Back to Journals » Clinical Interventions in Aging » Volume 11
Long noncoding RNAs and Alzheimer's disease
Received 24 February 2016
Accepted for publication 9 April 2016
Published 29 June 2016 Volume 2016:11 Pages 867—872
DOI https://doi.org/10.2147/CIA.S107037
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zhi-Ying Wu
Qiong Luo,1,2 Yinghui Chen1,2
1Department of Neurology, Jinshan Hospital, 2Department of Neurology, Shanghai Medical College, Fudan University, Shanghai, People’s Republic of China
Abstract: Long noncoding RNAs (lncRNAs) are typically defined as transcripts longer than 200 nucleotides. lncRNAs can regulate gene expression at epigenetic, transcriptional, and posttranscriptional levels. Recent studies have shown that lncRNAs are involved in many neurological diseases such as epilepsy, neurodegenerative conditions, and genetic disorders. Alzheimer’s disease is a neurodegenerative disease, which accounts for >80% of dementia in elderly subjects. In this review, we will highlight recent studies investigating the role of lncRNAs in Alzheimer’s disease and focus on some specific lncRNAs that may underlie Alzheimer’s disease pathophysiology and therefore could be potential therapeutic targets.
Keywords: lncRNA, Alzheimer’s disease, ncRNAs, amyloid β peptide, BACE1, BC200, BACE1-AS
Introduction
Alzheimer’s disease (AD) is one of the most common neurodegenerative disease and accounts for >80% of dementia cases in people aged older than 65 years.1 The disease is characterized by devastating symptoms such as apraxia, agnosia, aphasia, and emotional disturbance because of progressive mental and behavioral function decline.2–4 The 2015 Alzheimer’s Association report predicts that, by 2050, there will be a new diagnostic case every 33 seconds, corresponding to 1 million new AD patients every year.5 Given the disability and dependence of these patients, the increasing prevalence of AD will impose huge burdens on families and society. Long noncoding RNAs (lncRNAs) comprise a subgroup of noncoding RNAs (ncRNAs) longer than 200 nucleotides (nt), accounting for the largest proportion of the mammalian noncoding transcriptome. lncRNAs impact AD pathogenesis because of their diverse biochemical and functional effects such as chromatin modulation, posttranscriptional and posttranslational regulation, and protein complex organization.6,7
AD pathophysiology
Since the time of Dr Alois Alzheimer, neuropathologists have known that brain tissue of patients with AD contains extracellular senile plaques and intracellular neurofibrillary tangles composed of amyloid beta (Aβ) protein and hyperphosphorylated tau protein, respectively.8–15 Amyloid precursor protein (APP) is sequentially cleaved by β-site APP cleaving enzyme-1 (BACE1), and γ-secretase during Aβ biosynthesis, with γ-secretase initiating the “amyloid-cascade”.16 Aβ peptides aggregate into soluble oligomers that are proposed to be the activator of N-methyl-D aspartate receptor endocytosis, mitochondrial dysfunction, oxidative damage, excessive calcium influx, lipid dysregulation, synaptic dysfunction, neuronal stress, apoptosis, aberrant neurogenesis, and neuroinflammation. However, whether or not Aβ induces tau aggregation is still being debated.17–21 But most recent studies suggest that Aβ oligomer formation may be the essential step in the pathophysiology underpinnings of AD.17,22–24
lncRNA
ncRNAs can be divided on the basis of size into short ncRNAs (<200 nt in length) and lncRNAs.17,25 lncRNAs vary from 200 nt to over 100 kb and usually lack an obvious open reading frame.26–30 lncRNAs secondary structure connected to specific functions are evolutionarily conserved.31,32 They regulate dynamically, localizing at specific cell types and in subcellular compartments.26,33,34 lncRNAs regulate gene expression at different levels.35 Most lncRNAs are located in the nucleus, which is consistent with their major function of epigenetic regulation.26,36 lncRNAs are not considered to be the “dark matter”, rather they have essential roles in controlling transcription and translation, as well as during genetic imprinting, genome rearrangement, chromatin modification regulation of the cell cycle, transcription, splicing, messenger RNA (mRNA) decay, and translation.27,30,37 The pathomechanism and genetic factors of AD have been investigated for nearly 100 years. Research is ongoing, many studies have demonstrated that dysregulation of lncRNAs involved in cancer; epilepsy; and cardiovascular, neurodegenerative, and genetic diseases. Some have posited that lncRNAs may also have a major role in AD35,38,39 (Table 1; Figure 1).
 | Table 1 Dysregulated lncRNAs in Alzheimer’s disease |
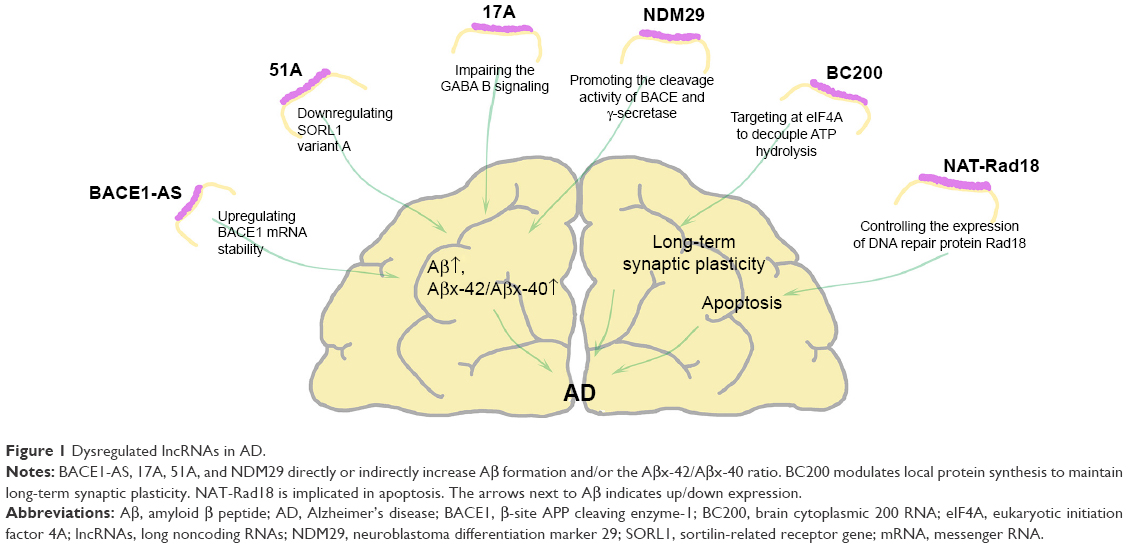 | Figure 1 Dysregulated lncRNAs in AD. |
BACE1-AS
β-site amyloid precursor protein cleaving enzyme-1 antisense transcript (BACE1-AS) is a conserved RNA transcribed from the positive strand of chromosome 11 on the opposite strand of the BACE1 locus (11q 23.3).16,17 BACE1-AS regulates BACE1 (β-site APP cleaving enzyme-1) expression at both the mRNA and protein levels. BACE1 is essential for the production of the toxic Aβ.40,41 AD pathogenesis has been implicated in many different cell stressors. Following exposure to high temperature, serum starvation, staurosporine, Aβ1–42, high glucose, BACE1-AS, and BACE1 mRNA are both upregulated. This suggests that cell stressors may alter BACE1-AS expression and subsequently BACE1 enzyme activity.16,42 Regardless of whether BACE1-AS is knocked down or overexpressed, both BACE1 mRNA and BACE1 protein are regulated in parallel, thereby reducing Aβ production and plaque deposition.16,17,42 In animals, loss of BACE1 results in numerous behavioral and physiological deficits, including memory loss, reduced synaptic plasticity,43 emotional deficits,44 and peripheral myelination defects.45–49 The delicate physiologic and pathologic boundaries indicate that BACE1 expression should be tightly regulated.16,49
In summary, cell stress increases BACE1-AS levels, which in turn stimulates BACE1 expression, which could enhance APP processing and Aβ1–42 production. Elevated Aβ1–42 levels can further promote BACE1-AS overexpression and the APP processing cascade in a feedforward manner.16,42,50 By forming an RNA duplex, BACE1-AS increases BACE1 mRNA stability.42,51,52 So, BACE1 and BACE1-AS may be potential biomarkers and treatment targets for AD.46,50,53,54
51A
The neuronal sortilin-related receptor gene (SORL1, also known as SORLA and LR11) has long been hypothesized to be involved in AD pathogenesis.55–58 Recent studies have posited that SORL1, as a sorting receptor for APP holoprotein, interacts with APP in endosomes and the trans-Golgi network where it affects trafficking and proteolytic processing.59 Decreased SORL1 expression might shift APP from the retromer recycling pathway to the β-secretase cleavage pathway, increasing secreted APP production and subsequent Aβ formation.59,60 51A is a novel ncRNA that maps in an antisense configuration to intron 1 of the SORL1 gene, the synthesis of which promotes the expression of alternatively spliced SORL1 variants. Notably, 51A is overexpressed in in vitro models and the AD brain. One possible mechanism by which 51A might increase AD susceptibility is by increasing amyloid formation via downregulating SORL1 variant A through alternative splicing.59,61
17A
17A is a 159-nt lncRNA synthesized by RNA polymerase III and maps in intron 3 of G-protein-coupled receptor 51 gene (GPR51); it undergoes alternative splicing, increasing the number of GABA B2 receptor isoforms. GABA B R2 splice variant B may affect GABA B biological function by regulating intracellular 3′–5′-cyclic adenosine monophosphate accumulation and the activation of specific potassium channels. These events would impair GABA B signaling, enhance Aβ secretion, and increase the Aβx-42/Aβx-40 ratio. 17A RNA is upregulated in AD compared with control tissues, suggesting that it could directly or indirectly be involved in the mechanism of AD.52,62,63
NDM29
Neuroblastoma differentiation marker 29 (NDM29) is an RNA polymerase III-transcribed ncRNA. NDM29 synthesis is dose-dependently induced by inflammatory stimulation. The upregulation of NDM29 RNA is accompanied by altered APP modulation. Meanwhile, it can promote the cleavage activities of BACE that, in turn, generates an enhanced amount of APP C-terminal fragments for further processing by the γ-secretase cleavage complex to increase Aβ formation and the Aβx-42/Aβx-40 ratio.12,63,64
Brain cytoplasmic 200 RNA (BC200)
BC200 is a translational regulator that targets eukaryotic initiation factor 4A, thus decoupling adenosine triphosphate hydrolysis from RNA duplex unwinding, modulating local protein synthesis in postsynaptic dendritic microdomains, and contributing to the maintenance of long-term synaptic plasticity.65
One postmortem study found that BC200 RNA levels in cortical areas are reduced by >60% between the ages of 49 and 86 years. Compared with age-matched normal brains, BC200 RNA is significantly upregulated in the AD brain. Moreover, the relative BC200 RNA levels in AD-involved brain areas increase in parallel with disease progression. Still, at least one study reported BC200 downregulation.66 The contradiction between studies may be due to differences in brain regions and varying disease severity, but aberrant BC200 RNA expression in AD is a possibility.67
Relative BC200 RNA levels decrease in dendrites but increase in somata. This divergent expression affects microtubule-dependent transport and could contribute to axonal and dendritic blockage that may be early events in AD. It could eventually contribute to local Aβ generation and subsequent amyloid deposition.24,68 Another group found that BC200 RNA is not affected under apoptotic conditions in vitro and hypothesized that BC200 is only involved in necrosis rather than apoptosis.22
NAT-Rad18
Apoptosis is the main form of programmed cell death, and excessive apoptosis causes progressive cell loss that contributes to many neurodegenerative disorders, including AD. Rad18 is a member of the Rad6 epistasis group, which is responsible for postreplication repair. NAT-Rad18 genes encode for natural antisense transcripts against Rad18, encoding a spectrum of DNA-damaging agents. There is a counterbalanced relationship between Rad18 and NAT-Rad18 in both mRNA and protein level, with Rad18 showing a low expression level. NAT-Rad18 is differentially up-regulated expressed in brain tissues especially cortical neurons following exposure to Aβ. Collectively, this evidence indicates that NAT-Rad18 may be involved in AD via its effects on DNA repair system.69
Conclusion
Almost all lncRNAs related to AD have been listed in this review, but investigation into this field is in the early stage. Since AD was first reported, a century passed before the discovery of basic molecular mechanism. Unquestionably, new information about lncRNAs may light a new beacon in the search for AD treatments. Depending on the mechanism of AD, BACE1-AS, 17A, 51A, and NDM29 directly or indirectly increase Aβ formation and/or the Aβx-42/Aβx-40 ratio. The roles of BC200 and NAT-Rad18 are different. BC200 modulates local protein synthesis to maintain long-term synaptic plasticity. NAT-Rad18 is implicated in apoptosis, while BC200 is only involved in necrosis. As the lncRNA field continues to develop, we still need to elucidate how lncRNAs operate at the molecular and cellular levels. Most recent studies suggest that lncRNAs are desirable candidates in the ongoing quest for AD biomarkers and could help identify rational therapeutic strategies. An enhanced understanding of lncRNA biology could open more avenues to early AD diagnosis and treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76 Pt A:27–50. | ||
Kalra J, Khan A. Reducing Abeta load and tau phosphorylation: emerging perspective for treating Alzheimer’s disease. Eur J Pharmacol. 2015; 764:571–581. | ||
Kumar A, Singh A, Ekavali. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep. 2015;67(2):195–203. | ||
Muller M, Kuiperij HB, Claassen JA, Kusters B, Verbeek MM. MicroRNAs in Alzheimer’s disease: differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol Aging. 2014;35(1):152–158. | ||
Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015;11(3):332–384. | ||
Zhang Z. Long non-coding RNAs in Alzheimer’s disease. Curr Top Med Chem. 2016;16(5):511–519. | ||
Kang MJ, Abdelmohsen K, Hutchison ER, et al. HuD regulates coding and noncoding RNA to induce APP-->Abeta processing. Cell Rep. 2014;7(5):1401–1409. | ||
Mattson MP. Degenerative and protective signaling mechanisms in the neurofibrillary pathology of AD. Neurobiol Aging. 1995;16(3):447–457; discussion 458–463. | ||
Hulstaert F, Blennow K, Ivanoiu A, et al. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology. 1999;52(8):1555–1562. | ||
Tarkowski E, Liljeroth AM, Nilsson A, et al. TNF gene polymorphism and its relation to intracerebral production of TNFalpha and TNFbeta in AD. Neurology. 2000;54(11):2077–2081. | ||
Algarzae N, Hebron M, Miessau M, Moussa CE. Parkin prevents cortical atrophy and Abeta-induced alterations of brain metabolism: (1)(3)C NMR and magnetic resonance imaging studies in AD models. Neuroscience. 2012;225:22–34. | ||
Massone S, Ciarlo E, Vella S, et al. NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid beta secretion. Biochim Biophys Acta. 2012;1823(7):1170–1177. | ||
Ramirez-Bermudez J. Alzheimer’s disease: critical notes on the history of a medical concept. Arch Med Res. 2012;43(8):595–599. | ||
Yamada K, Patel TK, Hochgrafe K, et al. Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol Neurodegener. 2015;10:55. | ||
Caillet-Boudin ML, Buee L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:28. | ||
Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. | ||
Modarresi F, Faghihi MA, Patel NS, Sahagan BG, Wahlestedt C, Lopez-Toledano MA. Knockdown of BACE1-AS nonprotein-coding transcript modulates beta-amyloid-related hippocampal neurogenesis. Int J Alzheimers Dis. 2011;2011:929042. | ||
Sun X, Chen WD, Wang YD. beta-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Front Pharmacol. 2015;6:221. | ||
Kawabata S, Higgins GA, Gordon JW. Amyloid plaques, neurofibrillary tangles and neuronal loss in brains of transgenic mice overexpressing a C-terminal fragment of human amyloid precursor protein. Nature. 1991;354(6353):476–478. | ||
Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clin Interv Aging. 2007;2(3):347–359. | ||
Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48. | ||
Liu Q, Sun S, Yu W, et al. Altered expression of long non-coding RNAs during genotoxic stress-induced cell death in human glioma cells. J Neurooncol. 2015;122(2):283–292. | ||
Herrera-Rivero M, Elena Hernandez-Aguilar M, Emiliano Aranda-Abreu G. A strategy focused on MAPT, APP, NCSTN and BACE1 to build blood classifiers for Alzheimer’s disease. J Theor Biol. 2015;376:32–38. | ||
Zhou X, Xu J. Identification of Alzheimer’s disease-associated long noncoding RNAs. Neurobiol Aging. 2015;36(11):2925–2931. | ||
Magistri M, Faghihi MA, St Laurent G 3rd, Wahlestedt C. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 2012;28(8):389–396. | ||
Knauss JL, Sun T. Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience. 2013;235:200–214. | ||
Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci. 2013;56(10):876–885. | ||
Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22(9):1760–1774. | ||
Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. | ||
Khorkova O, Hsiao J, Wahlestedt C. Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev. 2015;87:15–24. | ||
Iwakiri J, Hamada M, Asai K. Bioinformatics tools for lncRNA research. Biochim Biophys Acta. 2016;1859(1):23–30. | ||
Hamada M. RNA secondary structure prediction from multi-aligned sequences. Methods Mol Biol. 2015;1269:17–38. | ||
Wilk R, Hu J, Blotsky D, Krause HM. Diverse and pervasive subcellular distributions for both coding and long noncoding RNAs. Genes Dev. 2016;30(5):594–609. | ||
Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014;24(4):616–628. | ||
Melissari MT, Grote P. Roles for long non-coding RNAs in physiology and disease. Pflugers Arch. Epub 2016 Mar 5. | ||
Zeng C, Yu X, Lai J, Yang L, Chen S, Li Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J Hematol Oncol. 2015;8(1):126. | ||
Derrien T, Guigo R, Johnson R. The long non-coding RNAs: a new (P)layer in the “Dark Matter”. Front Genet. 2011;2:107. | ||
Kraus TF, Greiner A, Guibourt V, Lisec K, Kretzschmar HA. Identification of stably expressed incRNAs as valid endogenous controls for profiling of human glioma. J Cancer. 2015;6(2):111–119. | ||
Sun M, Nie FQ, Wang ZX, De W. Involvement of incRNA dysregulation in gastric cancer. Histol Histopathol. 2016;31(1):33–39. | ||
Mulder SD, van der Flier WM, Verheijen JH, et al. BACE1 activity in cerebrospinal fluid and its relation to markers of AD pathology. J Alzheimers Dis. 2010;20(1):253–260. | ||
Dash R, Emran TB, Uddin MM, Islam A, Junaid M. Molecular docking of fisetin with AD associated AChE, ABAD and BACE1 proteins. Bioinformation. 2014;10(9):562–568. | ||
Liu T, Huang Y, Chen J, et al. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1AS expression. Mol Med Rep. 2014;10(3):1275–1281. | ||
Ma H, Lesne S, Kotilinek L, et al. Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104(19):8167–8172. | ||
Laird FM, Cai H, Savonenko AV, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25(50):11693–11709. | ||
Hu X, Hicks CW, He W, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9(12):1520–1525. | ||
Decourt B, Sabbagh MN. BACE1 as a potential biomarker for Alzheimer’s disease. J Alzheimers Dis. 2011;24(Suppl 2):53–59. | ||
Borghi R, Patriarca S, Traverso N, et al. The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol Aging. 2007;28(7):1009–1014. | ||
Stockley JH, O’Neill C. Understanding BACE1: essential protease for amyloid-beta production in Alzheimer’s disease. Cell Mol Life Sci. 2008;65(20):3265–3289. | ||
Vassar R, Kandalepas PC. The beta-secretase enzyme BACE1 as a therapeutic target for Alzheimer’s disease. Alzheimers Res Ther. 2011;3(3):20. | ||
Dislich B, Lichtenthaler SF. The membrane-bound aspartyl protease BACE1: molecular and functional properties in Alzheimer’s disease and beyond. Front Physiol. 2012;3:8. | ||
Yuan J, Venkatraman S, Zheng Y, McKeever BM, Dillard LW, Singh SB. Structure-based design of beta-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2013;56(11):4156–4180. | ||
Wan P, Su W, Zhuo Y. The role of long noncoding RNAs in neurodegenerative diseases. Mol Neurobiol. Epub 2016 Feb 24. | ||
Evin G, Hince C. BACE1 as a therapeutic target in Alzheimer’s disease: rationale and current status. Drugs Aging. 2013;30(10):755–764. | ||
Perneczky R, Alexopoulos P. Cerebrospinal fluid BACE1 activity and markers of amyloid precursor protein metabolism and axonal degeneration in Alzheimer’s disease. Alzheimers Dement. 2014;10(5 Suppl):S425–S429.e421. | ||
Jacobsen L, Madsen P, Moestrup SK, et al. Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem. 1996;271(49):31379–31383. | ||
Yamazaki H, Bujo H, Saito Y. A novel member of the LDL receptor gene family with eleven binding repeats is structurally related to neural adhesion molecules and a yeast vacuolar protein sorting receptor. J Atheroscler Thromb. 1997;4(1):20–26. | ||
Lee JH, Barral S, Reitz C. The neuronal sortilin-related receptor gene SORL1 and late-onset Alzheimer’s disease. Curr Neurol Neurosci Rep. 2008;8(5):384–391. | ||
Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. | ||
Ciarlo E, Massone S, Penna I, et al. An intronic ncRNA-dependent regulation of SORL1 expression affecting Abeta formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis Model Mech. 2013;6(2):424–433. | ||
Khvotchev M, Sudhof TC. Proteolytic processing of amyloid-beta precursor protein by secretases does not require cell surface transport. J Biol Chem. 2004;279(45):47101–47108. | ||
Ma QL, Galasko DR, Ringman JM, et al. Reduction of SorLA/LR11, a sorting protein limiting beta-amyloid production, in Alzheimer disease cerebrospinal fluid. Arch Neurol. 2009;66(4):448–457. | ||
Massone S, Vassallo I, Fiorino G, et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol Dis. 2011;41(2):308–317. | ||
Gavazzo P, Vassalli M, Costa D, Pagano A. Novel ncRNAs transcribed by Pol III and elucidation of their functional relevance by biophysical approaches. Front Cell Neurosci. 2013;7:203. | ||
Vella S, Penna I, Longo L, et al. Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing over-expression of NDM29 ncRNA. Sci Rep. 2015;5:18144. | ||
Lin D, Pestova TV, Hellen CU, Tiedge H. Translational control by a small RNA: dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol Cell Biol. 2008;28(9):3008–3019. | ||
Mus E, Hof PR, Tiedge H. Dendritic BC200 RNA in aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104(25):10679–10684. | ||
Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull. 2013;97:69–80. | ||
Iacoangeli A, Bianchi R, Tiedge H. Regulatory RNAs in brain function and disorders. Brain Res. 2010;1338:36–47. | ||
Parenti R, Paratore S, Torrisi A, Cavallaro S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during beta-amyloid-induced apoptosis. Eur J Neurosci. 2007;26(9):2444–2457. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.