). Mean IOP in the study eye decreased from baseline (postoperative day 1) at all postbaseline visits in both groups. At the final on-treatment study visit (day 15), mean (SD) IOP was 14.4 (2.97) mm Hg in the LE gel 0.38% TID group and 13.9 (3.02) mm Hg in the vehicle group. One subject in the LE gel 0.38% TID group experienced a clinically significant increase (≥10 mm Hg) from screening in IOP in the study eye. The same subject had a treatment-emergent IOP of ≥30 mm Hg; IOP in the study eye was 20 mm Hg at screening and increased to 35 mm Hg at baseline (visit 3), with an IOP of 30 mm Hg recorded at day 3 (visit 4). The elevated IOP was recorded as an AE and was considered not related to study medication and not serious. Study medication was withdrawn, and IOP decreased to 15.5 mm Hg by the subsequent visit (day 8). No subjects in the vehicle group experienced an IOP elevation ≥10 mm Hg.
Tolerability
At baseline, 47.2% and 52.4% of LE gel 0.38%- and vehicle-treated subjects, respectively, had grade 0 (no) photophobia, while 75.1% and 78.6% had grade 0 (no) itching, 57.5% and 58.9% had grade 0 (no) tearing, and 90.5% and 89.7% had grade 0 (no) ocular discharge. Compared with the vehicle group, significantly fewer subjects in the LE gel 0.38% TID group had worsening of photophobia and tearing at days 3, 8, and 15 (P<0.02 for all). At day 8, 77.3% (266/344) and 81.0% (235/290) of subjects in the LE gel 0.38% TID and vehicle groups, respectively, reported no drop discomfort in the study eye. One subject in the LE gel 0.38% TID group reported experiencing severe drop discomfort at day 8, while moderate discomfort was reported by 3.2% (11/344) of subjects in the LE gel 0.38% TID group and 2.1% (6/290) of those in the vehicle group.
Discussion
LE (submicron) ophthalmic gel 0.38% is a topical corticosteroid formulation recently approved by the United States FDA for the treatment of postoperative inflammation and pain following ocular surgery.18 Results of this integrated analysis of data from two Phase III, randomized, double-masked, vehicle-controlled studies provide strong evidence that LE ophthalmic gel 0.38% is effective in treating postoperative inflammation and pain following cataract surgery when instilled TID beginning the day after surgery and continuing throughout the first 2 weeks after surgery. The superiority of LE gel 0.38% over vehicle was demonstrated on both primary outcome measures (complete resolution of AC cells at day 8 and complete resolution of ocular pain at day 8). Additionally, efficacy was observed with LE gel 0.38% TID compared with vehicle across nearly all secondary outcome measures, including resolution of pain as early as day 3 (2 days following treatment initiation).
This integrated analysis also confirms that LE gel 0.38% is safe and well tolerated when dosed TID. Ocular AEs were infrequent and likely resulted from the surgical procedure itself. Indeed, no AE occurred in more than 1% of subjects. With the exception of headache and bronchitis, all nonocular AEs occurred in a single subject in only one of the treatment groups, and none were considered treatment related. Biomicroscopy and VA findings in the LE gel 0.38% group were similar or improved relative to the vehicle-treated group; fundoscopy findings were similar between treatment groups. As observed in previous studies of LE 0.5% formulations,8,12–15 the mean IOP was not increased among subjects using LE gel 0.38%, and only 1 subject in the LE gel 0.38% group had a clinically significant transient elevation (≥10 mm Hg) from screening in IOP in the study eye. LE gel 0.38% was well tolerated based on symptoms and drop sensation/discomfort, and there were no reports of blurred vision with LE gel 0.38%.
As expected, for resolution of AC cells and ocular pain at postoperative day 8, the magnitude of difference between LE gel 0.38% TID and vehicle in this integrated analysis was similar to that reported in each of the 2 original studies.20,21 Findings were also similar to those reported with a higher concentration, micronized LE gel formulation (LE gel 0.5%) administered QID.14 The difference compared with vehicle in the proportion of subjects with complete resolution of AC cells at day 8 was 14.6% with LE gel 0.38% TID in the current integrated analysis compared with 15.7% in a pooled analysis of data from the 2 multicenter, randomized, double-masked trials of LE 0.5% gel QID.14 Likewise, the differences relative to vehicle in the proportion of subjects with complete resolution of ocular pain at day 8 reported with LE gel 0.38% TID and LE gel 0.5% QID were 25.6% and 30.5%, respectively. Findings were also comparable to those of another newly developed LE formulation—a suspension formulation with comparable smaller LE drug particles (0.2–0.4 µm vs 0.4–0.6 µm), but with a 2.6-fold higher drug concentration (1% vs 0.38%) approved with BID dosing.22,23 In this study, differences relative to vehicle in the proportion of LE suspension 1%-treated subjects with complete resolution of AC cells and ocular pain, respectively, at day 8 and maintained through day 15 were 10.9% and 19.9%. Future head-to-head studies are needed to explore the potential differences between formulations. Notably, the effects of LE gel 0.38% TID on resolution of AC cells and ocular pain following cataract surgery in this integrated analysis were also similar to those reported in studies evaluating the efficacy of difluprednate 0.05% dosed BID or QID.24,25
The clinical rationale for the new LE (submicron) gel 0.38% formulation is to allow for improved penetration of LE into ocular tissues while reducing drug concentration compared with the micronized LE gel 0.5% formulation and also to allow for a reduction in dosing frequency.16 This was accomplished through reduction of the median particle size in LE (submicron) gel 0.38% compared to LE gel 0.5% while maintaining the mucoadhesive properties of the original gel formulation. Decreasing the size of drug particles is a common strategy employed to improve dissolution kinetics of drugs with low aqueous solubility. Dissolution at the ocular surface is a prerequisite to absorption, and fast dissolution is critical given rapid tear turnover. The approximately 5- to 10-fold decrease in median diameter of the LE drug particles in the current LE gel formulation equates to an increase in total surface area of approximately 5- to 10-fold for an equivalent concentration of drug, which, based on the Noyes-Whitney equation, would be expected to increase dissolution rate by ~5–10-fold.26,27 Indeed, in vitro dissolution studies confirmed that LE gel 0.38% had a markedly more rapid rate of dissolution as compared to LE gel 0.5% and resulted in improved (~2-fold greater) penetration into the aqueous humor, despite the reduction in drug concentration.16 Importantly, a reduction in drug concentration has the benefit of reducing overall exposure to the drug, while reducing the dosing frequency to TID (vs QID with LE gel 0.5%) could result in greater adherence by the patient to the prescribed dosing regimen.19 The formulation retains the advances of the previous LE gel formulation; namely in that it is a nonsettling, shear-thinning, mucoadhesive gel with a pH close to that of tears and with a low level of the preservative benzalkonium chloride (0.003%), while also containing propylene glycol and glycerin,18 known moisturizers which may improve drop comfort.
In conclusion, the results of this integrated analysis of two Phase III studies demonstrate that LE (submicron) gel 0.38% TID is safe and effective for the treatment of ocular inflammation and pain following cataract surgery with minimal risk of IOP elevation.
Data sharing statement
There are no plans to share individual deidentifeid participant data from the individual studies pooled herein. No other data or study-related documents will be made available/accessible.
Acknowledgments
This integrated analysis was conducted by Bausch & Lomb Incorporated, which designed and conducted the original studies. Statistical assistance was provided by Howard M Proskin & Associates, Inc., Rochester, NY, and editorial assistance was provided by Churchill Communications, Maplewood, NJ, USA; both were funded by Bausch + Lomb, a division of Bausch Health US, LLC. The authors acknowledge the expert assistance of Gary Mosehauer of Bausch + Lomb in reviewing the statistical analyses. The study data were presented in part at the 2018 annual meeting of the American Academy of Optometry, November 7-10, San Antonio, TX.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
MEC, HHD, and JLV are employees of Bausch Health US, LLC. The authors report no other conflicts of interest in this work.
References
1. Grob SR, Gonzalez-Gonzalez LA, Daly MK. Management of mydriasis and pain in cataract and intraocular lens surgery: review of current medications and future directions. Clin Ophthalmol. 2014;8:1281–1289.
2. McGhee CN, Dean S, Danesh-Meyer H. Locally administered ocular corticosteroids: benefits and risks. Drug Saf. 2002;25(1):33–55. doi:10.2165/00002018-200225010-00004
3. Pleyer U, Ursell PG, Rama P. Intraocular pressure effects of common topical steroids for post-cataract inflammation: are they all the same? Ophthalmol Ther. 2013;2(2):55–72. doi:10.1007/s40123-013-0020-5
4. Comstock TL, DeCory HH. Advances in corticosteroid therapy for ocular inflammation: loteprednol etabonate. Int J Inflam. 2012;789623.
5. Comstock TL, Sheppard JD. Loteprednol etabonate for inflammatory conditions of the anterior segment of the eye: twenty years of clinical experience with a retrometabolically designed corticosteroid. Expert Opin Pharmacother. 2018;19(4):337–353. doi:10.1080/14656566.2018.149920
6. Sheppard JD, Comstock TL, Cavet ME. Impact of the topical ophthalmic corticosteroid loteprednol etabonate on intraocular pressure. Adv Ther. 2016;33(4):532–552. doi:10.1007/s12325-016-0315-8
7. Lotemax [package insert]. Bridgewater, NJ; Bausch & Lomb Incorporated; 2016.
8. Lane SS, Holland EJ. Loteprednol etabonate 0.5% versus prednisolone acetate 1.0% for the treatment of inflammation after cataract surgery. J Cataract Refract Surg. 2013;39(2):168–173. doi:10.1016/j.jcrs.2012.10.039
9. Lotemax gel [package insert]. Bridgewater, NJ; Bausch & Lomb Incorporated; 2018.
10. United States Food and Drug Administration. Drug approval package 2012. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202872_lotemax_toc.cfm.
11. Coffey MJ, DeCory HH, Lane SS. Development of a non-settling gel formulation of 0.5% loteprednol etabonate for anti-inflammatory use as an ophthalmic drop. Clin Ophthalmol. 2013;7:299–312.
12. Abessi B, Brooksby L, Schultze RL. Comparison of efficacy of difluprednate 0.05% and loteprednol gel 0.5% after cataract surgery. Eye Contact Lens. 2018;44(Suppl 2):S37–S42. doi:10.1097/ICL.0000000000000407
13. Fong R, Leitritz M, Siou-Mermet R, Erb T. Loteprednol etabonate gel 0.5% for postoperative pain and inflammation after cataract surgery: results of a multicenter trial. Clin Ophthalmol. 2012;6:1113–1124. doi:10.2147/OPTH.S32643
14. Rajpal RK, Fong R, Comstock TL. Loteprednol etabonate ophthalmic gel 0.5% following cataract surgery: integrated analysis of two clinical studies. Adv Ther. 2013;30(10):907–923. doi:10.1007/s12325-013-0059-7
15. Rajpal RK, Roel L, Siou-Mermet R, Erb T. Efficacy and safety of loteprednol etabonate 0.5% gel in the treatment of ocular inflammation and pain after cataract surgery. J Cataract Refract Surg. 2013;39(2):158–167. doi:10.1016/j.jcrs.2012.09.013
16. Cavet ME, Glogowski S, Lowe ER, Phillips E. Rheological properties, dissolution kinetics, and ocular pharmacokinetics of loteprednol etabonate (submicron) ophthalmic gel 0.38%. J Ocul Pharmacol Ther. 2019;35:291–300. doi:10.1089/jop.2018.0136
17. Google Patents. Loteprednol acetate suspension eye drops. Available from: https://patents.google.com/patent/CN103565740A/en.
18. Lotemax SM [package insert]. Bridgewater, NJ, Baush & Lomb Incorporated; 2019.
19. Steil CF, Covington TR. Pharmaceutical and regulatory aspect of ophthalmic drug administration. In: Bartlett JD, Januus SD, editors. Clinical Ocular Pharmacology.
20. Fong R, Silverstein BE, Peace JH, Williams JI, Vittitow JL. Submicron loteprednol etabonate ophthalmic gel 0.38% for the treatment of inflammation and pain after cataract surgery. J Cataract Refract Surg. 2018;44(10):1220–1229. doi:10.1016/j.jcrs.2018.06.056
21. Vittitow JL, LoBue T, Martel J. Safety and efficacy of a novel submicron loteprednol etabonate gel in the treatment of inflammation and pain post-cataract surgery. Invest Ophthalmol Vis Sci. 2018;59(9):2235. doi:10.1167/iovs.17-23678
22. Kim T, Sall K, Holland EJ, Brazzell RK, Coultas S, Gupta PK. Safety and efficacy of twice daily administration of KPI-121 1% for ocular inflammation and pain following cataract surgery. Clin Ophthalmol. 2018;13:69–86. doi:10.2147/OPTH.S185800
23. Inveltys [package insert]. Waltham, MA; Kala Pharmaceuticals, Inc; 2018.
24. Korenfeld MS, Silverstein SM, Cooke DL, Vogel R, Crockett RS; Difluprednate Ophthalmic Emulsion 0.05% (Durezol) Study Group. Difluprednate ophthalmic emulsion 0.05% for postoperative inflammation and pain. J Cataract Refract Surg. 2009;35(1):26–34. doi:10.1016/j.jcrs.2008.09.024
25. Smith S, Lorenz D, Peace J, McLeod K, Crockett RS, Vogel R. Difluprednate ophthalmic emulsion 0.05% (Durezol) administered two times daily for managing ocular inflammation and pain following cataract surgery. Clin Ophthalmol. 2010;4:983–991. doi:10.2147/OPTH.S10696
26. Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–934. doi:10.1021/ja02086a003
27. Jambhekar SS, Breen PJ. Drug dissolution: significance of physicochemical properties and physiological conditions. Drug Discov Today. 2013;18(23–24):1173–1184. doi:10.1016/j.drudis.2013.08.013
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.